

Abstract
Small leucine-rich proteoglycans (SLRPs) are a unique class of proteins that exist in the extracellular matrix, playing key roles in cell proliferation and function. In bone, SLRPs such as bigly can and decorin affect osteogenesis and bone remodeling. Their essential role in this organ system has created the need to isolate these proteins for study. Bone presents unique obstacles to the study of proteins; however, through the use of demineralizing agents, efficient methods of the purification of proteoglycans have been developed. Additionally, methods have been developed that allow for the production and isolation of proteoglycans from conditioned media, which opens the door to a wide array of in vitro and in vivo assays. In stride with the purification and utilization of proteoglycans is the need to insure proteoglycan identity and purity, which is accomplished through enzymatic deglycosylation and blot analysis.
1. INTRODUCTION
Bone is formed by two unique pathways—intramembranous and endochondral ossification. Intramembranous ossification involves the proliferation of osteogenic cells within bone islands, which produces the flat bones of the face and clavicles. Endochondral ossification is the process by which the long bones of the body form, and is most commonly the way in which damaged adult bone heals (i.e., fracture healing). In endochondral ossification, cartilage serves as a precursor or template for future bone. Chondrocytes initially secrete the extracellular matrix that constitutes cartilage, but cellular hypertrophy and environmental constraints trigger the production of alkaline phosphatase, which allows for the mineralization of the cartilage. Blood vessels penetrate the calcifying cartilage, providing the cells necessary for ossification. Infiltrating osteoblasts use the mineralized cartilage as scaffolding, laying down layers of osteoid that will calcify and produce trabecular bone. These processes, and the processes required for maintenance and repair of the created bone, require tight orchestration of complex cellular functions. The proteins within bone are critical in this orchestration, with one group of proteins called proteoglycans that are increasingly being proven to be important.
Small leucine-rich proteoglycans (SLRPs) are a family of proteoglycans comprised of a small protein core containing a leucine-rich domain, as well as single or multiple attached glycosaminoglycan (GAG) chains (Iozzo, 1997). SLRPs are predominantly secreted, extracellular proteins that have structural and regulatory functionality. Chemically, the GAG chains attract water molecules, maintaining hydration and osmotic pressure in tissue. This is useful in tissues like cartilage, which have high SLRP content, where osmotic pressure assists in the compressive strength and nutrient supply is predominantly through diffusion. At the cellular level, SLRPs are able to interact with cytokines and cell-surface receptors, affecting cell signaling and function.
SLRPs play a significant role in osteogenesis and bone remodeling (Nikitovic et al., 2012). This is achieved through interactions not only with the collagen framework, which will later become mineralized, but also with the cytokines and receptors that regulate cell proliferation, apoptosis, osteogenesis, and remodeling. Biglycan and decorin are class I SLRPs that contain either dermatan or chondroitin sulfate GAG chains, depending on the tissue source. Biglycan has been shown to promote ERK phosphorylation, BMP2 binding to ALK6, BMP4 activity, and Wnt3a binding to LRP6 which all stimulate osteogenesis (Berendsen et al., 2011; Chen, Fisher, Robey, & Young, 2004; Mochida, Parisuthiman, & Yamauchi, 2006; Wang et al., 2010). Biglycan has been shown to be particularly important in the process of fracture healing and more recently has been shown to affect angiogenesis in fracture healing (Myren et al., 2016). Decorin has been shown to bind to TGF-beta, modulating its binding to TGFBR1 and regulating receptor activation (Hildebrand et al., 1994). Whereas bigly can has been shown to facilitate angiogenesis, decorin has proven to be an inhibitor of angiogenesis (Neill et al., 2012).
The critical role of these proteins necessitates their evaluation for clinical importance. One obstacle in this evaluation is the host environment of bone. The mineralized portion of bone is comprised of calcium and phosphate, predominantly existing as calcium hydroxyapatite; this gives bone the compression strength and its structural rigidity. The organic portion of bone provides tensile and compressive strength, as well as cell signaling and function mediation, and is comprised of collagens, glycoproteins, proteoglycans, and hyaluronic acid. While this makes bone ideal for its role in the body, it impedes most scientific techniques aimed at studying it. This is especially true from a perspective of protein extraction. Protein extraction in soft tissue uses powerful denaturing solutions to solubilize the protein. In bone, this direct approach will not work as the protein is encased in the mineralized shell that gives bone its unique characteristic. The calcium must be removed in order to isolate protein from bone. This is the experimental challenge: How to demineralize bone without damaging or degrading the native protein. This chapter describes (1) a method to extract nondegraded small proteoglycans from mineralized matrix, (2) a simple method to produce and isolate small proteoglycans from the conditioned media of osteogenic cells forced to maximize their expression, and (3) a method to deglycosylate and analyze recovered protein.
2. ISOLATION OF PROTEIN FROM BONE
2.1. BACKGROUND
The isolation of protein from bone is complicated by its high mineral content. Classic approaches to protein isolation result in low protein yield, as much of the protein remains locked away by the calcified matrix of bone. Demineralizing agents have been utilized to remove this obstacle. The general approach to purification is to first demineralize the bone with agents such as EDTA or hydrochloric acid. Typically, the bone is mechanically broken down prior to demineralization to increase the surface area and therefore the efficiency of demineralization. Once demineralized, the protein can be solubilized using denaturing agents such as guanidinium chloride, urea, or ammonium bicarbonate. The high salts in these latter solutions can impair subsequent protein analysis and disrupt protein function. Therefore, the samples must be dialyzed in order to remove the salts. This process can be extensive and repetitious, resulting in loss of product and damage to the protein. Below is a tested method for extraction of protein that results in efficient yield with minimal protein degradation.
2.2. MATERIALS
2.2.1. Equipment
Liquid nitrogen
Dry ice
Mortar and pestle
1 mL glass tissue homogenizer (Wheaton Dounce Tissue Grinder)
Metal spatula
Amicon Ultra-4 centrifugal filters 10–30K MW
Assorted protease-free pipette tips
Assorted protease-free polypropylene tubes
Centrifuge tube rotisserie (SCILOGEX Analog Rotisserie Tube Rotator MX-RL-E, 82422001)
Sonicator
High-speed centrifuge (capable of 15,000 × g)
Swivel-bucket centrifuge (capable of 4000 × g)
BCA protein assay kit (Pierce)
2.2.2. Solutions and reagents
Bulk protein extraction buffer: 8 M urea, 0.5 M EDTA, 50 mM Tris–HCl, pH 7.5
Demineralizing buffer: 0.5M EDTA, 50 mM Tris–HCl, pH 7.5
Solubilizing buffer: 8M urea, 50mM Tris–HCl, pH 7.5
Tris-buffered saline (TBS) 1 ×: 50mM Tris–HCl, 150mM NaCl, pH 7.5
2.3. METHOD
2.3.1. Preparation of sample
Protein is easily degraded by proteases endogenous to tissue, impairing future analysis. Fresh samples should be used and samples should be kept cold throughout the process.
Bone should be surgically isolated with care taken to remove muscles and tendons superficial to the periosteum, insuring sample purity.
The bone section of interest should be isolated (i.e., isolation of the fracture callus) and immediately snap frozen in liquid nitrogen.
Store bone fragments at −80°C until protein extraction.
2.3.2. Pulverization of bone
The bone fragments must be pulverized to increase protein yield efficiency. The mortar used should be kept on dry ice, and the pestle and spatula used for this step should be cooled with liquid nitrogen. This will prevent protein degradation and will maintain the pulverized bone as a powder (see Fig. 1).
To pulverize the sample, transfer the frozen bone fragments to a cooled mortar and add liquid nitrogen to keep the sample frozen. Grind the sample in a small amount of liquid nitrogen with a cooled pestle until the bone is a fine powder.
With a cooled spatula, transfer the bone powder to a glass tissue homogenizer.
FIG. 1.
Equipment used for pulverizing bone fragments. Top left is a mortar and pestle; top right is the glass homogenizer; bottom is a spatula.
2.3.3. Protein extraction (bulk extraction)
Note: This method extracts all protein (matrix bound and nonmatrix bound) in one sample. If the desire is to isolate the fractions separately, follow the method described in Section 2.3.4. Fig. 2 represents extracted protein collected by the fractionated method.
- Add 1 mL bulk protein extraction buffer (0.5 M EDTA/8 M urea/50 mM Tris–HCl) to the homogenizer with sample. Apply the solution to the sides of the homogenizer to collect sample that may have stuck to the wall.
-
○Consider adding protease inhibitor at this stage if protein degradation is a concern. However, the urea should denature any proteases, which will not refold until the sample is desalted.
-
○
- Homogenize the sample first with the “loose” pestle, then the “tight pestle.” Movement of the pestle should be smooth and fluid when the sample is appropriately homogenized.
-
○Note: This technique is optimized for 0.2g samples. For larger samples, a larger homogenizer may be appropriate.
-
○
- Transfer solution to a polypropylene tube with volume to accommodate for demineralization buffer. Bring to volume with 0.5 M EDTA/8M urea/50 mM Tris–HCl (~10mL/g bone). Use the demineralization buffer to wash out the glass homogenizer, minimizing lost sample.
- ○ Note: Polypropylene tubes are preferred, as protein will bind the walls of polystyrene tubes, reducing protein yield (Goebel-Stengel, Stengel, Taché, & Reeve, 2011).
Sonication is useful at this stage to increase yield. Leave samples on ice and perform sonication at low power for 3 s. Repeat for a total of three cycles with time for cool-down between cycles.
The samples should be set to rotate at 4°C for 48 h at 30 RPM. A centrifuge tube rotisserie is recommended for this step.
Spin down the sample at ~15,000 × g, 4°C, 30 min. This may require aliquoting of the sample among tubes suitable for high-speed centrifugation.
Transfer the supernatant to a clean tube for desalting. The pellet should be stored for future study if protein yield is inadequate.
FIG. 2.
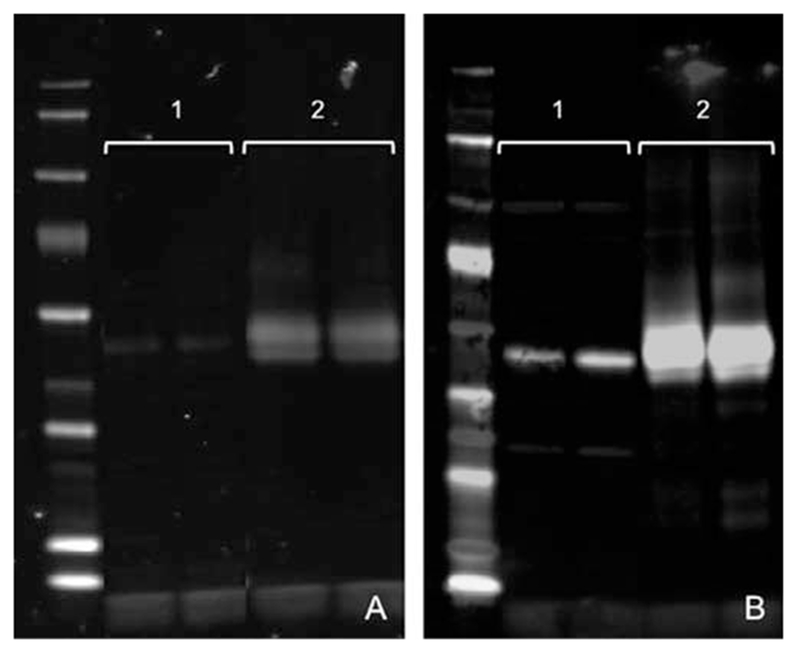
Bone protein extracts analyzed via Western blot with stain for decorin (A) and biglycan (B). On both gels, lanes labeled “1” are protein samples extracted via urea alone; lanes labeled “2” are the same samples with subsequent extraction via urea/EDTA. While both extract fractions contain SLRPs, significantly more SLRPs are present in the EDTA-treated fraction, indicating that SLRPs are significantly encased in mineralized matrix. Mouse biglycan antisera LF-159 and decorin antisera LF-114 at a 1:2000 dilution were used to probe this membrane.
2.3.4. Protein extraction (fractionated)
- Add 1 mL solubilizing buffer (8 M urea/50 mM Tris-HCl) to the homogenizer with sample. Apply the solution to the sides of the homogenizer to collect sample that may have stuck to the wall.
-
○Consider adding protease inhibitor at this stage if protein degradation is a concern. However, the urea should denature any proteases, which will not refold until the sample is diluted.
-
○
- Homogenize the sample first with the “loose” pestle, then the “tight pestle.” Movement of the pestle should be smooth and fluid when the sample is appropriately homogenized.
-
○Note: This technique is optimized for 0.2 g samples. For larger samples, a larger homogenizer may be appropriate.
-
○
- Transfer solution to a polypropylene tube with volume to accommodate for demineralization buffer. Bring to volume with 8 M urea/50 mM Tris–HCl (~10mL/g bone). Use the same buffer to wash out the glass homogenizer, minimizing lost sample.
-
○Note: Polypropylene tubes are preferred, as protein will bind the walls of polystyrene tubes, reducing protein yield (Goebel-Stengel et al., 2011).
-
○
Sonication is useful at this stage to increase yield. Leave samples on ice and perform sonication at low power for 3 s. Repeat for a total of three cycles with time for cool-down between cycles.
The samples should be set to rotate at 4°C for 24 h at 30 RPM. A centrifuge tube rotisserie is recommended for this step.
Spin down the sample at ~15,000 × g, 4°C, 30 min. This may require aliquoting of the sample among tubes suitable for high-speed centrifugation.
Transfer and store the supernatant at 4°C. This is the nonmatrix-bound fraction, which will require desalting prior to further analysis.
Re suspend the pellet in bulk extraction buffer (0.5 M EDTA/8 M urea/50 mM Tris–HCl ~10mL/g bone) and set to rotate at 4°C for 48 h at 30 RPM.
Spin down the sample at ~15,000 × g, 4°C, 30 min.
Transfer the supernatant for desalting. The pellet should be stored for future study if protein yield is inadequate.
2.3.5. Desalting of protein
At this stage, the samples have high concentrations of calcium, EDTA, and urea, all of which will impede protein analysis. Removal of these salts can be accomplished through dialysis or through utilization of centrifugal filters. The goal is [EDTA] <10mM and [Urea] <1 M, as this is required for protein concentration calculation with the Bradford assay. Described below is the use of centrifugal filters (Fig. 3).
- Transfer samples to Amicon centrifugal tubes (10–30K MW filter) set on ice.
-
○Note: Filter size choice is dependent upon the smallest protein of interest. SLRPs are small proteins (~40K MW); therefore, a smaller filter pore size is chosen.
-
○
Centrifuge at 4000 × g, 4°C, 30 min. Discard flow through.
- Note volume remaining in upper column (typically 100–200 μL). Bring this protein extract to 4 mL with ice-cold 1 × TBS. Pipette solution up and down to insure homogenous mixture.
-
○Note: A rough estimation of sample dilution can now be obtained. For example, if 100 μL remained in the column after centrifugation, and 3.9 mL of TBS was added, the sample underwent roughly a 40-fold dilution. The concentration of urea at this point, which was initially 8 M, would now be around 0.2 M.
-
○
Repeat spin at 4000 × g, 4°C, 30 min. Discard flow through.
Repeat dilution with 1 × TBS through filter centrifugation until appropriate salt dilution is achieved.
Transfer samples to sterile protease-free tubes. If protein degradation is a concern, add protease inhibitor cocktail at this point. Store at −20°C.
FIG. 3.

Amicon centrifugal filter in tube. 30K MW is preferable when working with SLRPs.
2.3.6. Protein concentration quantification
Protein should be quantified according to BCA protein assay instructions.
3. ISOLATION OF PROTEOGLYCANS FROM CONDITIONED MEDIA
3.1. BACKGROUND
To further explore the function of proteoglycans in skeletal cells, it is important to obtain high-quality non denatured functional form testing. While commercial sources are available for bigly can and decorin, they are in fact suboptimal in that they (1) are isolated using highly denaturing conditions that are not conducive for proper folding and normal secondary structure configuration and (2) are often contaminated with copurified materials such as collagen or aggrecan (Brown et al., 2017). For this reason we devised a simple and effective procedure that uses nonimmortalized bone marrow stromal cells to serve as “factories” for large-scale bigly can and decorin production and isolation.
Proteoglycans are unique because they contain chains of GAGs that are highly charged. This property can be used to aid in their isolation. In our experimental approach, we took advantage of the biological rigor of primary cells isolated from bone marrow stroma that, when transduced with adenovirus encoding either biglycan or decorin, can be further maintained in serum-free media and is subsequently the source of high amounts of secreted proteoglycans. The conditioned media containing highly enriched biglycan or decorin are then purified using an anion (negatively charged) exchange column with Sepharose Q beads. These beads can bind to the positively charged GAG chains on the proteoglycans that are covalently connected to the core protein. The SLRPs, bound through their GAGs with their attached core, are then gently removed by increasing concentrations of salt using physiological buffers. This is a simple procedure that can be performed with common reagents and lab equipment.
3.2. MATERIALS
3.2.1. Equipment
Tissue culture dishes
12-in. disposable chromatography columns
Assorted disposable pipettes
15 and 50 mL disposable test tubes with caps
Dialysis cassettes (Slide-A-Lyzer G2, 3.0 mL capacity, 100K MWCO)
3.2.2. Solutions and reagents
Alpha MEM with and without 10% FBS (with pen–strep/glutamate)
Phenol red-free alpha MEM (with pen–strep/glutamate)
Human bone marrow stromal cells (Lonza)*
Adenovirus with expressing decorin or biglycan**
Sepharose Q beads (Fast Flow 7732-185)
2 M solution of NaCl in 1 × PBS (58.4g NaCl/500mL 1 × PBS)
10%, 20%, 30%, 40%, and 50% of the 2M NaCl solution diluted with 1 × PBS (10% would be 5 mL 2M solution plus 45 mL PBS in a total volume of 50 mL, etc.)
Coomassie blue stain
Stains-ALL***
*Note: Human bone marrow stromal cells can be passaged and frozen for future use.
**Note: Adenoviruses work best when using human cells. They are extremely efficient at transduction of human BMSC and can obtain 100% cell transduction (Sommer et al., 1999). However, because they do not integrate into the genome, they are transient in nature. They are generated by subcloning a cDNA encoding the entire coding region of the desired protein (Fisher, Termine, & Young, 1989) into adenoviral vectors; in our case, pACCMV.pLpA that contained a CMV promoter was used. The vector with the subcloned cDNA is then recombined in HEK cells specifically engineered for this purpose. The creation of active adenoviruses can be done with help from commercial sources (www.viraquest.com).
***Note: Stains-ALL is made with a stock composed of 2.5 mL of 0.1% Stains-ALL in fresh formamide (stock can be stored in foil, RT), 2.5 mL formamide, 12.5 mL isopropanol, 0.5 mL 1.5 M Tris, pH 8.8 (bring to total of 50 mL with water). Before staining, the SDS in the gel needs to be removed. This can be done by incubating gels in 25% isopropanol three times for 1 h each and then overnight at RT with shaking. GAG chains will appear blue/purplish, while the protein will be red.
3.3. METHOD
3.3.1. Preparation of cells for adenovirus transduction
Plate human bone marrow stromal cells using standard procedures and allow them to come to 70% confluence in the presence of alpha MEM plus 10% FBS, pen/strep/glutamine in 5% CO2 at 37°C.
Add increasing doses of adenovirus (saturating levels up to 100 μL of 1 × 1010 PFU/150mm flask of human BMSC) by adding and then gently swirling the media.
Allow transduction to proceed for up to 1 day. Remove media and replace with serum- and phenol red-free media (plus pen–strep/glutamate). Collect serum-free media every 2 days replacing with fresh serum-free media. Collect the conditioned media up to 14days and store at 4°C (or −20°C for long term).
Test each of the collections by gel electrophoresis to determine which time points have maximum expression and pool them for further purification (Western blot analysis is not needed at this point as the expression should be so high it is visible by gel staining alone).
3.3.2. Purification of biglycan and decorin from conditioned media
Pool and filter the biglycan/decorin-containing media prepared as described above.
Shake bottle of Sepharose Q before pipetting out 10 mL of the bead slurry and rinse three times with water in a 50 mL test tube.
Add the media (up to 200 mL) to the Sepharose Q into a conical-bottomed disposable test tube and gently rotate for at least 1 h at room temperature and then centrifuge at 100 RPM.
Pipette the incubated Sepharose solution into a disposable 12-in. column and insert a small porous disc on top to prevent disturbing the packed column when subsequent salt solutions are added.
Add the 10% solution to the column and collect four fractions 10 mL each numbering them A, B, C, and D in sequence. Repeat this for the 20%, 30%, 40%, and 50% solutions. You will end up with 20 test tubes of fractionated media.
Take 10 μL from each fraction and run on a protein gel. Stain the gel. Your desired material will elute mainly in the 40% B (with minor smaller-sized material in 40% A and larger in the 50% of 2 M solution of NaCl in 1 × PBS) (see Fig. 4 as an example).
Dialyze the desired fractions in ABCase buffer (or water depending on the next use, see formula for ABCase buffer in the next section). Freeze or freeze dry (if in water) for long-term storage. Purified PGs do not work well for conventional protein concentrations assays (like BCA), the GAGs interfere. Thus, weighing is the best way to determine proteoglycan content.
FIG. 4.
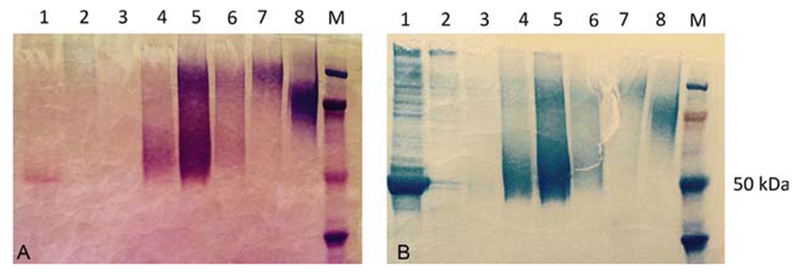
Representative picture of biglycan purification from Sepharose Q chromatography column. Panel A shows the Stains-ALL-stained material (note the reddish stain of the core in lane 1); panel B shows the same material using Coomassie blue. Lanes by salt elution percent: 1%–10% A, 2%–20% A, 3%–30% A, 4%–40% A, 5%–40% B, 6%–40% C, 7%–50% A. Lane 8 is decorin reference. The highest concentration of PG eluted in the lane 5 (40% B) with small forms in lane 4 (40% A; less GAGs, weaker binding, less salt required) and larger forms in 50% (lane 7; more GAGS, tighter binding, more salt required).
4. TREATMENT WITH CHONDROITINASE AND ASSESSMENT OF SLRPs
4.1. BACKGROUND
The functionality of SLRPs varies with the presence or absence of the attached GAG chains. Some protein interactions only occur if the GAG chains are attached, while some of SLRP’s functions are actually increased by removal of the GAG chains. For example, inhibition of fibrillogenesis by decorin is independent of the presence of the GAG chains, yet disruption of the core protein secondary structure of the core protein results in loss of inhibition (Ramamurthy, Hocking, & McQuillan, 1996; Vogel, Koob, & Fisher, 1987). Prior studies have demonstrated that removal of the chondroitin/dermatan sulfate chains from decorin and biglycan results in increased affinity for TGF-beta (Hildebrand et al., 1994) and TNF-alpha (Tufvesson & Westergren-Thorsson, 2002). Alternatively, biglycan and decorin have been shown to have their interaction with WISP-1 abolished by selectively removing dermatan sulfate chains (Desnoyers, Arnott, & Pennica, 2001).
When studying SLRPs, it is important to determine the proportion of SLRPs with attached GAG chains, and to have a way to remove the GAG chains. This provides the investigator with the ability to determine how the GAG chains are affecting the subject system being studied. Chondroitinase treatment is an effective method to remove chondroitin sulfate and dermatan sulfate GAG chains, without disrupting the core protein. This enzyme is ideal for the deglycosylation of biglycan and decorin because their GAGs are primarily made of chondroitin or dermatan sulfate (CS/DC) that are both sensitive to Abase action. By combining the technique of deglycosylation with gel electrophoresis and blot analysis, proteoglycan samples can be easily identified and manipulated according to the requirements of the investigation. Below are described the techniques required to achieve this.
4.2. MATERIALS
4.2.1. Equipment
Small disposable protease-free test tubes
Vortex test tube mixer
Dry bath
Gel electrophoresis and Western blotting system
4.2.2. Solutions and reagents
Chondroitinase ABC enzyme (ABCase), protease free (Proteus vulgaris, Sigma, #9024-13-9, BSA free)*
Chondroitinase ABC buffer 5 ×: 200mM sodium acetate, 200mM Tris–HCl, pH 8.0
Protease inhibitor cocktail 50 × (cOmplete Cocktail, Sigma-Aldrich)
Rabbit polyclonal antibodies to mouse biglycan (LF-159) or mouse decorin (LF-114)**
*Note: Removes chondroitin sulfate and dermatan sulfate without disturbing the core protein.
**Note: Antibodies specific for SLRPs and many other ECM components can be obtained from Larry Fisher, NIDCR, NIH by going to the following website: https://www.nidcr.nih.gov/research/NIDCRLaboratories/CranioSkeletal/Antisera.htm. It should be noted commercial sources are also an option but not used here.
4.3. METHOD
4.3.1. Removal of GAG chains
Required reaction components should be calculated according to the reaction mixture described below. This requires protein sample concentration to be known (complete BCA protein assay prior to starting).
- Reaction mixture:
Component Volume Notes MilliQ water 38-X μL Chondroitinase ABC buffer (5 ×) 10 μL Protease inhibitor (50 ×) 1 μL Sample X μL 50 μg of protein Chondroitinase ABC 1 μL Add components in order listed using protease-free pipette tips. Vortex to insure mixture, then spin down components on desktop mini centrifuge.
Set tubes in a dry bath at 37°C for 2h.
Spin down components on desktop mini centrifuge, briefly vortex, then spin down again. This will insure sample homogeneity and minimize loss.
Store sample at −20°C for further studies.
4.3.2. Assessment by Western blot analysis
As Western blot is a common laboratory technique, please use standard Western blot protocols. This section carries suggestion for optimized Western blot analysis. An example Western blot of biglycan is shown in Fig. 5.
Load 20–25 μg of protein per well.
After transfer of protein to the membrane, stain the gel with a Coomassie dye. This will allow you to visualize how the protein has run. A smear or abnormal gel may indicate high salt in the sample (requiring further desalting of sample), or that the protein has been degraded (requiring protease inhibitors and greater care with isolation).
- Dr. Larry Fisher has developed antibodies to human, monkey, murine, rat, and bovine biglycan and decorin, which have been previously demonstrated as effective in Western blot analysis for SLRPs (https://www.nidcr.nih.gov/research/NIDCRLaboratories/CranioSkeletal/Antisera.htm). Recommended antibodies (see Fig. 3):
-
○Murine biglycan LF-159 antisera (1:2000 dilution)
-
○Murine decorin LF-114 antisera (1:2000 dilution)
-
○
FIG. 5.
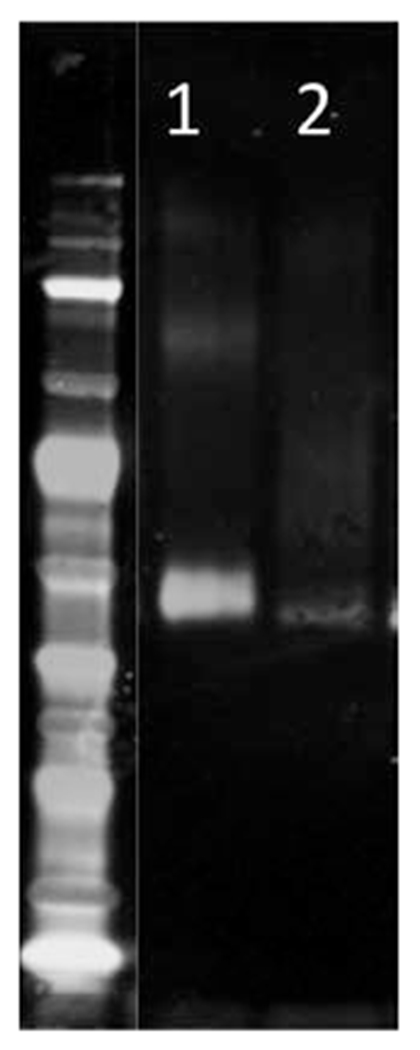
Representative gel of biglycan after (Berendsen et al., 2011) and before (Brown et al., 2017) treatment with chondroitinase ABC. In lane 2, a smear is present representing the size variation attributable to GAG chain length. The band at 40 kDa represents deglycosylated biglycan, and has increased signal in lane 1 after treatment with chondroitinase ABC.
5. SUMMARY
Bone is a unique organ system that plays a critical role in mammals. Critical in its role are the proteins within bone that facilitate both form and function. Biglycan and decorin are two proteins, both proteoglycans with highly ordered secondary structure, that allow for normal bone growth, development, and maintenance. However, due to the hard mineralized bone in which these proteins reside, unique methods have been developed to study them. The proteins can be optimally purified from bone using a method that uses EDTA to remove the mineral from bone, and urea to solubilize the proteins present. The salts are then removed and the sample is ready for analysis. This is an ideal method as it is simple and provides a protein sample suitable for function studies. Additionally, methods have been developed to culture and purify large quantities of specific proteoglycans, creating a source for further in vitro and in vivo studies. Finally, through the use of deglycosylating enzymes such as chondroitinase, it is possible to selectively remove GAG chains while maintaining the protein core, providing the means to analyze the functionality of members of the proteoglycans.
REFERENCES
- Berendsen AD, Fisher LW, Kilts TM, Owens RT, Robey PG, Gutkind JS, et al. (2011). Modulation of canonical Wnt signaling by the extracellular matrix component biglycan. Proceedings of the National Academy of Sciences of the United States of America, 108, 17022–17027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown SJ, Fuller HR, Jones P, Caterson B, Shirran SL, Botting CH, et al. (2017). Contaminants in commercial preparations of ‘purified’ small leucine-rich proteoglycans may distort mechanistic studies. Bioscience Reports, 37, BSR20160465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XD, Fisher LW, Robey PG, & Young MF (2004). The small leucine-rich proteoglycan biglycan modulates BMP4-induced osteoblast differentiation. The FASEB Journal, 18, 948–958. [DOI] [PubMed] [Google Scholar]
- Desnoyers L, Arnott D, & Pennica D (2001). WISP-1 binds to decorin and biglycan. The Journal of Biological Chemistry, 276, 47599–47607. [DOI] [PubMed] [Google Scholar]
- Fisher LW, Termine JD, & Young MF (1989). Deduced protein sequence of bone small proteoglycan I (biglycan) shows homology with proteoglycan II (decorin) and several non-connective tissue proteins in a variety of species. The Journal of Biological Chemistry, 264, 4571–4576. [PubMed] [Google Scholar]
- Goebel-Stengel M, Stengel A, Taché Y, & Reeve JR Jr. (2011). The importance of using the optimal plastic ware and glassware in studies involving peptides. Analytical Biochemistry, 414, 38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand A, Romarís M, Rasmussen LM, Heinegård D, Twardzik DR, Border WA, et al. (1994). Interaction of the small interstitial proteoglycans biglycan, decorin, and fibro-modulin with transforming growth factor-β. The Biochemical Journal, 302, 527–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo RV (1997). The family of the small leucine-rich proteoglycans: Key regulators of matrix assembly and cellular growth. Critical Reviews in Biochemistry and Molecular Biology, 32, 141–174. [DOI] [PubMed] [Google Scholar]
- Mochida Y, Parisuthiman D, & Yamauchi M (2006). Biglycan is a positive modulator of BMP2-induced osteoblast differentiation. Advances in Experimental Medicine and Biology, 585, 101–113. [DOI] [PubMed] [Google Scholar]
- Myren M, Kirby DJ, Noonan ML, Maeda A, Owens RT, Ricard-Blum S, et al. (2016). Biglycan potentially regulates angiogenesis during fracture repair by altering expression and function of endo statin. Matrix Biology, 52–52, 141–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neill T, Painter H, Buraschi S, Owens RT, Lisanti MP, Schaefer L, et al. (2012). Decorin antagonizes the angiogenic network. The Journal of Biological Chemistry, 287, 5492–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitovic D, Aggelidakis J, Young MF, Iozzo RV, Karamanos NK, & Tzanakakis GN (2012). The biology of small leucine-rich proteoglycans in bone pathophysiology. The Journal of Biological Chemistry, 287, 33926–33933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramamurthy P, Hocking AM, & McQuillan DJ (1996). Recombinant decorin glycoforms. Purification and structure. The Journal of Biological Chemistry, 271, 19578–19584. [DOI] [PubMed] [Google Scholar]
- Sommer B, Kuznetsov SA, Robey PG, O’Connell B, Cristiano RJ, & Young MF (1999). Efficient gene transfer into normal human skeletal cells using recombinant adenovirus and conjugated adenovirus-DNA complexes. Calcified Tissue International, 64, 45–49. [DOI] [PubMed] [Google Scholar]
- Tufvesson E, & Westergren-Thorsson G (2002). Tumour necrosis factor-alpha interacts with biglycan and decorin. FEBS Letters, 530, 124–128. [DOI] [PubMed] [Google Scholar]
- Vogel KG, Koob TJ, & Fisher LW (1987). Characterization and interactions of a fragment of the core protein of the small proteoglycan (PGII) from bovine tendon. Biochemical and Biophysical Research Communications, 148, 658–663. [DOI] [PubMed] [Google Scholar]
- Wang X, Harimoto K, Xie S, Cheng H, Liu J, & Wang Z (2010). Matrix protein biglycan induces osteoblast differentiation through extracellular signal-regulated kinase and Smad pathways. Biological & Pharmaceutical Bulletin, 33, 1891–1897. [DOI] [PubMed] [Google Scholar]
FURTHER READING
- Fisher LW, Hawkins GR, Tuross N, & Termine JD (1987). Purification and partial characterization of small proteoglycans I and II, bone sialoproteins I and II, and osteonectin from the mineral compartment of developing human bone. The Journal of Biological Chemistry, 262, 9702–9708. [PubMed] [Google Scholar]