

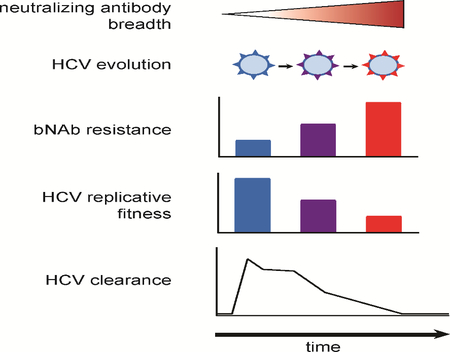
SUMMARY
The role that broadly neutralizing antibodies (bNAbs) play in natural clearance of human hepatitis C virus (HCV) infection and the underlying mechanisms remain unknown. Here, we investigate the mechanism by which bNAbs, isolated from two humans who spontaneously cleared HCV infection, contribute to HCV control. Using viral gene sequences amplified from longitudinal plasma of the two subjects, we found that these bNAbs, which target the front layer of the HCV envelope protein E2, neutralized most autologous HCV strains. Acquisition of resistance to bNAbs by some autologous strains was accompanied by progressive loss of E2 protein function, and temporally associated with HCV clearance. These data demonstrate that bNAbs can mediate clearance of human HCV infection by neutralizing infecting strains and driving escaped viruses to an unfit state. These immunopathologic events distinguish HCV from HIV-1 and suggest that development of an HCV vaccine may be achievable.
Keywords: Hepatitis C virus, HCV clearance, Neutralizing Antibodies, Binding Sites, Immunologic Memory, Antibody Specificity
eTOC blurb
Kinchen et al. demonstrate that antibodies isolated from humans who spontaneously cleared HCV infection blocked many different HCV strains. Viruses that evolved resistance to these antibodies lost the ability to replicate, suggesting that antibodies are important for HCV clearance, and an HCV vaccine may be attainable.
Graphical Abstract
INTRODUCTION
Hepatitis C virus (HCV) infection is a global health crisis, with approximately 71 million people infected worldwide (WHO, 2017). Direct-acting antiviral (DAA) therapy has revolutionized care for patients with HCV, but infection incidence rates remain high globally and are rising in the United States (Suryaprasad et al., 2014). The WHO estimates that there were 1.75 million new HCV infections in 2017, and incident infection is likely to remain an issue in the absence of a preventative HCV vaccine. Only a small minority of those people who are aware of their infection have access to treatment, and those who are untreated remain at risk for transmitting the infection to others and developing HCV-related complications. For these reasons, development of a prophylactic vaccine remains a critical component of HCV eradication efforts.
While most individuals who are infected with HCV remain persistently infected, approximately 25% clear the infection without treatment (Micallef et al., 2006; Osburn et al., 2010). Notably, clearance of primary human HCV infection is associated with the early development of broadly-neutralizing plasma antibodies against HCV (Osburn et al., 2014; Pestka et al., 2007). However, there is also an association between robust T cell responses and clearance (Chang et al., 2001; Rehermann, 2009), and bNAb neutralization of autologous viruses has never been analyzed in persons who spontaneously cleared HCV infection. Therefore, it has remained unclear whether bNAbs play a direct role in natural clearance of human HCV infection.
To address this question, we studied the antibody responses of two human subjects, designated C117 and C110, who cleared one or multiple HCV infections without treatment. We recently described bNAbs isolated from the B cells of both subjects (Bailey et al., 2017). We demonstrated that these bNAbs neutralized broadly across diverse heterologous HCV strains, despite having surprisingly few somatic mutations. A bNAb isolated from subject C117, designated HEPC3, showed reduced binding affinity for autologous envelope proteins (E1E2) of viral strains circulating immediately prior to clearance of infection, suggesting that HEPC3 may have selected resistant viral strains. However, it remained unclear whether this selection pressure played a role in clearance of infection. The accompanying study by Flyak et al. describes the Xray crystal structures of HEPC3 as well as a bNAb designated HEPC74, which was isolated from subject C110, demonstrating that these bNAbs are structurally homologous. The structural similarity of these bNAbs, as well as our previous data suggesting that HEPC3 drove evolution of autologous viruses, engendered interest in determining whether these bNAbs mediated HCV clearance in vivo.
In this study, we define the mechanism by which C117 and C110 bNAbs mediated clearance of HCV infection. Using HCV gene sequences amplified from longitudinal plasma isolates of these subjects, we found that bNAbs neutralized most autologous HCV strains. Remarkably, when bNAb resistance developed, it was accompanied by progressive loss of E2 protein function, which was temporally associated with clearance of HCV infection. These data demonstrate that bNAbs targeting the front layer of E2 can mediate clearance of human HCV infection both by neutralization and by driving bNAb resistant viruses to an unfit state.
RESULTS
Early development of bNAbs in persons with spontaneous clearance of HCV.
Subjects C110 and C117 were persons who inject drugs (PWID) enrolled in the Baltimore Before and After Acute Study of Hepatitis (BBAASH) cohort who were identified before or very early during acute HCV infection and subsequently followed with frequent sampling of plasma and peripheral blood mononuclear cells (PBMC) for over six years (Fig 1A). C117 was aviremic and HCV seronegative until three months after entry into the cohort, when he became infected with a genotype 1a virus. He cleared this infection without treatment after 388 days and subsequently remained aviremic over more than three years of follow-up. We isolated bNAbs designated HEPC3 and HEPC98 from C117 B cells immediately after clearance of infection, as previously described (Bailey et al., 2017).
Figure 1. Longitudinal neutralizing antibody responses of two subjects with HCV clearance and a third with persistence of infection.

(A) HCV RNA was measured at initial infection and then periodically for more than six years. Dashed line indicates limit of detection (LOD) of the HCV RNA assay. (B) Neutralizing breadth of longitudinal plasma samples or bNAbs HEPC3 and HEPC98 (isolated from C117), bNAbs HEPC74 and HEPC43 (isolated from C110), or CBH-2 (control bNAb). Values are the number out of nineteen HCVpp neutralized at least 50% by a 1:50 dilution of plasma, or by 10 μg/mL of mAb, tested in duplicate. (C) Spearman correlations were measured between neutralization profiles (i.e., patterns of relative neutralization potency across a panel of 19 HCV strains) of C117 or C110 plasma samples and autologous or negative control (CBH-2) bNAbs (see also Fig S1). Significant r values are colored with a yellow to green heatmap.
C110 was infected acutely with a genotype 1a virus on entry into the cohort (‘Infxn 1’). Sequencing of Core and E1 genes of plasma viruses 178 days later indicated that the Infxn 1 had been replaced by a distinct genotype 1a HCV strain (‘Infxn 2’). Subject C110 cleared this second infection without treatment 436 days after initial infection, then exhibited recurrent viremia 665 days later with a third genetically distinct genotype 1a HCV strain (‘Infxn 3’). Infxn 3 persisted for 774 days, with low-level viremia, brief intervals of intervening aviremia, transient coinfection with a genotype 1b strain (‘Infxn 4’), and ultimately clearance without treatment 1875 days after initial infection. Clearance was confirmed with frequent HCV RNA testing without detection of virus over the subsequent five years. We isolated bNAbs designated HEPC74 and HEPC43 from C110 B cells immediately after clearance of Infxn 3, as previously described (Bailey et al., 2017).
A subject with persistent infection, designated P29, was selected as a control for this study because he also was identified prior to HCV infection with a genotype 1a strain, and then sampled frequently during acute and chronic infection. P29 remained persistently HCV infected over more than six years of follow-up.
We measured neutralizing breadth of antibodies in plasma obtained at longitudinal time points during infection of subjects C117, C110, and P29, as well as neutralizing breadth of bNAbs HEPC3, HEPC98, HEPC74, and HEPC43, and of control bNAb CBH-2, using a panel of 19 genotype 1a and 1b HCV pseudoparticles (HCVpp) (Fig 1B). This panel comprises 94% of the amino acid polymorphisms present at greater than 5% frequency in a reference panel of 643 genotype 1 HCV isolates from GenBank. Consistent with prior studies of subjects with HCV clearance, both C117 and C110 displayed very early development of bNAbs, with detectable neutralizing activity against heterologous HCV strains in plasma isolated 17 or 178 days after infection of C117 or C110, respectively. Neutralizing breadth of the plasma of both subjects generally increased over time, reaching a peak of twelve of nineteen HCVpp neutralized by C117 plasma and thirteen of nineteen HCVpp neutralized by C110 plasma. In contrast, plasma isolated from P29 failed to neutralize any HCVpp, which is typical of early infection plasma from subjects who subsequently develop persistent infection (Osburn et al., 2014; Pestka et al., 2007). Neutralizing breadth of bNAbs HEPC3, HEPC74, and HEPC43 at 10 μg/mL concentration was similar to peak neutralizing breadth of plasma from the same donors, neutralizing fourteen, seventeen, and eleven out of nineteen HCVpp, respectively. HEPC98 and the control bNAb CBH-2 each neutralized five of nineteen HCVpp. Together, these data confirm that bNAbs developed very early during acute infection of C117 and C110, and the peak neutralizing breadth of plasma from these donors was similar to that of bNAbs isolated from the B cells of the same donors.
We previously demonstrated that a neutralization profile (i.e., pattern of relative neutralization potency across a panel of diverse heterologous HCV strains) is unique for each bNAb (Bailey et al., 2015), and others have demonstrated that neutralization profiles can be used to identify the activity of virus-specific bNAbs present in plasma (Georgiev et al., 2013). Therefore, to determine the time points at which specific autologous bNAbs were likely present in plasma, we used Spearman correlations to compare neutralization profiles of autologous bNAbs to the neutralization profiles of longitudinal plasma samples from C117 and C110 (Fig 1C and Fig S1). Remarkably, the neutralization profile of each C117 plasma sample correlated significantly with the neutralization profile of autologous bNAb HEPC3. The strength of this correlation generally increased with successive plasma samples (D17 r=0.55, p=0.015; D388 r=0.66, p=0.002). Correlations with the autologous bNAb HEPC98 were positive but not statistically significant. The neutralization profile of each C110 plasma sample correlated significantly with the neutralization profiles of autologous bNAbs HEPC74 and HEPC43. Correlation of HEPC43 was greatest with D436 (Infxn 2) plasma (r=0.64, p=0.003), and correlation of HEPC74 was greatest with D1875 (Infxn 3) plasma (r=0.68, p=0.002). As expected, correlations between plasma and the control bNAb, CBH-2, were not significant. Neutralization profiles of P29 plasma could not be analyzed due to a lack of detectable neutralizing activity. Taken together, these data demonstrate that HEPC3-like antibodies developed very early in infection of C117 (on or before D17) and were present in plasma through D388 when C117 cleared their infection. HEPC74 and HEPC43-like antibodies developed early in infection of C110 (on or before D178) and were present in plasma on D436 and D1875, when C110 cleared Infxn 2 and Infxn 3, respectively. P29, who remained persistently infected, did not develop bNAbs during the first 416 days of infection.
Phylogenetic analysis of HCV nucleotide sequences obtained at longitudinal time points reveals transmitted/founder strains, selection bottlenecks, and strains circulating immediately prior to clearance of infection.
We performed extensive longitudinal sequencing of the HCV quasispecies present in plasma samples collected over time from acute infection through clearance (C117 and C110) or through D416 of persistent infection (P29) (Fig 2). Plasma RNA was isolated and RT-PCR was performed with single-genome amplification of 10–45 (median 33) amplicons per time point in order to define viral diversity and viral evolution over time for each subject. For C117, near fullgenome amplification was performed from D17 to D354 plasma, and D388 genomes were amplified with overlapping 5′ and 3′ hemigenomic amplicons. For C110 and P29, approximately 5 kb hemigenomes spanning Core through NS3 genes were amplified from all plasma time points. As shown in Fig 2A, phylogenetic analysis of 5′ hemigenomes indicated that C117 was infected initially by at least three different transmitted/founder (T/F) viruses, which was consistent with our prior analysis limited to E1E2 genes from this subject (Bailey et al., 2017). T/F#1 and T/F#2 lineages were extinguished early in infection, but the T/F#3 lineage persisted and evolved throughout the course of infection, exhibiting a series of stringent population bottlenecks. This lineage was last detected at the final viremic time point (D388) just prior to its extinction, when the HCV RNA level had declined to 754 IU/mL.
Figure 2. Maximum likelihood phylogenetic trees of 5 kb hemigenomic HCV nucleotide sequences obtained by single-genome amplification from plasma at longitudinal time points throughout the course of infection.

Sequences are color-coded by duration of infection at the time of sampling. Transmitted/founder (T/F) sequences inferred by phylogeny and date of sampling are indicated. Identical sequences amplified in multiple independent reactions are boxed. Strains selected for further phenotypic analysis are indicated with an asterisk. All distances are drawn to scale, except distance to the outgroup. Nodes with bootstrap values >80 are indicated with black circles. (A) C117, clearance of a genotype 1a infection. (B) C110, clearance of three genotype 1a infections. (C) P29, persistence of a genotype 1a infection.
From D17 to D388 (immediately before clearance of infection), the T/F#3 lineage accumulated twelve substitutions in E2, with one substitution in NS5A and two in NS5B (Fig S2A and Table S1). Of these twelve E2 substitutions, six fell in the hypervariable region 1 (HVR1, aa 385–411), two in the front layer (aa 424–459), and four in the remainder of E2. The substitutions accumulated in E2 in a stepwise manner and were present in 100% of the viral genomes amplified from D388 plasma, indicating strong selective pressure favoring viruses with these substitutions.
Phylogenetic and genetic distance analyses of hemigenomic sequences confirmed that C110 was infected sequentially with at least three distinct genotype 1a strains (Fig S3). At the first sampled time point, when still HCV antibody negative, C110 was infected with a quasispecies of closely-related viruses (Infxn 1) (Fig 2B). By D178 of infection, Infxn 1 was extinguished and replaced by at least six T/F strains of a distinct genotype 1a infection (Infxn 2). The T/F#5 (D178-A31) lineage persisted and evolved over time. Like C117 viruses, C110 Infxn 2 viruses exhibited a series of stringent population bottlenecks until infection was cleared after D436. Infxn 3 already was established for five months when virus was sequenced on D1260, so T/F strains could not be identified, but Infxn 3 viruses also exhibited a series of stringent population bottlenecks over the sampled interval, until Infxn 3 was cleared after D1875.
We also analyzed the sequences of Infxn 1, 2 and 3 viruses together to identify polymorphisms that may have provided Infxn 2 and 3 strains with a selective advantage relative to the Infxn 1 viruses that were cleared rapidly (Fig S2B and Table S1). Core-NS3 sequences of D178-A31 (Infxn 2) and D0-A4 (Infxn 1) strains differed by sixty-six amino acids (4%). Given this large number of amino acid differences, we also compared the D178 virus to the Bole1a sequence, a phylogenetically reconstructed ancestor of genotype 1a viruses, because we previously found that amino acid differences from Bole1a are more likely to represent immune escape substitutions (Burke et al., 2012). We identified thirty-five polymorphisms in the D178-A31 sequence that differed from both the D0-A4 (Infxn 1) virus and the Bole1a sequence. Sixteen of these polymorphisms fell in E2, with six polymorphisms in HVR1 and ten in the remainder of E2. Seven additional substitutions arose subsequently in HVR1 of the D178-A31 lineage over the course of Infxn 2, along with two substitutions in NS2 and one in NS3. We performed similar analysis with Infxn 3 viruses and found that strain D1260-B3 (Infxn 3) virus had nineteen polymorphisms in E2 that did not match D0-A4 (Infxn 1) or Bole1a sequences. Over the course of Infxn 3, this lineage also acquired four new substitutions: three in E2 and one in NS3.
P29 was infected initially by at least two distinct T/F viruses (Fig 2C). Like C117 and C110, P29 viruses exhibited population bottlenecks over time. Over the 416 days of infection analyzed, P29 viruses acquired only four substitutions: two in E2 and two in NS2 (Fig S2C and Table S1). Unlike C117 and C110 viruses, P29 viruses did not accumulate any substitutions in HVR1.
Taken together, viral sequencing of the subjects who cleared infection indicated strong selective pressure favoring viruses with progressive substitutions in E2. Next, we investigated the hypothesis that antibodies provided this strong selective pressure.
Naturally selected substitutions in C117 E2 confer partial resistance to autologous plasma antibodies and to the autologous bNAb HEPC3, and complete escape from HEPC98.
To determine whether antibodies in C117 plasma could bind E1E2 of autologous viruses, we used an ELISA to measure plasma IgG binding to longitudinal C117 E1E2 proteins, which demonstrated progressive divergence from the T/F in both HVR1 and more conserved regions of E2 (Fig 3A). For these experiments, we used D388 plasma, which was isolated immediately prior to clearance of infection (Fig 3B). Remarkably, we observed progressively less binding of D388 plasma IgG to sequential C117 E1E2 strains (Fig 3B, left panel). For E1E2 strains that could be used to generate HCV pseudoparticles (HCVpp) with adequate hepatoma cell entry for neutralization assays, we also tested HCVpp neutralization by D388 plasma (Fig 3B, right panel). We observed potent neutralization of D17 HCVpp, with reduced neutralization of later strains D194-FB05 and D194-FC03. Together, these data suggest that NAbs present in D388 plasma selected NAb-resistant E1E2 strains.
Figure 3. Naturally selected substitutions in C117 E2 confer partial resistance to autologous plasma antibodies and autologous bNAbs.

(A) All substitutions arising longitudinally in E2 over the course of C117 infection. Dots indicate homology with the D17–1a53 (T/F) sequence. (B-D) Binding in an ELISA to longitudinal C117 E1E2 proteins (left) or neutralization of HCVpp expressing longitudinal C117 E1E2 proteins (right) by C117 D388 plasma IgG (B), HEPC3 (C), or HEPC98 (D) All assays were performed in duplicate, and values are means +/− SD. (See Fig S5 for additional independent experiments).
Next, we measured binding and neutralization of the same E1E2 strains by HEPC3, the bNAb that was likely predominant in C117 plasma. Remarkably, as with D388 plasma, we observed progressively less binding of HEPC3 to sequential C117 E1E2 proteins (Fig 3C, left panel). We also observed a similar progressive drop in HEPC3 binding to D17 E1E2 variants after stepwise introduction by site-directed mutagenesis (SDM) of only non-HVR1 E2 substitutions (N434D, H444Y, F465Y, P498S, D533N, and D610H), suggesting that these substitutions were mediators of the HEPC3 resistance observed with the longitudinal natural C117 E1E2 strains (Fig S4A). As with D388 plasma, we observed potent neutralization of D17 HCVpp by HEPC3, with reduced neutralization of later strains D194-FB05 and D194-FC03 (Fig 3C, right panel). Together these data suggest that HEPC3 selected NAb resistant E1E2 strains. Notably, however, later E1E2 strains still exhibited some sensitivity to HEPC3 binding and neutralization, suggesting that HEPC3 resistance was incomplete.
We also assessed binding and neutralization of the same E1E2 strains by HEPC98, another bNAb isolated from C117 (Fig 3D). We observed potent binding of D17 E1E2, with reduced binding of D194-FB05 E1E2. For all other variants, binding was not detected, suggesting complete escape from HEPC98 by later strains. HEPC98 neutralization results using HCVpp were concordant with the binding results. Together, these data suggest that HEPC98 selected progressively more resistant E1E2 strains early in infection, and by D285 of infection all circulating strains had escaped fully from HEPC98 binding and neutralization.
Naturally selected substitutions in C110 E2 confer partial resistance to autologous plasma antibodies and to autologous bNAbs HEPC43 and HEPC74.
To determine whether antibodies in C110 plasma neutralized autologous viruses, we first measured neutralization of HCVpp bearing E1E2 amplified from Infxn 1 or Infxn 2 viruses. Each of the Infxn 2 strains differed in HVR1 sequence but carried the same polymorphisms relative to Infxn 1 (D0) virus in the remainder of E2 (Fig 4A). First, we tested neutralization by plasma isolated on D436 of infection, immediately prior to clearance of Infxn 2 (Fig 4B). We observed potent neutralization of D0 HCVpp, with reduced neutralization of Infxn 2 T/F and intermediate time point viruses, and no detectable neutralization of contemporaneous D436 HCVpp. Together, these data suggest that NAbs present in D436 plasma selected progressively more NAb resistant E1E2 strains.
Figure 4. Naturally selected substitutions in C110 E2 confer partial resistance to autologous plasma antibodies and autologous bNAbs.

(A) E2 polymorphisms where the D178-A31 (Infxn 2 T/F) sequence differs from both D0-A4 (Infxn 1) and Bole1a (an inferred ancestral reference sequence), and substitutions arising over the course of Infxn 2. Homology to the D0-A4 sequence is indicated with a dot. (B-D) Neutralization of HCVpp expressing longitudinal C110 E1E2 strains from Infxn 1 and 2 by D436 C110 plasma (B), HEPC43 (C), or HEPC74 (D). (E) E2 polymorphisms where the D1260-B3 (Infxn 3) sequence differs from both D0-A4 (Infxn 1) and Bole1a, and substitutions arising over the course of Infxn 3. Homology to the D0-A4 sequence is indicated with a dot. (F-H) Neutralization of HCVpp expressing longitudinal C110 E1E2 strains from Infxns 1 and 3 by D1875 C110 plasma (F), HEPC43 (G), or HEPC74 (H). (I) Substitutions arising in longitudinal P29 E1E2 clones. (J) Neutralization of HCVpp expressing longitudinal P29 E1E2 strains by autologous D416 plasma. All assays were performed in duplicate, and values are means +/− SD. (See Fig S5 for additional independent experiments.)
Next, we measured neutralization of the same HCVpp by HEPC43 and HEPC74, since these bNAbs likely were present in D436 plasma at the time of clearance of Infxn 2 (Fig 4C-D). As expected, longitudinal E1E2 strains showed the same hierarchy of resistance to HEPC43 as they had with D436 plasma. D0 HCVpp was most neutralization sensitive, with Infxn 2 T/F and intermediate time point virus more resistant, and D436 HCVpp most resistant. We also measured HEPC74 neutralization of these same HCVpp. Like HEPC43, HEPC74 neutralized the D0 HCVpp most potently, with relatively reduced neutralization of all Infxn 2 HCVpp. Together, these data suggest that HEPC43 and HEPC74 selected NAb-resistant E1E2 strains during Infxn 2. Notably, later viral strains still exhibited partial sensitivity to HEPC43 and HEPC74, suggesting that resistance to these bNAbs was incomplete.
To better define the specific polymorphisms in Infxn 2 E1E2 clones conferring partial HEPC74/43 resistance, we evaluated the phenotype of six E2 front layer and variable region 2 (VR2, aa 460–485) polymorphisms present in Infxn 2 viruses (L438I, F442I, N445H, R461L, A466D, and A475T) (Fig S4B). We hypothesized that these polymorphisms were most likely to confer resistance to HEPC43 and HEPC74, given their proximity to the HEPC74/43 epitopes. We used SDM to introduce these six substitutions into the D0 E1E2 clone. As predicted, these substitutions conferred partial resistance to both HEPC43 and HEPC74, suggesting that they contributed to the partial HEPC43 and HEPC74 resistance observed in natural Infxn 2 strains.
We also assessed plasma and autologous bNAb neutralization of HCV strains isolated during C110 Infxn 3. These viruses differed in both HVR1 sequence and at polymorphisms in the remainder to E2 (Fig 4E). In contrast with D436 plasma, D1875 plasma, which was isolated immediately prior to clearance of Infxn 3, potently neutralized all strains tested (Fig 4F). HEPC43 also neutralized these HCVpp (Fig 4G). Relative neutralization of these HCVpp by HEPC74 was strikingly similar to neutralization by plasma (Fig 4H). Together, these data suggest that C110 Infxn 3 viruses did not escape from HEPC74 and HEPC43 neutralization and that these bNAbs contributed to the suppression of viremia and ultimate clearance of Infxn 3.
Finally, we measured neutralization of autologous HCVpp by P29 plasma. P29 viruses acquired only two substitutions in E2 over 416 days of infection (Fig 4I). We did not observe neutralization of HCVpp with T/F, intermediate time point, or contemporaneous autologous E1E2 by D416 plasma (Fig 4J), demonstrating that this plasma lacked the capacity to neutralize autologous viruses from any time point in the first 416 days of infection in the subject who progressed to chronic infection.
HEPC3 and HEPC74/HEPC43 resistance substitutions lead to progressive loss of viral fitness.
We next assessed the mechanism of clearance of viral strains that acquired partial resistance to bNAb neutralization. Since some E1E2 clones arising later in infection were relatively HEPC3 or HEPC74/43 resistant, it is less likely that these viruses were cleared through neutralization alone. Therefore, we next assessed the effect of HEPC3 and HEPC74/43 resistance substitutions on E1E2 function. First, we measured entry of HCVpp expressing longitudinal E1E2 clones isolated over the course of C117 infection (Fig 5A). Entry of D17 HCVpp was greatest, with a progressive drop in entry of each longitudinal HCVpp. Remarkably, entry of HCVpp expressing E1E2 isolated immediately prior to clearance of C117 infection (D388) was only 1.3% of entry of HCVpp expressing D17 E1E2. We observed a similar progressive drop in entry of HCVpp expressing D17 E1E2 after stepwise introduction by SDM of only non-HVR1 E2 substitutions (N434D, H444Y, F465Y, P498S, D533N, and D610H), suggesting that these substitutions were the principal mediators of the fitness loss observed with the longitudinal natural C117 E1E2 strains (Fig 5B).
Figure 5. Naturally selected HEPC3 and HEPC74/43 resistance substitutions lead to progressive loss of viral fitness.

Relative entry into hepatoma cells of HCVpp expressing: (A) Longitudinal natural C117 E1E2 strains (B) C117 D17 E1E2 before and after introduction by site directed mutagenesis (SDM) of the substitutions that arose outside of HVR1. Substitutions present in each SDM are indicated, with homology to the D17 sequence indicated with a dot. (C) Longitudinal natural C110 E1E2 strains (D) C110 D0 E1E2 before and after introduction by SDM of polymorphisms present in the front layer and VR2 of Infxn 2 viruses. (E) Longitudinal natural P29 E1E2 strains. HCVpp entry values were normalized to the entry of the chronologically earliest strain from each subject. Each symbol represents an independent experiment. Horizontal lines indicate means. (F-G) Infectivity of serial dilutions of chimeric replication competent cell culture virus (HCVcc) expressing longitudinal C117 (F) or C110 (G) E1E2 strains, measured as focus forming units/mL (FFU/mL) in Huh7.5.1 cells 48 hours after inoculation. Values are normalized for input HCV viral copy number. (See Fig S6 for second independent experiment). All assays were performed in duplicate, and values are means +/− SD.
We performed similar experiments with longitudinal E1E2 clones isolated over the course of C110 Infxn 1 and 2 (Fig 5C). Infxn 3 viruses were not included in this analysis because all strains were potently neutralized by autologous plasma and bNAbs. As with C117 HCVpp, we observed a progressive decline in entry of longitudinal C110 HCVpp. Entry of HCVpp expressing E1E2 isolated immediately prior to clearance of C110 Infxn 2 (D436) was 18% of HCVpp expressing D0 E1E2. We observed a similar reduction in D0 HCVpp infectivity after introduction by SDM of only the Infxn 2 E2 front layer and VR2 polymorphisms (L438I, F442I, N445H, R461L, A466D, A475T), suggesting that these polymorphisms were the principal mediators of the reduced function of Infxn 2 strains relative to Infxn 1 (Fig 5D). As a control, we compared entry of HCVpp expressing longitudinal E1E2 clones isolated over more than a year of infection of P29, the subject who remained persistently infected (Fig 5E). In contrast to the reduction in entry observed over time with C117 and C110 E1E2 clones, HCVpp expressing E1E2 isolated on D416 of P29’s infection displayed two-fold greater entry than HCVpp expressing T/F virus E1E2. Thus, E1E2 evolution in the subject who progressed to chronic infection suggests selection of substitutions that enhanced entry.
We confirmed HCVpp entry results by cloning representative longitudinal C117 and C110 E1E2 genes into chimeric, full-length, replication competent HCV (HCVcc) (Fig 5F-G). Infectivity of these HCVcc followed the same hierarchy as HCVpp entry. Of chimeric C117 HCVcc, D17 was most infectious, D285 less infectious, and D388 least infectious (D388 maximum FFU/mL=110, 154-fold less infectious than D17). Similarly, of chimeric C110 HCVcc, D0 was most infectious, D178 less infectious, and D436 least infectious (D436 maximum FFU/mL=5,420, 3-fold less infectious than D0). Overall, these results suggest that HEPC3 and HEPC74/43 resistance substitutions in E2 conferred a progressive loss of E1E2 function, while E1E2 in the subject with persistence of infection gained function over time.
HEPC3 and HEPC43/HEPC74 resistance substitutions reduced E2 binding affinity for CD81 and SR-B1.
To investigate the mechanism of the loss of E1E2 function associated with acquisition of bNAb resistance substitutions, we expressed longitudinal E2 strains from C117 and C110 as soluble E2 proteins and measured binding of these proteins to two principal HCV cell surface receptors, CD81 or scavenger receptor B1 (SR-B1), expressed on the surface of Chinese hamster ovary (CHO) cells (Fig 6). Longitudinal C117 sE2 clones displayed progressively lower binding to CD81, with the same ranking that they demonstrated in HCVpp and HCVcc fitness (Fig 6A). Maximal CD81 binding of D388 sE2 was only 17% of D17 sE2 binding. SR-B1 binding of D388 sE2 was similar to that of D17 sE2 (Fig 6B). We used site-directed mutagenesis to introduce only non-HVR1 E2 substitutions into D17 sE2. One mutant D17 clone (D17-SDM3) was generated expressing N434D, F465Y, and D610H, since these substitutions were present in most E1E2 strains by D285 of infection. A second mutant D17 clone (D17-SDM5) was generated expressing N434D, H444Y, F465Y, P498S, D533N, and D610H, since these substitutions were present in all HCV isolates circulating at D388 immediately prior to clearance of infection. As with the natural sE2 clones, these site-directed mutants displayed dramatically reduced binding to CD81. For example, maximal CD81 binding of D17-SDM5 was 13% of maximal binding of wild-type D17 sE2 (Fig 6C). Binding of D17 sE2 and the site-directed mutant sE2s to SR-B1 were similar (Fig 6D). Overall, these studies demonstrate that HEPC3 resistance substitutions reduced E1E2 function by reducing E2 binding affinity for CD81.
Figure 6. HEPC3 and HEPC74/43 resistance substitutions reduced E2 binding affinity for CD81 and SR-B1.

Binding of serial dilutions of soluble E2 (sE2) protein to CD81 or SR-B1 expressed on the surface of Chinese hamster ovary (CHO) cells, measured as median fluorescent intensity (MFI) of cells after incubation with concentration-normalized, His-tagged sE2 and a fluorescently labelled anti-His antibody. MFI values were normalized to binding of the chronologically earliest strain from each subject. (A-B) Longitudinal naturally occurring sE2 strains from C117. (C-D) C117 D17–1a53 sE2 or site-directed mutants D17-SDM3 (N434D, F465Y, D610H) or D17-SDM5 (N434D, H444Y, F465Y, P498S, D533N, D610H). (E-F) Longitudinal naturally occurring sE2 strains from C110. (G-H) C110 D0-A4 sE2 or site-directed mutant D0-SDM (L438I, F442I, N445H, R461L, A466D, A475T).
We performed similar experiments to measure CD81 and SR-B1 binding of longitudinal C110 sE2 clones from Infxn 1 and Infxn 2. CD81 binding of both Infxn 2 strains was reduced relative to binding of the D0 strain, again following the same ranking as in HCVpp entry and HCVcc fitness (Fig 6E). Notably, in contrast with C117 sE2 clones, SR-B1 binding of C110 Infxn 2 clones also was reduced dramatically relative to D0 sE2 (Fig 6F). Remarkably, introduction by SDM of only the six Infxn 2 E2 front layer and VR2 polymorphisms (L438I, F442I, N445H, R461L, A466D, A475T) into D0 sE2 also led to a dramatic drop in both CD81 and SR-B1 binding (Fig 6G-H). Notably, this loss of SR-B1 binding occurred even though no substitutions were introduced into HVR1, the best characterized SR-B1 binding domain. Overall, these studies demonstrate that the HEPC74/43 resistance polymorphisms in the front layer and VR2 of E2 reduced E1E2 function by reducing E2 binding affinity for both CD81 and SR-B1.
BNAb resistance/loss of fitness substitutions selected in C117 and C110 E2 overlap with HEPC3/74 binding epitopes and the CD81 binding site.
To investigate the link between bNAb resistance and loss of E1E2 function, we mapped C117 and C110 bNAb resistance/loss of fitness substitutions onto the E2ecto1b09 crystal structure, along with bNAb and CD81 long extracellular loop (CD81LEL) contact residues and/or critical binding residues (Fig 7). The accompanying study by Flyak et al. describes the X-ray crystal structures of HEPC3 and HEPC74 in complex with E2 protein ectodomains, demonstrating that E2 contact residues of HEPC3 and HEPC74 fall in the front layer (aa 424–459) and the CD81 binding loop (aa 519–535) of the E2 protein (Table S2).
Figure 7. BNAb resistance/loss of fitness substitutions selected in C117 and C110 E2 overlap with HEPC3 and HEPC74 binding epitopes, as well as the CD81 binding site.

Binding and resistance substitution positions are indicated with colored spheres superimposed on the HEPC3-E2ecto1b09 structure. E2 structure is gray. (A) Main chain atoms of critical binding residues and/or contact residues of HEPC3 (yellow spheres) and C117 resistance/loss of fitness substitutions (magenta spheres). (B) Main chain atoms of critical binding residues and/or contact residues of HEPC74 (orange spheres) and C110 resistance/loss of fitness substitutions (blue spheres). (C) Main chain atoms of critical binding residues of CD81LEL (cyan spheres) and resistance/loss of fitness substitutions of C117 (magenta) or C110 (blue). Resistance/loss of fitness substitutions are indicated that fall at contact or critical binding residues (**) or within two amino acids of a contact or critical binding residue (*).
Critical E2-binding residues for HEPC3, HEPC43, HEPC74, HEPC98, and CD81LEL, a required HCV cell surface receptor, were previously reported based upon relative binding of these bNAbs (Bailey et al., 2017) or CD81LEL (Gopal et al., 2017) to strain H77 E1E2 protein or a comprehensive panel of alanine scanning mutants spanning the full H77 E1E2 sequence. We re-analyzed these alanine scanning-binding data to more comprehensively define residues where mutation reduced binding of these bNAbs and CD81LEL (see Methods and Table S2).
Four of six HEPC3 resistance substitutions arising in C117 E2 fell at or within two amino acids of a HEPC3 contact residue and/or a HEPC3 critical binding residue (N434D, H444Y, D533N, and P498S), suggesting that each of these substitutions could disrupt the contact interface between HEPC3 and E2 (Fig 7A). Notably, the site of one resistance substitution, N434, forms one hydrogen bond with the HEPC3 heavy chain complementarity determining region 3 (CDRH3) and one hydrogen bond with heavy chain framework region 1 (FRH1). Similarly, as shown in Fig 7B, three of six HEPC74/43 resistance polymorphisms in C110 E2 fell at a HEPC74 contact and/or a critical binding residue (L438I, F442I, and N445H), making it likely that they would disrupt the contact interface between HEPC74 and E2. In order to evaluate the role of these same substitutions in the loss of E2 binding affinity for CD81, we also mapped C117 and C110 E2 substitutions relative to critical CD81LEL binding residues (Fig 7C). Five of twelve substitutions fell at or within two amino acids of a CD81LEL critical binding residue (L438I, F442I, H444Y, N445H, D533N). Notably, L438 is a critical binding residue for HEPC3, HEPC74, HEPC43, and CD81LEL, and F442 is a critical binding residue for both HEPC3 and CD81LEL. Taken together, this analysis suggests that, given the remarkable similarity between the HEPC3/74/43 binding epitopes and the binding site of CD81, as well as the critical role of CD81 in HCV entry, acquisition of resistance to these bNAbs is tightly linked to loss of viral replicative fitness.
T cell responses.
To assess the potential contribution of T cell responses to viral evolution and clearance of infection in these subjects, we used an ELISpot assay to measure T cell interferon gamma secretion in response to overlapping peptides spanning the entire HCV polyprotein, using previously described methods (Cox et al., 2005b) (Table S3). Surprisingly, C117 had no detectable T cell responses at any of the four time points tested, ranging from D46 to D285 of infection. For C110, sufficient numbers of cells were not available to perform this analysis using Infxn 2 time points, but responses to six different epitopes in NS3, NS5A, or NS5B proteins were detected using D1267 (Infxn 3) cells. None of the epitopes in the sequenced amplicon acquired substitutions over the course of Infxn 3. P29, the control subject with persistent infection, had detectable T cell responses at each of three time points tested, D58, D114, and D135 of infection. These responses targeted one E2 epitope and two NS3 epitopes. Only the E2 epitope mutated over the course of infection. Taken together, these results demonstrate that C117 cleared HCV despite an undetectable HCV-specific T cell response, while P29 infection persisted despite detectable responses against multiple epitopes. For C110, we could not determine whether T cell responses were present during Infxn 2, but a relatively broad T cell response during Infxn3 likely contributed to clearance of this infection.
DISCUSSION
Here, we demonstrate that bNAbs targeting the front layer of E2 mediated clearance of human HCV infection. These bNAbs neutralized most autologous HCV strains, and when bNAb resistance developed, it was accompanied by progressive loss of E2 protein function that was associated temporally with clearance of HCV infection. We found that the loss of E1E2 function in both bNAb donors could be attributed to a loss of E2 binding affinity for CD81, and that the link between bNAb resistance and loss of E1E2 fitness could be explained by the extraordinary degree of overlap between the bNAb epitopes and the CD81 receptor binding site of E2.
It is likely that T cells and bNAbs each contributed to HCV clearance in subjects C110 and C117, given extensive literature documenting an association between robust T cell responses and clearance (Rehermann, 2009; Schulze zur Wiesch et al., 2012), and evidence for T cell responses against multiple epitopes in subject C110. Clearance of infection by subject C117 was particularly remarkable given the lack of detectable HCV-specific T cells in peripheral blood, although responses may have been detected if assays were performed using autologous peptides or intrahepatic T cells.
These findings contrast notably with similar studies of another clinically important human RNA virus, HIV-1. Potent and broadly-neutralizing antibodies can develop relatively late in HIV-1 infection, and some studies have demonstrated fitness loss of autologous viruses resulting from acquisition of resistance to these bNAbs (Lynch et al., 2015; Reh et al., 2018). However, adequate replication capacity of these escaped viruses is maintained, since all HIV-1 bNAbs identified to date were isolated from individuals who failed to control viremia (Bunnik et al., 2008; Euler et al., 2010). In the case of the potent HIV-1-specific bNAb VRC01, infection persisted despite bNAb escape-associated viral fitness loss because the virus rapidly acquired compensatory fitness substitutions in the env gene (Lynch et al., 2015). Even if full suppression of replication were achieved, it is unlikely that bNAbs could clear the latent reservoir of integrated HIV-1 proviruses in resting CD4+ T cells (Sengupta and Siliciano, 2018). In contrast, there is no latent integrated form of HCV, and we show here that HCV-specific bNAbs arose early in infection, selecting viruses with profound deficits in replicative fitness, without acquisition of compensatory fitness substitutions, leading to natural clearance of infection.
We described in a prior study (Bailey et al., 2017) and in Flyak et al. the structural and functional similarities among HEPC3, HEPC74, and a bNAb designated AR3C, which was isolated by Law et al. from a person with chronic HCV infection, along with closely related mAbs AR3A, AR3B and AR3D (Law et al., 2008). Notably, infusion of AR3A or AR3B partially protected human liver chimeric mice from HCV challenge, and infusion of AR3A, AR3B and a third bNAb abrogated established HCV infection in a mouse model (de Jong et al., 2014). Together, these results support our findings here that bNAbs in this class can be protective in vivo.
It is interesting to note that, unlike C117 and C110, the donor of the AR3 bNAbs did not spontaneously clear HCV infection. There are multiple possible explanations for this observation. First, structural differences between HEPC3, HEPC74 and AR3C described in Flyak et al. may have important functional implications. Second, unlike for C117 and C110, it has not been demonstrated that bNAbs were expressed at significant titers in the serum of the AR3 donor. Third, the timing of development of the AR3 bNAbs is unclear. In contrast, HEPC3, HEPC74, and HEPC43 likely developed very early in acute infection. Timing of bNAb development may be crucial, if it limits opportunities for acquisition of resistance and compensatory fitness substitutions and maximizes opportunities for synergy with the CD8+ T cell response, which tends to wane during the transition from acute to chronic infection (Cox et al., 2005a; Rehermann, 2009). The development of neutralizing antibodies in subject C117 only seventeen days after infection was somewhat surprising since seroconversion to HCV infection requires a median of thirty-six days (Cox et al., 2005c). Although multiple antibody tests prior to infection were negative, indicating that C117 was not previously infected, it is intriguing to consider that past low-level exposure to HCV through repeated injection drug use might have primed this individual to mount a very rapid antibody response. Finally, NAbs targeting other epitopes may also have contributed to HEPC3/74/43-mediated clearance. In the case of C117, we found that HEPC98 and HEPC3 exerted simultaneous selective pressure on different E2 epitopes early in infection. It would be of particular interest to determine whether mAbs specific for the E1E2 heterodimer were also expressed by these subjects, given recent studies suggesting that these mAbs can be synergistic with mAbs targeting the CD81 binding site of E2 (Carlsen et al., 2014; de Jong et al., 2014). Each of these hypotheses merits further investigation.
Despite dramatic improvements in HCV treatment, an effective prophylactic vaccine is also needed to eradicate the HCV pandemic. Vaccine development efforts to date have been hindered by the extraordinary genetic diversity of the virus. Extensive E2 diversity, strong evidence for a role of CD8+ T cells in control of HCV, and a relative lack of similar direct evidence for bNAb-mediated control, has tempered enthusiasm somewhat for development of a bNAb-inducing vaccine. Some recent vaccine trials, including the only vaccine trial in at-risk humans, have focused exclusively on induction of T cell responses (Clinicaltrials.gov NCT01436357, (Swadling et al., 2014)). Data here suggest that induction of bNAbs also may be critical for vaccine protection. These data also suggest that pan-neutralizing breadth may not be necessary for protection if bNAb-resistant strains are replication-deficient. This study provides strong in vivo evidence that bNAbs directed at the CD81 binding residues of E2 may result in infection clearance through a combination of neutralization and pressure that selects replication-deficient viruses with impaired CD81 binding. This study highlights the value of selection of vaccine antigens that induce bNAbs targeting the CD81 binding site as an important strategy in prophylactic HCV vaccine design.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further requests for information, resources and reagents should be directed to and will be fulfilled by Justin Bailey (jbailey7@jhmi.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects.
Plasma and PBMCs were obtained from the BBAASH cohort (Cox et al., 2005c). In this cohort, the incidence, immunology, and virology of HCV infection in young injection drug users is prospectively monitored. Ages at seroconversion of subjects C110, C117 and P29 were 28, 31, and 25, respectively. All subjects were HIV-negative, and none were treated with interferon or antiviral drugs over the course of sampling. The protocol was approved by the Institutional Review Board of the Johns Hopkins Hospital, and written informed consent was obtained from all study participants.
Cell Lines.
HEK293T/17 cells (sex: female) were obtained from ATCC (cat# CRL-11268) and maintained in Dulbecco’s Modified Eagle Medium and supplemented with sodium pyruvate, 10% heat inactivated fetal bovine serum, and glutamine. HEP3B cells (sex: male), were obtained from the ATCC (cat # HB-8064), and maintained in Modified Eagle Medium, supplemented with sodium pyruvate, 10% heat inactivated fetal bovine serum, nonessential amino acids and glutamine. Huh7.5.1 cells (sex: male) were obtained from Charles Rice (The Rockefeller University, New York City, New York, USA), and maintained in Dulbecco’s Modified Eagle Medium and supplemented with sodium pyruvate, 10% heat inactivated fetal bovine serum, and nonessential amino acids. Cells were cultured at 37°C in a humidified incubator with 5% CO2, and monolayers were disrupted at 80% to 100% confluence with Trypsin-EDTA.
Virus Strains.
All HCV strains used in this study were amplified by RT-PCR from plasma of HCV-infected individuals. Neutralizing breadth against heterologous strains was measured using a panel of HCVpp generated using genotype 1a and 1b E1E2 clones isolated from unrelated individuals. All viral strains amplified and analyzed for this study from subjects C117, C110, and P29 were genotype 1a. For each subject, E1E2 genes were cloned and expressed from transmitted/founder strains and strains isolated longitudinally over the course of infection.
METHODS DETAILS
Source of reference bNAbs.
CBH-2 (Keck et al., 2012) and HC33.1(Keck et al., 2013) were gifts from Steven Foung (Stanford University School of Medicine, Stanford, California, USA). HEPC3, HEPC43, HEPC74, and HEPC98 were isolated by AIF and JEC (Bailey et al., 2017).
HCV viral load and serology testing.
HCV viral loads (IU/mL) were quantified after RNA extraction with the use of commercial real-time reagents (Abbot HCV Real-time Assay) migrated onto a research-based real-time PCR platform (Roche 480 LightCycler). HCV seropositivity was determined using the Ortho HCV version 3.0 ELISA Test System (Ortho Clinical Diagnostics).
Identification of bNAb and CD81LEL critical binding residues.
Comprehensive alanine scanning mutagenesis of an expression construct for HCV E1E2 (genotype 1a, strain H77) changed each residue to alanine (with alanine residues changed to serine) to create a library of clones, each representing an individual point mutant, covering 552 of 555 target E1E2 residues. Binding of each mAb and CD81LEL to wild type E1E2 and to each of these alanine mutants was quantitated as previously described (Bailey et al., 2017; Gopal et al., 2017). Binding residues for HEPC3, HEPC43, HEPC74, HEPC98 (Bailey et al., 2017), and CD81LEL (Gopal et al., 2017) based on this binding data were previously reported, but revised for this study using new analysis of alanine scanning-binding results. Substitutions inducing global misfolding of E1E2 were identified and excluded as binding residues using all alanine scanning mutagenesis mapping data from Integral Molecular, Philadelphia PA published in Gopal et al. (Gopal et al., 2017), and Bailey et al. (Bailey et al., 2017) as follows: residues at which alanine substitution reduced E1E2 binding of all conformational E2-binding and E1E2-binding mAbs to <75% of binding to wild type protein were excluded as potential binding residues. For each mAb, after exclusion of these global misfolding substitutions, E1E2 residues were ranked from greatest to least effect of alanine substitution on mAb binding. Up to 20 substitutions with greatest effect on binding that also reduced binding by at least 50% were selected as critical binding residues. The maximum number of likely binding residues was chosen based on studies suggesting that the average epitope includes 15+/−4 contact residues (Kringelum et al., 2013).
HCVpp production, infectivity, and neutralization assays.
HCVpp were produced by lipofectamine-mediated transfection of HCV E1E2 and pNL4–3.Luc.R-E-plasmids into HEK293T cells as previously described (Hsu et al., 2003; Logvinoff et al., 2004). For infectivity testing, HCVpp were serially diluted 10-fold and incubated on Hep3B target cells for 5 to 6 hours before media was removed. Values from HCVpp dilutions within the linear range of the luminometer were compared. The panel of 19 heterologous genotype 1 HCVpp has been described previously (Bailey et al., 2015; Osburn et al., 2014). Neutralization assays were performed as described previously (Dowd et al., 2009). For breadth testing, HCVpp were incubated for 1 hour with heat-inactivated plasma at a 1:100 dilution or mAb at 10 μg/mL and then added in duplicate to Hep3B target cells for 5 to 6 hours before medium was changed. Infection was determined after 3 days by measurement of luciferase activity of cell lysates in RLU. All HCVpp used in neutralization assays produced RLU values at least 10-fold above background entry by mock pseudoparticles. To assess neutralization of autologous variants, mAbs were serially diluted five-fold, starting at a concentration at 100 ug/mL. To assess neutralization of autologous variants by plasma, heat-inactivated plasma was serially diluted 2.5fold, starting at a 1:50 dilution.
Generation of HCVcc chimeras.
HCVcc chimeras were generated as previously described (Wasilewski et al., 2016a; Wasilewski et al., 2016b). Briefly, after digestion of the HCVcc backbone with AfeI (New England Biolabs), c110 and c117 E12 genes were inserted in frame using In-Fusion cloning (Clontech). Plasmid DNA was linearized using XbaI (New England Biolabs) then used for in vitro RNA transcription using the T7 MEGAscript kit (Ambion). RNA clean-up was performed using RNeasy mini kit (Qiagen), quantified using a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). 10 μg of RNA was transfected into 1.8 × 106 Huh7.5.1 cells (a gift of Charles Rice, The Rockefeller University, New York City, New York, USA) using Nucleofector Kit T (Amaxa) and plated in a 6-cm plate. Medium was changed at 24 hours and supernatants were collected 48 hours later. To control for transfection efficiency, FFU values were adjusted for relative input copy numbers of HCVcc.
HCVcc infectivity assays.
Human hepatoma Huh7.5.1 cells (a gift from Charles Rice, The Rockefeller University, New York City, New York, USA) were maintained in DMEM supplemented with 10% fetal bovine serum and non-essential amino acids. 8,000 Huh7.5.1 cells per well were plated in flat-bottom 96-well tissue culture plates and incubated overnight at 37°C. The following day, HCVcc were normalized for viral load then serially diluted 2-fold and added to Huh7.5.1 cells in duplicate and incubated overnight, after which the HCVcc were removed and replaced with 100 μL of medium and incubated for 24 hours at 37°C. After 24 hours, medium was removed, and cells were fixed and stained with primary anti-NS5A antibody 9E10 (a gift from Charles Rice, The Rockefeller University, New York City, New York, USA) at a 1:2,000 dilution for 1 hour at room temperature, and with secondary antibody Dylight 488– conjugated goat anti-mouse IgG (Abcam) at a 1:500 dilution for 1 hour at room temperature. Images were acquired and spot-forming units (SFU) were counted using an AID iSpot Reader Spectrum operating AID ELISpot Reader version 7.0.
Single HCV genome amplification.
HCV hemigenomes from plasma virus were amplified by RT-PCR after limiting dilution to ensure single-genome amplification, using previously described methods (Li et al., 2012). PCR products were gel extracted and directly Sanger sequenced. E1E2 was PCR amplified from single-genome amplification amplicons of interest and cloned as previously described (Osburn et al., 2014). Sequences of all E1E2 clones were confirmed after cloning. All original sequence data were deposited in GenBank.
Site directed mutagenesis.
Substitutions were introduced into E1E2 and sE2 plasmids using the QuikChange Lightning Multi Site-Directed Mutagenesis Kit (Agilent) and confirmed by Sanger sequencing.
Phylogenetic analysis.
Nucleotide sequences were trimmed and aligned using MUSCLE, with the alignment manually adjusted in BioEdit. Phylogenetic trees were inferred from nucleotide sequences using the maximum likelihood method based on the Tamura 3-parameter model (Tamura and Kumar, 2002), gamma distributed. The tree with highest log likelihood is shown with branches drawn to scale. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor-joining and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood approach and then selecting the topology with superior log likelihood value. 500 bootstrap tests were performed. Analyses were implemented in the Mega7 program (http://www.megasoftware.net). T/F genomes were inferred as previously described (Li et al., 2012; Li et al., 2016). Highlighter plots were generated using aligned E1E2 amino acid sequences and the Highlighter tool at the Los Alamos HIV database (http://www.lanl.gov/).
HCV E1E2 ELISA.
MAb binding to E1E2 was quantitated using an ELISA as previously described (Keck et al., 2009). Briefly, 293T cells were transfected with E1E2 expression constructs. Cell lysates were harvested at 48 hours. Plates were coated with 500 ng Galanthus nivalis lectin (EY Labs) and blocked with PBS containing 0.5% Tween-20, 1% nonfat dry milk, and 1% goat serum, and E1E2–containing cell lysates were added. MAbs were assayed in duplicate at 5-fold serial dilutions, starting at 100 μg/mL, and binding was detected with HRPconjugated anti-human IgG secondary antibody (Vector Laboratories PI-3000). Equivalent protein concentrations in each E1E2 lysate preparation were confirmed using serial dilutions of control mAb, HC33.4 (Fig S7).
Expression of soluble E2.
The E2 ectodomain of s110 and s117 HCV strains were cloned into a mammalian expression vector (phCMV3-Ig Kappa-HIS, a gift of Leopold Kong, The Scripps Research Institute, La Jolla, California, USA). This truncated, soluble form of E2 ectodomain (sE2) retains antigenicity and function as previously described (Kong et al., 2013), encompassing residues 384–645. The vector allows expression of E2 protein with a C-terminal His tag as well as an N-terminal murine Ig Kappa leader signal for efficient protein secretion. Each E2 construct was co-transfected with pAdvantage (Promega) into HEK293T cells and incubated for 72 hours at 37°C. Supernatant was collected at 48 and 72 hours, passed through a 0.2 μm filter, and concentrated using a regenerated cellulose centrifugal filter with a 10 kDa cutoff (Amicon). Quantitation of relative sE2 protein concentration (Fig S7). Serial 5-fold dilutions of each sE2 supernatant were immobilized onto ELISA wells pre-coated with 500 ng Galanthus nivalis lectin (Ey Labs) and blocked with PBS containing 0.5% Tween 20, 1% nonfat dry milk, and 1% goat serum. Wells were probed with 0.5 μg of a mouse monoclonal anti-6× His-tag antibody (Thermo Fisher Scientific) and quantified using an HRP-conjugated goat anti-mouse IgG secondary antibody (Abcam). The EC50 value for each sE2 construct was calculated by nonlinear regression analysis and fold differences in EC50 values used to normalize sE2 concentration in subsequent experiments.
sE2 binding to CHO cells.
CHO-CD81 and CHO-SR-B1 binding experiments were carried out as previously described (Sabo et al., 2011). CHO cells expressing recombinant human CD81 or SR-B1 (a gift from Dr. Matthew Evans, Icahn School of Medicine, Mount Sinai, New York) were detached using PBS supplemented with 4mM EDTA and 10% FBS and washed in PBS containing 1% BSA. Cells (2 × 105) were pelleted in a 96-well u-bottom plate and resuspended in 2-fold serial dilutions of each normalized sE2 construct. Following a 30-minute incubation on ice, the cells were washed twice and incubated with 0.5 μg of AF647-conjugated mouse anti-6× His-tag antibody (Thermo Fisher Scientific) for another 20 minutes on ice. After a final wash, the cells were fixed with 1% paraformaldehyde and analyzed on a LSRII flow cytometer (Becton-Dickinson).
Interferon-γ ELISpot assay.
HCV-specific CD8+ T-cell responses were quantified by interferon-γ ELISpot assay as previously described (Cox et al., 2005a). Briefly, PBMC were screened for recognition of HCV-specific antigens using overlapping peptides and previously described optimal HCV epitopes. Pooled cytomegalovirus, Epstein-Barr virus, and influenza antigens and phytohemagglutinin were used as positive controls. Responses were considered positive if the number of spots per well minus the background was at least 25 spot forming cells (SFC) per million PBMC.
Experimental Design.
Each neutralization and ELISA assay was performed in duplicate with values reported as means +/− SD. Experiments shown in Figures 3B-D, 4B, 4J, and 5F-G were repeated in duplicate experiments (See Fig S5). Entry of HCVpp was tested in three independent experiments, with all data shown in Fig 5A-D. Infectivity of HCVcc was tested in two independent experiments (Fig 5F-G, Fig S6). 10,000 events were recorded for each flow cytometry measurement (Fig 6).
QUANTIFICATION AND STATISTICAL ANALYSIS.
Neutralization profiles of plasma samples were compared pairwise to neutralization profiles of HEPC3, HEPC98, HEPC74, HEPC43, and CBH-2 mAbs (Fig 1) using Spearman correlation in Prism v7.0 (Graphpad). Correlations with p-values <0.05 were deemed statistically significant. Neutralization curves for HCVpp and ELISA binding curves were fit by nonlinear regression in Prism v7.0 (Graphpad). Flow cytometry data were analyzed using FlowJo v10 software (Tree Star). Mean fluorence intensity (MFI) values were calculated from 10,000 events.
Supplementary Material
Highlights.
Broadly neutralizing antibodies (bNAbs) were isolated from donors who cleared HCV
Most autologous HCV strains were sensitive to these bNAbs
HCV strains that evolved resistance to these bNAbs lost replicative fitness
bNAbs targeting the E2 front layer contributed directly to HCV clearance
ACKNOWLEDGEMENTS
The authors would like to thank Michelle Colbert and Alexander Gooden for technical support. This research was supported by the National Institutes of Health grants R01AI127469 (to J.R.B. and P.J.B.), U19 AI088791 (to J.R.B, G.M.S., and A.L.C), and NIH contract HHSN272201400058C (to B.J.D.) A.I.F. is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute. Content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
DATA AND SOFTWARE AVAILABILITY.
Viral sequences generated for this study are accessible from Genbank (accession numbers: MH834650-MH835192). All other data that support the findings of this study are available from the Lead Contact upon request.
DECLARATION OF INTERESTS
A.I.F., J.E.C., G.M.S., and J.R.B. are inventors of patents submitted pertaining to some of the antibodies and antigens presented in this paper. J.E.C. has served as a consultant for Takeda Vaccines, Sanofi Pasteur, Pfizer, and Novavax, is on the Scientific Advisory Boards of CompuVax, GigaGen, Meissa Vaccines, PaxVax, and is Founder of IDBiologics, Inc. The other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bailey JR, Flyak AI, Cohen VJ, Li H, Wasilewski LN, Snider AE, Wang S, Learn GH, Kose N, Loerinc L, et al. (2017). Broadly neutralizing antibodies with few somatic mutations and hepatitis C virus clearance. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JR, Wasilewski LN, Snider AE, El-Diwany R, Osburn WO, Keck Z, Foung SK, and Ray SC (2015). Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. The Journal of clinical investigation 125, 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnik EM, Pisas L, van Nuenen AC, and Schuitemaker H (2008). Autologous neutralizing humoral immunity and evolution of the viral envelope in the course of subtype B human immunodeficiency virus type 1 infection. J. Virol 82, 7932–7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke KP, Munshaw S, Osburn WO, Levine J, Liu L, Sidney J, Sette A, Ray SC, and Cox AL (2012). Immunogenicity and cross-reactivity of a representative ancestral sequence in HCV infection. J.Immunol 188, 5177–5188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsen TH, Pedersen J, Prentoe JC, Giang E, Keck ZY, Mikkelsen LS, Law M, Foung SK, and Bukh J (2014). Breadth of neutralization and synergy of clinically relevant human monoclonal antibodies against HCV genotypes 1a, 1b, 2a, 2b, 2c, and 3a. Hepatology 60, 1551–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KM, Thimme R, Melpolder JJ, Oldach D, Pemberton J, Moorhead-Loudis J, McHutchison JG, Alter HJ, and Chisari FV (2001). Differential CD4(+) and CD8(+) T-cell responsiveness in hepatitis C virus infection. Hepatology 33, 267–276. [DOI] [PubMed] [Google Scholar]
- Cox AL, Mosbruger T, Lauer GM, Pardoll D, Thomas DL, and Ray SC (2005a). Comprehensive analyses of CD8+ T cell responses during longitudinal study of acute human hepatitis C. Hepatology 42, 104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AL, Mosbruger T, Mao Q, Liu Z, Wang XH, Yang HC, Sidney J, Sette A, Pardoll D, Thomas DL, et al. (2005b). Cellular immune selection with hepatitis C virus persistence in humans. J Exp.Med 201, 1741–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AL, Netski DM, Mosbruger T, Sherman SG, Strathdee S, Ompad D, Vlahov D, Chien D, Shyamala V, Ray SC, et al. (2005c). Prospective evaluation of community-acquired acute-phase hepatitis C virus infection. Clin.Infect.Dis 40, 951–958. [DOI] [PubMed] [Google Scholar]
- de Jong YP, Dorner M, Mommersteeg MC, Xiao JW, Balazs AB, Robbins JB, Winer BY, Gerges S, Vega K, Labitt RN, et al. (2014). Broadly neutralizing antibodies abrogate established hepatitis C virus infection. Science translational medicine 6, 254ra129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd KA, Netski DM, Wang XH, Cox AL, and Ray SC (2009). Selection Pressure from Neutralizing Antibodies Drives Sequence Evolution during Acute Infection with Hepatitis C Virus. Gastroenterology 136, 2377–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler Z, van Gils MJ, Bunnik EM, Phung P, Schweighardt B, Wrin T, and Schuitemaker H (2010). Cross-reactive neutralizing humoral immunity does not protect from HIV type 1 disease progression. J. Infect. Dis 201, 1045–1053. [DOI] [PubMed] [Google Scholar]
- Georgiev IS, Doria-Rose NA, Zhou T, Kwon YD, Staupe RP, Moquin S, Chuang GY, Louder MK, Schmidt SD, Altae-Tran HR, et al. (2013). Delineating antibody recognition in polyclonal sera from patterns of HIV-1 isolate neutralization. Science 340, 751–756. [DOI] [PubMed] [Google Scholar]
- Gopal R, Jackson K, Tzarum N, Kong L, Ettenger A, Guest J, Pfaff JM, Barnes T, Honda A, Giang E, et al. (2017). Probing the antigenicity of hepatitis C virus envelope glycoprotein complex by high-throughput mutagenesis. PLoS Pathog. 13, e1006735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, and McKeating JA (2003). Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc.Natl.Acad.Sci.U.S.A 100, 7271–7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck Z, Wang W, Wang Y, Lau P, Carlsen TH, Prentoe J, Xia J, Patel AH, Bukh J, and Foung SK (2013). Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J.Virol 87, 37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck ZY, Li SH, Xia J, von Hahn T, Balfe P, McKeating JA, Witteveldt J, Patel AH, Alter H, Rice CM, et al. (2009). Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J.Virol 83, 6149–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY, Patel AH, Lemon SM, et al. (2012). Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS pathogens 8, e1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Giang E, Nieusma T, Kadam RU, Cogburn KE, Hua Y, Dai X, Stanfield RL, Burton DR, Ward AB, et al. (2013). Hepatitis C virus E2 envelope glycoprotein core structure. Science 342, 10901094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kringelum JV, Nielsen M, Padkjaer SB, and Lund O (2013). Structural analysis of B-cell epitopes in antibody:protein complexes. Mol. Immunol 53, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, et al. (2008). Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat.Med 14, 25–27. [DOI] [PubMed] [Google Scholar]
- Li H, Stoddard MB, Wang S, Blair LM, Giorgi EE, Parrish EH, Learn GH, Hraber P, Goepfert PA, Saag MS, et al. (2012). Elucidation of hepatitis C virus transmission and early diversification by single genome sequencing. PLoS pathogens 8, e1002880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Stoddard MB, Wang S, Giorgi EE, Blair LM, Learn GH, Hahn BH, Alter HJ, Busch MP, Fierer DS, et al. (2016). Single-genome sequencing of hepatitis C virus in donor-recipient pairs distinguishes modes and models of virus transmission and early diversification. J. Virol 90, 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, Feinstone SM, Alter H, Rice CM, and McKeating JA (2004). Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc.Natl.Acad.Sci.U.S.A 101, 10149–10154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch RM, Wong P, Tran L, O’Dell S, Nason MC, Li Y, Wu X, and Mascola JR (2015). HIV-1 fitness cost associated with escape from the VRC01 class of CD4 binding site neutralizing antibodies. J. Virol 89, 4201–4213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micallef JM, Kaldor JM, and Dore GJ (2006). Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. Journal of viral hepatitis 13, 34–41. [DOI] [PubMed] [Google Scholar]
- Osburn WO, Fisher BE, Dowd KA, Urban G, Liu L, Ray SC, Thomas DL, and Cox AL (2010). Spontaneous Control of Primary Hepatitis C Virus Infection and Immunity Against Persistent Reinfection. Gastroenterology 138, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osburn WO, Snider AE, Wells BL, Latanich R, Bailey JR, Thomas DL, Cox AL, and Ray SC (2014). Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology 59, 2140–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka JM, Zeisel MB, Blaser E, Schurmann P, Bartosch B, Cosset FL, Patel AH, Meisel H, Baumert J, Viazov S, et al. (2007). Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc.Natl.Acad.Sci.U.S.A 104, 6025–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reh L, Magnus C, Kadelka C, Kuhnert D, Uhr T, Weber J, Morris L, Moore PL, and Trkola A (2018). Phenotypic deficits in the HIV-1 envelope are associated with the maturation of a V2-directed broadly neutralizing antibody lineage. PLoS Pathog. 14, e1006825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehermann B (2009). Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence. The Journal of clinical investigation 119, 1745–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo MC, Luca VC, Prentoe J, Hopcraft SE, Blight KJ, Yi M, Lemon SM, Ball JK, Bukh J, Evans MJ, et al. (2011). Neutralizing monoclonal antibodies against hepatitis C virus E2 protein bind discontinuous epitopes and inhibit infection at a postattachment step. J.Virol 85, 7005–7019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze zur Wiesch J, Ciuffreda D, Lewis-Ximenez L, Kasprowicz V, Nolan BE, Streeck H, Aneja J, Reyor LL, Allen TM, Lohse AW, et al. (2012). Broadly directed virus-specific CD4+ T cell responses are primed during acute hepatitis C infection, but rapidly disappear from human blood with viral persistence. J.Exp.Med 209, 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, and Siliciano RF (2018). Targeting the Latent Reservoir for HIV-1. Immunity 48, 872–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suryaprasad AG, White JZ, Xu F, Eichler BA, Hamilton J, Patel A, Hamdounia SB, Church DR, Barton K, Fisher C, et al. (2014). Emerging epidemic of hepatitis C virus infections among young nonurban persons who inject drugs in the United States, 2006–2012. Clin Infect Dis 59, 1411–1419. [DOI] [PubMed] [Google Scholar]
- Swadling L, Capone S, Antrobus RD, Brown A, Richardson R, Newell EW, Halliday J, Kelly C, Bowen D, Fergusson J, et al. (2014). A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci. Transl. Med 6, 261ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, and Kumar S (2002). Evolutionary distance estimation under heterogeneous substitution pattern among lineages. Mol. Biol. Evol 19, 1727–1736. [DOI] [PubMed] [Google Scholar]
- Wasilewski L, Ray S, and Bailey JR (2016a). Hepatitis C virus resistance to broadly neutralizing antibodies measured using replication competent virus and pseudoparticles. J Gen Virol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasilewski LN, El-Diwany R, Munshaw S, Snider AE, Brady JK, Osburn WO, Ray SC, and Bailey JR (2016b). A Hepatitis C Virus Envelope Polymorphism Confers Resistance to Neutralization by Polyclonal Sera and Broadly Neutralizing Monoclonal Antibodies. J Virol 90, 3773–3782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2017). Global hepatitis report, 2017. (World Health Organization; ). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.