

Abstract
Cerebral white matter injury (WMI) contributes to cognitive dysfunction associated with pathological aging. Because reactive astrocyte-related factors contribute to remyelination failure after WMI, we sought accurate, cost-effective, and reproducible histopathological approaches for quantification of morphometric features of reactive astrogliosis in aged human white matter in patients with vascular brain injury (VBI). We compared 7 distinct approaches to quantify the features of glial fibrillary acidic protein (GFAP)-labeled astrocytes in the prefrontal white matter of brains from patients with VBI (n = 17, mean age 88.8 years) and controls that did not exhibit VBI (n = 11, mean age 86.6 years). Only modern stereological techniques (ie, optical fractionator and spaceballs) and virtual process thickness measurements demonstrated significant changes in astrocyte number, process length, or proximal process thickness in cases with VBI relative to controls. The widely employed methods of neuropathological scoring, antibody capture assay (histelide), area fraction fractionator, and Cavalieri point counting failed to detect significant differences in GFAP expression between the groups. Unbiased stereological approaches and virtual thickness measurements provided the only sensitive and accurate means to quantify astrocyte reactivity as a surrogate marker of WMI in human brains with VBI.
Keywords: Dementia, Glial fibrillary acidic protein, Reactive astrogliosis, Stereology, Vascular brain injury, White matter injury.
INTRODUCTION
Cerebral white matter injury (WMI) contributes to cognitive dysfunction associated with both normative and pathological aging ( 1–3 ). In advanced normative aging, the burden of WMI is often greater than cumulative Alzheimer disease (AD) pathology and typically precedes the onset of mild cognitive impairment or dementia ( 4 ). In pathological aging, early AD is often comorbid with WMI, which suggests common pathogenic mechanisms that may overlap and involve disturbances to the neurovascular unit ( 5–8 ). However, mechanistic evidence linking vascular disease to WMI and AD is lacking and our understanding of the contribution of structural versus nonstructural dysfunction in AD and vascular dementia remains incomplete ( 9 ). These data underscore the need for a comprehensive, systematic characterization of the pathological hallmarks of age-related WMI to define its pathogenesis and contribution to cognitive impairment.
The strong associations between WMI and chronic hypertension as well as diabetic microvascular changes support a vascular etiology for age-related WMI ( 3 ). Moreover, biomarkers of free radical injury to axons and myelin are significantly elevated in WMI, independent of the burden of AD pathology ( 1 ). Neuroimaging studies have demonstrated that progressive WMI commonly localizes to the periventricular white matter as well as the frontal pathways that subserve executive functions ( 10 , 11 ). The prevalence of MRI-defined white matter hyperintensities has approached 90% in several serial antemortem studies ( 12–14 ). In postmortem high-field MRI studies, reduced fractional anisotropy was significantly associated with coincident AD and vascular pathology ( 1 , 15 ).
Despite the high prevalence and apparent impact of age-related WMI on cognitive decline in both normative and pathological aging, efforts to define the pathogenesis of WMI have been hindered by the absence of systematic quantitative measures that describe both the magnitude and distribution of human white matter pathology. Commonly employed histological indicators of WMI (eg, routine hematoxylin and eosin stains) or stains for myelin (eg, Luxol fast blue histochemistry or myelin basic protein immunohistochemistry) are subject to observer bias and are not amenable to rigorous quantitative analysis. In particular, these stains do not permit investigators to quantify changes in specific cell populations or changes to fine cellular structure, which would help to define mechanistic endpoints.
In the present study, we sought to identify a robust, cost-effective, and reproducible histopathological approach to quantify accurately the magnitude of chronic vascular WMI. We evaluated reactive astrogliosis because it is an early and persistent pathological sequela of cerebral WMI and manifests as astrocyte proliferation and/or extensive ramification of existing astrocyte processes ( 16–19 ). In response to WMI, reactive astrocytes contribute to extensive remodeling of the extracellular matrix with pathological accumulation of proteoglycans and hyaluronic acid (HA) that disrupt white matter regeneration and repair ( 20 ). Reactive astrocytes produce excess hyaluronan in the prefrontal cortex of aged nonhuman primates. In several conditions, hyaluronan levels are significantly elevated in diffuse white matter lesions in association with pronounced expansion of the total pool of oligodendrocyte lineage cells ( 1 , 20–22 ).
Here, we employed several quantitative approaches to address the limitations of prior studies that used nonquantitative analyses to assess the contribution of WMI to age-related cognitive decline. We applied 3 quantification methods (area fraction fractionator, Cavalieri point counting, and histelide) to measure general threshold values of the glial fibrillary acidic protein (GFAP) signal in tissue sections. We employed 2 modern, 3-dimensional, design-based stereological quantification methods to obtain accurate and unbiased estimates of astrocyte number and total process length. Lastly, we applied systematic, random sampling and a virtual measuring tool to estimate average process caliber. We determined the optimal approaches for sensitive detection of significant differences in the burden of reactive astrogliosis.
MATERIALS AND METHODS
Subjects
Written informed consent was obtained from all individual participants included in this study. All procedures involving human subjects were approved by the University of Washington Institutional Review Board in accordance with the Helsinki Declaration of the World Medical Association. We utilized 28 human white matter samples at the level of the olfactory bulb that were collected in the University of Washington Neuropathology Core at autopsy from participants in the Adult Changes in Thought (ACT) study, an ongoing population-based study of aging and cognitive decline among men and women in Seattle, WA. Demographic information and clinical and pathological diagnoses are indicated in Table 1 . Inclusion criteria consisted of a postmortem interval of less than 8 hours, Braak neurofibrillary tangle distribution stage of III or less, absence of Lewy body pathology, and absence of gross hemorrhages. Cases that exhibited cerebral microinfarcts (CMIs) within the cerebral cortex and/or deep cerebral nuclei (neostriatum and thalamus) were stratified into the vascular brain injury (“VBI”) group. Cases that lacked cerebral cortical or deep CMIs were placed in the “No VBI” group ( Fig. 1 ). Collection of the prefrontal white matter (WM) samples relied on landmarks that included the rostral and caudal tips of the olfactory bulbs, as previously described ( 1 ). Frontal white matter was collected in a coronal slice anterior to the lateral ventricle at the level of the olfactory bulb in order to sample prefrontal white matter. The sampling protocol included white matter deep to Brodmann areas 8 and 9 (lateral components) and 32 (medial). Medial frontal cortex that contained the orbitofrontal cortical gray matter was retained for purposes of orientation for other studies not reported here. This prefrontal white matter block was immersion fixed in buffered 4% paraformaldehyde, pH 7.4, for 48 hours and transferred to phosphate-buffered saline (PBS) containing 0.01% sodium azide for storage at 2 °C–4 °C.
TABLE 1.
Patient Demographics Stratified by Presence or Absence of Vascular Brain Injury
| No vascular brain injury | |
|---|---|
| No. | 11 |
| Age, y | 86.6 ± 8 |
| F:M | 4:7 |
| PMI, h | 4.54 ± 1.7 |
| Brain weight, g | 1,251 ± 98 |
| Vent., cm | 1.01 ± 0.95 |
| Fr. CAA | 0 |
| Fr. Plq. | 1 |
| Vascular brain injury | |
| No. | 17 |
| Age, yr | 88.8 ± 7 |
| F:M | 7:10 |
| PMI, h | 4.45 ± 1.6 |
| Brain weight, g | 1,233 ± 130 |
| Vent., cm | 1.25 ± 1.08 |
| Fr. CAA | 0 |
| Fr. Plq. | 0 |
No vascular brain injury is defined as no infarcts (territorial, lacunar, or cerebral microinfarcts [CMIs]). Vascular brain injury is defined as any CMIs. No territorial or lacunar infarcts were observed in the VBI cohort.
No., number of subjects; Age, average age at death (mean ± SD); F, female; M, male; PMI, postmortem interval (mean ± SD); Vent, left ventricular diameter at temporal tips (mean ± SD); Fr. CAA, frontal cerebral amyloid angiopathy density score (median); Fr. Plq., frontal amyloid plaque density score (median); h, hours; yr, years.
FIGURE 1.
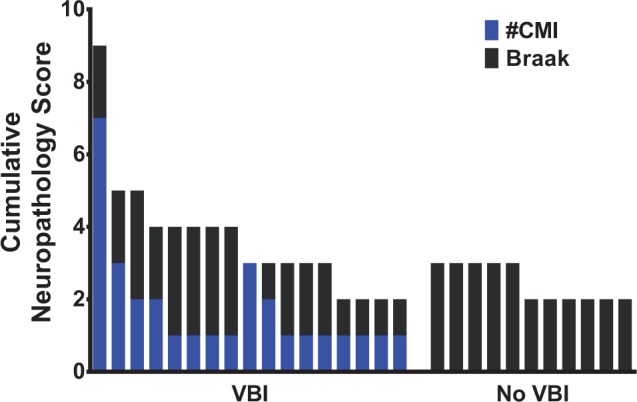
The burden of Alzheimer disease (AD) and vascular brain injury (VBI) pathology for each of the 28 cases. Schematic representation of a cumulative neuropathology score, as previously reported by us ( 1 ), provides a comparison of the mixed VBI and AD pathology in the patients studied. The ordinal scores are depicted in a cumulative fashion to illustrate the relative burdens of cerebral microinfarcts (CMIs) and neurofibrillary tangles, as defined by Braak staging for each case. Because CMI number and Braak stages are potentially related forms of pathology, the cumulative score should not be interpreted as an additive score of total pathology. All VBI cases had at least 1 CMI (range: 1–7) and Braak stages ≤3 (range: 0–3). CMIs are absent from cases in the No VBI group, which had Braak stages ≤3 (range: 2–3) and were similar to those of the VBI group.
Pathological Evaluation
Neuropathological examinations of autopsied brains were performed as previously described ( 1 ) and identified the overall VBI burden based on the number of CMIs. Evaluations of VBI and AD neuropathologic changes, including assessment of neuritic plaques (CERAD score) and neurofibrillary tangles (Braak stage), were performed according to recently revised consensus guidelines ( 23 ).
Immunohistochemistry
For each tissue block, coronal sections (50 µm) were cut free-floating in PBS using a Leica VTS-1000 vibrating microtome (Leica Microsystems Inc., Bannockburn, IL). We employed a binning approach for tissue collection wherein 5 consecutive sections per well were collected in multiwell culture plates. Sections underwent antigen retrieval for 10 minutes at 95 °C in EDTA-tris buffer, pH 9.0, with 0.5% Tween-20, and were rinsed in PBS. They were incubated for 10 minutes in 3% hydrogen peroxide and rinsed in PBS. Each section was blocked in 5% normal goat serum with 1.25% Triton x-100 for 1 hour. Sections were then incubated in monoclonal mouse anti-human GFAP primary antibody (1:500; Merck Millipore, Bedford, MA) for 48 hours at 2 °C–4 °C and then exposed to biotinylated goat anti-mouse IgG secondary antibody (1:200; Vector Laboratories, Burlingame, CA) for 3 hours at room temperature. Thereafter, sections were incubated in avidin-biotin peroxidase complex (ABC Vectastain, Vector Laboratories) for 4 hours, and immunolabeling was visualized using a nickel enhanced 3,3′-diaminobenzidine (DAB) peroxidase substrate solution (SK-4100; Vector Laboratories).
Tissue blocks used in ordinal rating of gliosis were collected from prefrontal white matter in fixed brains after coronal sectioning, adjacent to those used in all other analyses, and were processed for paraffin wax embedding and sectioning using standard protocols on a Leica automated processor (Leica Biosystems, Buffalo Grove, IL). Sections were cut using a Leica microtome at 5-μm thickness and mounted on charged glass slides. GFAP immunostains were performed using a Leica Bond Autostainer with the Bond Polymer Refine Detection system and polyclonal rabbit anti-GFAP (Dako, Glostrup, Denmark) at 1:1,000 dilution according to protocols recommended by the manufacturer. Briefly, the process included deparaffinization followed by antigen retrieval in citrate buffer (ER1 Solution, Leica Biosystems), endogenous peroxidase quench with 3%–4% hydrogen peroxide, incubation with primary antibody (15 minutes), followed by Polymer Refine Detection using goat antirabbit-Poly-HRP-IgG in buffer with 10% normal goat serum, and DAB Refine complex with appropriate washes between each step. Counterstain was achieved using hematoxylin staining prior to dehydration and coverslipping. All reagents were purchased from Leica Biosystems unless otherwise noted.
Histopathological Analysis of Injury by Ordinal Rating
Paraffin-embedded tissues sections (5 μm) from prefrontal white matter were immunostained for GFAP. We employed an ordinal rating scale to estimate the burden of reactive astrogliosis in the white matter using a protocol modified from Riddle et al ., wherein astrogliosis was scored by a blinded neuropathologist (CDK) as follows: 0 (none); 1 (focal, < 25% of tissue involved); 2 (patchy, 25%–75% of tissue involved); and 3 (diffuse ≥75% of tissue involved) ( Fig. 2D ) ( 24 ).
FIGURE 2.

Neuropathologic assessment of white matter astrogliosis after vascular brain injury (VBI). Neuropathologic scoring of gliosis defined by glial fibrillary acidic protein (GFAP) immunohistochemistry was analyzed in formalin-fixed, paraffin-embedded white matter samples. (A) A No VBI case with minimal white matter astrogliosis. (B) A No VBI case with diffuse white matter astrogliosis. (C) A VBI case with diffuse white matter astrogliosis. (D) Comparison of ordinal gliosis index scores for VBI and No VBI cases demonstrates that this approach did not identify significant differences between the 2 groups (median score: 3 for both groups). (E) GFAP immunolabeling in white matter after histelide preparation in a No VBI case with minimal white matter astrogliosis. (F) A No VBI case with diffuse white matter astrogliosis. (G) A VBI case with diffuse white matter astrogliosis. (H) Graph of the mean absorbance between No VBI and VBI groups determined by histelide. There was no significant difference between the calculated means for the 2 groups (p = 0.79) by Student t -test. Box and whiskers plot shows minimum, 25th percentile, median, 75th percentile, and maximum. Scale bars = 100 µm.
Histelide Quantification
We employed histelide as previously described ( 25 ). We evaluated blocks from prefrontal white matter in 27 of the 28 cases collected for this study. Tissue blocks were embedded in paraffin and cut at 5 μm on a microtome. Antigen retrieval was accomplished by boiling the sections in 0.1 M citrate buffer for 20 minutes. Tissue sections were rinsed 3 times in a tris-buffered saline tween solution (TBST), and blocked in 5% normal goat serum with 2% BSA in TBST followed by rabbit anti-GFAP (Dako; 1:1,000) or the rabbit IgG isotype control antibody (Abcam, Cambridge, UK; 1:1,000) for 2 hours. The slides were rinsed 3 times in blocking solution and incubated with an alkaline phosphatase-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA; 1:400). Slides were washed 3 times in TBST and once in DEA (diethanolamine) solution. Each slide was separately immersed in 5 ml of p-nitrophenyl phosphate (PNPP) solution (Sigma-Aldrich, St. Louis, MO) and incubated for at least 2 hours. The PNPP solution was sampled at 30, 60, and 90 minutes to determine absorbance values. Sampling continued at 30-minute intervals until 2 consecutive absorbance values for the slide exceeded 0.5 units. Afterwards, the tissue was transferred to a TBST solution and rinsed twice with a NTM (sodium/magnesium/tris) solution. Slides were placed in NBT (nitroblue tetrazolium)/BCIP (5-bromo-4-chloro-3-indolyl phosphate) solution (Sigma-Aldrich) for 30 minutes at 4 °C, rinsed in PBS, dried at room temperature, and coverslipped with Clear Mount mounting medium (Electron Microscopy Sciences, Hatfield, PA). After drying overnight, the tissue section on each slide was traced under a Nikon 90i microscope (Nikon Instruments, Tokyo, Japan) equipped with Stereo Investigator Software (MBF Biosciences, Williston VT), and the tissue area was calculated. Due to variation in section size, the total PNPP signal was normalized to the area of the tissue section. We calculated the final value (SI GFAP FINAL , Absorbance/cm 2 ) by subtracting the normalized signal (SI ISOTYPE , Absorbance/cm 2 ) produced by the isotype control section from the normalized signal obtained from the GFAP-stained slide (SI GFAP , Absorbance/cm 2 ) ( Fig. 2H ).
Area Fraction Fractionator Quantification
This analysis provided an estimate of the volume fraction of white matter occupied by GFAP-positive astrocytes. To implement the area fraction fractionator, we analyzed a single GFAP immunostained section selected at random. With Stereo Investigator software, a closed contour was drawn around the white matter using a 2.5× N Plan achromatic objective (Leica Microsystems, Wetzlar, Germany). We used a 63×/NA = 1.4 Plan apochromatic oil immersion objective (Leica Microsystems) to perform a systematic random sample of sites within the closed white matter contour. We selected an 80 × 80 µm 2 counting frame that was contained within the field of view. The program superimposed a 5 × 5 µm 2 grid over the field of view. At each predetermined site, a specific marker was placed over GFAP-labeled somata and a separate marker was placed over the surrounding white matter ( Fig. 3A ). This counting procedure was repeated until all sites had been quantified.
FIGURE 3.

Immunohistochemical visualization of glial fibrillary acidic protein (GFAP) labeling in prefrontal white matter samples with and without vascular brain injury (VBI). (A) Representative GFAP labeling of astrocytes in a No VBI case and a VBI case (B) , both taken at low power, demonstrate the homogenous distribution of GFAP-positive cells within the underlying white matter. Dashed line demarcates the cerebral gray/white matter borders. (C) A higher power image of GFAP labeling in the white matter in the same No VBI case as in (A) . Note the presence of heavy GFAP labeling of the ramified processes of many of the astrocytes. Overall, the labeled processes are indicative of normal astrocyte morphology. (D) GFAP labeling of astrocytes in the same VBI case as shown in (B) . Morphology of the cells is representative of astrocytes, but a slight increase in proximal process thickness and ramification of more distal processes indicates that these likely are reactive. The subtle visual differences between these panels underscore the challenge of differentiating between moderately reactive and nonreactive astroglial phenotypes when evaluated by low-power, 2-dimensional fields or through similar conventional histopathologic methods. Scale bar: A = 500 µm and also applies to B; C = 50 µm and also applies to D.
Cavalieri Point Counting Quantification
This technique also provided an estimate of the white matter volume occupied by GFAP-positive somata. The Cavalieri point counting method was described previously ( 21 ). Briefly, we used the same GFAP-immunostained single section as for the area fraction fractionator. Labeled astrocytes were viewed under brightfield illumination with a 20× N Plan achromatic objective (Leica Microsystems). The fractionator probe in Stereo Investigator was used to collect images from a systematic, random sample of sites within the white matter. At each predetermined site, a 400 × 305 µm 2 digital image was captured using a high-acquisition Hamamatsu Orca-ER monochrome camera (Hamamatsu Photonics, Hamamatsu, Japan) (≥30 images per case). Each image was opened in Fiji, an expanded module of ImageJ (NIH, Bethesda, MD) and a 12.5 × 12.5 µm 2 grid was superimposed onto the image. Grid vertices that landed over GFAP-labeled somata where counted with a specific marker ( Fig. 3C ). This counting procedure was repeated for each systematic random site throughout the white matter. The GFAP area fraction within the white matter was calculated as the ratio of total markers on GFAP-labeled somata to total grid vertices falling on white matter.
Stereological Quantification of Numerical Cell Densities (N v ) and Process Length Densities (Lv)
Design-based stereological estimation of the difference in N v and L v between VBI and No VBI cases was accomplished with the optical fractionator (for N v ) and spaceballs (for L v ) probes, respectively. Applications of the optical fractionator and spaceballs to estimate N v and L v have been described ( 26–30 ). Specifically, the optical fractionator is a local 3D stereological probe designed to estimate the population of discreet objects, in this study GFAP-immunoreactive somata. Spaceballs is also a local 3D probe but estimates length, in this study the length of GFAP-immunoreactive processes.
All stereological analyses were accomplished on an upright Leica DMRA microscope (Leica Microsystems), which was coupled to a high-precision ProScan III motorized stage (Prior Scientific Instruments, Cambridge, UK) and equipped with a high-acquisition Hamamatsu Orca ER cooled monochrome CCD camera. These components were integrated with Stereo Investigator. As described below, we employed several sampling parameters that included a defined region of interest (ROI), thickness of the tissue section, construction of a counting brick, systematic random sampling of the ROI, and rigorous counting procedures, as previously shown ( 26 , 31 ). All data were analyzed with GraphPad Prism version 6.0 (GraphPad Software, La Jolla, CA). To compare group means, unpaired 2-tailed Student t -tests were performed. Correlation analyses were performed using the Pearson Product Coefficient.
Delineation of ROI
In immunohistochemically reacted sections, the gray-white matter boundary was macroscopically discernable ( Fig. 3A, B ). The slide was visualized under a 2.5× N Plan objective and viewed in Stereo Investigator using the Hamamatsu Orca ER monochrome CCD camera. Using the contour tool, a closed line was drawn that followed both the cut edges of the white matter and the gray-white matter border. This contour was defined as the white matter ROI.
Measuring Section Thickness
Because of shrinkage in the Z plane during tissue processing, we made an empirical estimate of the tissue thickness. To determine specific parameters necessary for estimating N v and L v accurately, we computed the mean thickness of the white matter ROI in each tissue section by measuring the thickness of at least 7 separate white matter sites. We used a 63×/NA = 1.4 Plan apochromatic oil immersion objective along with a Heidenhain MT 1271 microcator (Johannes Heidenhain GmbH, Traunreut, Germany), coupled to the microscope stage and integrated with Stereo Investigator software. We focused on the top surface of the section and set the microcator to zero. Focusing through the tissue until the lowest focal plane fell out of complete focus yielded a thickness measurement in micrometers. The value from the microcator at this bottom plane was recorded and used to compute the mean thickness for each case ( Tables 2 and 3 ).
TABLE 2.
Stereological Parameters and Results for Calculation of Numerical Cell Densities for Glial Fibrillary Acidic Protein-Positive Astrocytes
| Case | Number of sections | Counting frame w/h (µm) | X,Y grid placement (µm) | Number of sites | Guard zone (µm) | Disector height (µm) | Disector volume (mm³) | Mean thickness (µm) | Number of counts (∑Q̄) | Estimated N v | CE (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 07-042 | 3 | 80 | 2357.50, 1894.10 | 133 | 2 | 16 | 1.024E-04 | 23.03 | 111 | 8,150 | 11 |
| 07-079 | 3 | 80 | 2017.70, 1206.60 | 144 | 2 | 16 | 1.024E-04 | 21.47 | 268 | 18,175 | 7 |
| 07-089 | 3 | 80 | 2262.00, 2116.20 | 158 | 2 | 16 | 1.024E-04 | 21.77 | 278 | 17,183 | 7 |
| 07-123 | 3 | 80 | 2437.70, 1347.90 | 149 | 2 | 16 | 1.024E-04 | 21.35 | 176 | 11,535 | 8 |
| 08-002 | 3 | 80 | 2207.10, 1742.60 | 146 | 2 | 16 | 1.024E-04 | 21.30 | 141 | 9,431 | 9 |
| 08-013 | 3 | 80 | 1613.55, 1928.51 | 176 | 2 | 16 | 1.024E-04 | 23.86 | 232 | 12,873 | 7 |
| 08-017 | 3 | 80 | 2417.86, 2548.32 | 173 | 2 | 16 | 1.024E-04 | 20.84 | 192 | 10,838 | 8 |
| 08-053 | 3 | 80 | 2370.20, 1320.00 | 164 | 2 | 16 | 1.024E-04 | 20.87 | 221 | 13,160 | 7 |
| 08-077 | 3 | 80 | 2134.80 1683.30 | 152 | 2 | 16 | 1.024E-04 | 20.49 | 230 | 14,777 | 7 |
| 08-111 | 3 | 80 | 1970.00, 1191.10 | 165 | 2 | 16 | 1.024E-04 | 20.92 | 201 | 11,896 | 8 |
| 08-123 | 3 | 80 | 1598.30, 1127.50 | 148 | 2 | 16 | 1.024E-04 | 21.50 | 305 | 20,125 | 6 |
| 08-134 | 3 | 80 | 1809.60, 1473.20 | 144 | 2 | 16 | 1.024E-04 | 21.87 | 261 | 17,700 | 7 |
| 10-016 | 3 | 80 | 1615.80, 1750.90 | 127 | 2 | 16 | 1.024E-04 | 20.19 | 163 | 12,534 | 9 |
| 10-035 | 3 | 80 | 1923.40, 1246.90 | 126 | 2 | 16 | 1.024E-04 | 22.03 | 136 | 10,541 | 9 |
| 10-071 | 3 | 80 | 1738.00, 1634.20 | 137 | 2 | 16 | 1.024E-04 | 21.62 | 146 | 10,407 | 9 |
| 10-085 | 3 | 80 | 1622.60, 1160.60 | 144 | 2 | 14 | 8.960E-05 | 18.46 | 251 | 19,454 | 7 |
| 10-117 | 3 | 80 | 1770.60, 1404.80 | 139 | 2 | 16 | 1.024E-04 | 20.32 | 196 | 13,770 | 8 |
| 11-034 | 3 | 80 | 1506.21, 1027.20 | 158 | 2 | 16 | 1.024E-04 | 21.53 | 326 | 20,149 | 6 |
| 11-070 | 3 | 80 | 1207.10, 773.50 | 165 | 2 | 14 | 8.960E-05 | 21.50 | 185 | 12,514 | 8 |
| 11-098 | 3 | 80 | 2573.00, 1306.00 | 146 | 2 | 16 | 1.024E-04 | 19.22 | 189 | 12,642 | 10 |
| 11-101 | 3 | 80 | 2582.40, 1692.30 | 158 | 2 | 16 | 1.024E-04 | 26.41 | 232 | 14,339 | 7 |
| 11-104 | 3 | 80 | 1639.20, 1470.20 | 149 | 2 | 16 | 1.024E-04 | 20.61 | 168 | 11,011 | 8 |
| 12-072 | 3 | 80 | 1936.20, 1080.40 | 154 | 2 | 16 | 1.024E-04 | 20.92 | 208 | 13,190 | 8 |
| 12-078 | 3 | 80 | 2273.40, 1357.30 | 168 | 2 | 16 | 1.024E-04 | 20.44 | 215 | 12,498 | 8 |
| 12-120 | 3 | 80 | 2720.80, 1992.90 | 117 | 2 | 16 | 1.024E-04 | 19.98 | 135 | 11,268 | 9 |
| 12-130 | 3 | 80 | 1916.70, 1254.20 | 163 | 2 | 14 | 8.960E-05 | 19.02 | 216 | 14,790 | 7 |
| 13-002 | 3 | 80 | 1336.40, 1361.40 | 152 | 2 | 14 | 8.960E-05 | 18.25 | 179 | 13,143 | 8 |
| 13-013 | 3 | 80 | 1874.00, 1222.40 | 141 | 2 | 14 | 8.960E-05 | 19.18 | 225 | 17,810 | 8 |
CE, coefficient of error.
TABLE 3.
Stereological Parameters and Results for Calculation of Length Densities for Glial Fibrillary Acidic Protein-Positive Astrocyte Processes
| Case | Number of sections | Sphere radius (µm) | X,Y grid placement (µm) | Number of sites | Guard zone (µm) | Surface area of sphere (mm 2 ) | Mean thickness (µm) | Number of intersections (∑Q̄) | Estimated L v | CE (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 07-042 | 3 | 8 | 4196.0, 3358.0 | 44 | 2 | 8.00E-04 | 23.03 | 165 | 9,429 | 8 |
| 07-079 | 3 | 8 | 2815.0,2270.0 | 52 | 2 | 8.00E-04 | 21.47 | 211 | 10,293 | 8 |
| 07-089 | 3 | 8 | 4272.0,4196.0 | 40 | 2 | 8.00E-04 | 21.77 | 171 | 10,688 | 9 |
| 07-123 | 3 | 8 | 3987.0,2256.0 | 54 | 2 | 8.00E-04 | 21.35 | 259 | 12,047 | 8 |
| 08-002 | 3 | 8 | 3428.0,3795.0 | 40 | 2 | 8.00E-04 | 21.30 | 288 | 18,000 | 7 |
| 08-013 | 3 | 8 | 2125.2,1829.5 | 165 | 2 | 8.00E-04 | 23.86 | 479 | 7,258 | 6 |
| 08-017 | 3 | 8 | 3027.8,3191.2 | 108 | 2 | 8.00E-04 | 20.84 | 570 | 13,256 | 6 |
| 08-053 | 3 | 8 | 4877.0,2272.0 | 46 | 2 | 8.00E-04 | 20.87 | 315 | 17,500 | 6 |
| 08-077 | 3 | 8 | 3648.0,2842.0 | 56 | 2 | 8.00E-04 | 20.49 | 318 | 14,455 | 7 |
| 08-111 | 3 | 8 | 3472.0,2085.0 | 55 | 2 | 8.00E-04 | 20.92 | 429 | 19,500 | 6 |
| 08-123 | 3 | 8 | 2792.0,1992.0 | 38 | 2 | 8.00E-04 | 21.50 | 237 | 15,800 | 7 |
| 08-134 | 3 | 8 | 3107.0,2527.0 | 47 | 2 | 8.00E-04 | 21.87 | 313 | 16,919 | 6 |
| 10-016 | 3 | 8 | 2804.0,3017.0 | 45 | 2 | 8.00E-04 | 20.19 | 375 | 20,833 | 6 |
| 10-035 | 3 | 8 | 3040.0,2337.0 | 46 | 2 | 8.00E-04 | 22.03 | 150 | 8,333 | 9 |
| 10-071 | 3 | 8 | 3088.0,2686.0 | 42 | 2 | 8.00E-04 | 21.62 | 131 | 7,939 | 9 |
| 10-085 | 3 | 7 | 1929.0,2430.0 | 56 | 2 | 6.00E-04 | 18.46 | 468 | 28,364 | 5 |
| 11-034 | 3 | 8 | 1494.1,2731.0 | 59 | 2 | 8.00E-04 | 21.53 | 424 | 18,043 | 6 |
| 11-070 | 3 | 7 | 2283.0,1449.0 | 46 | 2 | 6.00E-04 | 21.50 | 192 | 14,222 | 8 |
| 11-098 | 3 | 8 | 3238.0, 1638.0 | 86 | 2 | 8.00E-04 | 19.22 | 387 | 11,382 | 6 |
| 11-101 | 3 | 8 | 4458.0,2749.0 | 51 | 2 | 8.00E-04 | 26.41 | 327 | 16,350 | 7 |
| 11-104 | 3 | 8 | 2957.0,2649.0 | 40 | 2 | 8.00E-04 | 20.61 | 114 | 7,125 | 10 |
| 12-078 | 3 | 8 | 3404.0,1996.0 | 77 | 2 | 8.00E-04 | 20.44 | 430 | 14,098 | 6 |
CE, coefficient of error.
Construction of the Counting Brick and Virtual Sphere
Once the ROI and average thicknesses were calculated, we used these parameters to construct virtual 3D counting bricks and spheres for the optical fractionator and spaceballs, respectively. We designed these probes to meet several specifications. The width and height (X, Y) of each disector and the diameter of each sphere had to fit within the field of view of our 63× oil immersion lens. The top and bottom guard zones were established and excluded from the analysis to prevent over- or underestimation related to surface artifact. The virtual height of either the 3D counting brick or sphere was always less than the minimum measured tissue thickness such that these virtual constructs fit entirely within the z-depth of the tissue section.
The X-Y dimensions of the 3D counting brick are provided in Table 2 . For all cases that were quantified using the optical fractionator, the width and height of the counting brick were determined to be 80 × 80 µm 2 . The disector height was typically 16 µm with the exception of 5 cases that necessitated a 14-µm counting brick due to lower minimum section thickness ( Table 2 ). The top guard zone for all cases was 2 µm and the bottom guard zone was the remaining thickness below the counting frame. The counting brick was completed with Stereo Investigator software by setting inclusion and exclusion zones at the edges of the counting brick ( Fig. 4 ).
FIGURE 4.

Area fraction fractionator and Cavalieri point counting assessment of astrogliosis. (A) Image showing the superimposed grid vertices over a heavily ramified astrocyte during implementation of the area fraction fractionator probe. The green circles represent points that do not fall on the labeled astrocyte somata; blue circles are points that fell on the labeled astrocyte somata. (B) Graph of the group means calculated using ratios of points occurring on a labeled astrocyte to points occurring in the surrounding white matter generated by the area fraction fractionator. The calculated ratio reflects the fraction of the volume of tissue occupied by labeled astrocyte somata. There was no significant difference between the groups by Student t -test (p = 0.500). (C) Image of a stained area of white matter demonstrating the Cavalieri point counting method. The green circles cover the entire field of view at vertices of a superimposed grid. The blue marker identifies labeled grid vertices that fall atop astrocyte somata. (D) Graph of astrocyte soma area fraction in the white matter for VBI and No VBI groups calculated by Cavalieri point counting. There was no significant difference between the group means by Student t -test (p = 0.254). Box and whiskers plot shows minimum, 25th percentile, median, 75th percentile, and maximum. Scale bars for A and C = 10 µm.
Our process length estimate relied on the spaceballs probe. A primary requisite for stereological estimation of length is that the probe must permit isotropic orientation of the tissue block before sectioning ( 28 , 30 ). That is, every angle of the tissue must have the same probability of being analyzed. Since we sectioned exclusively along the anteroposterior axis, our tissue orientation was inherently anisotropic. To circumvent this anisotropy, we used virtual 3D spheres whose outer edges are inherently isotropic, thereby giving all angles of a process equal probability of being measured. The radii of the 3D spheres were 8 µm, with the exception of 2 cases that necessitated a 7 µm diameter due to a lower minimum tissue thickness as calculated for the optical fractionator ( Table 3 ). The top guard zones for every case were set at 2 µm.
Systematic Random Sampling of ROIs
After the counting brick and virtual sphere were defined, we determined the fraction of the total area of white matter to be quantified with the stereological probes. We employed a systematic random sample of the white matter area in which the Stereo Investigator software randomly placed the first counting brick or sphere and then spaced these constructs in a systematic fashion throughout the frontal white matter based upon the number of sites we chose to sample. For the optical fractionator, the number of sites sampled ranged from 117 to 176 ( Table 2 ). For the spaceballs probe, the number of sites sampled ranged from 40 to 165 ( Table 3 ).
Counting Procedures for Optical Fractionator and Spaceballs
Quantification of astrocyte number followed the counting rules of Gundersen ( 27 ). Astrocytes were counted by focusing through the top guard zone and into the counting brick using a 63×/NA = 1.4 Plan apochromatic oil immersion objective. Objects were counted if they fell within the counting brick or contacted the green inclusion lines, and objects were not counted if they fell outside the counting brick or contacted the red exclusion lines ( Fig. 4 ). To compute N v , the density of GFAP-positive cells in the sampled tissue volume, we used the formula where Q - is the number of GFAP-positive cells for all sampling sites within the 3 sections under analysis ( 31 ). The precision of the estimate was determined by calculating a coefficient of error (CE). The CE computes the total variance, which includes inter- and intrasectional variation of the overall sampling design. This was calculated as , where Vtot is the total variability and N represents the total number of sections analyzed ( 32 ).
Our estimate of astrocyte process length using the spaceballs probe was derived from the number of intersections between a labeled process and the circumference of a virtual isotropic sphere. The same sections were employed for the optical fractionator and the spaceballs probe; however, 6 of the 28 cases analyzed by optical fractionator exhibited incomplete labeling in the fine secondary astroglial processes and did not meet our criteria for quantification using spaceballs. Therefore, our final spaceballs cohort consisted of 22 cases. The top guard zone thickness was the same as for the optical fractionator. The diameter of the sphere ( Table 3 ) was the same as the thickness of the counting bricks ( Table 2 ). Focusing through the tissue with the 63× oil immersion objective, cross-sections of the sphere enlarge in diameter as a function of the depth along the Z-axis until the virtual equator is reached, then progressively decrease toward the bottom of the tissue. Each intersection between a process and the virtual sphere was marked. Once the entire extent of the sphere had been examined, the stage was moved to a new site and the procedure was repeated. Estimation of the process length using only the volume of each sphere (L v ) was computed with the formula , where Q i is the number of intersections counted, and A is the area of the sphere in cubic millimeter (mm 3 ) ( 28 ). Precision of the estimated total length was calculated as a CE according to the equation , where CE is the coefficient of error, Vtot is the variance across all sections, and s2 the variance due to noise ( 32 , 33 ).
Analysis of Astrocytic Proximal Process Caliber
To characterize the response of white matter astrocytes to VBI further, we quantified the caliber of astrocyte processes. We measured the thickness of proximal processes with a quick measure line tool in Stereo Investigator. We sampled astrocytes throughout the white matter of each section from each of the 22 cases already subjected to spaceballs. This approach followed the sampling procedure conducted for estimation of astrocyte number and process length described above. Astrocytes were only included if at least the cell body, proximal processes, and secondary processes were labeled. Using a 63×/NA = 1.4 Plan apochromatic oil immersion objective, we measured process thickness at its apposition with and parallel to the astrocyte soma.
RESULTS
Our previous studies with the ACT cohort demonstrated that AD and VBI are commonly associated ( 1 , 34 ). In order to study cases with a primary burden of vascular pathology and investigate the influence of VBI on white matter lesions independent of AD pathology, we excluded cases with neurofibrillary tangle burden consistent with Braak stages above III. The burden of VBI pathology was defined as the total number of CMIs. We did not select cases based on clinical diagnosis. The cumulative neuropathology scores for each of the 28 cases included in this study based on Braak stage and number of CMIs are plotted in Figure 1 . We identified 17 individuals with CMIs that fulfilled the criterion of VBI ( Table 1 ). Eleven individuals in the ACT cohort with Braak Stage of III or less lacked CMIs and lacunar infarcts, and were identified as pathological controls (No VBI). Both groups were similar in terms of age at autopsy, proportion of women, postmortem interval, and degree of amyloid pathology ( Table 1 ). We did not detect any significant differences in brain weight (p = 0.70; unpaired Student t -test), ventricular diameter (p = 0.56; unpaired Student t -test), frontal amyloid plaque density (p = 0.82; Mann-Whitney U test), or frontal cerebral amyloid angiopathy (p = 0.50; Mann-Whitney U test), between the 2 groups.
Neuropathologic Injury Assessment Is Insensitive to the Full Spectrum of White Matter Astrogliosis Associated With VBI
To define the spectrum of WMI in VBI versus No VBI cases, we adapted an ordinal rating scale to estimate the burden of astrogliosis based upon neuropathological review of GFAP-immunostained, paraffin-embedded tissue sections (24). Using this scale, we observed a broad spectrum of astrocyte reactivity across the cohort of No VBI cases. Some cases exhibited relatively mild GFAP immunoreactivity that corresponded to a low density of astrocytes with limited process ramification ( Fig. 2A ), whereas others exhibited a much higher density of astrocytes with more elaborate process ramification ( Fig. 2B ). This latter more reactive-appearing phenotype was common in the VBI group ( Fig. 2C ). Despite the greater range of astrocyte reactivity observed among cases without VBI, the median gliosis score was 3 for both groups ( Fig. 2D ). Hence, this subjective ordinal rating scale lacked sufficient sensitivity to discriminate between the VBI and No VBI cases.
Histelide Detection of White Matter Astrogliosis Does Not Discriminate Between VBI and No VBI Cases
We next evaluated a novel antibody capture assay (histelide), which we previously used to detect the accumulation of β-amyloid and paired helical filament-tau in paraffin-embedded tissue ( 25 ). We adapted this assay to estimate the absorbance signal detected for GFAP-labeled cellular elements in paraffin-embedded tissue sections ( Fig. 2E–H ) from 27 of the 28 cases evaluated with our ordinal rating scale (one of the oldest cases in the series was excluded due to insufficient tissue available for the assay). This method calculates an absorbance factor per square centimeter (cm 2 ) in immunohistochemically reacted tissue sections. We calculated a mean absorbance factor of 1.79 ± 0.12 SEM per cm 2 for control cases versus 1.80 ± 0.15 SEM for VBI cases ( Fig. 2H ), which was not significantly different by group means comparison (p = 0.99; unpaired, 2-tailed Student t -test).
GFAP Area Fraction Estimators Are Ineffective in Detecting White Matter Astrogliosis Associated With VBI
We next analyzed a series of methods that measured threshold levels of GFAP in immunohistochemically stained sections. Representative GFAP staining from a control ( Fig. 3A, C ) and an individual with VBI ( Fig. 3B, D ) illustrates that the distribution and overall appearance of astrocytes in the cerebral white matter were similar for both groups. Astrocyte reactivity appeared lower among cases without VBI ( Fig. 3C ) relative to the VBI group ( Fig. 3D ), whereas the extent of astrogliosis ranged from moderate to severe among VBI cases.
To quantify astrocyte reactivity in the 2 groups, we first tested 2 similar methods that estimate the white matter volume occupied by GFAP-positive somata, the area fraction fractionator ( Fig. 4A, B ), and the Cavalieri point counting method ( Fig. 4C, D ). Both methods provide a calculated ratio of labeled cells to unlabeled white matter. Immunohistochemical visualization of GFAP-labeled astrocytes demonstrated a homogenous distribution of staining within the cell. A dense, high-contrast precipitate was observed in astrocyte somata and processes surrounded by low background labeling ( Fig. 4A ). GFAP immunoreactivity in astrocyte processes exhibited a heavy amount of label located within larger, more proximal processes with a gradual decrease in intensity among distal processes. The amount of label within these fine distal processes was clearly detectable in many cases ( Fig. 4A ). However, 6 of the 28 cases exhibited incomplete labeling in the fine primary and secondary processes (data not shown). The area fraction fractionator yielded a calculated mean ratio of 0.015 ± 0.0013 SEM for the control cases and a mean ratio of 0.014 ± 0.0011 SEM for VBI cases ( Fig. 4B ). We found no significant group difference between these mean ratios (p = 0.50). From the Cavalieri point counting method, we obtained a similar mean ratio of 0.015 ± 0.0017 SEM for the control cases and 0.013 ± 0.0008 SEM for the VBI cases ( Fig. 4D ), which was also not significant (p = 0.25). Hence, 2 probes that estimate the white matter volume occupied by GFAP-positive somata yielded similar mean ratios and were not sufficiently sensitive to detect differences in astrocyte reactivity between the VBI and No VBI groups.
Design-Based Stereological Estimators of Astrocyte Soma Density and Process Length Density Reliably Quantify the Magnitude of VBI
As an alternative strategy, we implemented 2 design-based stereology probes to estimate 2 independent measures of astrocyte reactivity: the density of astrocyte cell bodies and the length density of astrocyte processes ( 16–19 ). We employed the optical fractionator probe to estimate changes in numerical cell density (for N v ; Fig. 5 ) and the spaceballs probe to estimate changes in process length density (for L v ; Fig. 6 ). Using the optical fractionator, we detected a significantly higher density of astrocytes (14,896 ± 794; SEM) in the VBI group relative to the controls (12,044 ± 765 SEM; p = 0.0214; unpaired, 2-tailed Student t -test) ( Fig. 7A ). With the spaceballs probe, we were able to detect GFAP labeling of fine distal astrocyte processes in 22 cases from the cohort. We note that 6 of the original 28 cases did not exhibit adequate labeling of fine distal processes and could not be included in our spaceballs analysis. Among the remaining 22 cases (13 VBI and 9 No VBI), we found a significant increase in the length density of astrocyte process for the VBI group (16,284 ± 1,451 SEM) relative to the control group (11,127 ± 1,137 SEM; p = 0.0175) ( Fig. 7B ). We saw greater variability in the process length density measurements from the VBI subgroup (range: 7,250 to 28,364 µm/mm 3 ; SD: 5,233 µm/mm 3 ) relative to the No VBI subgroup (range: 7,125 to 17,500 µm/mm 3 ; SD: 3,412 µm/mm 3 ).
FIGURE 5.

Measurement of astrocyte density using the optical fractionator. Four panels illustrating the implementation of the optical fractionator at a single site within the white matter. The counting brick is composed of a counting frame with a red exclusion line, a green inclusion line, and a fraction of the volume of the measured thickness of the tissue. (A) At the top of the section, visible astrocytes are not yet counted as they are outside the focal plane. (B) As the probe descends through the z dimension, several astrocytes come into fine focus and are counted. Arrows show that 2 cells can be counted: 1 is in the counting frame and 1 is touching the green inclusion line. The cell marked by the arrowhead is not counted because it contacts the red exclusion line. (C, D) The labeled cells are not in focus and are not counted. Repeating these procedures at each site within the white matter produces an unbiased estimate of the number of astrocytes within the counting bricks. Scale bar for A = 10 µm.
FIGURE 6.

Measurement of astrocyte process length using spaceballs. (A) Implementation of spaceballs requires analysis of only a fraction of the total measured thickness of the tissue section. At the top of the tissue thickness, a 2-µm guard zone is set to prevent erroneous counts of processes that may have been affected during sectioning. At this depth, no processes can be marked. (B) Descending through the tissue, a cross-section of the virtual sphere appears and expands (red circle). Markers (green crosses) are placed at all points where the circle is intersected by labeled processes. (C) Focusing down, all profiles of labeled processes are out of focus and are not counted. (D) Near the bottom of the tissue section, 1 process intersects the virtual sphere surface and is counted (green cross). Application of these rules to each site allows the investigator to calculate an unbiased estimate of process length. Scale bar = 10 µm.
FIGURE 7.

Quantification of astrogliosis using the optical fractionator, spaceballs, and process thickness measurement. Also shown is the comparison of groups for thickness of proximal processes, absorbance values, and the correlation between astrocyte number and process length. (A) Graph of the estimated mean astrocyte density for No vascular brain injury (VBI) and VBI groups collected using the optical fractionator. A Student t -test found a significant increase in astrocyte density for the VBI group when compared to the No VBI group (asterisk) (p = 0.0214). (B) Graph of the estimated mean process length density for No VBI and VBI groups using the spaceballs probe. A Student t -test found a significant increase in process length for the VBI groups when compared to the No VBI group (asterisk) (p = 0.0175). (C) Graph of the calculated means of the thickness of the proximal process between No VBI and VBI groups (p = 0.0001). (D) Analysis of the relationship between astrocyte soma density and astrocyte process density found a significant association between astrocyte N v and astrocyte L v (r = 0.43, p = 0.0477). (E) Analysis of the relationship between astrocyte process length and ordinal gliosis score found no relationship between these 2 variables (p = 0.46). (F) Analysis of the relationship between astrocyte soma density and ordinal gliosis score found no relationship between these 2 variables (p = 0.49). Box and whiskers plot shows minimum, 25th percentile, median, 75th percentile, and maximum.
Systematic, Random Assessment of Astrocyte Process Thickness Reliably Detects White Matter Astrogliosis Associated With VBI
To estimate the thickness of proximal astrocyte processes, we employed a sampling scheme similar to that applied by the optical fractionator and spaceballs probes. We analyzed at least 7 sites throughout the white matter in each of 3 sections from the same cases used for our spaceballs probe. We measured 405 proximal processes in the control group and 451 processes in the VBI group. There was a significantly greater mean thickness of proximal astrocyte processes in the VBI group (2.76 ± 0.05 SEM) versus the controls (2.23 ± 0.11 SEM; p = 0.0001, unpaired, 2-tailed Student t -test; n = 23) ( Fig. 7C ).
We examined the association between astrocyte density and process length and hypothesized that as the number of astrocytes increased, length density of astrocyte processes should also increase. We observed a modest but significant interaction between these 2 variables (r = 0.43, p = 0.048) ( Fig. 7D ). In contrast, we did not detect significant associations between our ordinal gliosis scores ( Fig. 2 ) and astrocyte density (p = 0.49) or process length density (p = 0.46) ( Fig. 7E, F ).
Hence, 2 modern, design-based stereological probes described a similar magnitude of astrocyte reactivity among a cohort of human autopsy cases diagnosed with VBI, and these 2 metrics trended together across the entire cohort of VBI and No VBI cases. A separate approach that relied on systematic random sampling to determine process thickness also showed significant changes consistent with reactive astrogliosis in frontal white matter from patients with VBI.
DISCUSSION
The cellular and molecular mechanisms related to cognitive decline in individuals with VBI remain poorly defined. The majority of chronic pathology in VBI localizes to cerebral white matter, suggesting that disturbances in one or more glial populations contribute to the clinical presentation and progression of cognitive impairment ( 8 ). We previously reported that VBI is associated with a significant increase in the total pool of oligodendrocytes, a relationship that is independent of the burden of AD pathology ( 1 ). Prior studies have described dissociation between white matter hyperintensities and cerebrospinal levels of amyloid-β, suggesting that the pathological processes related to AD and VBI are partially distinct ( 35 ), despite growing evidence that AD is frequently comorbid with VBI ( 4 , 8 , 36–38 ). Neuroimaging studies have shown that frontal WM tracts are particularly vulnerable to white matter hyperintensities presumed to be of vascular origin ( 10 , 11 ). In postmortem high-field MRI studies, reduced fractional anisotropy was significantly associated with coincident AD and vascular pathology in the frontal white matter ( 1 , 15 ), but the specific microstructural abnormalities underlying these changes have not been fully defined at the cellular and molecular level. To determine whether MR signal abnormalities are caused by reduced myelin integrity, altered glial populations, and/or aberrant cell morphology, rigorous and unbiased histological methods are needed to quantify accurately cellular disturbances related to VBI.
Although reactive astrogliosis is a widely recognized pathological feature of VBI, there is a paucity of research aimed at rigorous quantification of the spectrum of astrocyte reactivity in the white matter after vascular injury. While conventional methods of astrogliosis scoring are clinically useful in detecting the difference between mild gliosis and moderate or severe lesions in early and midlife, these approaches appear less useful to discriminate between moderate to severe astrocyte reactivity in the context of brain aging and cognitive decline. For example, astrocyte reactivity in the nonhuman primate brain increases with normative aging ( 39 ), which may partly account for the lack of sensitivity of our ordinal scales in discriminating between the relative burden of astrogliosis in VBI and No VBI cases. Our results indicate that there are relatively modest differences between the changes in astrogliosis associated with normative aging and VBI, which underscores the need for more rigorous quantitative approaches.
Here we demonstrate that unbiased, 3-dimensional stereological or morphometric analysis of astrocytes is required to detect alterations in GFAP expression and/or astrocyte proliferation, increases in process length, and hypertrophy of proximal processes across the spectrum of astrogliosis that we encountered. We further demonstrate that, when quantified in this fashion, reactive astrogliosis is a sensitive surrogate marker of VBI severity. Astrocytes regulate vascular function in the gray matter, but their role in the pathogenesis of vascular injury within the white matter is not well understood ( 40 ). Our study allowed us to identify several novel quantitative approaches that detected differences in several distinct measures of reactive astrogliosis. These new approaches may accelerate progress toward defining the role of white matter astrocytes in human WMI by allowing unbiased identification of cases that carry a primary diagnosis of VBI.
We demonstrated that several approaches for quantifying astrocyte reactivity lacked sensitivity to discriminate across the spectrum of WMI or to distinguish VBI cases from cases without VBI. We previously reported that the Cavalieri point counting method is sensitive to regional differences in the magnitude of preterm human cerebral WMI between controls and cases with WMI ( 21 ). However, in our study of WMI in older adults, the Cavalieri point counting method, the area fraction fractionator, and histelide lacked sufficient sensitivity to identify cases with VBI. This lack of sensitivity may be related to a more complex lesion environment, greater baseline astrocyte reactivity, or a narrower dynamic range of astrogliotic changes in the aged white matter between control and VBI cases. Moreover, we encountered reduced GFAP staining of distal astrocyte processes, which may have reduced the sensitivity of some of our methods that relied upon quantification of changes in these fine distal processes.
The unbiased stereological protocols we employed provided continuous histopathological outcome measures for astrogliosis derived from the entire white matter region sampled. By contrast, although conventional neuropathological review of paraffin-embedded tissue sections is widely employed by neuropathologists for diagnostic purposes, this approach lacked the sensitivity to distinguish control from VBI cases. In experimental models of preterm WMI, we found that pathology scores have markedly reduced sensitivity when WMI is in the mild to moderate severity range and more closely resemble controls ( 24 ). The fact that we detected group differences using optical fractionator, spaceballs, and process thickness measurements (but not with other methodologies) supports the utility of high-magnification and 3-dimensional stereological approaches to quantify the burden of astrogliosis across an unidentified cohort of cases with variable age-related vascular injury and low AD burden.
Our studies support the assertion that white matter astrocytes undergo significant changes in density, soma hypertrophy, process length, and proximal process thickness in response to vascular injury. Most previous studies of astrogliotic responses to brain injury have focused on cortical astrocytes and optic nerve injury in various animal models, with limited data from human white matter astrocytes ( 41–46 ). Several of these studies employed 3D reconstruction of labeled astroglia to examine changes in the morphology of astrocytes following injury ( 41 ). Such approaches found that the reactive phenotype of cortical astrocytes involves structural and cellular responses that include changes in GFAP expression, astrocyte proliferation, process hypertrophy, and increases in distal process length and elaboration. In addition, reactive astrocytes regulate the extent of injury, calcium homeostasis, neuromodulation, and neurotransmitter maintenance. These structural and cellular responses enable astrocytes to sequester the injury site, reestablish the blood–brain barrier, and minimize excitotoxicity ( 17 , 45 ). Additionally, these studies have demonstrated that the responses of astrocytes to injury encompass a broad continuum of cellular changes ( 47 ), such that mild insults may result in the upregulation of GFAP expression in a subset of astrocytes that do not express GFAP at appreciable levels under physiologic conditions ( 17 , 44 ). Astrocytes responding to moderate insults may upregulate GFAP and undergo hypertrophy, whereas severe injury appears to involve all of the foregoing alterations along with proliferation and an increase in process length, producing the classical features of the glial scar ( 44 ).
Pathogenesis of WMI
Our subjects were drawn from a cross-sectional population-based study in which latent VBI and AD are prevalent and commonly comorbid in older adults with or without dementia ( 1 ). In order to define the spectrum of astrogliosis associated with relatively “pure” VBI, we selected for control and VBI cases with a similar low burden of AD pathology, as defined by neurofibrillary tangles. Although amyloid pathology in this population is highly prevalent, AD pathology was defined by the neurofibrillary tangle burden, which is the strongest pathologic correlate of dementia in AD ( 48 ). We nevertheless recognize the potential influence of Aβ neurotoxicity on VBI via parenchymal or vascular deposition as an important area of study ( 49 ). Here, we specifically evaluated parenchymal (plaque) and vascular (cerebral amyloid angiopathy) amyloid burden using Aβ immunohistochemistry and found no significant differences between groups. Further study is needed using quantitative assessments, such as unbiased stereology and biochemistry, to more thoroughly evaluate the role of Aβ neurotoxicity in WMI, but we did not observe specific Aβ pathology in our white matter samples.
White matter astrogliosis is a prominent feature in both AD and VBI, but the relative contributions of each to the total burden of astrogliosis requires further study. The contributors to astrogliosis in VBI are likely related to multiple pathogenic processes that contribute to diffuse WMI in VBI. We found that oxidative damage contributed significantly to the magnitude of VBI independent of the burden of AD pathology. Nevertheless, a similar magnitude of neuro-axonal oxidative damage was associated with either low or high AD burden, as defined by measurement of F4-neuroprostanes. Thus, Wallerian degeneration of axons could contribute to astrogliosis via neuronal death due to AD and/or vascular gray matter injury or via focal vascular injury to axons in the white matter. The low density of CMIs observed in the VBI group suggests that cortical neuronal damage was not the primary direct source of white matter astrogliosis in this cohort, although it is estimated that each microinfarct identified in an autopsy examination represents approximately 500 unidentified microinfarcts ( 50 ). Thus, it is likely that neuropathological surveys of CMIs underestimate the actual burden of ischemic neuronal injury and, therefore, Wallerian degeneration.
Our VBI cohort also excluded other forms of macroscopic cerebrovascular injury such as cystic infarcts, lacunar infarcts, or larger territorial infarcts. The etiology of microinfarcts appears to be only partly related to primary microvascular disease. A recent report from an aged population enriched for patients with AD and subcortical ischemic vascular disease demonstrated that microinfarcts strongly associated with cerebral atherosclerosis ( 51 ). This supports the notion that atheroemboli could be a significant source of microinfarcts.
We have been pursuing the hypothesis that reactive astrogliosis is mechanistically linked to disturbances in white matter regeneration and repair in VBI or developmental forms of WMI ( 20 , 52 ). We have observed that HA, a key component of the extracellular matrix derived from reactive astrocytes, accumulates to significantly higher levels in VBI cases where the total pool of oligodendrocyte lineage cells is also increased ( 1 , 39 ). The observation that HA accumulates in areas of white matter damage, demyelination, and reactive astrogliosis, and that specific molecular size ranges of HA block oligodendrocyte progenitor maturation, suggests a role for HA in disrupting white matter repair after VBI ( 21 , 22 , 53 , 54 ). Thus, quantitative studies of the temporal and spatial features of astrocyte reactivity in VBI may provide further mechanistic insights into the role of HA in myelination failure associated with chronic VBI.
An emerging role for microglia in myelination failure in models of multiple sclerosis may also be relevant to VBI. During the chronic phase of WMI, M2 microglia exhibiting an anti-inflammatory phenotype are necessary to promote oligodendrocyte differentiation and myelination ( 55 ). Because microglia also respond to astrocyte-derived factors by altering their activation phenotype and redox status ( 56 ), complex glial interactions may contribute to remyelination failure in VBI.
Technical Considerations
Stereological methods intended to quantify cellular parameters have focused on population estimates derived from a known volume of tissue ( 27 , 29–31 ). Because the tissue samples available in this study represented only a small proportion of the total white matter from a region without rigorously defined histological boundaries, we applied an alternative approach. Rather than population estimates, we relied on numerical cell density and process length density calculated using the sum of disector volumes rather than the volume of the tissue itself. Given these limitations, we took measures to ensure that calculated changes were due to differences in the number of quantified objects rather than differences in the reference volume, although we acknowledge that this is a risk inherent to any density quantification. To address potential pitfalls, we sought to minimize differential shrinkage by standardizing tissue preparation methods and attempted to maintain similar disector volumes among cases and between groups, which reduced the possibility that disector volume changes contributed significantly to the change in densities that we observed. There were several cases that required smaller counting bricks than the majority of the cohort due to variation in section cut thickness, but this discrepancy resulted in only minor differences in disector height of no more than 2 µm ( Table 2 ).
We also employed a nonstereological, morphometric approach to estimate the thickness of astrocyte proximal processes. We recognized a potential selection bias for larger astrocytes, which may arise from the relative ease of visualizing more robustly labeled cells with more intense proximal process labeling and thicker processes. To help reduce this potential bias, we analyzed astrocytes that had already been observed and counted during execution of the optical fractionator probe. Importantly, the optical fractionator allows quantification of cells regardless of their size, shape, or orientation due to its systematic random sampling; thus, it provided a mixed sample proximal process thickness analysis irrespective of cell size.
One limitation of GFAP staining that we encountered was the suboptimal detection of label in astrocyte processes in a subset of 6 of our 28 cases. This typically manifested as reduced detection of label or visible truncation in a substantial proportion of primary and/or secondary processes that precluded reliable analysis by the spaceballs probe and proximal process thickness measurement. This phenomenon did not appear to be related to postmortem interval or the extent of vascular injury, as defined by number of CMIs.
We found that stereological and morphometric approaches detected significant differences in the more moderate range of astrogliosis associated with diffuse WMI in VBI. Our cohort excluded more severe forms of necrotic WMI arising from macroscopic cerebrovascular injury. We also excluded more severe forms of AD pathology. More severe vascular WMI in association with a higher burden of AD pathology would likely result in more pronounced astrocyte reactivity. Thus, in a population with severe, coincident VBI and AD, other methods including point counting techniques or antibody capture assays may achieve sufficient sensitivity to distinguish the degree of astrogliosis between the VBI and No VBI groups.
CONCLUSIONS
We employed a combination of complementary approaches to measure astrocyte proliferation, process ramification, and proximal process thickness, which provided a comprehensive analysis of the major cellular changes that accompany astrocyte reactivity in human frontal white matter in response to VBI. We accomplished each of these measurements using stereological or morphometric analysis strategies that avoid bias through random and systematic sampling to yield results representative of large white matter regions. Our summed disector volume approach to the calculation of object and length density was also ideally suited to the analysis of human autopsy tissue samples where it is not typically feasible to obtain serial sections from large volumes of tissue. Our approaches provide quantitative measures of white matter astrocyte reactivity as an independent confirmation of the magnitude of VBI severity. These approaches will provide access to future studies intending to define the role of astrocyte-derived factors that may disrupt intrinsic regenerative and repair processes in response to VBI. Identification of such factors offers the potential for new therapeutic strategies intended to reduce or reverse the magnitude of cognitive decline associated with both normative aging and VBI.
ACKNOWLEDGMENTS
We gratefully acknowledge the ACT participants who donated their tissues to this study. We also acknowledge Kim Howard and Samantha Rice for their outstanding technical support, as well as Carol Arnold, Allison Beller, and Aimee Schantz for administrative support. We thank Drs Eric Larson, Paul Crane, and Randy Woltjer for their helpful discussions.
REFERENCES
- 1. Back SA, Kroenke CD, Sherman LS , et al. . White matter lesions defined by diffusion tensor imaging in older adults . Ann Neurol 2011. ; 70 : 465 – 76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ylikoski A, Erkinjuntti T, Raininko R , et al. . White matter hyperintensities on MRI in the neurologically nondiseased elderly. Analysis of cohorts of consecutive subjects aged 55 to 85 years living at home . Stroke 1995. ; 26 : 1171 – 7 [DOI] [PubMed] [Google Scholar]
- 3. Breteler MM, van Swieten JC, Bots ML , et al. . Cerebral white matter lesions, vascular risk factors, and cognitive function in a population-based study: The Rotterdam Study . Neurology 1994. ; 44 : 1246 – 52 [DOI] [PubMed] [Google Scholar]
- 4. Jellinger KA, Attems J. Prevalence and pathology of vascular dementia in the oldest-old . J Alzheimers Dis 2010. ; 21 : 1283 – 93 [DOI] [PubMed] [Google Scholar]
- 5. Canuet L, Pusil S, López ME , et al. . Network disruption and cerebrospinal fluid amyloid-beta and phospho-tau levels in mild cognitive impairment . J Neurosci 2015. ; 35 : 10325 – 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chui HC, Ramirez-Gomez L. Clinical and imaging features of mixed Alzheimer and vascular pathologies . Alzheimers Res Ther 2015. ; 7 : 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skoog I, Lernfelt B, Landahl S , et al. . 15-year longitudinal study of blood pressure and dementia . Lancet 1996. ; 347 : 1141 – 5 [DOI] [PubMed] [Google Scholar]
- 8. Iadecola C. The pathobiology of vascular dementia . Neuron 2013. ; 80 : 844 – 66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Love S, Miners JS. Cerebrovascular disease in ageing and alzheimer's disease . Acta Neuropathol 2015. Dec 28. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Salt A, Redshaw M. Neurodevelopmental follow-up after preterm birth: Follow up after two years . Early Hum Dev 2006. ; 82 : 185 – 97 [DOI] [PubMed] [Google Scholar]
- 11. Han Z, Ma Y, Gong G , et al. . White matter structural connectivity underlying semantic processing: Evidence from brain damaged patients . Brain 2013. ; 136 : 2952 – 65 [DOI] [PubMed] [Google Scholar]
- 12. de Leeuw FE, de Groot JC, Achten E , et al. . Prevalence of cerebral white matter lesions in elderly people: A population based magnetic resonance imaging study. The Rotterdam Scan Study . J Neurol Neurosurg Psychiatry 2001. ; 70 : 9 – 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Conklin J, Silver FL, Mikulis DJ , et al. . Are acute infarcts the cause of leukoaraiosis? Brain mapping for 16 consecutive weeks . Ann Neurol 2014. ; 76 : 899 – 904 [DOI] [PubMed] [Google Scholar]
- 14. Silbert LC, Nelson C, Howieson DB , et al. . Impact of white matter hyperintensity volume progression on rate of cognitive and motor decline . Neurology 2008. ; 71 : 108 – 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kochunov P, Williamson DE, Lancaster J , et al. . Fractional anisotropy of water diffusion in cerebral white matter across the lifespan . Neurobiol Aging 2012. ; 33 : 9 – 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lundgaard I, Osorio MJ, Kress BT , et al. . White matter astrocytes in health and disease . Neuroscience 2014. ; 276 : 161 – 73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation . Trends Neurosci 2009. ; 32 : 638 – 47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang L, Wu ZB, Zhuge Q , et al. . Glial scar formation occurs in the human brain after ischemic stroke . Int J Med Sci 2014. ; 11 : 344 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Buffo A, Rite I, Tripathi P , et al. . Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain . Proc Natl Acad Sci USA 2008. ; 105 : 3581 – 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sherman LS, Back SA. A ‘gag’ reflex prevents repair of the damaged CNS . Trends Neurosci 2008. ; 31 : 44 – 52 [DOI] [PubMed] [Google Scholar]
- 21. Buser JR, Maire J, Riddle A , et al. . Arrested preoligodendrocyte maturation contributes to myelination failure in premature infants . Ann Neurol 2012. ; 71 : 93 – 109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Preston M, Gong X, Su W , et al. . Digestion products of the ph20 hyaluronidase inhibit remyelination . Ann Neurol 2013. ; 73 : 266 – 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hyman BT, Phelps CH, Beach TG , et al. . National Institute on Aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease . Alzheimers Dement 2012. ; 8 : 1 – 13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Riddle A, Maire J, Cai V , et al. . Hemodynamic and metabolic correlates of perinatal white matter injury severity . PLoS One 2013. ; 8 : e82940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Postupna N, Rose SE, Bird TD , et al. . Novel antibody capture assay for paraffin-embedded tissue detects wide-ranging amyloid beta and paired helical filament-tau accumulation in cognitively normal older adults . Brain Pathol 2012. ; 22 : 472 – 84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McNeal DW, Darling WG, Ge J , et al. . Selective long-term reorganization of the corticospinal projection from the supplementary motor cortex following recovery from lateral motor cortex injury . J Comp Neurol 2010. ; 518 : 586 – 621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gundersen HJ, Bagger P, Bendtsen TF , et al. . The new stereological tools: Disector, fractionator, nucleator and point sampled intercepts and their use in pathological research and diagnosis . APMIS 1988. ; 96 : 857 – 81 [DOI] [PubMed] [Google Scholar]
- 28. Mouton PR, Gokhale AM, Ward NL , et al. . Stereological length estimation using spherical probes . J Microsc 2002. ; 206 : 54 – 64 [DOI] [PubMed] [Google Scholar]
- 29. West MJ. Basic Stereology for Biologists and Neuroscientists . 1st ed.Cold Spring Harbor, NY: : Cold Spring Harbor Laboratory Press; 2012 [Google Scholar]
- 30. Evans SM, Janson AM, Nyengaard JR . Quantitative Methods In Neuroscience: A Neuroanatomical Approach . 1st ed.New York, NY: : Oxford University Press; 2004 [Google Scholar]
- 31. Gundersen HJ. Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson . J Microsc 1986. ; 143 : 3 – 45 [PubMed] [Google Scholar]
- 32. Gundersen HJ, Jensen EB, Kieu K , et al. . The efficiency of systematic sampling in stereology—Reconsidered . J Microsc 1999. ; 193 : 199 – 211 [DOI] [PubMed] [Google Scholar]
- 33. Gundersen HJ, Jensen EB. The efficiency of systematic sampling in stereology and its prediction . J Microsc 1987. ; 147 : 229 – 63 [DOI] [PubMed] [Google Scholar]
- 34. Sonnen JA, Larson EB, Crane PK , et al. . Pathological correlates of dementia in a longitudinal, population-based sample of aging . Ann Neurol 2007. ; 62 : 406 – 13 [DOI] [PubMed] [Google Scholar]
- 35. Haight TJ, Landau SM, Carmichael O , et al. . Dissociable effects of Alzheimer disease and white matter hyperintensities on brain metabolism . JAMA Neurol 2013. ; 70 : 1039 – 45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Snowdon DA, Greiner LH, Mortimer JA , et al. . Brain infarction and the clinical expression of Alzheimer disease . The Nun Study. JAMA 1997. ; 277 : 813 – 7 [PubMed] [Google Scholar]
- 37. Vermeer SE, Longstreth WT, Jr, Koudstaal PJ. Silent brain infarcts: A systematic review . Lancet Neurol 2007. ; 6 : 611 – 9 [DOI] [PubMed] [Google Scholar]
- 38. Brundel M, de Bresser J, van Dillen JJ , et al. . Cerebral microinfarcts: A systematic review of neuropathological studies . J Cereb Blood Flow Metab 2012. ; 32 : 425 – 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cargill R, Kohama SG, Struve J , et al. . Astrocytes in aged nonhuman primate brain gray matter synthesize excess hyaluronan . Neurobiol Aging 2012. ; 33 : 830.e13 – 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iadecola C, Nedergaard M. Glial regulation of the cerebral microvasculature . Nat Neurosci 2007. ; 10 : 1369 – 76 [DOI] [PubMed] [Google Scholar]
- 41. Ostergaard PJ, Jensen MB. Histological quantification of astrocytosis after cerebral infarction: A systematic review . Int J Neurosci 2013. ; 123 : 439 – 43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Colangelo AM, Alberghina L, Papa M. Astrogliosis as a therapeutic target for neurodegenerative diseases . Neurosci Lett 2014. ; 565 : 59 – 64 [DOI] [PubMed] [Google Scholar]
- 43. Rodriguez JJ, Olabarria M, Chvatal A , et al. . Astroglia in dementia and Alzheimer’s disease . Cell Death Differ 2009. ; 16 : 378 – 85 [DOI] [PubMed] [Google Scholar]
- 44. Sofroniew MV, Vinters HV. Astrocytes: Biology and pathology . Acta Neuropathol 2010. ; 119 : 7 – 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pekny M, Wilhelmsson U, Pekna M. The dual role of astrocyte activation and reactive gliosis . Neurosci Lett 2014. ; 565 : 30 – 8 [DOI] [PubMed] [Google Scholar]
- 46. Sun D, Lye-Barthel M, Masland RH , et al. . Structural remodeling of fibrous astrocytes after axonal injury . J Neurosci 2010. ; 30 : 14008 – 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Anderson MA, Ao Y, Sofroniew MV. Heterogeneity of reactive astrocytes . Neurosci Lett 2014. ; 565 : 23 – 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nelson PT, Alafuzoff I, Bigio EH , et al. . Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature . J Neuropathol Exp Neurol 2012. ; 71 : 362 – 81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iadecola C, Gorelick PB. Converging pathogenic mechanisms in vascular and neurodegenerative dementia . Stroke 2003. ; 34 : 335 – 7 [DOI] [PubMed] [Google Scholar]
- 50. Westover MB, Bianchi MT, Yang C , et al. . Estimating cerebral microinfarct burden from autopsy samples . Neurology 2013. ; 80 : 1365 – 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zheng L, Vinters HV, Mack WJ , et al. . Cerebral atherosclerosis is associated with cystic infarcts and microinfarcts but not Alzheimer pathologic changes . Stroke 2013. ; 44 : 2835 – 41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Back SA, Miller SP. Brain injury in premature neonates: A primary cerebral dysmaturation disorder? Ann Neurol 2014. ; 75 : 469 – 86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sloane JA, Batt C, Ma Y , et al. . Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through tlr2 . Proc Natl Acad Sci USA 2010. ; 107 : 11555 – 60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Back SA, Tuohy TM, Chen H , et al. . Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation . Nat Med 2005. ; 11 : 966 – 72 [DOI] [PubMed] [Google Scholar]
- 55. Miron VE, Boyd A, Zhao JW , et al. . M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination . Nat Neurosci 2013. ; 16 : 1211 – 8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Min KJ, Yang MS, Kim SU , et al. . Astrocytes induce hemeoxygenase-1 expression in microglia: A feasible mechanism for preventing excessive brain inflammation . J Neurosci 2006. ; 26 : 1880 – 7 [DOI] [PMC free article] [PubMed] [Google Scholar]