

Abstract
The negative correlation between the rate of protein evolution and expression level of a gene has been recognized as a universal law of the evolutionary biology (Koonin 2011). In our study, we apply a population-based approach to systematically investigate the relative importance of unequal mutation rate, linkage, and selection in the origin of the expression-polymorphism anticorrelation. We analyzed the DNA sequence of protein coding genes of 24 Saccharomyces cerevisiae and 58 Schizosaccharomyces pombe strains. We found that highly expressed genes had a substantially decreased number of polymorphic sites when compared with genes transcribed less extensively. This expression-dependent reduction was especially strong in the nonsynonymous sites, although it was also present in the synonymous sites and untranslated regions, both up and down of a gene. Most importantly, no such trend was found in introns. We used these observations, as well as analyses of site frequency spectra and data from mutation accumulation experiments, to show that the purifying selection acting on nonsynonymous sites was the main, but not exclusive, factor impeding molecular evolution within the coding sequences of highly expressed genes. Linkage could not fully explain the observed pattern of polymorphism within the untranslated regions and synonymous sites, although the contribution of selection acting directly on synonymous variants was extremely small. Finally, we found that the impact of mutational bias was rather negligible.
Keywords: protein evolution, polymorphism, mutation bias, transcription, Saccharomyces cerevisiae, Schizosaccharomyces pombe
Introduction
The sequences of different proteins from the same species evolve at different rates. Studies comparing homologous sequences between species demonstrated that the best known predictor of the protein divergence rate is its expression level (Pál et al. 2001; Rocha and Danchin 2004; Drummond and Wilke 2008). This result counters some former expectations, especially those assuming that the functional importance of a protein is a chief determinant of the rate of evolution (Kimura and Ohta 1974). Several distinct explanations have been proposed to clarify why abundant proteins undergo slower evolution (for an extensive review see Zhang and Yang 2015). These include selection against erroneous translation and protein instability (Drummond et al. 2005; Yang et al. 2010), selection on protein synthesis efficiency and speed (Akashi 2001; Plotkin and Kudla 2011), selection on transcript stability (Park et al. 2013) and preservation of proper physical interactions of proteins (Vavouri et al. 2009; Yang et al. 2012). Thus, most of the currently top ranked hypotheses assume stronger purifying selection operating on final protein products of the highly expressed genes. The explanations linked to translational robustness and mRNA folding energy put additional constraints on transcripts.
The possible impact of an unequal mutation rate on expression-divergence anticorrelation is usually not considered. This can be justified by the findings of genome-wild studies of bacteria, yeast, and human (Park et al. 2012; Chen and Zhang 2013, 2014). All of these studies reported elevated mutation rates for intensively expressed genes. This in turn, implies that mutation bias alone should generate positive, not negative, correlation between gene expression and evolution rate. It should be noted though, that two of the above studies used data obtained for mutants with defective DNA repair system (Park et al. 2012; Chen and Zhang 2013). Relations found for such mutants might not accurately reflect those existing in the wild-type organisms. Moreover, the negative correlation between transcription and mutation rate was found in wild E. coli populations (Martincorena et al. 2012). All things considered, although mutation bias is unlikely to be the main evolutionary force leading to expression-divergence anticorrelation, its impact may differ between species, and thus its contribution is worth further investigation.
Here, we analyzed the distribution and frequencies of single nucleotide variants in previously published whole genome sequencing data sets for 24 Saccharomyces cerevisiae and 58 Schizosaccharomyces pombe strains isolated from their worldwide/extant populations. We reasoned that focusing on the genetic variation within populations could be a novel and promising way to estimate the relative contribution of purifying selection, linkage, and mutational pressure in creating the observed pattern of expression-divergence anticorrelation. In the case of S. cerevisiae, we assigned a total of 57,000 single nucleotide variants to the coding sequences, introns, 3′-UTR and 5′-UTR of ∼2,400 genes (for which the boundary of at least one UTR region is known). Analogously, we investigated ∼150,000 SNPs in 5,060 S. pombe genes. We applied two types of tests for all variants. First, we analyzed the relation between polymorphism density and gene expression level. Similar tests, regarding nucleotide or amino acid substitutions, had been previously used in comparisons between closely and distantly related species (Wall et al. 2005; Zhou et al. 2010). Nevertheless, their results are sensitive to background selection and their reliability critically depends on equal mutation rates for compared genes and genes’ regions. Therefore, we decided to extend our analyses to encompass the available data on the distribution of mutations accumulating under the laboratory propagation scheme, in which the operation of the natural selection is minimal. This allowed us to estimate the strength of the transcription driven mutational biases in both species. For the second test, we calculated the fractions of singletons within variants assigned to different genes and their regions, and checked whether these fractions depend on gene expression level and type of the mutated site. Although this test has lower power to detect negative selection, as it loses information about the most severe and thus not present in populations variants, its results are unaffected by background selection and mutation biases. Therefore, this test seems to be especially relevant to the analysis of polymorphism within untranslated regions and synonymous sites.
We found that in the natural populations the level of polymorphism was negatively correlated with gene expression only in the functional regions of protein coding genes. It is significant to note that such pattern was not found for the laboratory mutation accumulation lines. Notably, fraction of singletons within variants assigned to functional regions of genes increased with gene expression, although this trend was rather weak. Thus, we show that in populations of both yeast species the decline in the genetic variation within extensively expressed genes was most likely produced by the purifying selection operating on gene protein products and transcripts.
Materials and Methods
Saccharomyces cerevisiae Polymorphism
We analyzed the sequences of 23 Saccharomyces cerevisiae strains from Skelly et al. (2013) (http://www.yeastrc.org/g2p-data/raw-data/genome-sequences). The sequenced strains were haploids derived from natural diploidal isolates. Raw reads were aligned to the reference sequence (S288C, genome assembly: R64-1-1) using bowtie2. BAM files were manipulated with samtools programs (version 0.1.19). Variants were called separately for each strain with samtools mpileup and filtered with grep and custom python script. First, we removed all indels. For further analysis, we used only SNPs with minimal QUAL value of 30, with coverage >5 and with at least 80% of reads supporting the altered nucleotide. The mapping and filtering scripts (mapping.sh; readVCF.py) can be found in the supplementary file S1, Supplementary Material online.
The filtered variants were assigned to the coding sequences, introns, 5′-UTR and 3′-UTR regions of 2,432 protein coding Saccharomyces cerevisiae genes. In order to be analyzed further, genes were required to fulfill two criteria:
Known location of at least one UTR region (retrieved from the SGD YeastMine page https://yeastmine.yeastgenome.org/yeastmine): The UTRs locations were compiled from former studies (Zhang et al. 2005; Miura et al. 2006; Nagalakshmi et al. 2008; Xu et al. 2009; Yassour et al. 2009). In cases where boundaries of the UTR regions differed between studies, we decided to use ones involving the longest stretches of DNA.
Coding sequence nonoverlapping with any other gene
First, we assigned variants to the coding sequences and introns of the analyzed genes. The remaining SNPs were then assigned to the UTR regions. The very short (<9 bp) and nonpolymorphic UTR regions were excluded from further analysis. Variants in the coding sequence were split into synonymous and nonsynonymous groups based on the results from the Variant Effect Predictor tool (http://www.ensembl.org/index.html). In sum, we analyzed 56,939 SNPs. Next, we calculated the number of polymorphic sites for each gene and gene region (divided by the length of respective regions). In addition, the fraction of singletons was determined. The expected number of potential synonymous and nonsynonymous sites was calculated for the coding sequences of the reference strain. We used the Nei–Gojobori method under the assumption of equal mutation probabilities for all types of substitutions (Nei and Gojobori 1986). Obtained values were then used as the lengths of synonymous and nonsynonymous regions.
Data on the mRNA level (mRNA molecules per haploid cell) were adopted from Csárdi et al. (2015). Data on gene indispensability was retrieved from SGD YeastMine page https://yeastmine.yeastgenome.org/yeastmine). The compiled data is available in the supplementary file S2, Supplementary Material online.
The fraction of singletons expected in neutrality for the folded site frequency spectrum was based on one million simulated samples of size 24. For the coalescent simulations, we used program ms (Hudson 2002), with the command: ms 24 1000000 -s 1. Then, we counted cases where the derived allele was present in only one or in 23 copies per sample.
Schizosaccharomyces pombe Polymorphism
Data on S. pombe variants were downloaded from the web page: https://figshare.com/articles/SNP_calls_for_all_161_strains_/3978303. These data were originally published by Jeffares et al. (2015) and represent single nucleotide polymorphism found in 161 natural isolates of Schizosaccharomyces pombe. The downloaded vcf file was already annotated. For the purpose of further analyses, we extracted from the original file information on variants present in 57 strains, described by authors of the original research paper as “nonredundant.” We filtered these variants using the same criteria as for S. cerevisiae. SNPs that passed the filters were then assigned to the different gene regions (synonymous coding, nonsynonymous coding, intron, 3′-UTR, 5′-UTR) on the basis of annotations provided in the original vcf file. In sum, we assigned 153,407 SNPs to 5,069 protein coding genes; out of those 2,496 contained introns. Data on gene expression was taken from Marguerat et al. (2012) (number of mRNA molecules per haploid cell). A list of essential genes was retrieved from PomBase (https://www.pombase.org/). Number of potential synonymous and nonsynonymous sites was obtained as described for S. cerevisiae. The compiled data set is available in the supplementary file S2, Supplementary Material online.
The expected fraction of singletons was determined analogously as described for S. cerevisiae, but the size of the simulated samples was set to 58.
Analysis of the S. cerevisiae Mutation Accumulation Experiment
We used results of the study by Zhu et al. (2014). The study describes 873 single nucleotide mutations that occurred in 145 yeast lines during the mutation accumulation experiment. Out of these, 636 were assigned to 577 Saccharomyces cerevisiae genes with mRNA level given in Csárdi et al. (2015). In this procedure, we used the Variant Effect Predictor tool. Next, we calculated EMA statistic: EMA=Σni·ln(mRNAi)/Σni, where ni stands for the number of mutations that were assigned to the i gene and mRNAi is the i gene mRNA level. Thus, the EMA statistic is the weighted average of the natural logarithms of the mRNA level, calculated for the genes that were found mutated in the mutation accumulation experiment. Compiled data can be found in the supplementary file S2, Supplementary Material online.
Simulation of Mutagenesis in S. cerevisiae
We used the genomic data on the mRNA expression (Csárdi et al. 2015) and gene GC content (Ensembl) to create two lists. The LAT list was composed of the ln(mRNAi) values for 5,850 Saccharomyces cerevisiae genes. Individual ln(mRNAi) values were represented in numbers equal to the numbers of AT pairs in respective genes (in sum 5,349,347 entries). The LGC list was composed according to the same pattern for GC pairs (3,484,611 entries in total). Then, we randomly chose 636 values from the above lists. To take account for the higher mutation probability of the GC nucleotides, 68% the ln(mRNAi) values were taken from the LGC list and 32% from the LAT list (as suggested by Zhu et al. 2014). The arithmetic mean of each random sample, ES was then calculated. The procedure was repeated 10,000 times. We considered the above procedure to simulate the mutational process in which the probability of mutation within any gene sequence is proportional to gene length and depends on gene GC content (simulated_data1). In another simulation, we proceeded in the same way, with the exception that the ln(mRNAi) values were drawn from the merged LAT and LGC lists. Thus, the second simulation represented the situation where the probability of any gene mutating depends only on its length and not on its individual GC content (simulated_data2). The script used to generate data is in the supplementary file S1, Supplementary Material online.
Analysis of the S. pombe Mutation Accumulation Experiments
We merged results from two mutation accumulation experiments: Behringer and Hall (2015) (422 single nucleotide mutations) and Farlow et al. (2015) (326 single nucleotide mutations). We annotated 615 of these mutations to 533 different S. pombe genes with mRNA level given in Marguerat et al. (2012) (http://fungi.ensembl.org/Schizosaccharomyces_pombe/Tools/VEP). The EMA statistic was calculated as described for S. cerevisiae. The corresponding data set is in the supplementary file S2, Supplementary Material online.
Simulation of Mutagenesis in Schizosaccharomyces pombe
Simulated data sets were created analogously as described for S. cerevisiae. We used the expression data from Marguerat et al. (2012) (5,059 protein coding genes) and data on gene GC content from the Ensembl fungi web page. The S. pombe LAT and LGC lists contained 6,907,927 and 4,071,402 entries accordingly. To obtain the simulated data dependent on both gene length and its GC content, 73.6% of values was taken from the LGC list because 457 of the 748 mutations found in both experiments related to the GC nucleotides. The whole S. pombe genome contains 4,538,978 GC and 8,052,273 AT base pairs. Thus, the probability of being mutated is 2.786 times higher for the GC nucleotides than for the AT nucleotides. This gives the relative mutation frequencies of 0.736 and 0.264 for the GC and AT nucleotides accordingly. At every of 10,000 iterations, an average of 615 randomly selected ln(mRNAi) values was calculated. The script used to generate data is in the supplementary file S1, Supplementary Material online.
Analysis of Codon Usage
We counted codons present within the coding sequences of all analyzed genes (for the reference strains of S. cerevisiae and S. pombe). These counts were then correlated (Spearman’s rank correlation) with the numbers of the matching tRNA genes present in the genomes of both species (multiplied by their wobble parameters). Correlation coefficients obtained for each gene constituted the rhotai statistics. In case of S. cerevisiae, tRNA gene copy numbers and wobble parameters were adopted from Weinberg et al. (2016). The gene copy numbers of tRNA genes present in S. pombe genome were taken from GtRNAdb (http://gtrnadb.ucsc.edu/). The wobble parameters were set as described in Curran and Yarus (1989) and Lim and Curran (2001). Correlation coefficients obtained for each gene along with tRNA copy numbers and wobble parameters used are given in the supplementary file S2, Supplementary Material online.
Data analysis
Data was analyzed in R (R Core Team 2015). We used the following functions: cor.test, glm, pcor.test from the ppcor package (Kim 2015). Correlation coefficients were compared with concor (http://comparingcorrelations.org/) (Diedenhofen and Musch 2015).
Results
We studied single nucleotide polymorphism within dozens of yeast strains isolated from extant populations of Saccharomyces cerevisiae and Schizosaccharomyces pombe. We found that the density of polymorphic sites within genes correlates negatively with the expression level measured as the number of mRNA molecules per haploid cell. In both yeast species, the relationship is especially clear for the nonsynonymous variants (fig. 1). A possible interpretation is that the purifying selection against changes in protein sequence operates and is more intense in the highly expressed genes. Indeed, the ratio of nonsynonymous to synonymous polymorphic sites densities (pN/pS) also correlates with the gene expression (S. cerevisiae: Spearman’s rank correlation rho=−0.35, P < 2.2×10−16, n = 2,373; S. pombe: Spearman’s rank correlation rho=−0.28, P < 2.2×10−16, n = 4,882).
Fig. 1.

—Relationship between gene expression (mRNA molecules per cell) and polymorphism (number of variants per region length).
In the next step, we analyzed minor site frequency spectra for both populations to test whether highly expressed genes have elevated fraction of singletons. We reasoned that deleterious mutations could be still present in populations if they were recent. Such mutations would be chiefly found among singletons, as they are removed from the population before reaching higher frequencies. This should lead to the elevated fraction of singletons within all variants. Indeed, we found higher than expected (for neutral polymorphism) fractions of nonsynonymous singletons in the analyzed data set: 0.40 versus expected 0.29 for S. cerevisiae and 0.40 versus 0.23 for S. pombe. Moreover, these fractions were also higher than fractions found for intronic variants (0.29 and 0.26 for S. cerevisiae and S. pombe accordingly), what indicates that the high percentage of nonsynonymous singletons cannot be entirely explained neither by the demographic process (e.g., population growth) nor by insufficient variant filtering procedure (fig. 2). More importantly, the fraction of nonsynonymous singletons correlates positively with gene expression level for both species (S. cerevisiae: rho = 0.06, P = 0.004, n = 2,133; S. pombe: rho = 0.11, P = 1.75×10−13, n = 4,593). Thus, the nonsynonymous site frequency spectra show clear signs of the negative selection in S. cerevisiae and S. pombe populations. Above all, higher fraction of newly arriving amino acid altering mutations is present in sequences coding for abundant proteins indicating stronger selection.
Fig. 2.
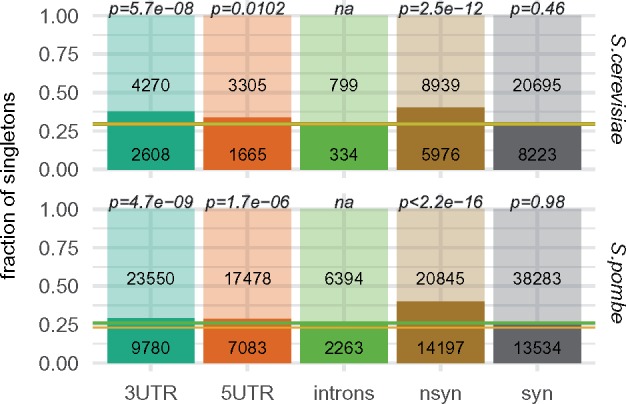
—Fraction of singletons within all analyzed variants (minor allele frequency spectra). Parts of the bars corresponding to singletons are filled with the opaque colors. Numbers inside bars indicate number of sites within each category (singletons/more frequent SNPs). Green line shows fraction of singletons found in introns—used as an approximation of the neutral value (S. cerevisiae: 0.29; S. pombe: 0.26). Yellow line indicates fraction of singletons expected for Wright–Fisher population (S. cerevisiae: 0.29; S. pombe: 0.23). Significance of χ2 tests comparing fraction of singletons within SNPs in a given region with fraction recorded for introns are given above the bars.
We also asked whether the observed low polymorphism in highly expressed genes could be ascribed to some specific forces, such as selection acting on transcripts or codon composition. In both yeast species, the negative correlation between gene expression and variant density is visible not only for nonsynonymous but also for synonymous sites and the UTR regions. In the two latter regions, however, the correlation is much weaker (fig. 1). Such pattern may indicate that abundant transcripts are also more constrained. An alternative, but not mutually exclusive explanation, is that the regions of highly expressed genes are depleted of polymorphism because they are linked to more constrained nonsynonymous sites. We addressed this issue using three distinct approaches described below.
First, we performed partial correlations (mRNA vs variant density) while controlling for the nonsynonymous variant densities. The purifying selection acting on nonsynonymous variant causes a reduction in the effective population sizes of the adjacent regions. Therefore, if the drop in variant densities in UTR regions and synonymous sites results exclusively from linkage, then the partial correlations should turn insignificant. This is not the case, as most of the correlations remain valid (table 1). Interestingly, the partial correlation between synonymous polymorphism density and gene expression (controlling for nonsynonymous polymorphism) remains significant for S. cerevisiae but disappears for S. pombe. This may suggest that the selection on codon usage is present only in the former species. However, even in S. cerevisiae the observed correlation is restricted to a small subset of genes expressed at a very high level. The partial correlation is no longer significant, when removing 118 genes with the highest expression (i.e., >40 mRNA molecules per cell) (Spearman’s rank correlation rho=−0.035, P = 0.09, n = 2,314). It should be also noted here, that partial correlations performed on noisy data may produce spuriously significant results (Drummond et al. 2006).
Table 1.
Results of the Spearman’s Rank Partial Correlations between mRNA Level and Number of Polymorphic Sites (per region length), Controlling for Nonsynonymous SNPs Density
| Region | Rho | P value | n |
|---|---|---|---|
| Saccharomyces cerevisiae | |||
| Synonymous sites | −0.0795 | 8.76e-05 | 2,432 |
| 5′-UTR | −0.0299 | 0.14 | 2,398 |
| 3′-UTR | −0.0634 | 0.0018 | 2,425 |
| Schizosaccharomyces pombe | |||
| Synonymous sites | 0.0085 | 0.55 | 5,069 |
| 5′-UTR | −0.1275 | 2.39e-18 | 4,664 |
| 3′-UTR | −0.1251 | 6.26e-18 | 4,718 |
Our second test uses the observation that background selection should remove polymorphism not only from functional regions of genes but also from introns. Therefore, if correlations between SNPs density and mRNA level, visible for UTRs and synonymous sites, were only byproducts of selection acting on nonsynonymous variants, then similarly strong correlations should be seen in introns, when taking into account smaller sample size (i.e., smaller number of genes with introns). To this end, we randomly drew sample of 162 genes for S. cerevisiae and 2,496 genes for S. pombe from the originally analyzed data sets. These numbers equal numbers of genes with introns used in our analyses. Next, we performed Spearman’s rank correlations between mRNA level and variant densities for the restricted data. This procedure was repeated 500 times. The distributions of obtained correlation coefficients for all functional regions are shown in figure 3A and in supplementary file S2, Supplementary Material online. Notably, these distributions are shifted toward negative values and, with the exception of synonymous variants in S. pombe, their 95 confidence intervals lie below the value obtained for introns (supplementary file S2, Supplementary Material online). Thus, results of these tests seem to indicate that purifying selection acts directly on variants that do not change amino acid sequence and that such selection is more important for highly expressed genes. It is important to note, however, that this test may not be stringent enough in case of the synonymous polymorphism, as on an average, the selected nonsynonymous variants lie in closer proximity to the synonymous sites than to introns.
Fig. 3.
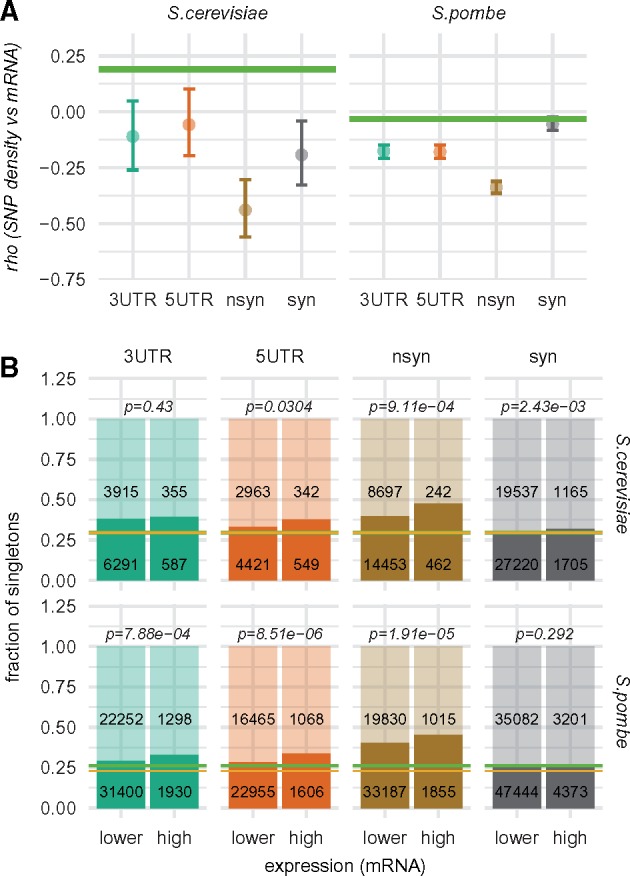
—(A) Distribution of the Spearman’s rank correlation coefficients (mRNA level vs variant density) obtained in the subsampling analyses (mean ± 95ci). Green line shows value of the correlation coefficient obtained for all introns; (B) Fractions of singletons within variants present in highly and less extensively expressed genes (see Results for the more detailed description of the used expression categories). Singletons—opaque colors, more frequent variants—transient colors. Numbers of variants within each group are given inside bars. Green line shows fraction of singletons found in introns, yellow line indicates fraction of singletons expected for ideal Wright–Fisher population. Significance of χ2 tests comparing fractions of singletons within variants found in genes belonging to the two expression categories are shown above the bars.
For the third analysis, we divided all considered genes into two groups according to their expression. The “high” group contained ∼ 10% of genes with the highest mRNA level (S. cerevisiae: 244 genes having >22 mRNA molecules per cell; S. pombe: 515 genes with >12 mRNA molecules per cell). The remaining genes formed the “lower” groups. We used χ2 tests to compare the frequency of singletons within variants found in these groups of genes. The results of these comparisons are summarized in figure 3B. Clearly, SNPs localized within sequences of genes from the “high” group are more likely to be singletons. This trend holds for all functional regions of genes, however, the differences are not always significant and are extremely small in case of the synonymous sites. Moreover, the frequencies of singletons calculated for the synonymous variants present in all analyzed genes do not differ from the frequencies obtained for introns (fig. 2). To summarize, the results of the three different tests point to the hypothesis that both untranslated regions of genes are under weak purifying and expression-dependent selection, distinct from the selection on protein products. On the contrary, we found little (for S. cerevisiae) or no (in case of S. pombe) evidence for the purifying selection acting directly on the synonymous sites. Even if such selection does contribute to the drop of polymorphism in highly expressed genes, its effect is rather weak and restricted to very abundant transcripts.
Finally, we asked whether the negative correlation between expression level and polymorphism density can be, at least partially, caused by unequal mutation rates. We started with introns, as we reasoned that variants in introns should not be influenced by the purifying selection. We found that introns of highly expressed genes in Saccharomyces cerevisiae may have an elevated number of variants per one nucleotide (rho = 0.19, P = 0.015, n = 162) (fig. 1). However, only a small portion of genes in this species has introns (162 in our data set). Moreover, these genes are generally highly expressed and may not appropriately represent all genes present in this species (Skelly et al. 2009). An analogous analysis involving almost 2,500 S. pombe genes provided contradictory results. In this species, the expression level of the gene did not influence the number of variants present in the intronic sequences (rho=−0.033, P = 0.097, n = 2,496) (fig. 1).
As the above results might appear insufficiently conclusive, we turned to studies on mutation accumulation experiments. Mutations found in these kind of studies are likely to represent the mutational spectrum largely unaffected by selection. We used simulations to test available empirical data for the presence of mutational bias. Simulations represented the situations in which the chance of occurring of new mutation within any gene depended on both the gene length and its GC content (fig. 4, upper panel: simulated_data1) or on the gene length only (fig. 4, lower panel: simulated_data2). Then, we compared the simulated distribution of possible results with the EMA statistic calculated for the actual data (in brief: EMA represents the average expression of genes that were found mutated in the mutation accumulation experiment; see Materials and Methods for full description). We found that in S. cerevisiae mutations are not less but more likely to occur in highly transcribed genes (EMA=1.1916, P = 0.0006; fig. 4). In this species, expression is likely to be mutagenic itself as the observed effect did not depend on the gene’s GC content (fig. 4). However, in S. pombe, the actual mutation spectrum appeared shifted toward genes of low expression (EMA=0.801, P = 0.036), but not when neglecting the GC content of the gene (P = 0.094, fig. 4). Thus, transcription does not influence the mutation rate in this yeast species, although the highly expressed genes may have slightly lower mutation rates due to their nucleotide composition. As the results of the mutation accumulation, data analyses differ between the two considered yeast species, we confirmed our conclusions by building several Poisson regression models (supplementary file S2, Supplementary Material online). Taken together, both the analysis of polymorphism in introns of extant populations and the test of distribution of mutations in accumulation lines provide results consistent with each other. They indicate that mutational process itself is unlikely to produce (S. pombe) or even acts against (S. cerevisiae) the observed decline in the polymorphism level associated with gene expression.
Fig. 4.

—Comparison between the expression of genes mutated in mutation accumulation experiments (EMA statistics, red arrows) and values obtained for simulated data; simulated_data1: probability of mutation within a gene proportional to its length and higher for G and C nucleotides, simulated_data2: probability of mutation proportional to gene length and equal for all nucleotides (See Materials and Methods for details).
Discussion
Studies involving comparisons between close and distant taxa showed that the rate of molecular evolution of a protein depends on the intensity of its expression with the abundant proteins being more conserved. We asked whether the predicted negative relationship between the level of gene expression and its genetic polymorphism can be observed also in current populations of a single species. We chose S. cerevisiae and S. pombe due to the fact that populations of these two yeast species are large enough, Ne∼107 (Skelly et al. 2009; Farlow et al. 2015), to enable accumulation of a sufficient number of mutations so that their distribution within a genome could be used to detect the work of an even relatively weak selection. Indeed, we observed lower variant densities in regions coding for highly expressed genes. Relevant correlation coefficients turned out to roughly match those reported for the between species comparisons (Zhang and Yang 2015).
We believe that the observed pattern of mutation accumulation could not result from the different rate of mutation at the compared sites. Firstly, we focused on isolates from extant populations and found no correlation (S. pombe), or possibly a positive correlation (S. cerevisiae), between the expression level and variant density within introns. This result suggests that the frequently transcribed genes have a mutation rate that is equal or even higher than those transcribed rarely. We then analyzed the distribution of new mutations accumulated in experimental populations and found further support for our claim. The probability of being mutated was higher for genes with high mRNA level in S. cerevisiae. In S. pombe, we detected a weak trend in the opposite direction which probably resulted from a bias in the GC content. We are aware that our analyses of mutation accumulation experiments are indirect in the sense that we used simulated data to form the null distribution. In this simulated data set, a gene underwent a mutation at the rate proportional to its length and GC content, neglecting a possible role of gene location, chromatin status, or other factors. What is important, we additionally confirmed these conclusions using general linear modelling. One could also argue that although introns are generally not exposed to selection, they may, nevertheless, mutate at a rate different than they do in exons due to the fact that different repair systems are thought to operate along the coding and noncoding sequences (Frigola et al. 2017). Regardless of the above, we are reassured by the fact that our analyses involving two completely different approaches gave consistent results. An overall pattern, a roughly equal or even higher polymorphism within introns of strongly expressed genes appears robust and therefore the reduction of polymorphism known for highly expressed exons is unlikely to result from the differences in mutation rate.
Some previous results appear to accord with our finding that polymorphism is not lower but higher in introns of highly expressed genes in Saccharomyces cerevisiae. One study used mutation-accumulation lines within a DNA repair defective (ung1Δ) strain and found an elevated mutation rate in intensively transcribed genes (Park et al. 2012). Data from the mutation accumulation experiments carried out with a wild-type S. cerevisiae strain and reanalyzed here (Zhu et al. 2014) had been already examined for the dependence between expression and mutation probability (Chen and Zhang 2014; Zhu et al. 2014). While authors of the original study found no such relation, conclusions similar to ours were reached by Chen and Zhang (2014). Thus, our analyses provided additional evidence that intensive transcription is associated with elevated mutation in S. cerevisiae. What is important, we applied different analytical methods to those used in the above studies. An analogous analysis based on both mutation accumulation data sets published for S. pombe, failed to detect any relation between mutation rate and expression level. The same conclusions were reached in one of the original mutation accumulation studies carried out for this species (Behringer and Hall 2015). Thus, while it is tempting to conclude that in S. cerevisiae the purifying selection in exons is strong enough to revert the mutational bias, in S. pombe transcription does not lead to elevated mutation probability. Transcription affects mutation rates through the processes of transcription associated mutagenesis and transcription-coupled DNA repair (Hanawalt and Spivak 2008; Kim and Jinks-Robertson 2012). It is possible that the final outcome of these antagonistic processes is not the same for all species. However, more intensive work on the experimental estimation of mutation rate in different genome regions is needed before its results can be used in quantitative tests of evolutionary hypotheses.
Apart from the clear difference in the distribution of polymorphism between introns and exons, there are other, more direct, arguments that selection operates most efficiently in highly expressed coding regions. First, the negative correlation between gene mRNA level and the density of polymorphism in exons was stronger for the nonsynonymous polymorphism. It is remarkable that this result was statistically significant given that the density of variants recorded for nonsynonymous sites in both species was generally low. Second, the fraction of rare nonsynonymous variants tended to increase with gene expression. Which is why, new mutations are most likely to be deleterious if they change the final protein products of an intensively transcribed gene. It is also worth noting that in both species the expression–divergence correlations for essential and nonessential genes have similar strength (S. cerevisiae: Fisher’s z=−1.42, P = 0.16; S. pombe: Fisher’s z=−0.71, P = 0.48) (supplementary file S2, Supplementary Material online). Thus, the functional importance of proteins is unlikely to be an important factor underlying the considered relation.
The negative correlation between gene expression level and polymorphism density appears to be considerably stronger for the surveyed populations of S. cerevisiae. This finding might suggest that the purifying selection is more effective in this species. However, gene expression, one of the analyzed factors, may have been estimated with different accuracy in the two species. The data set for S. cerevisiae is possibly more reliable as based on a vast number of independent RNA expression studies (Csárdi et al. 2015). Moreover, it is imaginable that in this species the expression data gathered under laboratory conditions are more indicative of “natural” gene expression. Regarding the polymorphism data, the effect of selection can be much more difficult to detect in the case of S. pombe because of the stronger linkage disequilibrium reported for this species (Jeffares et al. 2015). Therefore, although the correlation coefficients relating the fraction of nonsynonymous sites and expression level differ significantly between S. cerevisiae and S. pombe (Fisher’s z = 5.0179, P < 10−6) this result does not necessarily reflect differences in the selection pressure.
With regards to the synonymous polymorphism, it was also negatively correlated with the mRNA level in both yeast species. These correlations, however, were much weaker that those found for the nonsynonymous sites. Moreover, results of three distinct and independent tests (see Results) indicated that these correlations were generated mainly by the background selection. This interpretation is straightforward in the case of S. pombe, as all three tests failed to detect signatures of negative selection acting directly on the synonymous variants in this species. The results obtained for Saccharomyces cerevisiae are more challenging to explain. Both tests utilizing information on variant densities (i.e., subsampling analysis and partial correlation controlling for nonsynonymous polymorphism density) gave results that supported the role of the direct purifying selection. On the other hand, the fraction of singletons within the synonymous sites was not any higher than fraction expected for neutrally evolving sites. This fraction was elevated for the 10% of genes having the highest expression, however, the effect was not substantial. One possible explanation is that synonymous variants are effectively neutral also in S. cerevisiae population (with the exception of sites within the small number of very intensively transcribed genes) and that two out of three applied tests were too permissive. The other explanation is that synonymous variants may be either very deleterious and rapidly selected against or neutral. The paucity of synonymous variants with low/moderate selection coefficients may explain the neutral-like shape of the frequency spectrum. Interestingly enough, such dichotomous fitness effects of the synonymous mutations were described for Drosophila melanogaster (Lawrie et al. 2013).
In light of the above findings, it is interesting to note, that in both yeast species strong codon bias exists, where the sequences of highly expressed genes are enriched in the major codons (Kurland 1991; Hiraoka et al. 2009). Such codon pattern is usually explained as a result of the selection for translation efficiency and accuracy, which should be more significant for more abundant mRNAs and proteins (Rocha 2004; Plotkin and Kudla 2011). Such selection should lead to adjustments between abundance of tRNA molecules and the usage of appropriate codons (Tuller et al. 2010). To estimate this adjustment, we calculated Spearman’s rank correlations coefficients relating these two quantities for the coding sequences of all analyzed genes—rhotai statistics (see Materials and Methods). As expected, obtained rhotai coefficients are positively correlated with gene’s expression (S. cerevisiae: r = 0.696, P < 2.2·10−16, n = 2,432; S. pombe: r = 0.653, P < 2.2·10−16, n = 5,069; supplementary fig. S1, Supplementary Material online). Later, we checked if they correlate with the density of the synonymous variants. While we found negative correlation for the S. cerevisiae data (rho=−0.11, P = 6.30·10−8, n = 2,432), which turned insignificant after controlling for the mRNA level (rho = 0.022, P = 0.27, n = 2,432), no relation was visible for S. pombe (rho = 0.04, P = 0.06, n = 5,069). Thus, also this analysis fails to detect clear signals of selection related to tRNA abundance within the synonymous sites. It should be noted that, much stronger correlations were visible for the nonsynonymous SNPs for both species (S. cerevisiae: rho=−0.38, P < 2.2·10−16, n = 2,432; S. pombe: rho=−0.30, P < 2.2·10−16, n = 5,069). Partial correlations controlling for gene expression were also highly significant in this case (S. cerevisiae: rho=−0.13, P = 4.38·10−11, n = 2,432; S. pombe: rho=−0.12, P = 2.91·10−18, n = 5,069). This may imply that the nonsynonymous changes have greater impact on our measure of translation optimization. Indeed, many of the codon pairs that are separated by one synonymous change are decoded by the same tRNA species. Moreover, the correlations between gene’s codon composition and tRNA gene copy numbers may be also affected by differences in amino acid usage. Taken together, our analyses show that the contribution of the negative selection acting directly on synonymous sites to the investigated expression–divergence anticorrelation may be extremely small or even nonexistent. Our results also imply that in both analyzed populations such selection may be very weak, with the selection coefficients around 1/Ne or less. Notably, even such a very weak selection might be sufficient to generate the codon usage bias (McVean and Charlesworth 1999; Charlesworth 2009).
Weak correlations between polymorphism density and expression were also found for the 5′-UTR and 3′-UTR regions. Moreover, analyses performed to decipher the impact of the background and direct selection confirmed the attribution of the latter. Therefore, it is possible that some additional selection pressures, acting directly on transcripts, contribute to the decline in polymorphism observed for the highly expressed genes. One possible factor could be the stability of mRNA (Park et al. 2013), while another the rate of translation initiation and elongation (Kudla et al. 2009; Shah et al. 2013; Yang et al. 2014). Thus, in populations of both yeast species, the untranslated regions of the most abundant transcripts would be under more intense purifying selection.
Our analyses of the genomic data of S. cerevisiae and S. pombe show that the differences in the purifying selection strength underlie the negative correlation between gene expression and its evolution rate. Notably, these differences are big enough to be easily noticeable even in populations of a single species. In both examined populations, the expression-dependent negative selection affects mainly amino acid altering variants. Although to much weaker extent, it also acts directly on polymorphism within untranslated regions of highly expressed genes. This result may imply that the evolutionary constraint is related not only to biophysical/functional properties of proteins but also to some features of transcripts or translation initiation. Interestingly, we found that background selection can almost entirely explain the observed pattern of synonymous polymorphism. The strength of the purifying selection acting directly on silent sites seems to be just on the edge of what can be detected in both analyzed populations. Finally, the relation between expression and mutation rate differs between S. pombe and S. cerevisiae, as strong mutagenic effect of transcription is visible only in the latter species. Nevertheless, transcription associated mutation bias does not significantly contribute to the considered correlation in any of the analyzed yeasts.
Supplementary Material
Acknowledgments
The authors thank Ryszard Korona for reading and commenting our manuscript. This work was supported by the Polish National Science Center grant 2014/13/B/NZ8/04668 to K.T. (2014/13/B/NZ8/04668 to K.T.).
Literature Cited
- Akashi H. 2001. Gene expression and molecular evolution. Curr Opin Genet Dev. 11(6):660–666. [DOI] [PubMed] [Google Scholar]
- Behringer MG, Hall DW.. 2015. Genome-wide estimates of mutation rates and spectrum in Schizosaccharomyces pombe indicate CpG sites are highly mutagenic despite the absence of DNA methylation. G3 (Bethesda) 6:149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. 2009. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet. 10(3):195–205. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang J.. 2013. No gene-specific optimization of mutation rate in Escherichia coli. Mol Biol Evol. 30(7):1559–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang J.. 2014. Yeast mutation accumulation experiment supports elevated mutation rates at highly transcribed sites. Proc Natl Acad Sci U S A. 111(39):E4062.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csárdi G, Franks A, Choi DS, Airoldi EM, Drummond DA.. 2015. Accounting for experimental noise reveals that mRNA levels, amplified by post-transcriptional processes, largely determine steady-state protein levels in yeast. PLoS Genet. 11(5):e1005206.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran JF, Yarus M.. 1989. Rates of aminoacyl-tRNA selection at 29 sense codons in vivo. J Mol Biol. 209(1):65–77. [DOI] [PubMed] [Google Scholar]
- Diedenhofen B, Musch J.. 2015. cocor: a comprehensive solution for the statistical comparison of correlations. PLoS One 10(4):e0121945.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Bloom JD, Adami C, Wilke CO, Arnold FH.. 2005. Why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 102(40):14338–14343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond DA, Raval A, Wilke CO.. 2006. A single determinant dominates the rate of yeast protein evolution. Mol Biol Evol. 23(2):327–337. [DOI] [PubMed] [Google Scholar]
- Drummond DA, Wilke CO.. 2008. Mistranslation-induced protein misfolding as a dominant constraint on coding-sequence evolution. Cell 134(2):341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlow A, et al. 2015. The spontaneous mutation rate in the fission yeast Schizosaccharomyces pombe. Genetics 201(2):737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigola J, et al. 2017. Reduced mutation rate in exons due to differential mismatch repair. Nat Genet. 49(12):1684–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G.. 2008. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 9(12):958–970. [DOI] [PubMed] [Google Scholar]
- Hiraoka Y, Kawamata K, Haraguchi T, Chikashige Y.. 2009. Codon usage bias is correlated with gene expression levels in the fission yeast Schizosaccharomyces pombe. Genes Cells Devoted Mol Cell 14(4):499–509. [DOI] [PubMed] [Google Scholar]
- Hudson RR. 2002. Generating samples under a Wright–Fisher neutral model of genetic variation. Bioinformatics 18(2):337–338. [DOI] [PubMed] [Google Scholar]
- Jeffares DC, et al. 2015. The genomic and phenotypic diversity of Schizosaccharomyces pombe. Nat Genet. 47(3):235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Jinks-Robertson S.. 2012. Transcription as a source of genome instability. Nat Rev Genet. 13(3):204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. 2015. ppcor: an R package for a fast calculation to semi-partial correlation coefficients. Commun Stat Appl Methods 22(6):665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Ohta T.. 1974. On some principles governing molecular evolution. Proc Natl Acad Sci U S A. 71(7):2848–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV. 2011. Are there laws of genome evolution? PLoS Comput Biol. 7(8):e1002173.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla G, Murray AW, Tollervey D, Plotkin JB.. 2009. Coding-sequence determinants of gene expression in Escherichia coli. Science 324(5924):255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland CG. 1991. Codon bias and gene expression. FEBS Lett. 285(2):165–169. [DOI] [PubMed] [Google Scholar]
- Lawrie DS, Messer PW, Hershberg R, Petrov DA.. 2013. Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genet. 9(5):e1003527.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim VI, Curran JF.. 2001. Analysis of codon: anticodon interactions within the ribosome provides new insights into codon reading and the genetic code structure. RNA 7(7):942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marguerat S, et al. 2012. Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151(3):671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Seshasayee ASN, Luscombe NM.. 2012. Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature 485(7396):95–98. [DOI] [PubMed] [Google Scholar]
- McVean GAT, Charlesworth B.. 1999. A population genetic model for the evolution of synonymous codon usage: patterns and predictions. Genet Res. 74(2):145–158. [Google Scholar]
- Miura F, et al. 2006. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proc Natl Acad Sci U S A. 103(47):17846–17851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagalakshmi U, et al. 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320(5881):1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T.. 1986. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 3:418–426. [DOI] [PubMed] [Google Scholar]
- Pál C, Papp B, Hurst LD.. 2001. Highly expressed genes in yeast evolve slowly. Genetics 158(2):927–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Chen X, Yang J-R, Zhang J.. 2013. Differential requirements for mRNA folding partially explain why highly expressed proteins evolve slowly. Proc Natl Acad Sci U S A. 110(8):E678–E686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Qian W, Zhang J.. 2012. Genomic evidence for elevated mutation rates in highly expressed genes. EMBO Rep. 13(12):1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin JB, Kudla G.. 2011. Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet. 12(1):32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2015. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing; Available from: http://www.R-project.org/. [Google Scholar]
- Rocha EPC. 2004. Codon usage bias from tRNA’s point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 14(11):2279–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha EPC, Danchin A.. 2004. An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol Biol Evol. 21(1):108–116. [DOI] [PubMed] [Google Scholar]
- Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB.. 2013. Rate-limiting steps in yeast protein translation. Cell 153(7):1589–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly DA, et al. 2013. Integrative phenomics reveals insight into the structure of phenotypic diversity in budding yeast. Genome Res. 23(9):1496–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skelly DA, Ronald J, Connelly CF, Akey JM.. 2009. Population genomics of intron splicing in 38 Saccharomyces cerevisiae genome sequences. Genome Biol Evol. 1:466–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E.. 2010. Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci U S A. 107(8):3645–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavouri T, Semple JI, Garcia-Verdugo R, Lehner B.. 2009. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell 138(1):198–208. [DOI] [PubMed] [Google Scholar]
- Wall DP, et al. 2005. Functional genomic analysis of the rates of protein evolution. Proc Natl Acad Sci U S A. 102(15):5483–5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DE, et al. 2016. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 14(7):1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, et al. 2009. Bidirectional promoters generate pervasive transcription in yeast. Nature 457(7232):1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J-R, Chen X, Zhang J.. 2014. Codon-by-codon modulation of translational speed and accuracy via mRNA folding. PLoS Biol. 12(7):e1001910.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J-R, Liao B-Y, Zhuang S-M, Zhang J.. 2012. Protein misinteraction avoidance causes highly expressed proteins to evolve slowly. Proc Natl Acad Sci U S A. 109(14):E831–E840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J-R, Zhuang S-M, Zhang J.. 2010. Impact of translational error-induced and error-free misfolding on the rate of protein evolution. Mol Syst Biol. 6:421.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassour M, et al. 2009. Ab initio construction of a eukaryotic transcriptome by massively parallel mRNA sequencing. Proc Natl Acad Sci U S A. 106(9):3264–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang J-R.. 2015. Determinants of the rate of protein sequence evolution. Nat Rev Genet. 16(7):409–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Schroeder S, Fong N, Bentley DL.. 2005. Altered nucleosome occupancy and histone H3K4 methylation in response to ‘transcriptional stress’. EMBO J. 24(13):2379–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Gu W, Wilke CO.. 2010. Detecting positive and purifying selection at synonymous sites in yeast and worm. Mol Biol Evol. 27(8):1912–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA.. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci U S A. 111(22):E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.