

Summary
Overexpression of platelet-derived growth factor recep tor alpha (PDGFR-A) has been documented in association with primary tumors and metastasis in medulloblastoma. Tumors from our genetically engineered SHH-driven medulloblastoma mouse model overexpress PDGFR-A in primary tumors and thus this mouse model is a good platform with which to study the role of PDGFR-A in this central nervous system malignancy. We hypothesized that inhibition of PDGFR-A in medulloblastoma can slow or inhibit tumor progression in living individuals. To test our hypothesis, we targeted PDGFR-a mediated tumor growth in vitro and in vivo using the tyrosine kinase inhibitor, tandutinib (MLN-518), which strongly inhibits PDGFR-A. Although PDGFR-A inhibition by this agent resulted in reduced mouse tumor cell growth and increased apoptosis in vitro, and reduced tumor cell proliferation in vivo, tandutinib did reduce tumor volume at the doses tested (360 mg/kg) in vivo. Thus, tandutinib may be an agent of interest for Shh-driven medulloblastoma if a synergistic drug combination can be identified.
Keywords: medulloblastoma, tandutinib, platelet-derived growth factor receptor, virtual histology
Medulloblastoma is the most common malignant brain tumor of children.1 In average-risk patients older than 3 years, therapies with surgical resection, chemotherapy, and craniospinal radiation lead to >80% 5-year overall survival, whereas survival for infants is approximately 70%.2–4 However, radiation treatment also results in cognitive impairment, psychiatric effects, endocrine disorders, and growth retardation depending upon the age of the child.4 Thus, the major therapeutic challenge is to avoid radiation and replace current treatments with novel agents that cross the blood-brain barrier to target tumor cell-specific signaling pathways while avoiding toxicity to normal tissue.
Platelet-derived growth factor receptor alpha (PDGFR-A) has been shown to be expressed or activated in metastasis-prone human medulloblastoma in multiple studies.5–7 Whether PDGFR-A acts as a homodimer or a heterodimer in medulloblastoma remains to be shown, however, given that PDGFR-B is also expressed.8 In vitro studies suggest that these 2 receptors may act at least as independent homodimers given that PDGFR-A shows increased kinase activity in the presence of PDGFR-A homodimer specific ligand, PDGF-AA, and that PDGFR-B shows increased kinase activity in the presence of PDGFR-B homodimer specific ligand, PDGF-BB.9 In these studies, PDGF-AA and PDGF-BB both result in increased tumor cell proliferation in vitro,9 perhaps as an autocrine loop.10 The PDGFR-A specific ligand, PDGF-CC, is also present in human medulloblastoma.9
Tandutinib (MLN518, CT53518) is a second-generation type II kinase inhibitor that has strong activity against PDGFR-A (Kd = 2.4 nM) and the related kinases PDGFR-B, Flt-3, CSF1R, and c-Kit.[11–13] The original phase I studies of tandutinib investigated safety and efficacy in acute myelogenous leukemia,14 whereas more recent phase I trials for recurrent glioblastoma have also established that a reasonable brain tumor to plasma ratio of 0.33 can be achieved.15 Therefore, given the potential therapeutic target of PDGFR-A in medulloblastoma and the availability of an inhibitor with central nervous system (CNS) penetration, we tested tandutinib for activity in our genetically engineered Ptch1, p53 conditional preclinical mouse model of medulloblastoma.16
METHODS
Mice, Drug Administration, and Tumor Monitoring
All animal procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio. Medulloblastoma-prone mice were generated and genotyped as previously described in this model with 100% penetrance.16 Tumor-prone mice genotypes were Ptch1 F1-2m/WT Trp53 F2-10/F2-10 Pax7 ICNm/WT. Tandutinib (MLN518) was purchased from ChemieTek (Indianapolis, IN). Tandutinib structure and purity was verified by 1H-NMR at ChemieTek (data not shown).
Tandutinib was dissolved in 0.5% methylcellulose in water and administered at a dose of 360 mg/kg divided twice daily by oral gavage from postnatal day 21 (p21) to postnatal day 43 (p43), which is a similar experimental design to our previous studies whereby drug treatment starts when tumors are initiated but not substantially progressed.17,18 Mice were then humanely euthanized and processed for microCT-based virtual histology.
RNA Isolation and RT-PCR
RNA isolation and reverse transcription-polymerase chain reaction (RT-PCR) were performed as previously described.16 In quantitative RT-PCR experiments, PDGFR-A was measured relative to Pgk1. Primers were as follows: for mouse PDGFR-A, 5′-CCATGCAGTTGCCTTACGAC-3′ (KN46) and 5′-AGAGCCTGCTTTTCACTAGACC-3′ (KN47) were used. For mouse Pgk1, 5′-GAGCCCATAGC TCCATGGT-3′ (KN40) and 5′-CAGTAGCTTGGCCAGT CTTG-3′ (KN41) were used.
Western Blotting
For western blotting, tumor tissues were lysed in RIPA buffer containing a phosphatase inhibitor and a protease inhibitor (Thermo Fisher Scientific, Waltham, MA). Lysates were homogenized and centrifuged at 12,000 g to remove debris. The resulting supernatants were used for immunoblot analysis. Rabbit anti-PDGFR-A (Cell Signaling Technology, Beverly, MA) and rabbit antiphospho PDGFR-A (Y754) (Abcam, Cambridge, MA) were used at a concentration of 1:1000, whereas mouse anti-β-actin (Sigma, St Louis, MI) was used at 1:1,000,000. Appropriate horseradish peroxidase-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA) were used at 1:5000 and chemiluminescence was detected using Western Pico substrate (Pierce Biotechnology, Rockford, IL).
IC50 Determinations
For in vitro cell growth inhibition assay, medulloblastoma primary cell cultures19 were plated in 96-well plates (2×103 cells per well) and cultured overnight. The cells were treated with different concentrations of tanduti nib dissolved in 1% DMSO (0 to 100 μM). Cells were then incubated for an additional 72 hours, followed with CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI).
MicroCT-based Virtual Histology and Histopathology
The methodology for staining, scanning, and image postprocessing and analysis has been described previously.20 After scanning, the virtual histology stained brains were paraffin-embedded, sectioned, and stained with hematoxylin and eosin (H&E).
Immunocytochemistry
Cleaved Caspase 3 immunocytochemistry was performed per manufacturer’s protocol (Cell Signaling Technology). In brief, U26159 mouse medulloblastoma cells were plated on glass coverslips at a density of 5×104 cells/well of a 24-well plate. Cells were incubated under growth conditions with or without indicated pharmacological inhibitors for 24 hours. At 24 hours postinhibitor treatment, the cells were fixed with prewarmed 4% paraformaldehyde in PBS for 15 minutes at room temperature. For immunocytochemistry, the cells were rinsed with PBS and incubated with blocking buffer (5% normal goat serum, 0.3% Triton X-100 in 1X PBS) for 60 minutes at room temperature. Primary antibodies [Anti- Cleaved Caspase 3 (Cell Signaling cat# 9661) at 1:1600 and β1-tubulin (E7 ascites from Developmental Studies Hybridoma Bank) at 1:5000] were diluted in antibody dilution buffer (1% BSA, 0.3% Triton X-100 in 1X PBS). After blocking, cells were incubated overnight at 4°C with diluted primary antibody solution. Cells were rinsed 3 times, 5 minutes each with PBS at room temperature. Secondary antibodies were diluted in antibody dilution buffer (Alexa-488 conjugated goat antimouse and Alexa- 594 conjugated goat antirabbit; both 1:1000). Cells were incubated with secondary antibodies at room temperature for 80 minutes, followed by washing with 1X PBS (3 times, 10 minutes each). The final wash included Hoechst 33342 to stain nuclei. After 1 rinse with distilled water, the coverslips were mounted on slides with Elvanol. Fluorescent images were acquired using a cooled CCD camera (Hamamatsu Photonics) attached to a Zeiss Axioplan 2 (Carl Zeiss) inverted microscope with a ×10 or ×20 lens. For quantification of apoptotic cells captured images were processed using Image J and Adobe Photoshop CS2.
FACS Analysis
Vehicle (0.1% DMSO), 1 μM, or 10 μM tandutinibtreated cells were gently trypsinized for 10 minutes. After neutralization of tryspin, the cells were centrifuged at 300 g for 5 minutes at 4°C, washed with PBS and centrifuged at 300 g. The resulting washed cell pellet was resuspended in 300 μL PBS and fixed with by dropwise addition of 70 μL of prechilled (−20°C) 100% ethanol followed by 15-minute incubation at 4°C. The fixed cells were pelleted by centrifugation at 300 g for 5 minutes and rinsed once with PBS. The final washed pellet consisting of fixed U26159 cells was incubated with Propidium Iodide (20 μg/mL) plus RNase (200 μg/mL) for 30 minutes at room temperature. PI-stained cells were sorted using a Becton Dickinson FACS Calibur and data were analyzed for cell cycle statistics using FCS Express software. Graphing was performed using Microsoft Excel in the form of average plus standard error of means representing a minimum of 400 cells per condition.
Statistical Analysis
For comparison between wild type and tumor assay measurements, we used the unpaired Student t test.
RESULTS
PDGRF-A is Overexpressed in Mouse Medulloblastoma
To determine whether PDGFR-A was overexpressed in our preclinical model of medulloblastoma, we compared expression by quantitative RT-PCR in mice. Medulloblastoma tumor tissue expressed PDGFR-A at a 60-fold higher level than age-matched control 8-week-old wild type mouse cerebella, and 12-fold higher than 9-day postnatal (p9) wild type mouse cerebella (P<0.01) (Fig. 1A). To confirm that PDGFR-A expression was also reflected at the protein level, we performed western blotting. Mouse medulloblastoma tissues demonstrated both higher total PDGFR-A and elevated phospho-Pdgfr (activated form) expression compared with 8-week-old wild type cerebella (Fig. 1B). Thus, PDGFR-A is not only uniformly expressed in the medulloblastoma model, but this receptor tyrosine kinase is also activated.
FIGURE 1.
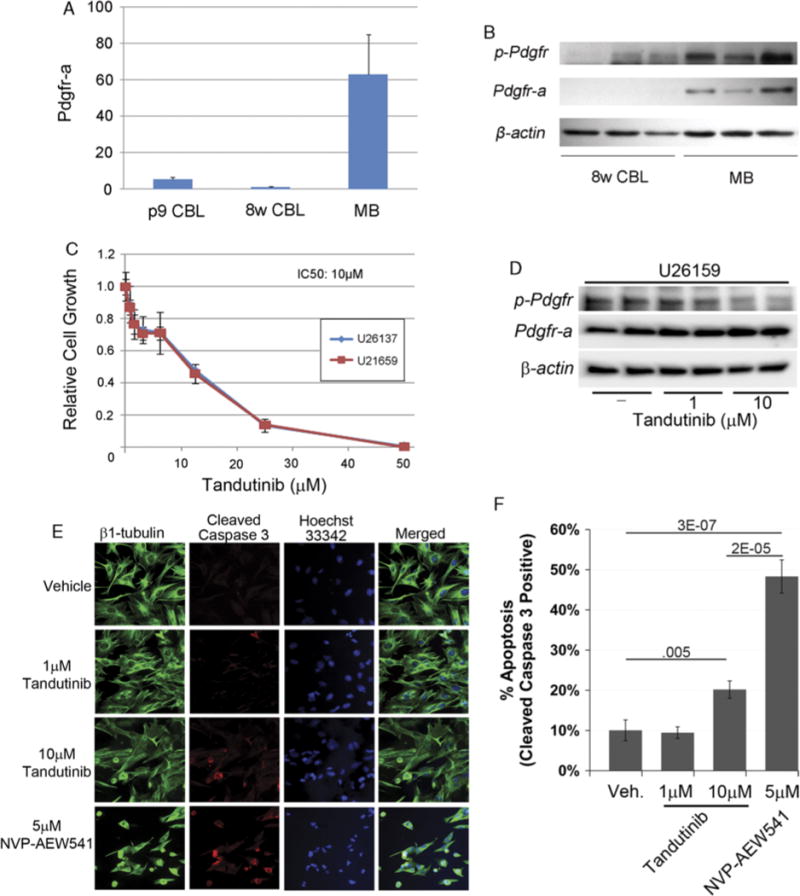
Validation of drug target and response to tandutinib in vitro and in vivo. A, Quantitative RT-PCR demonstrates overexpression of PDGFR-A in the preclinical medulloblastoma model tissue compared with nontumor control tissue. p9 WT CBL indicates postnatal 9 day wild type mouse cerebellum; 8w WT CBL, 8-week-old wild type mouse cerebellum; MB tumor, medulloblastoma mouse tumor, n = 5 each. B, Western blotting for PDGFR-A and phosphorylated Pdgfr shows high level of protein expression and activation in the preclinical model tumors relative to WT cerebellum. β-actin was used for internal control. C, Cell growth inhibition assay establishing the tandutinib IC50 in medulloblastoma. D, Western blotting for PDGFR-A and phosphorylated Pdgfr for medulloblastoma primary cell culture treated with medulloblastoma. E and F, Immunocytochemistry for Cleaved Caspase 3 at 1 and 10 μM tandutinib. The Igf1r inhibitor NVP-AEW541 is shown as an apoptosis control.
Downregulation of Phosphorylated Pdgfr Inhibits Mouse Tumor Cell Growth in Vitro
To investigate whether the partially selective PDGFR antagonist tandutinib inhibits growth of medulloblastoma, we treated mouse medulloblastoma primary cell cultures (U26137 and U21659) with tandutinib (0 to 100 μM). The growth of both cell lines was inhibited by 50% at drug concentrations of 10 μM at 72 hours (ie, IC50 = 10 μM) (Fig. 1C). We confirmed that tandutinib growth inhibition was coincident with a target-specific decrease in phospho-Pdgfr at 10 μM (Fig. 1D). We also tested whether tandutinib action was tumoristatic at this dose, and affirmed an increase in apoptosis at 10 μM as measured by Cleaved Caspase 3 immunocytochemistry (Figs. 1E–F). Of note, however, the statistically significant induction of Cleaved Caspase 3 was nonetheless 2-fold lower than the Igf1-r inhibitor, NVP-AEW541.
Tandutinib Does not Result in Therapeutic Efficacy in Medulloblastoma at the Doses Tested
The highest tolerated dose of tandutinib in vivo was taken from an earlier preclinical study.21 From a tolerability standpoint, no definite correlation existed between body weight and cerebellum volume for either the treatment or control group (Fig. 2A), although body weights and cerebellar volumes were noticeably lower in 2 instances for tandutinib-treated mice than mice from previous studies22 (see Discussion). From the point of view of therapeutic efficacy, tandutinib treatment over a 3-week period did not affect cerebellar volume, which we have established as a surrogate for tumor volume (Fig. 2B).22 However, the frequency of mitoses was significantly lower in the tandutinib-treated group (Fig. 2C). Representative virtual histology and histopathology images (for vehicle and tandutinib-treated mice) are given in Figs. 2D and E, and Figs. 2F and G, respectively.
FIGURE 2.
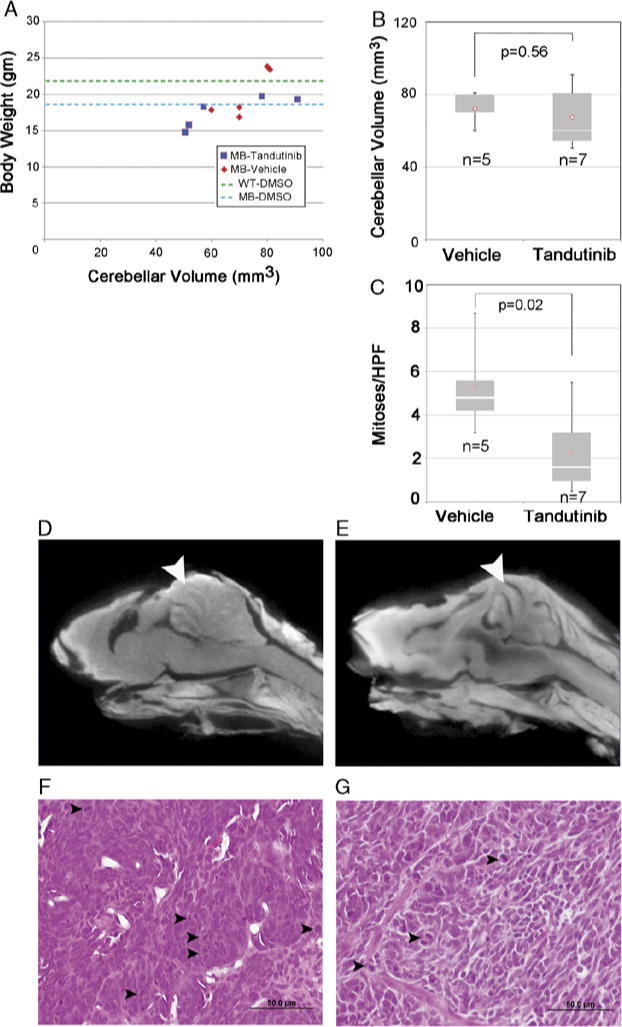
In vivo effect of tandutinib in medulloblastoma. A, Animal weights and cerebellar volume at the end of treatment. C, Bar graph of mitoses per high power field (HPF). Tandutinib treated tumor showed significantly fewer mitosis than vehicle treated tumor. P = 0.02. B, Bar graph of cerebellar volumes after 3 weeks treatment. Control mice were treated with 0.5% methylcellulose in water. D to E, Representative virtual histology for (D) control tumors and (E) tandutinib-treated tumors. F and G, Representative H&E histology for (F) control tumors and (G) tandutinib-treated tumors. No evidence of differentiation was seen. Arrowheads indicate mitotic figures. Scale Bar: 50 μm.
DISCUSSION
In our study we have shown that PDGFR-A is upregulated and activated in our Ptch1, p53 mouse medulloblastoma model as is true for metastasis-prone human medulloblastoma.5–7 We had hypothesized that inhibition of PDGFR-A in medulloblastoma could slow or inhibit tumor growth and metastasis in living individuals. Downregulation of PDGFR-A activation using tandutinib inhibited mouse tumor cell growth and induced apoptosis in vitro at modest concentrations (IC50: 10 μM). That PDGFR-A might play a role in cerebellar tumor biology is not a surprise given that PDGFR-A is required during embryogenesis and postnatal brain development, with essential roles in neural crest and CNS including astrocytes, oligodendrocyte, and subsets of developing neurons.23 In the hindbrain, inhibition of the PDGFR-A pathway causes hypomyelination in cerebellum,24 which in light of limited instances of small cerebellar volumes even relative to bodyweight in some of our tandutinib-treated mice (Fig. 1E) might be a consideration as a potential morbidity of PDGFR-A targeting therapeutic strategies.
We performed tandutinib treatment from postnatal day 21 (p21), when symptoms are not apparent, until p43 (a 3 wk course) at a dose of 360 mg/kg divided twice daily by oral gavage. There was no significant reduction in tumor growth between tandutinib-treated and tandutinib-untreated mice using cerebellar volume as a surrogate biomarker, but a significant decrease in tumor cell mitoses was seen in the tandutinib-treated group. The disparity between tumor volume and mitotic count could reflect either a tumoristatic role of tandutinib (consistent with a relatively low degree of apoptosis induction in vitro), or a failure of tandutinib crossing the blood-brain barrier to reach a critical threshold concentration that will sufficiently induce apoptosis (consistent with the narrow dose dependency of apoptosis induction demonstrated in our in vitro studies). From the perspective of therapeutic index and tolerability, Griswold et al21 analyzed the effect of tandutinib on murine hematopoiesis after chemotherapy-induced myelosuppression using the same dosage as our study and found tandutinib to have only mild toxicity. As tandutinib has been reported to have good brain penetration in humans and rodents with glioblastoma (brain:plasma ratio 0.33), a single-agent phase 2 study is currently underway.15 We speculate that if we increased treatment dose and period, efficiency in our preclinical model of medulloblastoma might be achieved.
Medulloblastoma has been classified as having 4 to 5 major molecular phenotypes, of which the Shh-driven phenotype may account for as much as 30% to 35% of cases.25 Loss of function of p53 is also documented in 7% to 16% of medulloblastomas,26,27 and of these p53 mutant tumors approximately 10% are Shh-driven. Because PDGFR-A is an established downstream target of Shh signaling in medulloblastoma,28 we speculate that our conditional Ptch1, p53 mouse model results apply to the larger fraction of Shh-driven medulloblastomas (30% to 35%). Our results may apply to these Shh-driven medulloblastoma, for which PDGFR-A has been shown to be a transcriptional target.28 Instead of a role only for tumor growth, PDGFR-A may also mediate leptomeningeal metastasis. Future experiments would be needed to test this hypothesis. Furthermore, in light of the recent report that p53 loss of function is a poor prognostic factor in medulloblastoma,29 and that PDGFR-A and Pdgfr-b transcription can be negatively regulated by p53,16 we speculate that Pdgf receptors may also, in part, mediate progression in medulloblastomas that lose p53 function.
In summary, tandutinib was efficacious in vitro and reduced the number of mitoses in vivo; however, tandutinib did not result in reduced tumor volume—a critical endpoint of single-agent efficacy. These results establish a biological basis for further study of tandutinib in medulloblastoma, perhaps in combination with other CNS-penetrating chemotherapy or molecularly targeted agents (1 example would be clinicaltrials.gov NCT00667394, tandutinib plus bevacizumab to treat recurrent brain tumors). As a prelude to studying combinations with tandutinib, pharmacokinetic optimization may be necessary, a closer examination of developmental toxicology as related to postnatal cerebellar development should be performed, and a closer examination of PDGFR-A heterodimeric partners may also be warranted. The combination of tandutinib with chemotherapy or a synergist targeted agent such as an Igf1r inhibitor may be another area of possible interest and investigation.
Acknowledgments
The authors thank Michelle Brady for histology services.
Supported in full by an $8600 Grant from the Rally Foundation in honor of Matthew Butterfield to the Pediatric Preclinical Testing Initiative.
Footnotes
C.K. received a honorarium to present at Research & Development grand rounds at Millennium Pharmaceuticals (a Takeda company). C.K. is a cofounder of Numira Biosciences, a preclinical imaging biotechnology company.
The other authors have no other conflicts of interest or financial disclosures.
References
- 1.Rutkowski S, Gerber NU, von Hoff K, et al. Treatment of early childhood medulloblastoma by postoperative chemotherapy and deferred radiotherapy. Neuro Oncol. 2009;11:201–210. doi: 10.1215/15228517-2008-084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Packer RJ, Gajjar A, Vezina G, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006;24:4202–4208. doi: 10.1200/JCO.2006.06.4980. [DOI] [PubMed] [Google Scholar]
- 3.Gajjar A, Chintagumpala M, Ashley D, et al. Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol. 2006;7:813–820. doi: 10.1016/S1470-2045(06)70867-1. [DOI] [PubMed] [Google Scholar]
- 4.Lafay-Cousin L, Strother D. Current treatment approaches for infants with malignant central nervous system tumors. Oncologist. 2009;14:433–444. doi: 10.1634/theoncologist.2008-0193. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald TJ, Brown KM, LaFleur B, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nat Genet. 2001;29:143–152. doi: 10.1038/ng731. [DOI] [PubMed] [Google Scholar]
- 6.Huber H, Eggert A, Janss AJ, et al. Angiogenic profile of childhood primitive neuroectodermal brain tumours/medulloblastomas. Eur J Cancer. 2001;37:2064–2072. doi: 10.1016/s0959-8049(01)00225-8. [DOI] [PubMed] [Google Scholar]
- 7.Chopra A, Brown KM, Rood BR, et al. The use of gene expression analysis to gain insights into signaling mechanisms of metastatic medulloblastoma. Pediatr Neurosurg. 2003;39:68–74. doi: 10.1159/000071317. [DOI] [PubMed] [Google Scholar]
- 8.Gilbertson RJ. Clifford SC: PDGFRB is overexpressed in metastatic medulloblastoma. Nat Genet. 2003;35:197–198. doi: 10.1038/ng1103-197. [DOI] [PubMed] [Google Scholar]
- 9.Andrae J, Molander C, Smits A, et al. Platelet-derived growth factor-B and -C and active alpha-receptors in medulloblastoma cells. Biochem Biophys Res Commun. 2002;296:604–611. doi: 10.1016/s0006-291x(02)00917-8. [DOI] [PubMed] [Google Scholar]
- 10.Whelan HT. Medulloblastoma cell line. Pediatr Neurol. 1990;6:282. doi: 10.1016/0887-8994(90)90127-m. [DOI] [PubMed] [Google Scholar]
- 11.Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- 12.Pandey A, Volkots DL, Seroogy JM, et al. Identification of orally active, potent, and selective 4-piperazinylquina-zolines as antagonists of the platelet-derived growth factor receptor tyrosine kinase family. J Med Chem. 2002;45:3772–3793. doi: 10.1021/jm020143r. [DOI] [PubMed] [Google Scholar]
- 13.Kelly LM, Yu JC, Boulton CL, et al. CT53518, a novel selective FLT3 antagonist for the treatment of acute myelo-genous leukemia (AML) Cancer Cell. 2002;1:421–432. doi: 10.1016/s1535-6108(02)00070-3. [DOI] [PubMed] [Google Scholar]
- 14.DeAngelo DJ, Stone RM, Heaney ML, et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108:3674–3681. doi: 10.1182/blood-2006-02-005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Supko JG, Grossman SA, Peereboom DM, et al. Feasibility and phase I trial of tandutinib in patients with recurrent glioblastoma. J Clin Oncol (Meeting Abstracts) 2009;27:2039. [Google Scholar]
- 16.Taniguchi E, Nishijo K, McCleish AT, et al. PDGFR-A is a therapeutic target in alveolar rhabdomyosarcoma. Oncogene. 2008;27:6550–6560. doi: 10.1038/onc.2008.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prajapati SI, Kilcoyne A, Samano AK, et al. MicroCT-based virtual histology evaluation of preclinical medulloblastoma. Mol Imaging Biol. 2010 doi: 10.1007/s11307-010-0372-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samano AK, Ohshima-Hosoyama S, Whitney TG, et al. Functional evaluation of therapeutic response for a mouse model of medulloblastoma. Transgenic Res. 2010;19:829–840. doi: 10.1007/s11248-010-9361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi E, Cho MJ, Arenkiel BR, et al. Bortezomib reverses a post-translational mechanism of tumorigenesis for patched1 haploinsufficiency in medulloblastoma. Pediatr Blood Cancer. 2009;53:136–144. doi: 10.1002/pbc.21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prajapati SI, Kilcoyne A, Samano AK, et al. Micro-CT based virtual histology evaluation of preclinical medulloblastoma. Mol Imaging Biol. 2010 doi: 10.1007/s11307-010-0372-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griswold IJ, Shen LJ, La Rosee P, et al. Effects of MLN518, a dual FLT3 and KIT inhibitor, on normal and malignant hematopoiesis. Blood. 2004;104:2912–2918. doi: 10.1182/blood-2003-05-1669. [DOI] [PubMed] [Google Scholar]
- 22.Samano AK, Ohshima-Hosoyama S, Whitney TG, et al. Functional evaluation of therapeutic response for a mouse model of medulloblastoma. Transgenic Res. doi: 10.1007/s11248-010-9361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoch RV, Soriano P. Roles of PDGF in animal development. Development. 2003;130:4769–4784. doi: 10.1242/dev.00721. [DOI] [PubMed] [Google Scholar]
- 24.Klinghoffer RA, Hamilton TG, Hoch R, et al. An allelic series at the PDGFalphaR locus indicates unequal contributions of distinct signaling pathways during development. Dev Cell. 2002;2:103–113. doi: 10.1016/s1534-5807(01)00103-4. [DOI] [PubMed] [Google Scholar]
- 25.Thompson MC, Fuller C, Hogg TL, et al. Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol. 2006;24:1924–1931. doi: 10.1200/JCO.2005.04.4974. [DOI] [PubMed] [Google Scholar]
- 26.Pfaff E, Remke M, Sturm D, et al. TP53 mutation is frequently associated with CTNNB1 mutation or MYCN amplification and is compatible with long-term survival in medulloblastoma. J Clin Oncol. 2010;28:5188–5196. doi: 10.1200/JCO.2010.31.1670. [DOI] [PubMed] [Google Scholar]
- 27.Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol. 2010;28:1345–1350. doi: 10.1200/JCO.2009.23.5952. [DOI] [PubMed] [Google Scholar]
- 28.Mao J, Ligon KL, Rakhlin EY, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res. 2006;66:10171–10178. doi: 10.1158/0008-5472.CAN-06-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tabori U, Baskin B, Shago M, et al. Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol. 28:1345–1350. doi: 10.1200/JCO.2009.23.5952. [DOI] [PubMed] [Google Scholar]