

Abstract
Purpose of review.
The current knowledge of pathophysiological and molecular mechanisms responsible for the genesis and development of heart failure (HF) is absolutely vast. Nonetheless, the hiatus between experimental findings and therapeutic options remains too deep, while the available pharmacological treatments are mostly seasoned and display limited efficacy. The necessity to identify new, non-pharmacological strategies to target molecular alterations led investigators, already many years ago, to propose gene therapy for HF. Here, we will review some of the strategies proposed over the past years to target major pathogenic mechanisms/factors responsible for severe cardiac injury developing into HF and will provide arguments in favor of the necessity to keep alive research on this topic.
Recent findings.
After decades of preclinical research and phases of enthusiasm and disappointment, clinical trials were finally launched in recent years. The first one to reach phase II and testing gene delivery of sarcoendoplasmic reticulum calcium ATPase did not yield encouraging results, however other trials are ongoing, more efficient viral vectors are being developed and promising new potential targets have been identified. For instance, recent research is focused on gene repair, in vivo, to treat heritable forms of HF, while strong experimental evidence indicates that specific microRNAs can be delivered to post-ischemic hearts to induce regeneration, a result that was previously thought possible only by using stem cell therapy.
Summary.
Gene therapy for HF is aging, but exciting perspectives are still very open.
Gene therapy: still an appealing option for the treatment of heart failure
Thanks to the joint effort of numerous investigators (1), the pharmacological treatment of heart failure (HF) is reaching a plateau that, save for possible major breakthroughs, will be hardly overcome in the near future. In fact, after decades of stalemate, only two drugs, namely sacubitril/valsartan and ivabradine, have been added to the seasoned pharmaceutical arsenal available to cardiologists for the treatment of this syndrome. (2) Unfortunately, severe HF is still characterized by very negative prognosis (1) and the therapeutic perspectives do not seem glaring, despite the cornucopia of studies that yielded a deep knowledge of cellular and molecular pathogenic mechanisms underlying this syndrome. The difficulty of pharmacologically targeting receptors and intracellular pathways involved in the pathogenesis of HF led investigators, many years ago (3), to propose cardiac gene therapy as an alternative therapeutic approach. The idea was simple and revolutionary at the same time: cell-directed delivery of exogenous genes (transgenes) would produce “curative” proteins able to compensate for pathological downregulations or to counteract detrimental molecular processes. Investigators in the field soon realized that the careful choice of proper targets and the availability of efficient gene carriers were critical requisites for the success of this therapeutic approach.
The history of cardiac gene therapy began with a seminal study published by Dr. Leiden’s group in 1990 (4). Those authors transfected rat cardiomyocytes, in vivo, by injecting plasmid DNA containing the β-galactosidase gene directly in left ventricular wall. β-galactosidase activity was then found in myocardium for up to 4 weeks. The 3 decades following that milestone report witnessed a real hype for HF gene therapy, which, after formidable efforts and persistence of courageous investigators, culminated with the first clinical trials in recent years.
HF is a complex, multifactorial syndrome. That is why authors have tested very diverse targets ranging from enzymes to structural proteins and cytoprotective factors. Here, we will review some of the strategies proposed over the past years to target major pathogenic mechanisms/factors responsible for severe cardiac injury developing into HF, and will provide arguments in favor of the necessity to keep alive research on this topic. We refer the reader to other reviews covering in detail the principles of gene therapy and its numerous applications in the cardiovascular field. (5–8)
Rescuing SERCA2a function
Sarcoendoplasmic reticulum calcium ATPase (SERCA2a) is a protein pump critically important for cytosolic Ca2+ homeostasis in cardiac myocytes. It works in coordination with ryanodine channels and Na+/Ca2+ exchangers, removing Ca2+ from the cytosol during the repolarization/relaxation phase. SERCA2a activity is modulated by the integral membrane protein phospholamban (PLN) via inhibitory phosphorylation. In turn, PLN is inhibited by phosphorylation with consequent release of the brake on SERCA2a and enhanced Ca2+ uptake by sarcoplasmic reticulum (9). There is evidence that SERCA2a is downregulated in failing hearts, leading to reduced Ca2+ re-uptake by the sarcoplasmic reticulum during diastole, hence impaired relaxation, higher energy requirements and predisposition to arrhythmias. Moreover, Ca2+ depletion in the sarcoplasmic reticulum has serious repercussions on the excitation/contraction coupling and therefore cardiac force generation (6, 10–11). Experimental models proved the concept that PLN gene deletion or forced expression of a constitutively phosphorylated form of PLN result in improved heart function. (12–14) But the hypothesis that ultimately led to a clinical trial was that SERCA2a gene transfer to cardiac cells might restore the physiological expression of this molecular pump and increase cardiac contractility in HF (15). After successful tests in isolated human cardiomyocytes (15), murine (16) and porcine models (17), this strategy was finally translated to the clinics, with the initiation in 2012 of CUPID (Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease), the first clinical trial of cardiac gene therapy for HF. Replication defective adeno-associated virus of serotype 1 (AAV1) was the vector chosen to carry SERCA2a transgenes. While the initial phases demonstrated safety and some beneficial effects (18), the final Phase 2b CUPID was unsuccessful. It involved 250 patients receiving intracoronary AAV1/SERCA2a or placebo. The treatment with AAV1/SERCA2a failed to prolong the time to the first terminal event. (19–20) Compared to placebo, treatment with AAV1/SERCA2a had no significant effect on any endpoints, including NYHA Functional class, 6-minute walk test distance or NT-proBNP levels (20).
Despite its failure, CUPID2b finally made gene therapy “palpable” in clinical cardiology after decades of preclinical research. The potential factors that caused the negative outcome, rather than discouraging the reiteration of similar attempts, should be carefully analyzed and spur further research and optimization. Possible problems relied in the selection of the patient population or/and of the endpoints, but also in the insufficient delivery or intracellular transfer of SERCA2a gene. In the CUPID2b trial, myocardial uptake of AAV vectors was ~1000 times lower than in animal models (20). Therefore, attempts should be made to improve transduction efficiency. For instance, the anterograde coronary delivery could perhaps be replaced by retrograde perfusion to prolong the transit time of the vector in capillaries. Another major issue is the low transduction efficiency of the AAV itself. Increasing the vector dose is not necessarily the right solution. Several teams tried to reengineer AAVs. For instance, some authors exploited the mechanism of “directed evolution”, a method of in vitro evolution followed by in vivo selection of AAV capsids targeting a certain organ, to generate an AAV mutant with cardiac-specific tropism (21). They obtained a capsid gene clone named M41 that displayed tenfold higher transduction efficiency in cardiac tissue compared with the liver. Another team of investigators transduced large animal hearts using reengineered AAVs, named BNP116, to deliver Protein Phosphatase Inhibitor-1 Gene (20). BNP116 is a synthetic AAV vector developed by site-directed mutagenesis on the heparan sulfate receptor footprint (22). This chimeric vector encompassing the serotypes AAV2 and 8 (AAV2i8) transduced a wide range of muscle groups in the murine forearms and legs as well as cardiac, intercostal and facial muscles (22). These encouraging results paved the way for an upcoming clinical gene therapy trial with AAV2i8 in 12 patients with advanced HF (7). AAV2i8 ability to efficiently transduce not only cardiac muscle, but also various skeletal muscle groups with 2–3 times higher transduction efficiency relative to the liver was definitely a major advancement in the field, particularly promising for the treatment of some forms of muscular dystrophy, as better detailed in the next paragraph.
Genetic correction of Duchenne muscular dystrophy/Duchenne cardiomyopathy
Duchenne muscular dystrophy (DMD) is the most common muscular dystrophy and is caused by gene mutations leading to deficiency of the protein dystrophin, a large structural protein that anchors the inner surface of muscle sarcolemma to F-actin (23). The structure of sarcolemma lacking dystrophin is unstable, causing muscle damage and triggering an inflammatory response, which leads to further damage, necrosis, and fibrosis. Dystrophin is mainly expressed in cardiac and skeletal muscle, which are the first organs to be affected by degeneration and weakening, yet DMD is a multi–system disorder resulting in severe physical disability and premature death (24–25).
The current therapy of DMD is merely palliative, based on corticosteroids and physical therapy (26). Given the monogenic origin of this disease, gene therapy is the obvious, ideal approach for a radical cure (27). However, despite the remarkable number of promising studies performed in animal models, the first clinical trials are still in the phase of planning and recruitment (28–29). As for other types of HF, AAVs are considered the vectors of choice also for DMD (30–32), but their key limitations emerge even more evident in this case, due to the necessity of whole body transduction and life-long transgene expression (29). To date, only one clinical trial has been completed, examining intramuscular injection of a hybrid AAV2.5 vector in six patients. This therapy proved safe, however the patients did not express significant levels of dystrophin (33,34). Despite the predictable difficulties, the extensive data accumulated in preclinical studies in non-human primates (35,36) and dogs with naturally occurring DMD (37–39) will likely prompt new clinical trials (34, 40–42).
A major hurdle for gene therapy of DMD is the large size of the dystrophin gene, which cannot be packaged as full-length coding sequence in AAVs (43). Clever solutions have been tested to overcome this obstacle. One of them is the trans-splicing of dual or triple AAV vectors, which consists of dividing a large gene and then packaging in and delivering it by two or three AAVs. The complete gene is then expressed by dual or triple trans-splicing (44–46). Although this approach has been established, the transduction efficiency is still too low to achieve therapeutic levels of functional proteins. More recently, the dual AAV therapy to deliver a miniaturized form of the dystrophin gene (mini-dystrophin) has been successfully tested in a canine model, thus providing the first clear evidence of clinical translation potential (38).
Another approach proposed to treat DMD is genome editing for repair of the defective gene at its locus. The cured gene would then retain its native regulation. To date, nucleases and clustered regularly interspaced short palindromic repeats (CRISPRs) systems have been tested in several models of disease. Nuclease-mediated gene editing by using zinc-finger nucleases, TALENs (transcription activator-like effector nucleases) and meganucleases have proved efficacious for the treatment of muscle disorders in experimental and even clinical studies (47–51). Very recently, the CRISPR/Cas9 system has been delivered via AAVs to mice with dystrophin deficiency caused by a spontaneous mutation of the dystrophin gene (43). The aim was to remove exon 23 from the dystrophin gene, leading to the expression of a partially functional dystrophin in skeletal myofibers and cardiac muscle and enhance muscle force (52–54).
Exon skipping is an alternative method of gene editing based on RNA splicing system, which can be briefly described as follows. Under normal conditions, the native RNA transcribed from a gene is processed by RNA splicing that involves the removal of the introns. The mature mRNA consists of the remaining sequences named exons. By removing an exon that lies near the mutation, the downstream reading frame can be corrected and partially functional dystrophin can be restored. Exon skipping can be manipulated by delivering antisense oligonucleotides (AONs) that target particular sequences in exons. This has been tested with satisfactory results in small and large animal DMD models, followed by the failure of a number of clinical trials (29). However, after phase II/III clinical trials resulting in 23% increase in dystrophin positive fibers, Eteplirsen (AON inducing exon 51 skipping) (55) was approved to by FDA in 2016. Sadly, Eteplirsen targets only 13% of DMD cases. Other clinical trials based on exon skipping are challenged by the higher inefficiency of this system (56). Interestingly, it was shown that Dantrolene, a drug currently used for the treatment of the malignant hyperthermia, can enhance antisense-directed exon skipping (57). This might be a promising strategy to improve AON treatment and reduce the dose of delivered oligonucleotides, therefore lowering costs and potential toxicity.
Despite promising results in animal models, clinical trials are not showing solid and consistent results. The patient phenotype is much more complex than experimental models. For instance, the sarcolemma integrity in muscle fibers, essential for efficient and long-term gene therapy, is severely and variously compromised in DMD patients (58,59). Procedural risks and observed toxicity (60) as well as potential side effects, such as myositis and contractures (61) following the gene delivery should be considered. Precise clinical characterization, individualization and combined therapies could prove key to overcome existing limitations (51).
Other strategies with promising translational potential
AC6 (Adenylyl cyclase) catalyzes the conversion of ATP to cyclic adenosine monophosphate (cAMP), a molecule that is essential for cardiac function. cAMP is generated in response to β-adrenoceptors stimulation by catecholamines and modulates excitation-contraction coupling leading to positive inotropic effect (62). In a swine model of HF, AC6 gene transfer by adenoviral vectors improved heart function and reversed pathological LV remodeling (63). Cardiac AC6 overexpression prolonged life in mice with genetic cardiomyopathy (64), didn’t cause unbridled cAMP generation and reduced arrhythmias (65). Recently, the safety and efficacy of intracoronary delivery of AC6 carried by adenoviral vectors has been assessed in patients with symptomatic HF (EF ≤40%) in a multicenter, double-blind, placebo-controlled, phase 2 study (66). This intervention improved LV function more than the standard HF therapy. Larger trials are expected.
S100A1 is a protein that modulates sarcoplasmic reticulum Ca2+ cycling and mitochondrial function through interactions with the ryanodine receptor, SERCA2 and mitochondrial F1-ATPase activity. It exerts anti-hypertrophic, inotropic and antiarrhythmic effects and attenuates energy depletion in HF (67,68). S100A1 is significantly downregulated in human as well as various animal models of HF (67–69), therefore this alteration is a potential target for corrective gene therapy. In fact, AAV9-S100A1 retrograde coronary gene transfer in myocardium rescued cardiac contractile function in a preclinical large animal model (69). Moreover, AAV6-S100A1 is safe (70). In 2016 the Dutch company UniQure has developed gene therapy with AMT-126 (AAV-S100A1) to selectively restore cardiac deficiency of S100A1 (71). MicroRNA-138 can be considered as another related target, since it causes hypoxia-induced endothelial cell dysfunction by downregulating S100A1 (72). Transfection of endothelial cells with a miR-138 mimics reduced S100A1–3’UTR reporter gene expression, while transfection with an anti-miR-138 prevented the hypoxia-induced downregulation of the reporter gene (72). Experiments in vivo will be necessary to test the real efficacy of this strategy.
VEGF-B (Vascular Endothelial Growth Factor-B) selectively binds VEGFR-1, one of the five receptors of the VEGF family (73). VEGFR-1 is not involved in angiogenesis, but mediates the strong cytoprotective/antiapoptotic effects of VEGF-B (73,74). Such characteristics render VEGF-B an ideal candidate for gene therapy of dilated cardiomyopathy, a form of HF characterized by a remarkable increase in cardiac cell apoptosis (75,76), which is directly proportional to the rapidity of clinical deterioration (77). Despite its lower prevalence compared to ischemic cardiomyopathy as a cause of HF, dilated cardiomyopathy is particularly malignant and refractory to pharmacological treatments, accounting for approximately 50% cases of heart transplantation in the US (78). The ideal cure of it would consist of halting cardiac apoptosis, while, different from ischemic HF, angiogenesis is not necessary or even detrimental. After the first encouraging results in a rat model of myocardial infarction (79), we have tested cardiac gene delivery of VEGF-B carried by AAV9 vectors in a preclinical dog model of tachypacing-induced dilated cardiomyopathy. Both of direct intramyocardial injection (80) and intracoronary infusion (81) of AAV-VEGF-B proved markedly beneficial, causing a delay in the onset of cardiac decompensation and opposing the development of numerous functional, histological and molecular alterations, including apoptosis. Such positive results in a well-established experimental large animal model prompted us to start a pilot veterinary clinical trial in dogs with spontaneous dilated cardiomyopathy (personal communication). The results of this ongoing study will provide indications about the real potential of VEGF-B gene therapy.
SDF-1 (stromal cell-derived factor-1) was shown to be critical in cardiac stem cell therapy, as it affects stem cell homing, cardiomyocyte survival and ventricular remodeling in animal models of acute myocardial infarction or chronic HF (82). Later evidence suggested that SDF-1 alone is sufficient to induce cardiac repair (83). A first non-randomized, open-label clinical trial tested SDF-1 direct trans-endocardial delivery with the plasmid vector in seventeen subjects with ischemic cardiomyopathy. (84) Besides the modality of delivery, i.e. intramyocardial instead of intracoronary infusion and naked DNA instead of AAV-carried transgene, this strategy differed from those adopted in the SERCA2a and AC6 trials relatively to the mechanism of action: SDF-1 is a cytokine with no direct effects on cardiac function, but operates by stimulating endogenous repair processes including angiogenesis. The initial promising results prompted a Phase 2 blinded, placebo-controlled, multicenter trial that included 93 subjects with stable symptomatic HF (ejection fraction <40%) of ischemic etiology (85). This study failed to achieve its primary endpoint of an improved composite score at 4 months after treatment. However, patients with the lowest ejection fraction, who received the highest dose of pSDF-1, showed a 7-unit increase in ejection fraction compared with a 4-unit decrease found in placebo patients at 12 months. These results led the authors to the conclusion that the treatment is more specifically effective in patients with very depressed ejection fraction, which might represent the target population in subsequent trials. The following paragraph will review more extensively other new strategies of gene therapy for cardiac regeneration.
A new frontier: gene therapy to stimulate cardiac regeneration
Cardiac regeneration has been a leading topic in cardiological research over the past 15 years. The long chased dream is to block the evolution of post-ischemic pathological remodeling and progressive dysfunction, i.e. the most frequent form of HF, by replacing areas of dead myocardium with fully functional and vascularized newly formed tissue. The main strategies proposed to date are based on transplantation of pluripotent stem cells, such as myoblasts deriving from skeletal muscle or cardiac and non-cardiac progenitor stem cells differentiating into cardiomyocytes or other mature cell lines (86–89). Extensive research in this field has already led to clinical applications; nonetheless, the effective role and retention of transplanted cells in injured hearts remain controversial (89,90).
An alternative approach would consist of stimulating the proliferation of the surviving/viable tissue in injured hearts. This idea seems much more arduous, since it is based on the assumption that mature cardiomyocytes, which have very low renewal rate soon after birth (91), and/or other resident cell types might proliferate and reconstruct functional myocardial tissue formed by muscular parenchyma, vessels, stroma and organized conduction system. However, over the past few years, parallel studies conducted in different laboratories on the overexpression of various cell cycle regulators indicated that the stimulation of mature endogenous cardiomyocyte proliferation is a realistic option (5) and targeting the intracellular signals involved in these processes might prove a novel strategy of gene therapy for HF. For example, transgenic overexpression of cyclin A2, which is normally silenced in postnatal hearts, was reported to stimulate cardiomyocyte proliferation (92) and to be beneficial after myocardial infarction (93). Forced overexpression of the cyclin-dependent kinase 2 also caused an increase in nuclear antigens associated with DNA replication in adult mouse transgenic hearts, followed by a transient increase in cardiomyocyte proliferation and the appearance of less differentiated mononuclear cardiomyocytes (94). Transgenic mice overexpressing the early G1 cyclins displayed sustained DNA synthesis, but abnormal patterns of multinucleation (95), whereas the overexpression of cyclin D2 induced regenerative growth and infarct regression (95,96). Taken together, these studies in transgenics indicate the potential to achieve postnatal cardiomyocyte DNA synthesis and nuclear division. However, they did not provide conclusive evidence that karyokinesis was followed by cytokinesis and, most importantly, by proliferation of these cells. On the other hand, it was reported that the deletion of the homeodomain transcription factor Meis1, a regulator of normal cardiac development, was sufficient to extend the postnatal proliferative window of cardiomyocytes and to re-activate cardiomyocyte mitosis in the adult heart, with no adverse effects on cardiac function (97). Moreover, it was recently shown that Tbx20, a T-box gene required for cardiomyocyte proliferation during heart development, can act as a transcriptional repressor of Mes1 (98). Tbx20 overexpression increased cardiomyocyte proliferation and preserved cardiac function maintenance after myocardial infarction in adult mice, therefore supporting Meis1 potential as a therapeutic target for cardiac regeneration (98).
An alternative research direction has focused on the role of miRNAs in cardiac cell proliferation. It was found in mice that the administration of antagonists of the microRNA-15 family was associated with an increased number of mitotic cardiomyocytes in the post-natal life (98). This study hinted at the possibility that micro-RNAs, important modulators of mRNAs translation, can be targeted to obtain cardiac regeneration. A functional screening of the whole human genome miRNA library led to the identification of specific miRNAs capable of promoting cardiomyocyte proliferation (99). The most effective ones, miRNA-590 and miRNA-199, were then delivered via AAV to mouse hearts in vivo, where they induced cardiac regeneration and consequent improvement of cardiac function after myocardial infarction (99). Therefore, this study showed for the first time that post-infarct cardiac regeneration could be obtained, in vivo, by delivering molecules (not cells) with a classical method of gene therapy. Subsequent investigations identified the Hippo pathway as a target of pro-regenerative miRNAs (Figure 1) (100). The Hippo pathway is a kinase cascade controlling organ size in mammalian organisms through the regulation of cell size, proliferation, apoptosis, survival, differentiation and migration in developing organs (101). This signaling pathway, which prevents adult cardiomyocyte proliferation and regeneration postnatally, is upregulated in human HF. (102) The main kinase of the Hippo pathway and the only one known to promote tissue growth is the transcriptional co-activator YAP (Yes-Activated Protein). Following phosphorylation of the Hippo pathway components Mst, Lats and Salv, the final step of activation is the phosphorylation and cytoplasmic sequestration of the transcriptional cofactor Yap. This leads to the binding of DNA by Tead factors (102). Recently, the Hippo pathway was “turned off” by deleting Salv in a mouse model of ischemic HF (103). Hippo-deficient cardiomyocytes displayed increased expression of pro-proliferative and stress response genes, such as the mitochondrial quality control gene Park2. Treated rodents presented reduced fibrosis and better recovery of heart function. Salvor Lats1 and Lats2 deletion can also improve heart function after myocardial infarction in small animal models (104–106).
Figure 1:
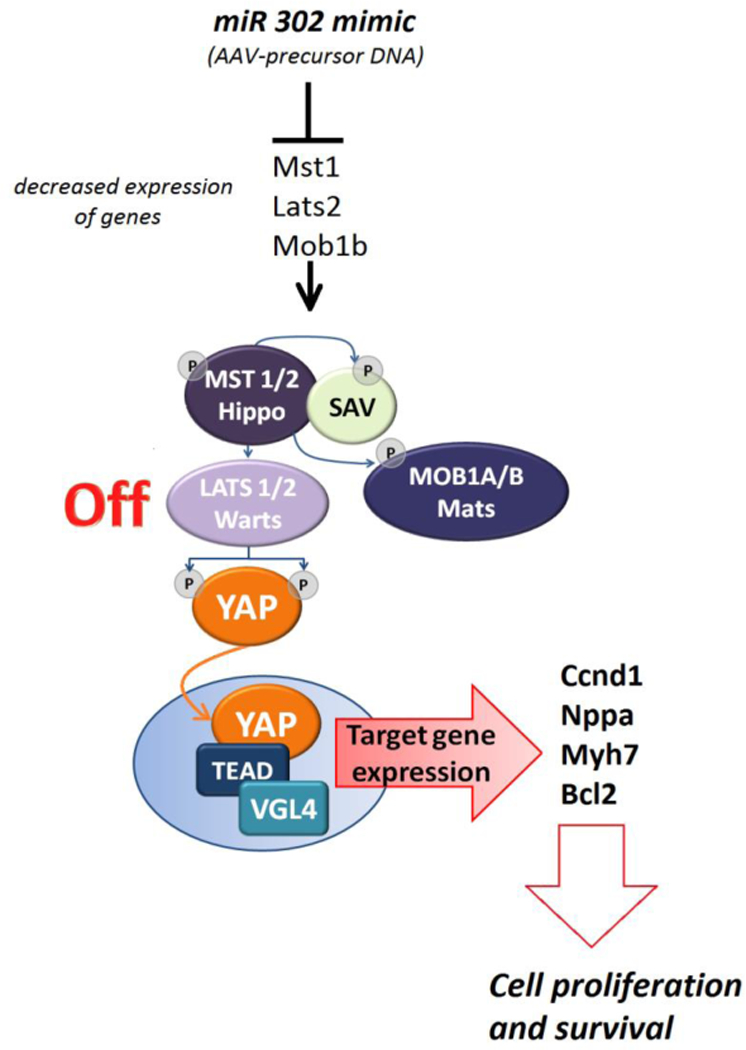
Several miRNAs may target the key kinases of hippo cascade: Mst, Lats, Mob. The loss of these essential components leads to transcriptional co-activator YAP (Yes-Activated Protein) translocation and accumulation in the nucleus, where it displaces VGL4 (the transcription cofactor vestigial-like protein 4), forms a complex with TEADs (TEA domain-containing sequence-specific transcription factors) and activates the transcription of its target pro-proliferative genes: Ccnd1 (cell proliferation), Nppa, Myh7 (fetal gene program), Bcl2 (anti-apoptosis).
Hippo pathway kinase phosphorylation cascade: Hippo (Mammalian sterile-20-like kinases type 1 and type 2 or MST1, MST2), Salvador (SAV), Warts (Large tumor suppressor-LATS 1/2) and Mob as tumor suppressor (Mps-one binder kinase activator-1; MOB1).
The capacity of various miRNAs to stimulate cardiomyocyte proliferation was further supported by later studies. It was shown that overexpression of the miRNA-17–92 cluster in transgenic mice induced cardiomyocyte proliferation either during embryonic or postnatal stages, resulting in high heart/body weight ratio and increased thickness of the left ventricular wall. Moreover, infarcted hearts of these transgenic mice displayed smaller scar size and improvement of cardiac function (107). Others reported that the treatment of neonatal cardiomyocytes with miR-29a, miR-30a or miR-141 inhibitors induces a higher percentage of cycling cells (108). The cluster miR-302–367 was identified as a driver of cardiomyocyte reentry in the cell cycle and inducer of cardiac regeneration after myocardial infarction, at least in part due to inhibition of the Hippo pathway (100). The miR-302–367 cluster was shown to repress the kinases Mst1, Lats2 and Mob1b: the loss of these essential components in the developing mouse heart caused increased proliferation in cardiomyocytes.
The role of miRNAs in the regulation of cardiomyocyte proliferation and the potential exploitation of the related mechanisms for future clinical applications is currently a hot topic (109–114) and a new exciting prospective for gene therapy of HF. Systematic tests in clinically relevant animal models are warranted. Our group has obtained very encouraging preliminary evidence in infarcted pigs (115).
CONCLUSIONS
Gene therapy for HF is rooted in pioneer studies published almost 30 years ago and has been characterized by phases of hype and disappointment. The failure of the first clinical trial launched in 2012 should not induce investigators to abandon this area of research. In fact, in 2016 4D Molecular Therapeutics made available the 3rd generation AAV vectors for gene delivery to treat HF (116). New molecular targets with remarkable curative potential have been and are being identified. The delivery of non-coding nucleic acids such as microRNAs is proving effective to induce cardiac regeneration, thus displaying a curative property so far attributed only to stem cells. HF is a complex, multi-factorial syndrome and, with the exception of the monogenic heritable forms, the potential therapeutic targets are numerous, hence it is difficult to identify the “magic” one. This explains in part the difficulties encountered by gene therapy for HF. It should be pointed out that also traditional therapeutic approaches are plagued by similar problems. Investigators in the area of gene therapy should persist in their efforts, especially because this therapeutic strategy can still reveal an unexpressed potential.
References
- 1.Yancy CW, Jessup M, Bozkurt B, et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of Amer. J Card Fail 2017;23(8):628–651. doi:10.1016/j.cardfail.2017.04.014. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen E, Weeda ER, White CM. A Review of New Pharmacologic Treatments for Patients With Chronic Heart Failure With Reduced Ejection Fraction. J Clin Pharmacol 2016;56(8):936–47. doi: 10.1002/jcph.677 [DOI] [PubMed] [Google Scholar]
- 3.Anderson WF. Human gene therapy. Science 1992;256(5058): 808–813. doi:10.1126/science.256.5058.808 [DOI] [PubMed] [Google Scholar]
- 4.Lin H, Parmacek MS, Morle G, Bolling S, Leiden JM. Expression of recombinant genes in myocardium in vivo after direct injection of DNA. Circulation 1990;82(6):2217–2221. doi:10.1161/01.CIR.82.6.2217. [DOI] [PubMed] [Google Scholar]
- 5.Zacchigna S, Giacca M. Extra- and intracellular factors regulating cardiomyocyte proliferation in postnatal life. Cardiovasc Res 2014;102(2):312–320. doi:10.1093/cvr/cvu057. [DOI] [PubMed] [Google Scholar]
- 6.Greenberg B Gene therapy for heart failure. J Cardiol 2015;66(3):195–200. doi:10.1016/j.jjcc.2015.02.006. [DOI] [PubMed] [Google Scholar]
- 7.Hulot J-S, Ishikawa K, Hajjar RJ. Gene therapy for the treatment of heart failure: promise postponed. Eur Heart J 2016;37(21):1651–1658. doi:10.1093/eurheartj/ehw019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rincon Melvin Y.,VandenDriessche Thierry, Chuah Marinee K.. Gene therapy for cardiovascular disease: advances in vector development, targeting, and delivery for clinical translation. Cardiovasc Res 2015. October 1; 108(1): 4–20. doi: 10.1093/cvr/cvv205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stammers AN, Susser SE, Hamm NC, Hlynsky MW, Kimber DE, Kehler DS, Duhamel TA. The regulation of sarco(endo)plasmic reticulum calcium-ATPases (SERCA). Can J Physiol Pharmacol 2015;93(10):843–54. doi: 10.1139/cjpp-2014-0463. [DOI] [PubMed] [Google Scholar]
- 10.Braz JC, Gregory K, Pathak A, et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med 2004;10(3):248–254. doi:10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 11.Kawase Y, Hajjar RJ. The cardiac sarcoplasmic/endoplasmic reticulum calcium ATPase: a potent target for cardiovascular diseases. Nat Clin Pract Cardiovasc Med 2008;5(9):554–565. doi:10.1038/ncpcardio1301. [DOI] [PubMed] [Google Scholar]
- 12.Pathak A, del Monte F, Zhao W, et al. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res 2005;96(7):756–766. doi:10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe S, Ishikawa K, Fish K, et al. Protein Phosphatase Inhibitor-1 Gene Therapy in a Swine Model of Nonischemic Heart Failure. J Am Coll Cardiol 2017;70(14):1744–1756. doi:10.1016/j.jacc.2017.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miyamoto MI, del Monte F, Schmidt U, et al. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci U S A 2000;97(2):793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Monte F del, Harding SE, Schmidt U, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 1999;100(23):2308–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lyon AR, Bannister ML, Collins T, et al. SERCA2a gene transfer decreases sarcoplasmic reticulum calcium leak and reduces ventricular arrhythmias in a model of chronic heart failure. Circ Arrhythm Electrophysiol 2011;4(3):362–372. doi:10.1161/CIRCEP.110.961615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawase Y, Ly HQ, Prunier F, et al. Reversal of cardiac dysfunction after long-term expression of SERCA2a by gene transfer in a pre-clinical model of heart failure. J Am Coll Cardiol 2008;51(11):1112–1119. doi:10.1016/j.jacc.2007.12.014 [DOI] [PubMed] [Google Scholar]
- 18.Jessup M, Greenberg B, Mancini D, et al. Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011;124(3):304–313. doi:10.1161/CIRCULATIONAHA.111.022889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenberg B, Yaroshinsky A, Zsebo KM, et al. Design of a phase 2b trial of intracoronary administration of AAV1/SERCA2a in patients with advanced heart failure: the CUPID 2 trial (calcium up-regulation by percutaneous administration of gene therapy in cardiac disease phase 2b). JACC Heart Fail 2014;2(1):84–92. doi:10.1016/j.jchf.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 20.Greenberg B, Butler J, Felker GM, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet (London, England) 2016;387(10024):1178–1186. doi:10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- 21.Yang L, Jiang J, Drouin LM, et al. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci U S A 2009;106(10):3946–3951. doi:10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asokan A, Conway JC, Phillips JL, et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat Biotechnol 2010;28(1):79–82. doi:10.1038/nbt.1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nowak Kristen J.,Davies Kay E.. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep 2004;5(9): 872–876. doi: 10.1038/sj.embor.7400221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yiu EM, Kornberg AJ. Duchenne muscular dystrophy. J Paediatr Child Health 2015;51(8):759–64. doi: 10.1111/jpc.12868. [DOI] [PubMed] [Google Scholar]
- 25.Manning J, O’Malley D. What has the mdx mouse model of Duchenne muscular dystrophy contributed to our understanding of this disease? J Muscle Res Cell Motil 2015;36(2):155–67. doi: 10.1007/s10974-015-9406-4. [DOI] [PubMed] [Google Scholar]
- 26.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 2010;9(2):177–189. doi:10.1016/S1474-4422(09)70272-8 [DOI] [PubMed] [Google Scholar]
- 27.Yue Y, Binalsheikh IM, Leach SB, Domeier TL, Duan D. Prospect of gene therapy for cardiomyopathy in hereditary muscular dystrophy. Expert Opin orphan drugs 2016;4(2):169–183. doi:10.1517/21678707.2016.1124039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blankinship MJ, Gregorevic P, Chamberlain JS. Gene therapy strategies for Duchenne muscular dystrophy utilizing recombinant adeno-associated virus vectors. Mol Ther 2006;13(2):241–249. doi:10.1016/j.ymthe.2005.11.001 [DOI] [PubMed] [Google Scholar]
- 29.Abdul-Razak H, Malerba A, Dickson G. Advances in gene therapy for muscular dystrophies. F1000Research 2016;5. doi:10.12688/f1000research.8735.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foster H, Popplewell L, Dickson G. Genetic therapeutic approaches for Duchenne muscular dystrophy. Hum Gene Ther 2012;23(7):676–687. doi:10.1089/hum.2012.099. [DOI] [PubMed] [Google Scholar]
- 31.Duan D Duchenne muscular dystrophy gene therapy: Lost in translation? Res Rep Biol 2011;2011(2):31–42. doi:10.2147/RRB.S13463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forbes SC, Bish LT, Ye F, Spinazzola J, Baligand C, Plant D, Vandenborne K, Barton ER, Sweeney HL, Walter GA et al. Gene transfer of arginine kinase to skeletal muscle using adeno-associated virus. Gene Ther 2014;21(4):387–92. doi: 10.1038/gt.2014.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bowles DE, McPhee SW, Li C.et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther 2012;20(2):443–55. doi: 10.1038/mt.2011.237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chamberlain JR, Chamberlain JS. Progress toward Gene Therapy for Duchenne Muscular Dystrophy. Mol Ther 2017; 25(5): 1125–1131. doi: 10.1016/j.ymthe.2017.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toromanoff A, Adjali O, Larcher T, Hill M. et al. Lack of immunotoxicity after regional intravenous (RI) delivery of rAAV to nonhuman primate skeletal muscle. Mol Ther 2010;18(1):151–60. doi: 10.1038/mt.2009.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodino-Klapac LR, Montgomery CL, Bremer WG. et al. Persistent expression of FLAG-tagged micro dystrophin in nonhuman primates following intramuscular and vascular delivery. Mol Ther 2010;18(1):109–17. doi: 10.1038/mt.2009.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kodippili K, Hakim CH, Pan X, et al. Dual AAV Gene Therapy for Duchenne Muscular Dystrophy with a 7-kb Mini-Dystrophin Gene in the Canine Model. Hum Gene Ther 2018;29(3):299–311. doi:10.1089/hum.2017.095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duan D Duchenne muscular dystrophy gene therapy in the canine model. Hum Gene Ther Clin Dev 2015. March;26(1):57–69. doi: 10.1089/humc.2015.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Storb R, Halbert CL, Banks GB, Butts TM, Finn EE, Allen JM, Miller AD, Chamberlain JS, Tapscott SJ. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: a preclinical model for human therapies. Mol Ther 2012;20(8):1501–7. doi: 10.1038/mt.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Systemic Gene Delivery Clinical Trial for Duchenne Muscular Dystrophy. https://clinicaltrials.gov/ct2/show/NCT03375164?cond=%22Muscular+Dystrophy%2C+Duc henne%22&lupd_s=11%2F13%2F2015&lupd_d=1000.
- 41.A Study to Evaluate the Safety and Tolerability of PF-06939926 Gene Therapy in Duchenne Muscular Dystrophy. https://clinicaltrials.gov/ct2/show/NCT03362502?cond=%22Muscular+Dystrophy%2C+Duc henne%22&lupd_s=11%2F13%2F2015&lupd_d=1000.
- 42.Gene Transfer Clinical Trial to Deliver rAAVrh74.MCK.GALGT2 for Duchenne Muscular Dystrophy. doi: 10.1016/j.omtm.2022.08.009. https://clinicaltrials.gov/ct2/show/NCT03333590?cond=%22Muscular+Dystrophy%2C+Duc henne%22&lupd_s=11%2F13%2F2015&lupd_d=1000. [DOI] [PMC free article] [PubMed]
- 43.Nance ME, Duan D. Perspective on Adeno-Associated Virus Capsid Modification for Duchenne Muscular Dystrophy Gene Therapy. Hum Gene Ther 2015;26(12):786–800. doi:10.1089/hum.2015.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh A, Duan D. Expanding adeno-associated viral vector capacity: a tale of two vectors. Biotechnol Genet Eng Rev 2007;24:165–177. [DOI] [PubMed] [Google Scholar]
- 45.Lai Y, Yue Y, Liu M, et al. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol 2005;23(11):1435–1439. doi:10.1038/nbt1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koo T, Popplewell L, Athanasopoulos T, Dickson G. Triple trans-splicing adeno-associated virus vectors capable of transferring the coding sequence for full-length dystrophin protein into dystrophic mice. Hum Gene Ther 2014;25(2):98–108. doi:10.1089/hum.2013.164. [DOI] [PubMed] [Google Scholar]
- 47.Chapdelaine P, Pichavant C, Rousseau J, Paques F, Tremblay JP. Meganucleases can restore the reading frame of a mutated dystrophin. Gene Ther 2010;17(7):846–858. doi:10.1038/gt.2010.26. [DOI] [PubMed] [Google Scholar]
- 48.Ousterout DG, Perez-Pinera P, Thakore PI, et al. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther 2013;21(9):1718–1726. doi:10.1038/mt.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ousterout DG, Kabadi AM, Thakore PI, et al. Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol Ther 2015;23(3):523–532. doi:10.1038/mt.2014.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Popplewell L, Koo T, Leclerc X, et al. Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in. Hum Gene Ther 2013;24(7):692–701. doi:10.1089/hum.2013.081. [DOI] [PubMed] [Google Scholar]
- 51.Cordova G, Negroni E, Cabello-Verrugio C, Mouly V, Trollet C. Combined Therapies for Duchenne Muscular Dystrophy to Optimize Treatment Efficacy. Front Genet 2018;9:114. doi:10.3389/fgene.2018.00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nelson CE, Hakim CH, Ousterout DG, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016;351(6271):403–407. doi:10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J-Z, Wu P, Shi Z-M, Xu Y-L, Liu Z-J. The AAV-mediated and RNA-guided CRISPR/Cas9 system for gene therapy of DMD and BMD. Brain Dev 2017;39(7):547–556. doi:10.1016/j.braindev.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 54.Zhu P, Wu F, Mosenson J, Zhang H, He T-C, Wu W-S. CRISPR/Cas9-Mediated Genome Editing Corrects Dystrophin Mutation in Skeletal Muscle Stem Cells in a Mouse Model of Muscle Dystrophy. Mol Ther Nucleic Acids 2017;7:31–41. doi:10.1016/j.omtn.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 2013;74(5):637–47. 10.1002/ana.23982 [DOI] [PubMed] [Google Scholar]
- 56.Nakamura A Moving towards successful exon-skipping therapy for Duchenne muscular dystrophy. J Hum Genet 2017;62(10):871–876. doi:10.1038/jhg.2017.57 [DOI] [PubMed] [Google Scholar]
- 57.Kendall GC, Mokhonova EI, Moran M, et al. Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Sci Transl Med 2012;4(164):164ra160. doi: 10.1126/scitranslmed.3005054 [DOI] [PubMed] [Google Scholar]
- 58.McElhanon KE, Bhattacharya S. Altered membrane integrity in the progression of muscle diseases. Life Sci 2018;192:166–172. doi:10.1016/j.lfs.2017.11.035. [DOI] [PubMed] [Google Scholar]
- 59.Le Hir M, Goyenvalle A, Peccate C, et al. AAV genome loss from dystrophic mouse muscles during AAV-U7 snRNA-mediated exon-skipping therapy. Mol Ther 2013;21(8):1551–1558. doi:10.1038/mt.2013.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stedman HH, Byrne BJ. Signs of Progress in Gene Therapy for Muscular Dystrophy Also Warrant Caution. Mol Ther 2012;20(2):249–51. doi: 10.1038/mt.2011.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kornegay JN, Li J, Bogan JR, Bogan DJ, Chen C, Zheng H, Wang B, Qiao C, Howard JF Jr, Xiao X. et al. Widespread muscle expression of an AAV9 human mini-dystrophin vector after intravenous injection in neonatal dystrophin-deficient dogs. Mol Ther 2010;18(8):1501–8. doi: 10.1038/mt.2010.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zaccolo Manuela. cAMP signal transduction in the heart: understanding spatial control for the development of novel therapeutic strategies Br J Pharmacol 2009;158(1): 50–60. doi: 10.1111/j.1476-5381.2009.00185.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lai NC, Roth DM, Gao MH, et al. Intracoronary adenovirus encoding adenylyl cyclase VI increases left ventricular function in heart failure. Circulation 2004;110(3):330–336. doi:10.1161/01.CIR.0000136033.21777.4D. [DOI] [PubMed] [Google Scholar]
- 64.Roth DM, Bayat H, Drumm JD, et al. Adenylyl cyclase increases survival in cardiomyopathy. Circulation 2002;105(16):1989–1994. [DOI] [PubMed] [Google Scholar]
- 65.Timofeyev V, He Y, Tuteja D, et al. Cardiac-directed expression of adenylyl cyclase reverses electrical remodeling in cardiomyopathy. J Mol Cell Cardiol 2006;41(1):170–181. doi:10.1016/j.yjmcc.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 66.Hammond HK, Penny WF, Traverse JH, et al. Intracoronary Gene Transfer of Adenylyl Cyclase 6 in Patients With Heart Failure: A Randomized Clinical Trial. JAMA Cardiol 2016;1(2):163–171. doi:10.1001/jamacardio.2016.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Most P, Seifert H, Gao E, et al. Cardiac S100A1 protein levels determine contractile performance and propensity toward heart failure after myocardial infarction. Circulation 2006;114(12):1258–1268. doi:10.1161/CIRCULATIONAHA.106.622415. [DOI] [PubMed] [Google Scholar]
- 68.Boerries M, Most P, Gledhill JR, et al. Ca2+ -dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol 2007;27(12):4365–4373. doi:10.1128/MCB.02045-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pleger ST, Shan C, Ksienzyk J, et al. Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med 2011;3(92):92ra64. doi:10.1126/scitranslmed.3002097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weber C, Neacsu I, Krautz B, et al. Therapeutic safety of high myocardial expression levels of the molecular inotrope S100A1 in a preclinical heart failure model. Gene Ther 2014;21(2):131–138. doi:10.1038/gt.2013.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.uniQure NV. Annual Report. 2017 http://www.uniqure.com/uniQure%20Annual%20Accounts%202017.pdf. [Google Scholar]
- 72.Sen A, Ren S, Lerchenmuller C, et al. MicroRNA-138 regulates hypoxia-induced endothelial cell dysfunction by targeting S100A1. PLoS One 2013;8(11):e78684. doi:10.1371/journal.pone.0078684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bry M, Kivelä R, Leppänen VM, Alitalo K. Vascular Endothelial Growth Factor-B in Physiology and Disease. Physiol Rev 2014;94:779–94. [DOI] [PubMed] [Google Scholar]
- 74.Li Y, Zhang F, Nagai N, et al. VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J Clin Invest 2008;118:913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Narula J, Haider N, Virmani R, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med 1996;335:1182–89. [DOI] [PubMed] [Google Scholar]
- 76.Olivetti G, Abbi R, Quaini F, et al. Apoptosis in the failing human heart. N Engl J Med 1997;336:1131–41. [DOI] [PubMed] [Google Scholar]
- 77.Saraste A, Pulkki K, Kallajoki M, et al. Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur J Clin Invest 1999;29:380–6. [DOI] [PubMed] [Google Scholar]
- 78.Khatiwala JR, Everly MJ. An Update on Cardiac Transplantation in the United States Based on an Analysis of the UNOS Registry. ClinTranspl 2015;31:27–34. [PubMed] [Google Scholar]
- 79.Zentilin L, Puligadda U, Lionetti V, et al. Cardiomyocyte VEGFR-1 activation by VEGF-B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB J 2010;24:1467–78. [DOI] [PubMed] [Google Scholar]
- 80.Pepe M, Mamdani M, Zentilin L, et al. Intramyocardial VEGF-B167 gene delivery delays the progression towards congestive failure in dogs with pacing-induced dilated cardiomyopathy. Circ Res 2010;106:1893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Woitek F, Zentilin L, Hoffman NE, Powers JC, Ottiger I, Parikh S, Kulczycki AM, Hurst M, Ring N, Wang T, Shaikh F, Gross P, Singh H, Kolpakov MA, Linke A, Houser SR, Rizzo V, Sabri A, Madesh M, Giacca M, Recchia FA. Intracoronary Cytoprotective Gene Therapy: A Study of VEGF-B167 in a Pre-Clinical Animal Model of Dilated Cardiomyopathy. J Am CollCardiol 2015;66:139–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Askari AT, Unzek S, Popovic ZB, et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet (London, England) 2003;362(9385):697–703. doi:10.1016/S0140-6736(03)14232-8. [DOI] [PubMed] [Google Scholar]
- 83.Penn MS, Pastore J, Miller T, Aras R. SDF-1 in myocardial repair. Gene Ther 2012;19(6):583–587. doi:10.1038/gt.2012.32. [DOI] [PubMed] [Google Scholar]
- 84.Penn MS, Mendelsohn FO, Schaer GL, et al. An open-label dose escalation study to evaluate the safety of administration of nonviral stromal cell-derived factor-1 plasmid to treat symptomatic ischemic heart failure. Circ Res 2013;112(5):816–825. doi:10.1161/CIRCRESAHA.111.300440. [DOI] [PubMed] [Google Scholar]
- 85.Chung ES, Miller L, Patel AN, et al. Changes in ventricular remodelling and clinical status during the year following a single administration of stromal cell-derived factor-1 non-viral gene therapy in chronic ischaemic heart failure patients: the STOP-HF randomized Phase II trial. Eur Heart J 2015;36(33):2228–2238. doi:10.1093/eurheartj/ehv254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Durrani S, Konoplyannikov M, Ashraf M, Haider KH. Skeletal myoblasts for cardiac repair. Regen Med 2010;5(6):919–932. doi:10.2217/rme.10.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Psaltis PJ, Schwarz N, Toledo-Flores D, Nicholls SJ. Cellular Therapy for Heart Failure. Curr Cardiol Rev 2016;12(3):195–215. doi: 10.2174/1573403X12666160606121858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ptaszek LM, Mansour M, Ruskin JN, Chien KR. Towards regenerative therapy for cardiac disease. Lancet (London, England) 2012;379(9819):933–942. doi:10.1016/S0140-6736(12)60075-0 [DOI] [PubMed] [Google Scholar]
- 89.Zhang Yiqiang, Mignone John, and Robb MacLellan W. Cardiac Regeneration and Stem Cells. Physiol Rev 2015;95(4): 1189–1204.doi: 10.1152/physrev.00021.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Steinhoff G, Nesteruk J, Wolfien M, Große J, Ruch U, Vasudevan P, Müller P. Stem cells and heart disease - Brake or accelerator? Adv Drug Deliv Rev 2017;120:2–24. doi: 10.1016/j.addr.2017.10.007 Epub 2017 Oct 18. [DOI] [PubMed] [Google Scholar]
- 91.Bergmann O, Bhardwaj RD, Bernard S, et al. Evidence for cardiomyocyte renewal in humans. Science 2009;324(5923):98–102. doi:10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chaudhry HW, Dashoush NH, Tang H, et al. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem 2004;279(34):35858–35866. doi:10.1074/jbc.M404975200. [DOI] [PubMed] [Google Scholar]
- 93.Woo YJ, Panlilio CM, Cheng RK, et al. Therapeutic delivery of cyclin A2 induces myocardial regeneration and enhances cardiac function in ischemic heart failure. Circulation 2006;114(1 Suppl):I206–13. doi:10.1161/CIRCULATIONAHA.105.000455. [DOI] [PubMed] [Google Scholar]
- 94.Liao HS, Kang PM, Nagashima H, et al. Cardiac-specific overexpression of cyclin-dependent kinase 2 increases smaller mononuclear cardiomyocytes. Circ Res 2001;88(4):443–450. [DOI] [PubMed] [Google Scholar]
- 95.Pasumarthi KBS, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res 2005;96(1):110–118. doi:10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- 96.Hassink RJ, Pasumarthi KB, Nakajima H, et al. Cardiomyocyte cell cycle activation improves cardiac function after myocardial infarction. Cardiovasc Res 2008;78(1):18–25. doi:10.1093/cvr/cvm101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mahmoud AI, Kocabas F, Muralidhar SA, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013;497(7448):249–253. doi:10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xiang F-L, Guo M, Yutzey KE. Overexpression of Tbx20 in Adult Cardiomyocytes Promotes Proliferation and Improves Cardiac Function After Myocardial Infarction. Circulation 2016;133(11):1081–1092. doi:10.1161/CIRCULATIONAHA.115.019357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Eulalio A, Mano M, Dal Ferro M, et al. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012;492(7429):376–381. doi:10.1038/nature11739 [DOI] [PubMed] [Google Scholar]
- 100.Tian Y, Liu Y, Wang T, et al. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci Transl Med 2015;7(279):279ra38. doi:10.1126/scitranslmed.3010841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Meng Zhipeng, Moroishi Toshiro, Guan Kun-Liang. Mechanisms of Hippo pathway regulation. Genes Dev 2016. January 1; 30(1): 1–17.doi: 10.1101/gad.274027.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Halder G, Johnson RL. Hippo signaling: growth control and beyond. Development 2011;138(1):9–22. doi:10.1242/dev.045500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Leach JP, Heallen T, Zhang M, et al. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017;550(7675):260–264. doi:10.1038/nature24045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Morikawa Y, Zhang M, Heallen T, et al. Actin cytoskeletal remodeling with protrusion formation is essential for heart regeneration in Hippo-deficient mice. Sci Signal 2015;8(375):ra41. doi:10.1126/scisignal.2005781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tao G, Kahr PC, Morikawa Y, et al. Pitx2 promotes heart repair by activating the antioxidant response after cardiac injury. Nature 2016;534(7605):119–123. doi:10.1038/nature17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heallen T, Morikawa Y, Leach J, et al. Hippo signaling impedes adult heart regeneration. Development 2013;140(23):4683–4690. doi:10.1242/dev.102798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen J, Huang Z-P, Seok HY, et al. mir-17–92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res 2013;112(12):1557–1566. doi:10.1161/CIRCRESAHA.112.300658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang Y, Matsushita N, Eigler T, Marban E. Targeted MicroRNA Interference Promotes Postnatal Cardiac Cell Cycle Re-Entry. J Regen Med 2013; :2. doi:10.4172/2325-9620.1000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Porrello ER, Johnson BA, Aurora AB, et al. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circ Res 2011;109(6):670–679. doi:10.1161/CIRCRESAHA.111.248880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Castellan RFP, Meloni M. Mechanisms and Therapeutic Targets of Cardiac Regeneration: Closing the Age Gap. Front Cardiovasc Med 2018;5:7. doi: 10.3389/fcvm.2018.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin Z, Pu WT. Releasing YAP from an α-catenin trap increases cardiomyocyte proliferation. Circ Res 2015;116(1):9–11. doi:10.1161/CIRCRESAHA.114.305496, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huynh K Basic research: Inhibition of Hippo pathway signalling reverses systolic heart failure. Nat Rev Cardiol 2017;14(12):697. doi: 10.1038/nrcardio.2017.166 [DOI] [PubMed] [Google Scholar]
- 113.Cahill TJ, Choudhury RP, Riley PR. Heart regeneration and repair after myocardial infarction: translational opportunities for novel therapeutics. Nat Rev Drug Discov 2017;16(10):699–717. doi: 10.1038/nrd.2017.106 [DOI] [PubMed] [Google Scholar]
- 114.Hill MC, Martin JF. Heart muscle regeneration: the wonder of a Cardio-Cocktail. Cell Res 2018;28(5):503–504. doi: 10.1038/s41422-018-0035-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gabisonia K, Prosdocimo G, Aquaro GD et al. Intramyocardial delivery of miR-199a reduces scar size and preserves contractile function in infarcted pig hearts. 2016 AHA Late-Breaking Basic Science Abstracts. Circ Res 2016;119:e160–e171.https://doi.org/10.1161/RES.0000000000000126 [Google Scholar]
- 116. https://www.4dmoleculartherapeutics.com/technology/aav-vectors/