

Abstract
The finding that hypoxia can induce cancer stemness in various experimental models is in agreement with the conceptual basis of cancer cell plasticity. Here, we aimed to gain insights into the molecular basis of hypoxia-induced cancer cell plasticity in triple negative breast cancer (TNBC). To achieve this goal, we employed our previously published in-vitro model of TNBC, in which a small subset of stem-like cells can be distinguished from the bulk cell population based on their responsiveness to a Sox2 reporter. In MDA-MB-231, a TNBC cell line, we observed that hypoxia significantly increased the expression of luciferase and green fluorescence protein (GFP), the readouts of the Sox2 reporter. Upon hypoxic challenge, the bulk, reporter unresponsive (RU) cells acquired stem-like features, as evidenced by the significant increases in the proportion of CD44high/CD24low cells, colony formation and resistance to cisplatin. Correlating with these phenotypic changes, RU cells exposed to hypoxia exhibited a substantial upregulation of the active/phosphorylated form of STAT3 (pSTAT3). This hypoxia-induced activation of STAT3 correlated with increased STAT3 transcriptional activity, as evidenced by increased STAT3-DNA binding and an altered gene expression profile. This hypoxia-induced STAT3 activation is biologically significant, since siRNA knockdown of STAT3 in RU cells significantly attenuated the hypoxia-induced acquisition of Sox2 activity and stem-like phenotypic features. In conclusion, our data have provided the proof-of-concept that STAT3 is a critical mediator in promoting the hypoxia-induced acquisition of cancer stemness in TNBC. Targeting STAT3 in TNBC may be useful in overcoming chemoresistance and decreasing the risk of disease relapse.
Electronic supplementary material
The online version of this article (10.1007/s12307-018-0218-0) contains supplementary material, which is available to authorized users.
Keywords: Hypoxia, STAT3, Cancer cell plasticity, Triple negative breast cancer, Cancer stemness
Introduction
Triple negative breast cancer (TNBC) is an aggressive type of breast cancer with limited treatment options and a poor prognosis. It has a high incidence of recurrence, metastasis and a short overall survival [1]. This can be partly attributed to the fact that targeted therapies are not available for this disease due to the lack of estrogen receptor, progesterone receptor and human epidermal growth factor receptor-2 [2–4]. The suboptimal therapeutic response of TNBC has also been linked to the existence of cancer stem cells (CSCs). CSCs are a small subpopulation of cancer cells that are known to be responsible for cancer recurrence and resistance to therapy. Purified CSCs derived from TNBC cell lines and patient samples have been shown to be 100-folds more tumorigenic than the bulk tumor cell population [5, 6]. Worse yet, tumor microenvironment factors, such as hypoxia, actively induce bulk cancer cells to assume the CSC phenotype [7] and TNBC tumors are known to carry a hypoxic phenotype [8]. These observations exemplify the concept of cancer cell plasticity, which has been recently reviewed [9–11]. In view of the importance of CSCs, it has been postulated that the eradication of CSCs and abrogation of acquired cancer stemness are important strategies in curing TNBC. Nonetheless, the molecular basis by which tumors acquire CSC-like features in the setting of hypoxia has not been extensively studied.
Our group has previously identified a novel level of intratumoral heterogeneity in different types of cancer cell lines including breast cancer, anaplastic large cell lymphoma, esophageal squamous cell carcinoma, and neuroblastoma, as well as patient samples by using a Sox2 regulatory region 2 (SRR2) reporter with green fluorescence protein (GFP) and luciferase as the readouts [12–16]. Specifically, we identified a relatively small subset of cells that are reporter-responsive (RR), and they express GFP and luciferase. In contrast, the majority of the cells are reporter unresponsive (RU), and they do not express GFP or luciferase [12–14]. Importantly, we found that RR cells are more CSC-like than RU cells, and this observation can be made in cell lines and primary patient samples [12, 13]. In this study, we have shown that hypoxia can efficiently convert the bulk RU cells into stem-like RR cells in MDA-MB-231 cells, a TNBC cell line. Using this experimental system, we aimed to gain insights into the molecular basis of cancer cell plasticity in this cell line. Our data suggests that STAT3 plays a key role in RU/RR conversion and conferring hypoxia-induced acquisition of cancer stemness in MDA-MB-231cells.
Materials and Methods
Cell Culture
The TNBC cell line, MDA-MB-231, was obtained from ATCC (Manassas, VA, USA) and maintained in Roswell Park Memorial Institute medium (RPMI) 1640 medium supplemented with 10% fetal bovine serum (Invitrogen, Karlsruhe, Germany), 100 units/mL penicillin, and 100 mg/mL streptomycin in a humidified incubator under 95% air and 5% CO2 at 37 °C. MDA-MB-231 cells were virally infected twice with mCMV (a minimal cytomegalovirus promoter) or SRR2 (Sox2 regulatory region 2 reporter) as previously described [13]. In brief, based on the GFP (green fluorescence protein) expression, RU and RR cells were purified and maintained in puromycin antibiotics. For stable cell lines overexpressing c-Myc (RU/Myc), 10 million RU cells derived from MDA-MB-231 were initially transfected with 15 μg pcDNA 3.3-Myc (a gift from Derrick Rossi (Addgene plasmid #26818)). RU cells carrying empty vector (RU/EV) were initially transfected with 15 μg pcDNA 3.3 plasmids Addgene (Cambridge, MA, USA). Both cell subsets then were cultured for 3–4 weeks in selection medium with increasing concentration of G418 up to 400 ng/mL. Cells were plated at 25–30% confluence and cultured until they reached 60–70% confluence for different treatments. For hypoxic conditions, cells were cultured in a CO2 incubator maintained at 94% N2, 5% CO2 and 1% O2. The O2 pressure was chosen based on our previous study with unsorted MDA-MB-231 cells [17]. The chemotherapeutic agent cisplatin (cis-diamminedichloroplatinum(II) (CDDP) (purity 99%), #H878, AK Scientific Inc., Union City, CA, USA) was freshly prepared in water as a stock solution (3.3 mM) and further diluted with the RPMI 1640 medium to reach the indicated concentrations.
Small Interfering RNAs (siRNAs) Complex Preparation
HIF-1α siRNAs (Hs_HIF1α_5 FlexiTube siRNA, #SI02664053, Qiagen, Hilden, Germany), scrambled (Scr) siRNAs (Negative Control siRNA, #1027310, Qiagen), STAT3 siRNAs (Hs_STAT3_7 FlexiTube siRNA, #SI02662338, Qiagen), Myc siRNAs (SMARTpool: ON-TARGET plus Myc siRNA, Dharmacon, Fisher Scientific, ON, Canada) and Lipofectamine™ 2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA), were used to make complexes (siRNA:polymer) at a ratio of 1:1 (weight/weight) in OptiMEM media (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s instructions. Complexes were added to the cells at 50 nM siRNA concentrations. Cells were transfected with siRNA complexes at 50–60% confluence.
SRR2 Luciferase Reporter Assay and Flow Cytometry
Luciferase reporter assay was performed using luciferase assay system kit (#E4530, Promega, Corporation, Madison, USA) according to the manufacturer’s protocol, on costar white polystyrene opaque 96-well plates (#3912, Corning, NY, USA) and analyzed on the FLUOstar Omega multi-mode microplate reader (BMG Labtech, Ortenburg, Germany). Flow cytometry analysis was performed as described previously [13].
MTT Assay
RU MDA-MB-231 cells (1 × 104 cells/well) were seeded in 96-well plates overnight and then exposed to increasing concentration of cisplatin (3.32–332 μM) for 48 h under hypoxia and normoxia. For siRNA transfection studies, RU cells (7 × 104 cells/well) were seeded in 24-well plates overnight. At 50–60% confluence, cells were transfected with siRNA complexes for 24 h under normoxia prior to cisplatin treatment. Cells were then exposed to cisplatin (25–30 μM) under hypoxia for 48 h. Cellular viability was assessed by the reduction of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolim bromide, Sigma–Aldrich, Oakville, ON, Canada) to formazan crystals. Briefly, MTT solution (5 mg/mL) was added to incubated cells for 4 h at 37 °C. Then the medium was replaced by DMSO (N,N,dimethyl sulfoxide) to dissolve the crystals formed. Optical density was measured spectrophotometrically using a plate reader (Synergy H1 Hybrid Reader, Biotek, Winooski, VT, USA) at 570 nm. The cellular activity ratio was represented relative to control.
Colony Formation Assay
RU and RR MDA-MB-231 cells (3.5 × 105 cells/flask) were seeded in 25 cm2 flasks overnight. The day after, cells were incubated either under normoxia or hypoxia for 24 h. Cells were then washed once in 1X phosphate buffered saline (PBS), harvested by trypsinization, counted using a hemocytometer, and then re-plated at three different densities of 500, 1000, and 2000 cells/well in duplicate in six-well plates under normoxia. After an additional 5–7 days of culture, cells were stained with a crystal violet solution (#HT90132, Sigma-Aldrich, St. Louis, MO, USA), and surviving colonies consisting of ~50 or more cells were counted with Alpha Imager HP, Protein Simple, CA, USA. In another set of experiments, RU cells were first transfected with scrambled or STAT3 siRNAs complexes for 24 h under normoxia. Cells were then subjected to 24 h hypoxia and then re-plated under normoxia as outlined above. Colony ability formation of RU/EV or RU/Myc MDA-MB-231 cells was also assessed after 24 h exposure to hypoxia and re-plating under normoxia as outlined above.
Mammosphere Assay
In a mammosphere assay, cells were seeded and cultured as previously described [13]. Briefly, RU MDA-MB-231 cells were transfected with HIF-1α/Scr siRNAs under normoxia for 24 h and then kept under hypoxia for 48 h. In the next step, cells were trypsinized and passed through a 40 μm cell strainer (BD, Franklin Lakes, New Jersey, USA) and 10,000 cells were seeded into ultra-low adherent plates (Corning, NY, USA) in Mammocult media (StemCell Technologies, Vancouver, BC, Canada) as per manufacturer’s instructions. Mammosphere larger than 60 μm were counted 5–7 days after seeding.
Flow Cytometry Analyses for CD44high/CD24low Expression
Single cell suspensions for flow cytometry were achieved by passing cells through a 40 μm cell strainer (BD Falcon) and staining with CD44-APC (#559942) and CD24-PerCP-Cy5.5 (#561647) from BD Pharmingen in Hanks’ buffer supplemented with 2% FBS according to the manufacturer’s instructions. All stained cells were run in a BD FACS Canto II (BD Biosciences, San Jose, CA, USA), and results were analyzed using FCS Express 5.0 software (De Novo Software, Glendale, CA, USA). To assess the changes in the expression of CD44high/CD24low under hypoxia, as compared to normoxia, gates were first established for positivity stained normoxic (native) RU cells with antibodies of CD44 and CD24 using unstained normoxic RU cells as a negative control. Then, the stained normoxic RU group was chosen as a control and the same gating was adopted to measure the expression of CD44high/CD24low in hypoxic RU cells or normoxic RR cells.
Western Blot
To measure the expression level of different proteins, RU or RR MDA-MB-231 cells (2.0 × 105 cells/well) were seeded in 6-well plates overnight. Then cells either left untreated or were transfected with siRNAs for 24 h under normoxia. Then after 48 h of incubation under hypoxia, cells were washed with cold 1X PBS and lysed using radioimmunoprecipitation assay buffer (RIPA) lysis buffer that was supplemented with 0.1 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich), Protease Inhibitor Cocktail Set III, Animal-Free - Calbiochem (#535140, Millipore, MA, USA), and Phosphatase Inhibitor Cocktail Set II (#524625, Millipore). The lysate was then incubated on ice for 30 min, followed by centrifugation at 17,000 g for 20 min to remove genomic DNA. Protein quantification was determined by the bicinchoninic acid (BCA) Protein Assay Kit (Pierce, Rockford, IL, USA), and equal amounts of protein (35–40 μg) were loaded in 4 − 15% Tris-Glycine gradient gel (#456-1084, Biorad, CA, USA). After gel electrophoresis, proteins were transferred to a nitrocellulose membrane. Membranes were probed with antibodies against HIF-1α (#3716 s, Cell Signaling Technologies), survivin (#2808 s, Cell Signaling Technologies, MA, USA), phospho-STAT3 (Tyr705) (p-STAT3) (#9131, Cell Signaling Technologies), Total-STAT3 (T-STAT3) (#8768 s, Cell Signaling Technologies), c-Myc (#5605 s, Cell Signaling Technologies), myeloid cell leukemia-1 (MCL-1) (#sc-819, Santa Cruz Biotechnology, Texas, USA), GAPDH (# sc-47,724, Santa Cruz Biotechnologies) and ß-actin (#sc-47,778, Santa Cruz Biotechnology). Proteins were then detected using peroxidase-conjugated anti-mouse IgG (#7076, Cell Signaling Technologies) or anti-rabbit IgG (#7074, Cell Signaling Technologies) and visualized by enhanced chemiluminescence (Pierce ECL Western Blotting Substrate, #32106, Thermo Scientific, Rockford, IL, USA).
Nuclear-Cytoplasmic Fractionation
Nuclear and cytoplasmic proteins of cells were extracted using the NE-PER Protein Extraction Kit (#78833, Thermo Scientific, USA) according to the manufacturer’s protocol. For nuclear fraction, histone deacetylase 1 (HDAC-1, Santa Cruz Biotechnology) was used as a loading control for western blotting experiments.
SRR2-Probe Pull Down Assay
A biotinylated SRR2 probe was synthesized from Integrated DNA Technologies, USA. The SRR2 sequence is 5′-AAGAATTTCCCGGGCTCGGGCAGCCATTGTGATGCATATAGGATTATTCACGTGGTAATG-3′, in which the STAT3 consensus sequence is underlined. 400 μg of nuclear protein was incubated with 3 pmole of either with or without the biotin-labeled SRR2 probe for 30 min at room temperature. Then 75 μL streptavidin beads were added and the samples were incubated overnight by rotation at 4 °C. The next day beads were washed 3 times with cold PBS and protein was eluted at 100 °C in 4X protein loading dye and loaded on SDS-PAGE gels and then processed for western blotting.
Human Cancer Pathway Finder PCR Array
The human cancer pathway finder super array (PAHS-033Z; Qiagen) was used to analyze mRNA levels of 84 genes related to cell proliferation, apoptosis, cell cycle, angiogenesis, invasion, and metastasis. All genes represented by the array showed a single peak on the melting curve characteristic to the specific products. Data analysis of gene expression was performed using Excel-based PCR Array Data Analysis Software provided by manufacturer (Qiagen). Fold-changes in gene expression were calculated using the 2-ΔΔCT method, and five stably expressed housekeeping genes (β2 microglobulin, hypoxanthine phosphoribosyltransferase 1, ribosomal protein large P0, GAPDH, and β-actin) were used for normalization of the results.
RNA Extraction, cDNA Synthesis, Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA extraction was performed with the Qiagen RNeasy Kit (#74104 Qiagen) according to the manufacturer’s protocol. 1 μg of RNA was reverse transcribed using oligo-dT and superscript II (Life Technologies, Grand Island, NY, USA) according to the manufacturer’s protocol. 1 μL of the resulting cDNA mixture was added to the Platinum SYBR Green qPCR SuperMix-UDG with Rox (Life Technologies, Grand Island, NY, USA) and amplified with target gene-specific primers (Table S1) on the Applied Biosystems 7900HT (Carsbad, CA, USA; The Applied Genomics Centre, Edmonton, Alberta, Canada). All genes of interest were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcript expression levels. For analysis of changes in gene expression under hypoxia, fold changes in gene expression were calculated using the 2-ΔΔCT method. Individual fold-changes for each of the hypoxic samples were calculated by subtracting the ΔCT (gene expression CT normalized to the endogenous control, GAPDH) for each sample from the average ΔCT for the normoxic group to obtain ΔΔCT and was entered into the formula 2-ΔΔCT to obtain the fold changes. For analysis of changes in gene expression under hypoxia after STAT3 knockdown, fold changes in gene expression were calculated using the 2-ΔΔCT method. Individual fold-changes for each of the hypoxic samples subjected to STAT3 siRNA were calculated by subtracting the ΔCT (gene expression CT normalized to the endogenous control, GAPDH) for each sample from the average ΔCT for the hypoxic samples subjected to scrambled siRNA to obtain ΔΔCT and was entered into the formula 2-ΔΔCT to obtain the fold changes.
Statistical Analysis
The statistical analysis was performed by Graphpad Prism (version 5.00, Graphpad Software Inc., La Jolla, CA, USA). Statistical analysis was performed either using unpaired Student’s t test or one-way ANOVA (analysis of variance) with Tukey post-test analysis. Statistical significance is denoted by (p < 0.05). All graphs represent the average of at least three independent experiments with triplicates, unless mentioned otherwise in the text, or graphs. Results were represented as mean ± standard deviation (SD).
Results
Hypoxia Induced Acquisition of the RR Phenotype in RU Cells
Purified RU cells derived from MDA-MB-231 showed an appreciable increase in GFP expression, with the observed median GFP expression level being 120 ± 35, as compared to 72 ± 17 in native RU cells (p = 0.01) (Fig. 1a). Of note, the median GFP expression level in hypoxic RU cells remained to be substantially lower than that of native RR cells (120 ± 35 versus 509 ± 28) (Fig. 1a). Since the Sox2 reporter also carries the luciferase reporter gene, we compared the luciferase activity between native RU and hypoxic RU cells. As shown in Fig. 1b, RU cells cultured under hypoxia showed a significant increase in luciferase activity (1.6 fold, p = 0.03); again, the luciferase activity level of hypoxic RU cells was appreciably lower than that of native RR cells. Overall, our flow cytometric results were in parallel with the luciferase assay results.
Fig. 1.

Hypoxia significantly increased tumorigenic and stem-like properties in RU cells. The effect of 48 h incubation under hypoxia, compared to RU and RR cells incubated under normoxia, in RU cells on: a GFP expression as illustrated by flow cytometry histogram plots, b SRR2 luciferase activity, and c Proportion of CD44high/CD24low cells as illustrated by flow cytometry dot plots. d Colony formation ability was assessed for RU cells after 24 h incubation under hypoxia. Cells were then re-plated at density of 1000 cells/well in duplicate in six-well plates under normoxia. The number of colonies formed from 1000 cells after 7 days was graphed. e Cellular viability was measured by MTT assay after 48 h exposure to increasing concentration of cisplatin (3.32–332 μM). (*) denotes a significant difference between hypoxic and native RU cells (one-way ANOVA followed by a post-hoc Tukey test, p < 0.05). Data are represented as mean ± SD (n = 3)
Hypoxia Significantly Increased Tumorigenic and Stem-like Properties in RU Cells
Next, we asked if the hypoxia-induced RU/RR conversion correlates with any phenotypic changes. RU cells cultured under hypoxia showed a significant increase in the percentage of cells expressing the CD44high/CD24low immunophenotype, a well-known marker to be associated with cancer stemness in TNBC [18]. Specifically, with triplicate experiments, the mean percentage of these stem cells in native RU cells was 1.7 ± 0.9%, as compared to 4.8 ± 0.8% in hypoxic RU cells (p = 0.03) (illustrated in Fig. 1c). As a comparison, the median percentage of cells expressing the CD44high/CD24low immunophenotype in native RR cells was 3.8 ± 0.6%. We then assessed the tumorigenicity using colony formation assay. Compared to native RU cells, hypoxic RU cells showed a significant increase in the number of colonies (330 ± 10 versus 414 ± 25, p = 0.01) (Fig. 1d). In the same assay, the median number of colonies formed by native RR cells was 545 ± 40, which is significantly different from that of hypoxic RU cells. As shown in Fig. 1e, hypoxia significantly increased the resistance of RU cells to cisplatin, with the inhibitory concentration at 50% (IC50) increasing from 16 μM to 300 μM, (p < 0.001) at 48 h.
STAT3 Activation Drives Hypoxia-Induced RU/RR Conversion
Since we have recently shown that signal transducer and activator of transcription 3 (STAT3) activation is important in conferring cancer stemness and chemoresistance in MDA-MB-231 under hypoxia [17], we hypothesized that the STAT3 signaling pathway may be important in regulating the hypoxia-induced RU/RR conversion. In support of this hypothesis, our previous bioinformatics studies have shown that the Sox2 regulatory region 2 (SRR2) sequence carries the STAT3 consensus binding sequence (data not shown). As shown in Fig. 2a, compared to native RU cells, hypoxic RU cells had a substantial upregulation of phospho-STAT3 (pSTAT3), the active form of STAT3, at 24 and 48 h. In comparison, the total STAT3 level was not appreciably changed. We then performed a DNA pull-down experiment. As shown in Fig. 2b, a biotinylated DNA probe with the Sox2 binding sequence (i.e. SRR2) pulled down a substantial amount of STAT3 protein in hypoxic RU cells. In contrast, only a relatively small amount of STAT3 was captured in native RU cells. We then treated these hypoxic RU cells with Stattic, a STAT3 inhibitor [19], and found that the STAT3-SRR2 interaction was dramatically decreased at 20 μM of Stattic (Fig. 2c).
Fig. 2.

STAT3 activation drives hypoxia-induced RU/RR conversion. a Western blot is depicting the effect of 24 and 48 h incubation under hypoxia on the protein expression profile of RU cells as compared to native RU and native RR cells. b Higher amount of STAT3 was pulled down with SRR2 probe, compared to native RU cells, in hypoxic RU cells after 48 h hypoxia. c Lower amount of STAT3 was pulled down with SRR2 probe in hypoxic RU cells in the presence of STAT3 inhibitor (20 μM Stattic)
To provide further evidence that STAT3 activation plays a direct role in the observed biological changes induced by hypoxia in RU cells, we knocked down the expression of STAT3 using siRNA. As shown in Fig. 3a, successful knockdown of STAT3 and its downstream target genes (i.e., survivin, Mcl-1 and c-Myc) was achieved. Importantly, STAT3 silencing in hypoxic RU cells hindered the hypoxia-induced conversion of RU/RR cells, as evidenced by a lower median GFP expression level in hypoxic RU cells subjected to STAT3 siRNA (85 ± 13), as compared to hypoxic RU cells subjected to scrambled siRNA (115 ± 13) (p = 0.01). Of note, in the same experiment, the median GFP expression level of native RU cells was (78 ± 15), which is not significantly different from that of hypoxic RU cells treated with STAT3 siRNA (Fig. 3b). Similar results were obtained when we measured the luciferase activity as the readout of the reporter (Fig. 3c). Overall, our results strongly suggest that STAT3 is a key contributor to the hypoxia-induced RU/RR conversion in MDA-MB-231 cells.
Fig. 3.
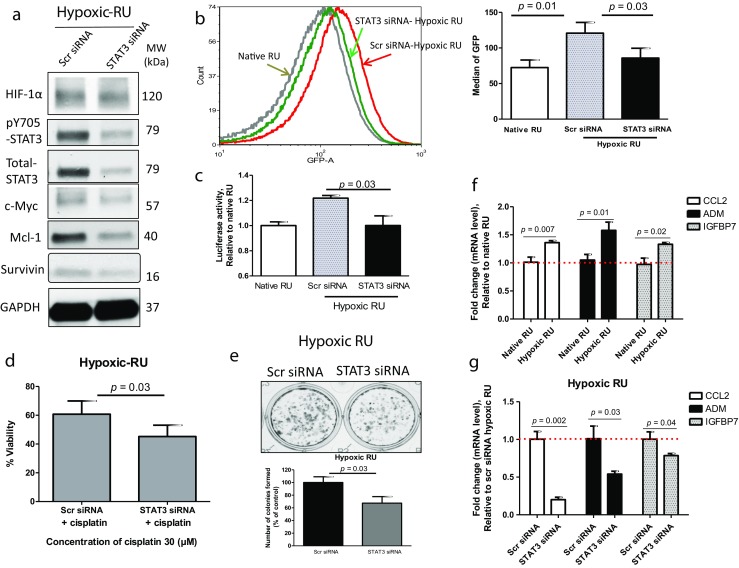
Suppression of STAT3 effectively inhibits RU/RR conversion. The effect of STAT3 silencing using siRNA in hypoxic RU cells (48 h hypoxia) on: a Protein expression profile as depicted by Western blot, b GFP expression as illustrated by flow cytometry histograms, and c SRR2 luciferase activity. d Cellular viability was measured after 48 h cisplatin (30 uM) exposure in hypoxic RU cells transfected with either scrambled or STAT3 siRNAs. e Clonogenic survival assay was conducted in hypoxic RU cells transfected with either scrambled or STAT3 siRNA. Cells were then re-plated at density of 1000 cells/well in duplicate in six-well plates under normoxia. The number of colonies formed from 1000 cells was graphed. f qRT-PCR results of CCL2, ADM and IGFBP7 genes expression in hypoxic RU cells (24 h hypoxia) normalized to GAPDH, and further normalized to native RU cells, and g qRT-PCR results of CCL2, ADM and IGFBP7 genes expression after STAT3 silencing using siRNA in hypoxic RU cells (24 h hypoxia) normalized to GAPDH, and further normalized to hypoxic RU cells subjected to scrambled siRNA
To further substantiate the importance of STAT3 in hypoxia-induced acquisition of stem-like phenotype, we found that STAT3 knockdown was efficient in abrogating hypoxia-induced cisplatin chemoresistance, as compared to hypoxic RU cells subjected to scrambled siRNA (p = 0.03) (Fig. 3d). Hypoxic RU cells also showed a significant decrease in colony formation when subjected to STAT3 siRNA, as compared to cells subjected to scrambled siRNA (124 ± 12 versus 84 ± 24, p = 0.03) (Fig. 3e).
To decipher the mechanism by which STAT3 promotes hypoxia-induced cancer stemness in TNBC, we performed oligonucleotide quantitative RT-PCR (qRT-PCR) array studies comparing native RU cells and hypoxic RU cells, as described in Materials and Methods. Of the 84 genes included in the array, all of which carry known functions in cancer proliferation, cell survival, hypoxia responses, cancer invasiveness and metastasis, we identified 37 gene targets showing ≥1.5 fold changes (Table S2 and S3). Seven of these 37 gene targets were found to be known STAT3 downstream targets via our literature search (Table 1). We then repeated the same qRT-PCR oligonucleotide array comparing hypoxic RU cells transfected with scrambled siRNA and cells transfected with STAT3 siRNA. As shown in Table 1, we found that all of the seven STAT3 targets showed >30% changes in their expression levels. Using quantitative RT-PCR, we were able to validate three of these seven genes, including ADM, CCL2 and IGFBP7) (Fig. 3f–g). Notably, native RR cells showed substantially higher expression levels of these 3 genes as compared to those in native RU cells or hypoxic RU cells (Fig. S1).
Table 1.
Genes regulated by STAT3
c-Myc Is Not a Contributing Factor in Hypoxia-Induced RU/RR Conversion
We also asked if c-Myc plays a role in the hypoxia-induced RU/RR conversion of MDA-MB-231 cells, since c-Myc has been implicated in cancer stemness in TNBC [28, 29] and SRR2 also has the c-Myc consensus binding site [30]. Furthermore, we recently found that c-Myc is an important mediator of the RR/RU dichotomy at the steady state [30]. As shown in Fig. 2a, we found that the expression of c-Myc in RU cells was dramatically downregulated under hypoxia. Accordingly, the DNA binding assay also showed a substantially lower level of c-Myc protein pulled down with the SRR2 probe in hypoxic RU cells as compared to native RU cells (Fig. 4a). Moreover, siRNA knockdown of c-Myc did not reverse the hypoxia-induced chemoresistance to cisplatin in hypoxic RU cells (Fig. 4b–c). Lastly, when we abrogated the hypoxia-mediated suppression of c-Myc by enforced expression of c-Myc in RU cells (Fig. 4d), we did not observe any significant change in the colony formation ability of these c-Myc-transfected cells as compared to RU cells transfected with an empty vector (Fig. 4e). Overall, our collected data suggest that c-Myc is not implicated in the acquisition of hypoxia-induced cancer stemness in MDA-MB-231 cells.
Fig. 4.

Hypoxia-induced conversion of RU/RR cells is independent of c-Myc. a Lower amount of c-Myc was pulled down with SRR2 probe, compared to native RU cells, in hypoxic RU cells after 48 h hypoxia. b Successful knockdown of c-Myc in hypoxic RU cells as depicted by Western blot after 48 h hypoxia. c siRNA knockdown of c-Myc did not reverse the chemoresistance in hypoxic RU cells after 48 h cisplatin exposure. d Hypoxia-mediated suppression of c-Myc, compared to RU cells carrying empty vector (RU/EV), was abrogated in c-Myc overexpressing RU cells (RU/Myc) as depicted by Western blot after 48 h hypoxia. e Abrogation of c-Myc suppression under hypoxia in overexpressing c-Myc RU cells (RU/Myc), compared to RU/EV cells, did not enhance their clonogenic potential. Colony formation ability was assessed for RU/Myc and RU/EV cells after 24 h incubation under hypoxia. Cells were then re-plated at density of 1000 cells/well in duplicate in six-well plates under normoxia. The number of colonies formed from 1000 cells after 7 days was graphed
Discussion
Malignant tumors are often made up of a heterogeneous population of cancer cells displaying a spectrum of tumor initiating potential or cancer stemness [31]. More recently, it has been recognized that acquisition of cancer stemness can be achieved spontaneously [32] or in response to specific stimuli such as hypoxia [33]. These observations are believed to be encompassed under the concept of cancer cell plasticity, which has been recently reviewed [9–11]. The acquisition of cancer cell stemness induced by hypoxia correlates with the de-differentiation of cancer cells and a significant increase in their malignant aggressiveness [34]. In keeping with these findings, evidence of cancer cell plasticity was found most frequently in the hypoxic regions of human tumors [35]. In a breast cancer study, hypoxia was found to correlate with an increase in the nuclear-cytoplasmic ratio, loss of cell polarity, downregulation of estrogen receptor-α, and upregulation of the epithelial breast stem cell marker CK19 [36]. In a study of neuroblastoma, highly tumorigenic cells were found preferentially located in the hypoxic regions [37]. Lastly, upregulation of CD133, another CSC marker, had been found preferentially in the hypoxic regions of medulloblastomas [38].
Hypoxic challenge has been used to induce cancer cell plasticity in a number of published studies. In many of these study models, cancer cells were found to exhibit evidence of de-differentiation, expressed CSC markers [37, 39] and showed a significant increase in their clonogenic potential in-vitro [40, 41]. Hypoxia has been shown to induce the epithelial-mesenchymal transition [42, 43]. Exposure of cancer cells to hypoxia in-vitro also was found to result in enhanced tumorigenic potential in a mouse xenograft model [39]. To study hypoxia-induced acquisition of cancer stemness, we employed an experimental model that is different from those used in most of the previously studies. Specifically, cancer stemness was detected based on the differential responses to a Sox2 reporter, rather than the presence or absence of cell-surface proteins. In this regard, we and others have previously identified and published similar intratumoral heterogeneity in different types of cancer, including estrogen receptor-positive breast cancer, esophageal squamous cell carcinoma and ALK-positive anaplastic large cell lymphoma [12–15]. Most recently, similar observations were made in neuroblastoma [16]. In all of these studies, RR cells were found to be consistently more tumorigenic and stem-like than RU cells. Importantly, evidence of the RU/RR dichotomy was identified in human samples, strongly arguing against the notion that this is not a cell-type specific phenomenon, and supporting the concept this is a valid and clinically relevant experimental model to study cancer cell plasticity. Unlike some of the previously published study models, the application of the RU/RR model is technically straightforward, since RU and RR cells can be easily detected and/or purified based on their differential expression of GFP/luciferase. In the case of MDA-MB-231 cells, we found that hypoxia consistently upregulates the expression of luciferase and GFP in hypoxia-treated RU cells, which correlated with their acquisition of stem-like features. Specifically, hypoxia-treated RU cells exhibited significantly higher colony formation capacity, chemoresistance to cisplatin, and a higher proportion of cells carrying the CD44high/CD24low immunophenotype, a well-known marker to be associated with cancer stemness in TNBC [18].
Although hypoxia-inducible factor-1 alpha (HIF-1α) is believed to be a key mediator of the biological changes in response to hypoxia in cancer cells [44], we did not find evidence that this transcription factor plays an important role in the hypoxia-induced RU/RR conversion in our experimental model. Specifically, siRNA knockdown of HIF-1α in RU cells did not result in significant changes to the hypoxia-induced increases in chemoresistance to cisplatin, the proportion of CD44high/CD24low cells and mammosphere formation ability (Fig. S2). Of note, HIF-1α was also found to be relatively irrelevant in promoting hypoxia-induced chemoresistance in other cancer models [17, 45–48]. Similarly, c-Myc, another well-known stem cell factor that can interact with SRR2 in our Sox2 reporter, was also found to be irrelevant to the hypoxia-induced RU/RR conversion in our model, as this protein was downregulated in response to hypoxia. Similar observations regarding hypoxia-induced downregulation of c-Myc have been demonstrated in different experimental models [49, 50]. Our studies have led us to discover that STAT3, for which the SRR2 contains the consensus binding sequence, is a key regulator for the hypoxia-induced RU/RR conversion in MDA-MB-231 cells. In this regard, RU cells exposed to hypoxia exhibited a substantial upregulation of pSTAT3 and its transcriptional activity, as evidenced by the results of our STAT3-DNA binding assay and oligonucleotide array. In support of the relevance of STAT3 activation in this context, we found that siRNA knockdown of STAT3 in RU cells significantly inhibited RU/RR conversion and the acquisition of associated stem-like features.
While HIF, histone 3 lysine 4 (H3K4) demethylase JARID1B and histone methyltransferase mixed-lineage leukemia 1 (MLL1) have been shown as to be key mediators of hypoxia-induced cancer stemness in a number of experimental models [51], the role of STAT3 in hypoxia-induced cancer stemness has not been previously described. Of note, we have previously shown that hypoxia-induced upregulation of STAT3 in MDA-MB-231 cells is HIF-independent [17], a finding also shared by another group of investigators [52]. This finding is consistent with our current observation that HIF is not relevant in promoting hypoxia-induced stemness in MDA-MB-231.
Results from the oligonucleotide array studies have served two purposes. Firstly, the results allow us to confirm that STAT3 transcription activity was upregulated upon hypoxic challenge, since we identified seven known STAT3 target genes showing ≥1.5 fold changes in response to hypoxia. Secondly, these results have shed insight into the mechanism by which STAT3 might promote hypoxia-induced cancer stemness. For instance, these seven STAT3 downstream target genes highlighted in our oligonucleotide array studies are known to be involved in angiogenesis and metastasis (ADM, ANGPT1, and CCL2) [20–23], cell growth (IGFBP5, and IGFBP7) [24, 25], and metabolism (LPL, and PFKL) [26, 27]. Of note, hypoxia-induced upregulation of ADM (adrenomodullin) in various cell lines was found in another study [53]. The expression of ADM has been reported in different types of cancer including that of lung, colon, ovary and breast [54]. ADM plays an important role in tumor progression and metastasis in breast cancer by upregulating a number of oncogenes such as RAS, RAF, PKC (protein kinase C) and MAPK (mitogen-activated protein kinase) [55]. Regarding CCL2 (CC-chemokine ligand 2), it has been reported that this molecule can stimulate stem-like features, such as mammosphere capacity and self-renewal ability in breast cancer cells [56]. IGFBP5 (insulin-like growth factor binding protein 5) is known to play a crucial role in carcinogenesis by regulating cell growth, migration, and invasion in different types of cancer [57]. PFK1 (phosphofructokinase 1) is a major regulatory enzyme in the glycolytic pathway, and hypoxia is known to confer growth advantage and tumorigenicity through induction of PFK1-associated glycosylation in lung cancer [58]. LPL (lipoprotein lipase) is another enzyme involved in metabolism which catalyzes hydrolysis of triglycerides into free fatty acids. It has been shown that LPL is aberrantly expressed in chronic lymphocytic leukemia and regulates the oxidative metabolic capacity of these leukemic cells [26].
We would like to point out that the major shortcoming of our study is that we described the results of only one cell line. In this regard, we did perform experiments using another TNBC cell line, SUM149, but the generated results were conflicting at times, resulting in major difficulties in presenting our findings. We speculated that the discrepancies in the results generated in two different TNBC cell lines are likely due to the fact that TNBC is a biologically and molecularly heterogeneous disease [59, 60]. In spite of this shortcoming, we believe that our results and conclusions are valid, and our studies have provide proof-of-principle that STAT3 is relevant and important in the context of hypoxia-induced RU/RR conversion and cancer cell plasticity, probably in a subset of TNBC. Further investigations using a large panel of TNBC cell lines and primary patient samples are warranted.
Conclusion
To conclude, we have provided evidence to support that STAT3 plays an important role in conferring hypoxia-induced acquisition of cancer stemness in MDA-MB-231 cells. Additional studies in other TNBC cell lines and primary samples are required to validate targeting of STAT3 as a useful therapeutic approach to overcome treatment-induced cancer stemness.
Electronic supplementary material
(DOCX 652 kb)
Acknowledgements
This work was financially supported by grants from Canadian Institutes of Health Research (CIHR) MOP 137153 and Canadian Breast Cancer Foundation (CBCF) awarded to A.L and R.L. H.S.A was awarded the Women and Children’s Health Research Institute (WCHRI) and Alberta Cancer Foundation (ACF) Graduate Studentships. N.G was funded by CBCF. The authors would like to thank Amir Soleimani, Department of Pharmacy and Pharmaceutical Sciences, University of Alberta, for critical reading of the manuscript.
Authors’ Contributions
H.S.A designed the research plan, carried out experiments and wrote the manuscript. N.G contributed to the design and performance of the experiments and data analysis and critical reading of the manuscript. A.A contributed to the design and data analysis of oligonucleotide arrays experiment and critical reading of the manuscript. K.G assisted with the flow cytometric detection of RU/RR conversion. A.L and R.L conceived and designed the research plan and critical reading of the manuscript. All authors read and approved the final manuscript.
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
Contributor Information
Hoda Soleymani Abyaneh, Email: hoda1@ualberta.ca.
Nidhi Gupta, Email: nidhi2@ualberta.ca.
Abdulraheem Alshareef, Email: al15@ualberta.ca.
Keshav Gopal, Email: keshav.gopal@ualberta.ca.
Afsaneh Lavasanifar, Email: afsaneh@ualberta.ca.
Raymond Lai, Phone: +1 780-432-8457, Email: rlai@ualberta.ca.
References
- 1.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 2.Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 4.Smid M, Wang Y, Zhang Y, Sieuwerts AM, Yu J, Klijn JG, Foekens JA, Martens JW. Subtypes of breast cancer show preferential site of relapse. Cancer Res. 2008;68(9):3108–3114. doi: 10.1158/0008-5472.CAN-07-5644. [DOI] [PubMed] [Google Scholar]
- 5.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100(7):3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res. 2008;10(2):R25. doi: 10.1186/bcr1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heddleston JM, Li Z, Lathia JD, Bao S, Hjelmeland AB, Rich JN. Hypoxia inducible factors in cancer stem cells. Br J Cancer. 2010;102(5):789–795. doi: 10.1038/sj.bjc.6605551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O'Reilly EA, Gubbins L, Sharma S, Tully R, Guang MH, Weiner-Gorzel K, McCaffrey J, Harrison M, Furlong F, Kell M, et al. The fate of chemoresistance in triple negative breast cancer (TNBC) BBA Clin. 2015;3:257–275. doi: 10.1016/j.bbacli.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doherty Mary, Smigiel Jacob, Junk Damian, Jackson Mark. Cancer Stem Cell Plasticity Drives Therapeutic Resistance. Cancers. 2016;8(1):8. doi: 10.3390/cancers8010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee G, Hall RR 3rd, Ahmed AU (2016) Cancer stem cells: cellular plasticity, niche, and its clinical relevance. J Stem Cell Res Ther 6(10) [DOI] [PMC free article] [PubMed]
- 11.Cabrera MC, Hollingsworth RE, Hurt EM. Cancer stem cell plasticity and tumor hierarchy. World J Stem Cells. 2015;7(1):27–36. doi: 10.4252/wjsc.v7.i1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu F, Zhang J, Wang P, Ye X, Jung K, Bone KM, Pearson JD, Ingham RJ, McMullen TP, Ma Y, et al. Identification of two novel phenotypically distinct breast cancer cell subsets based on Sox2 transcription activity. Cell Signal. 2012;24(11):1989–1998. doi: 10.1016/j.cellsig.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 13.Jung K, Gupta N, Wang P, Lewis JT, Gopal K, Wu F, Ye X, Alshareef A, Abdulkarim BS, Douglas DN, et al. Triple negative breast cancers comprise a highly tumorigenic cell subpopulation detectable by its high responsiveness to a Sox2 regulatory region 2 (SRR2) reporter. Oncotarget. 2015;6(12):10366–10373. doi: 10.18632/oncotarget.3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gelebart P, Hegazy SA, Wang P, Bone KM, Anand M, Sharon D, Hitt M, Pearson JD, Ingham RJ, Ma Y, et al. Aberrant expression and biological significance of Sox2, an embryonic stem cell transcriptional factor, in ALK-positive anaplastic large cell lymphoma. Blood Cancer J. 2012;2:e82. doi: 10.1038/bcj.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang HF, Wu C, Alshareef A, Gupta N, Zhao Q, Xu XE, Jiao JW, Li EM, Xu LY, Lai R. The PI3K/AKT/c-MYC Axis promotes the acquisition of cancer stem-like features in esophageal squamous cell carcinoma. Stem Cells. 2016;34(8):2040–2051. doi: 10.1002/stem.2395. [DOI] [PubMed] [Google Scholar]
- 16.Alshareef A, Gupta N, Zhang HF, Wu C, Haque M, Lai R. High expression of beta-catenin contributes to the crizotinib resistant phenotype in the stem-like cell population in neuroblastoma. Sci Rep. 2017;7(1):16863. doi: 10.1038/s41598-017-17319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soleymani Abyaneh H, Gupta N, Radziwon-Balicka A, Jurasz P, Seubert J, Lai R, Lavasanifar A. STAT3 but not HIF-1alpha is important in mediating hypoxia-induced chemoresistance in MDA-MB-231, a triple negative breast cancer cell line. Cancers (Basel) 2017;9(10):137. doi: 10.3390/cancers9100137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaggupilli A, Elkord E. Significance of CD44 and CD24 as cancer stem cell markers: an enduring ambiguity. Clin Dev Immunol. 2012;2012:708036. doi: 10.1155/2012/708036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13(11):1235–1242. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 20.Lim SY, Ahn SH, Park H, Lee J, Choi K, Choi C, Choi JH, Park EM, Choi YH. Transcriptional regulation of adrenomedullin by oncostatin M in human astroglioma cells: implications for tumor invasion and migration. Sci Rep. 2014;4:6444. doi: 10.1038/srep06444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snyder M, Huang X-Y, Zhang JJ. Identification of novel direct Stat3 target genes for control of growth and differentiation. J Biol Chem. 2008;283(7):3791–3798. doi: 10.1074/jbc.M706976200. [DOI] [PubMed] [Google Scholar]
- 22.Singha B, Gatla HR, Vancurova I. Transcriptional regulation of chemokine expression in ovarian cancer. Biomolecules. 2015;5(1):223–243. doi: 10.3390/biom5010223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen W, Gao Q, Han S, Pan F, Fan W. The CCL2/CCR2 axis enhances IL-6-induced epithelial-mesenchymal transition by cooperatively activating STAT3-twist signaling. Tumour Biol. 2015;36(2):973–981. doi: 10.1007/s13277-014-2717-z. [DOI] [PubMed] [Google Scholar]
- 24.Kojima H, Kunimoto H, Inoue T, Nakajima K. The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle. 2012;11(4):730–739. doi: 10.4161/cc.11.4.19172. [DOI] [PubMed] [Google Scholar]
- 25.Couto JP, Daly L, Almeida A, Knauf JA, Fagin JA, Sobrinho-Simões M, Lima J, Máximo V, Soares P, Lyden D, et al. STAT3 negatively regulates thyroid tumorigenesis. Proc Natl Acad Sci. 2012;109(35):E2361–E2370. doi: 10.1073/pnas.1201232109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rozovski U, Grgurevic S, Bueso-Ramos C, Harris DM, Li P, Liu Z, Wu JY, Jain P, Wierda W, Burger J, et al. Aberrant LPL expression, driven by STAT3, mediates free fatty acid metabolism in CLL cells. Mol Cancer Res. 2015;13(5):944–953. doi: 10.1158/1541-7786.MCR-14-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang TX, Shi XY, Cong Y, Zhang ZQ, Liu YH. Prosapogenin A inhibits cell growth of MCF7 via downregulating STAT3 and glycometabolism-related gene. Yao Xue Xue Bao. 2013;48(9):1510–1514. [PubMed] [Google Scholar]
- 28.Moumen M, Chiche A, Decraene C, Petit V, Gandarillas A, Deugnier MA, Glukhova MA, Faraldo MM. Myc is required for beta-catenin-mediated mammary stem cell amplification and tumorigenesis. Mol Cancer. 2013;12(1):132. doi: 10.1186/1476-4598-12-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moumen M, Chiche A, Deugnier MA, Petit V, Gandarillas A, Glukhova MA, Faraldo MM. The proto-oncogene Myc is essential for mammary stem cell function. Stem Cells. 2012;30(6):1246–1254. doi: 10.1002/stem.1090. [DOI] [PubMed] [Google Scholar]
- 30.Gupta N, Jung K, Wu C, Alshareef A, Alqahtani H, Damaraju S, Mackey JR, Ghosh S, Sabri S, Abdulkarim BS, et al. High Myc expression and transcription activity underlies intra-tumoral heterogeneity in triple-negative breast cancer. Oncotarget. 2017;8(17):28101–28115. doi: 10.18632/oncotarget.15891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marusyk A, Almendro V, Polyak K. Intra-tumour heterogeneity: a looking glass for cancer? Nat Rev Cancer. 2012;12(5):323–334. doi: 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 32.Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO, Brooks M, Reinhardt F, Su Y, Polyak K, et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci. 2011;108(19):7950–7955. doi: 10.1073/pnas.1102454108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8(12):967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN. The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle. 2009;8(20):3274–3284. doi: 10.4161/cc.8.20.9701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hill RP, Marie-Egyptienne DT, Hedley DW. Cancer stem cells, hypoxia and metastasis. Semin Radiat Oncol. 2009;19(2):106–111. doi: 10.1016/j.semradonc.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 36.Helczynska K, Kronblad A, Jogi A, Nilsson E, Beckman S, Landberg G, Pahlman S. Hypoxia promotes a dedifferentiated phenotype in ductal breast carcinoma in situ. Cancer Res. 2003;63(7):1441–1444. [PubMed] [Google Scholar]
- 37.Das B, Tsuchida R, Malkin D, Koren G, Baruchel S, Yeger H. Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells. 2008;26(7):1818–1830. doi: 10.1634/stemcells.2007-0724. [DOI] [PubMed] [Google Scholar]
- 38.Platet N, Liu SY, Atifi ME, Oliver L, Vallette FM, Berger F, Wion D. Influence of oxygen tension on CD133 phenotype in human glioma cell cultures. Cancer Lett. 2007;258(2):286–290. doi: 10.1016/j.canlet.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Jogi A, Ora I, Nilsson H, Lindeheim A, Makino Y, Poellinger L, Axelson H, Pahlman S. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc Natl Acad Sci U S A. 2002;99(10):7021–7026. doi: 10.1073/pnas.102660199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desplat V, Faucher JL, Mahon FX, Dello Sbarba P, Praloran V, Ivanovic Z. Hypoxia modifies proliferation and differentiation of CD34(+) CML cells. Stem Cells. 2002;20(4):347–354. doi: 10.1634/stemcells.20-4-347. [DOI] [PubMed] [Google Scholar]
- 41.Kim Y, Lin Q, Zelterman D, Yun Z. Hypoxia-regulated delta-like 1 homologue enhances cancer cell stemness and tumorigenicity. Cancer Res. 2009;69(24):9271–9280. doi: 10.1158/0008-5472.CAN-09-1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, Liu J, Wang Q, Zhu J, Feng X, et al. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor -1alpha in hepatocellular carcinoma. BMC Cancer. 2013;13:108. doi: 10.1186/1471-2407-13-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Renaud S, Guenot D, Falcoz P-E, Massard G, Beau-Faller M. Role of hypoxia in epithelial-to-mesenchymal transition (EMT) in non-small cell lung cancer (NSCLC) Eur Respir J. 2014;44(Suppl 58):P814. [Google Scholar]
- 44.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29(5):625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adamski J, Price A, Dive C, Makin G. Hypoxia-induced cytotoxic drug resistance in osteosarcoma is independent of HIF-1Alpha. PLoS One. 2013;8(6):e65304. doi: 10.1371/journal.pone.0065304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piret JP, Cosse JP, Ninane N, Raes M, Michiels C. Hypoxia protects HepG2 cells against etoposide-induced apoptosis via a HIF-1-independent pathway. Exp Cell Res. 2006;312(15):2908–2920. doi: 10.1016/j.yexcr.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Biju MP, Wang MH, Haase VH, Dong Z. Cytoprotective effects of hypoxia against cisplatin-induced tubular cell apoptosis: involvement of mitochondrial inhibition and p53 suppression. J Am Soc Nephrol. 2006;17(7):1875–1885. doi: 10.1681/ASN.2005121371. [DOI] [PubMed] [Google Scholar]
- 48.Hao J, Song X, Song B, Liu Y, Wei L, Wang X, Yu J. Effects of lentivirus-mediated HIF-1alpha knockdown on hypoxia-related cisplatin resistance and their dependence on p53 status in fibrosarcoma cells. Cancer Gene Ther. 2008;15(7):449–455. doi: 10.1038/cgt.2008.4. [DOI] [PubMed] [Google Scholar]
- 49.Wong WJ, Qiu B, Nakazawa MS, Qing G, Simon MC. MYC degradation under low O2 tension promotes survival by evading hypoxia-induced cell death. Mol Cell Biol. 2013;33(17):3494–3504. doi: 10.1128/MCB.00853-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8(1):51–56. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 51.Yun Z, Lin Q. Hypoxia and regulation of cancer cell stemness. Adv Exp Med Biol. 2014;772:41–53. doi: 10.1007/978-1-4614-5915-6_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pawlus MR, Wang L, Hu CJ. STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene. 2014;33(13):1670–1679. doi: 10.1038/onc.2013.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sena JA, Wang L, Pawlus MR, Hu CJ. HIFs enhance the transcriptional activation and splicing of adrenomedullin. Mol Cancer Res. 2014;12(5):728–741. doi: 10.1158/1541-7786.MCR-13-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jean Miller M, Martínez A, Unsworth EJ, Thiele CJ, Moody TW, Elsasser T, Cuttitta F. Adrenomedullin expression in human tumor cell lines. Its potential role as an autocrine growth factor. J Biol Chem. 1996;271(38):23345–23351. doi: 10.1074/jbc.271.38.23345. [DOI] [PubMed] [Google Scholar]
- 55.Martinez A, Vos M, Guedez L, Kaur G, Chen Z, Garayoa M, Pio R, Moody T, Stetler-Stevenson WG, Kleinman HK, et al. The effects of adrenomedullin overexpression in breast tumor cells. J Natl Cancer Inst. 2002;94(16):1226–1237. doi: 10.1093/jnci/94.16.1226. [DOI] [PubMed] [Google Scholar]
- 56.Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, Luu T, Li AX, Wu X, Ye W, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72(11):2768–2779. doi: 10.1158/0008-5472.CAN-11-3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gullu G, Karabulut S, Akkiprik M. Functional roles and clinical values of insulin-like growth factor-binding protein-5 in different types of cancers. Chin J Cancer. 2012;31(6):266–280. doi: 10.5732/cjc.011.10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, 3rd, Peters EC, Driggers EM, Hsieh-Wilson LC. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337(6097):975–980. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehmann BD, Pietenpol JA. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast. 2015;24(Suppl 2):S36–S40. doi: 10.1016/j.breast.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bianchini G, Balko JM, Mayer IA, Sanders ME, Gianni L. Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat Rev Clin Oncol. 2016;13(11):674–690. doi: 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 652 kb)