

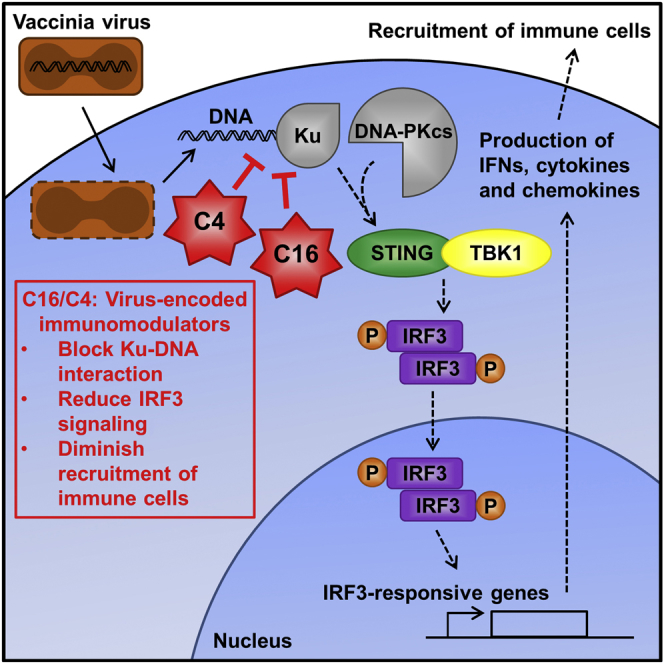
Summary
Virus infection is sensed by pattern recognition receptors (PRRs) detecting virus nucleic acids and initiating an innate immune response. DNA-dependent protein kinase (DNA-PK) is a PRR that binds cytosolic DNA and is antagonized by vaccinia virus (VACV) protein C16. Here, VACV protein C4 is also shown to antagonize DNA-PK by binding to Ku and blocking Ku binding to DNA, leading to a reduced production of cytokines and chemokines in vivo and a diminished recruitment of inflammatory cells. C4 and C16 share redundancy in that a double deletion virus has reduced virulence not seen with single deletion viruses following intradermal infection. However, non-redundant functions exist because both single deletion viruses display attenuated virulence compared to wild-type VACV after intranasal infection. It is notable that VACV expresses two proteins to antagonize DNA-PK, but it is not known to target other DNA sensors, emphasizing the importance of this PRR in the response to infection in vivo.
Keywords: DNA sensing, DNA protein kinase, pattern recognition receptor, IRF3 signaling, vaccinia virus, immune evasion, protein C4, virulence factor
Graphical Abstract
Highlights
-
•
DNA-PK is a pattern recognition receptor that binds cytosolic DNA
-
•
Vaccinia virus proteins C4 and C16 antagonize DNA-PK by blocking DNA binding
-
•
C4 and C16 inhibit IRF3 signaling, cytokine production, and immune cell recruitment
-
•
C4 and C16 share redundant and non-redundant functions in vivo
DNA-PK is a pattern recognition receptor (PRR) that binds cytosolic DNA and stimulates IRF3 signaling. Scutts et al. show that vaccinia virus antagonizes this DNA sensor with two proteins, C4 and C16, which both block DNA binding.
Introduction
When confronted by a pathogen, the host’s innate immune system is activated by pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) (Brubaker et al., 2015). For viruses, the detection of viral nucleic acids is a critical element of this recognition (Luecke and Paludan, 2017, Pichlmair and Reis e Sousa, 2007). There are several intracellular DNA sensors, such as DNA-dependent activator of interferon (IFN)-regulatory factor (DAI) (Takaoka et al., 2007), absent in melanoma 2 (AIM2) (Bürckstümmer et al., 2009, Dombrowski et al., 2011, Fernandes-Alnemri et al., 2009, Hornung et al., 2009, Roberts et al., 2009), RNA polymerase III (Ablasser et al., 2009, Chiu et al., 2009), LRRFIP1 (Yang et al., 2010), DExH-box helicase (DHX)9/DHX36 (Kim et al., 2010), double-strand break repair protein MRE11 (MRE11) (Kondo et al., 2013), polyglutamine binding protein-1 (PQBP1) (Yoh et al., 2015), gamma-IFN inducible protein 16 (IFI16) (Unterholzner et al., 2010), cyclic guanosine monophosphate-AMP synthase (cGAS) (Sun et al., 2013), and DNA-dependent protein kinase (DNA-PK) (Ferguson et al., 2012, Zhang et al., 2011). However, which DNA sensors detect specific viruses, whether these PRRs act independently or cooperatively, and their relative importance in vivo require further study.
Poxviruses have double-stranded DNA (dsDNA) genomes, yet they replicate within the cytoplasm. Poxviruses that cause human disease include monkeypox virus (Reynolds et al., 2004), molluscum contagiosum virus (Hanson and Diven, 2003) and variola virus (VARV), the cause of smallpox, a disease eradicated by vaccination with vaccinia virus (VACV) (Fenner et al., 1988). VACV is a useful expression vector (Mackett et al., 1982, Panicali and Paoletti, 1982) and has applications as vaccines against other pathogens (Panicali et al., 1983, Smith et al., 1983a, Smith et al., 1983b) and as an oncolytic agent (Buller et al., 1985, Heo et al., 2013, Kirn and Thorne, 2009). In addition, interest in VACV endures because it is an excellent model to study host-pathogen interactions and cell biology (Smith et al., 2013).
The detection of DNA by PRRs triggers the production of type I IFN, cytokines, and chemokines (Stetson and Medzhitov, 2006) via a pathway that requires stimulator of IFN genes (STING) (Ishikawa et al., 2009), TANK-binding kinase 1 (TBK1), and IFN regulatory factor 3 (IRF3) (Ishii et al., 2008, Ishii et al., 2006, Tanaka and Chen, 2012). For example, the detection of VACV DNA induced IFN-β independently of Toll-like receptors (TLRs) and RNA polymerase III but was dependent on STING, TBK1, and IRF3 (Unterholzner et al., 2010).
DNA-PK is a heterotrimeric complex consisting of the catalytic subunit DNA-PKcs and a heterodimer of Ku70 and Ku80. DNA-PK binds dsDNA breaks and functions in non-homologous end joining (NHEJ) (Pannunzio et al., 2018) and also in DNA sensing, upregulating type I IFN and cytokines via the STING pathway (Ferguson et al., 2012). The kinase activity of DNA-PKcs is essential for DNA repair (Kurimasa et al., 1999) but not for innate immune signaling. Cells and mice lacking DNA-PK components show impaired response to infection with VACV and herpes simplex virus 1 (HSV-1) (Ferguson et al., 2012). Ku70 also induces the expression of type III IFN in response to DNA (Sui et al., 2017, Zhang et al., 2011).
The evolution of pathogens with their hosts has produced intriguing strategies for both host detection and pathogen subversion. VACV is a good example of this, and between one-third and one-half of its 200 proteins modulate the immune response (Bowie and Unterholzner, 2008, Elde et al., 2012, Gubser et al., 2004). However, for dsDNA-binding PRRs, the only known direct inhibitor is protein VACV C16, an inhibitor of DNA-PK signaling (Peters et al., 2013). Mechanistically, the C-terminal region of C16 binds directly to the Ku heterodimer to block its binding to DNA. C16 also induces a hypoxic response by binding to PHD2 via its N-terminal domain (Mazzon et al., 2013) and, in doing so, reprograms cellular energy metabolism (Mazzon et al., 2015). In a murine intranasal (i.n.) model of infection, the VACV strain Western Reserve (WR) that lacks C16 caused an increase in infiltrating leukocytes, less weight loss with fewer signs of illness, and greater cytokine synthesis (Fahy et al., 2008, Peters et al., 2013).
Here, another VACV protein, C4, is shown to target DNA-PK and inhibit DNA sensing. C4 shares sequence similarity to C16 and, like C16, binds to Ku to diminish DNA binding. This function mapped to the C-terminal domain of C4, and mutagenesis of three residues in both C4 and C16 abrogated binding to Ku. The infection of mice by viruses lacking C4 or C16 singly or together showed that these two proteins have both redundant and non-redundant functions. The loss of C4 increased recruitment and activation of cells involved in both innate and adaptive immunity. This phenotype is attributed to the suppression of cytokine production by C4 both in vitro and in vivo. Overall, this study identifies a second DNA-PK inhibitor encoded by a virus, adding to the evidence that DNA-PK is important to innate immunity. The complexity of host-pathogen interactions is highlighted by the finding that VACV encodes two multifunctional proteins that inhibit the same PRR to directly evade the detection of its genome, but, as yet, there are no reported inhibitors of other DNA sensors.
Results
C4 Interacts with the Ku Heterodimer
VACV proteins C4 and C16 share 54.4% identity in their C-terminal region of 150 amino acids (aa) (Figure S1A). Because this region of C16 binds the Ku heterodimer, we hypothesized that C4 might also share this function. Interestingly, C4 and C16 are conserved in orthopoxviruses, with some viruses encoding both proteins (VACV, cowpox virus [CPXV], and VARV), whereas others encode only C16 (camelpox virus [CMLV], monkeypox virus [MPXV], and taterapox virus [TATV]) or C4 (ectromelia virus [ECTV] and horsepox virus [HSPV]) (Figure S1B), but all encode at least one.
To identify C4 binding partners, an inducible HEK293 TRex cell line was constructed that, upon addition of doxycycline, expresses C4 fused with Strep and FLAG tags (Gloeckner et al., 2007), termed C4-TAP (Figure S2). C4-TAP was purified by tandem affinity purification and analyzed by SDS-PAGE and silver staining. This revealed two proteins of 70 and 80 kDa that were more abundant than from uninduced cells (Figure 1A) and were confirmed as Ku70 and Ku80 by immunoblotting (Figure 1B). The third component of the DNA-PK complex DNA-PKcs did not bind to C4 (Figure 1B).
Figure 1.

C4 Co-immunoprecipitates with the Ku Heterodimer
(A) Tandem affinity purification of TAP-tagged C4. The C4-C′TAP HEK293 TRex cell line was induced with 2 μg/ml doxycycline for 24 hr. Protein lysates from induced and uninduced cells were then subjected to tandem affinity purification. After elution, proteins from beads and supernatant (final elution) were separated by SDS-PAGE and visualized by silver staining. Red arrows point to bands of interest observed in the final elution.
(B) Whole cell lysates (inputs) and final elution proteins from (A) were analyzed by immunoblotting with antibodies against the indicated proteins.
(C) Tandem affinity purification of TAP-tagged C4 in the context of infection. HEK293T cells were infected with 2 plaque-forming units (PFU)/cell of vC4-TAP or vC6-TAP for 16 hr, and protein lysates were subjected to tandem affinity purification, separated by SDS-PAGE, and visualized by silver staining. Red arrows point to bands of interest observed in the final elution.
(D) Protein samples from (C) were analyzed by immunoblotting with antibodies against the indicated proteins.
(E and F) Confirmation of the C4 interaction with Ku70 and Ku80. HEK293T cells were infected with 2 PFU/cell of vC4-TAP or vC6-TAP for 16 hr. Pre-cleared lysates were then immunoprecipitated with (E) anti-Ku70, (F) anti-Ku80, or isotype control antibodies (Iso). Input and precipitated protein complex (IP) samples were analyzed by SDS-PAGE and immunoblotting. The positions of molecular mass markers are shown (kDa).
To examine if C4 binds Ku during infection and to test if C4 binds VACV proteins, HEK293T cells were infected with a VACV expressing TAP-tagged C4 (vC4-TAP) at the endogenous level (Ember et al., 2012), and C4-TAP was purified as before. A VACV expressing TAP-tagged C6 (vC6-TAP) was used as a control (Maluquer de Motes et al., 2014). As before, C4-TAP co-purified with Ku70 and Ku80 (Figure 1C) but not DNA-PKcs (Figure 1D). Immunoblotting for C16 showed that C16 did not associate with C4 (Figure 1D), indicating that C4 and C16 do not interact and that a Ku heterodimer does not bind C4 and C16 simultaneously to form a C4:Ku:C16 complex.
Immunoprecipitation of endogenous Ku70 (Figure 1E) and Ku80 (Figure 1F) from HEK293T cells infected with vC4-TAP or vC6-TAP confirmed that C4 interacts with the Ku heterodimer at endogenous levels during VACV infection.
C4 Blocks the Binding of Ku to DNA and Inhibits DNA Sensing
To test if C4 affects binding of Ku to DNA, hemagglutinin (HA)-tagged C4 or C16 were expressed by transfection in HEK293T cells, and 24 hr later cells were transfected for 1 hr with biotinylated immunostimulatory DNA (ISD). The cytoplasmic DNA was then purified, and associated proteins were separated by SDS-PAGE. C4 was absent, confirming that C4 does not bind DNA (Figure 2A). However, the level of Ku70 precipitated by DNA was reduced by C4. Quantification showed that C4 significantly reduced Ku-DNA binding to levels similar to that of C16 (Figure 2B).
Figure 2.

C4 Inhibits DNA Sensing
(A) DNA affinity purification. HEK293T cells were transfected with 2 μg/ml of EV or plasmids encoding HA-tagged C4 or C16 for 24 hr, followed by 7.5 μg/ml of 45-base pair (bp) biotinylated immunostimulatory DNA (ISD) for 1 hr. Cells were lysed, and streptavidin beads were incubated with cytoplasmic fractions to purify the biotinylated DNA and associated proteins. Samples were analyzed by SDS-PAGE and immunoblotting with Ku70 and HA antibodies.
(B) Integrated intensity of Ku70 AP from (A) from three experimental replicates was calculated by infrared imaging by using a Li-Cor Odyssey scanner.
(C) P-IRF3 immunoblotting. HeLa cells were transfected for 24 hr with EV or C4-TAP plasmid and then stimulated by transfection with 7.5 μg/ml of 180-bp ISD at the time point indicated. Whole cell lysates were analyzed by immunoblotting with antibodies against the indicated proteins.
(D) Integrated intensity of P-IRF3 from (C) was normalized to corresponding α-tubulin levels from three experimental replicates by using a Li-Cor Odyssey scanner.
(E and F) MEFs were mock transfected (NS), transfected with 80 ng/ml EV or plasmids encoding TAP-tagged C4 or C16, and cotransfected with 750 ng/ml ISD or 100 ng/ml poly (I:C) for 24 hr. Levels of (E) IL-6 and (F) CXCL10 in supernatants were measured by ELISA, performed in quadruplicate.
(G) MEFs were mock transfected (NS), transfected with 80 ng/ml EV or plasmids encoding TAP-tagged C4 or C16, and cotransfected with 750 ng/ml EV pcDNA4/TO linearized previously with BamHI and NotI. After 24 hr, levels of CXCL10 in supernatants were measured by ELISA in quadruplicate.
(H) Prkdc−/− MEFs were mock transfected (NS), transfected with 80 ng/ml EV or TAP-tagged C4 plasmid, and cotransfected with 750 ng/ml ISD or 100 ng/ml poly (I:C) for 24 hr. Levels of CXCL10 in supernatants were measured by ELISA, performed in quadruplicate.
Data are represented as ±SD. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, n.s. = non-significant. Each experiment was performed three times and one representative experiment is shown.
Because DNA-PK is a DNA sensor, the ability of C4 to alter IRF3 activation was tested by immunoblotting for phosphorylated IRF3 (P-IRF3) after transfection with ISD in HeLa cells. C4 reduced P-IRF3 levels (Figure 2C), and this was quantified by normalization to α-tubulin (Figure 2D). ELISAs showed that mouse embryonic fibroblasts (MEFs) co-transfected with ISD and a plasmid expressing C4 produced less interleukin-6 (IL-6) (Figure 2E) and C-X-C motif chemokine 10 (CXCL10) (Figure 2F) compared with cells transfected with an empty vector (EV), but no reduction was observed in response to the double-stranded RNA (dsRNA) analog poly (I:C) (Figures 2E and 2F). In addition, a pcDNA plasmid was digested with BamHI and NotI to produce dsDNA breaks that could be recognized by DNA-PK. C4 also reduced production of CXCL10 in response to this stimulus (Figure 2G). MEFs lacking DNA-PK produce much lower levels of cytokines and chemokines in response to ISD (Ferguson et al., 2012). To confirm that the antagonism of DNA sensing by C4 was dependent on DNA-PK, a CXCL10 ELISA was performed with DNA-PKcs null cells (Prkdc−/− MEFs). Stimulation with ISD resulted in a low level of CXCL10 production, as expected, and this was unaffected by C4 (Figure 2H).
Overall, these data demonstrate that C4 inhibits DNA sensing but not RNA sensing and reduces the synthesis of cytokines and chemokines. Mechanistically, C4 binds to the Ku heterodimer and diminishes its ability to bind DNA in the cytoplasm, antagonizing activation of the IRF3 pathway.
Inhibition of Ku Is Mediated by the C-Terminal Region of C4
To identify the region of C4 needed to bind Ku, TAP-tagged truncation mutants of C4 (Figure 3A) were expressed in HEK293T cells, and their ability to bind to Ku was assessed. The C-terminal fragments 157–331 and 167–331 co-immunoprecipitated with Ku70, but further truncation (177–331 and 187–331) caused a loss of binding (Figure 3B). This suggested that residues between 167 and 176 are needed for the interaction. The fragments 1–206 and 1–156 were unable to bind, and because the 1–206 fragment contains residues 167–176, this shows that this ten-residue region is necessary but not sufficient for the interaction (Figure 3B).
Figure 3.

The C-Terminal Domain of C4 Is Sufficient for the Inhibition of DNA Sensing
(A) Schematic of C4 truncation mutants showing C4 fragments capable (gray bars) and not capable (white bars) of binding Ku70.
(B) Affinity purification of C4 truncations. TAP-tagged full-length C4 (a) and amino acid residues 1–206 (b), 1–156 (c), 157–316 (d), 167–316 (e), 177–316 (f), and 187–316 (g) were expressed by transfection in HEK293T cells and affinity purified with Strep-Tactin beads. Whole cell lysates (input) and affinity purified proteins (AP) were separated by SDS-PAGE and immunoblotted with anti-FLAG and anti-Ku70 antibodies.
(C and D) MEFs were mock transfected (NS), transfected with 80 ng/ml EV or plasmids encoding TAP-tagged full-length C4 or C4 amino acid residues 167–316 or 177–316, and cotransfected with 750 ng/ml ISD. After 24 hr, levels of (C) IL-6 and (D) CXCL10 in supernatants were measured by ELISA.
Data are represented as ±SD. ∗∗p < 0.01, ∗∗∗p < 0.001, n.s. = non-significant. Each experiment was performed three times and one representative experiment is shown.
These mutants were then tested for inhibition of CXCL10 and IL-6 production. In MEFs, 167–316, which binds Ku, reduced levels of IL-6 and CXCL10 in response to ISD, whereas 177–316, which does not bind Ku, did not (Figures 3C and 3D). This shows that the C-terminal domain of C4 causes an inhibition of the DNA-PK-mediated DNA sensing and that it is dependent on binding to Ku.
Site-Directed Mutagenesis of C4 and C16 Abrogates Binding to Ku
Because residues needed to bind Ku might lie between C4 residues 167 and 176, these residues from different orthologs were compared with the corresponding residues in C16 (Figure 4A). Residues C174, Y175, and C176 were conserved in all C4 orthologs and also in C16 (C16 residues C187, Y188, and C189).
Figure 4.

Site-Directed Mutagenesis of Three Conserved Residues in C16 and C4 Abrogates Binding to Ku
(A) Amino acid residues CYC are conserved between C4 (C174, Y175, and C176) and C16 (C187, Y188, and C189). Protein sequence alignment of C4 residues 167–177 from VACV strain WR and corresponding residues of C16 and other C4 orthologs of various orthopoxviruses. Black shading represents the sequence others are aligned to. Red and yellow shading indicates identical and highly similar residues, respectively. GenBank accession numbers: VACV-WR C16, YP_232892.1; VACV-WR C4L, YP_232906.1; VACV-Lister 021, ABD52470.1; VACV-COP C4L, AAA47996.1; CPXV-BR 033, NP_619822.1; CPXV-GRI 030, CAA64101.1; RBPX-UTR 016, AAS49729.1; VARV-IND3 011, NP_042055.1; VARV-GAR B17L, CAB54611.1; CMLV-CMS 22L, AAG37478.1; MPXV-ZAR D13L, NP_536443.1; and TATV 025, YP_717332.1.
(B) Mutagenesis of C4 residues CYC alters binding to Ku. TAP-tagged wild-type C4 (CYC) and C4 mutants as indicated were expressed in HEK293T cells and immunoprecipitated with anti-FLAG agarose beads. Proteins were separated by SDS-PAGE and analyzed by immunoblotting with anti-Ku70 and imaged with high and low intensity, anti-FLAG, and anti-C4 antibodies.
(C) Mutagenesis of CYC to AAA abrogates binding to Ku. Different quantities of wild-type C4 plasmid were transfected into HEK293T cells to obtain comparable expression with the C4 AAA mutant, and then lysates were subjected to immunoprecipitation with anti-FLAG agarose beads and analyzed by immunoblotting.
(D and E) The aromatic ring of tyrosine in CYC of (D) C4 and (E) C16 is a critical component of this interaction. Plasmids encoding indicated TAP-tagged proteins were transfected into HEK293T cells and protein complexes were immunoprecipitated with anti-FLAG agarose beads. Immunoblotting was performed with the indicated antibodies. Immunoblots are representative of three independent experiments.
The CYC motif was then mutated, and the binding of these mutants was assessed. Substitution of either cysteine residue to alanine did not reduce the amount of co-precipitated Ku; however, a Y175A mutation decreased binding, suggesting that the tyrosine was important for the interaction (Figure 4B). Further mutation with double and triple alanine substitutions showed that only CYC to AAA fully abrogated binding to Ku, demonstrating that each residue plays a role in the interaction (Figure 4B). Notably, the expression of the mutants was reduced, indicating that these residues are important for stability. To ensure that the lack of binding was not due to less prey protein, the dose of wild-type (WT) C4 plasmid was reduced to obtain similar levels as the C4 AAA mutant. Immunoprecipitation showed that WT C4 bound to Ku70 and C4 AAA did not, confirming that mutagenesis abrogated binding (Figure 4C).
Substitution of tyrosine by phenylalanine (AFA) retained binding to Ku, indicating that the aromatic ring is important for binding (Figure 4D). Consistent with this, the mutants AFC, CFA, and CFC all co-precipitated similar levels of Ku as those of WT C4 (Figure 4D). Given that these residues are conserved in C16, the CYC residues of C16 were mutated to AAA and AFA. The C16 AAA mutant did not bind Ku, whereas AFA did. This demonstrated that these residues are important for binding Ku in both C4 and C16 and that the tyrosine aromatic ring is critical for the C16 interaction (Figure 4E). Importantly, the C16 mutants were stable, and immunoblotting with an anti-prolyl hydroxylase domain-containing protein 2 (PHD2) antibody showed that they still bound to PHD2, another C16 binding partner.
Collectively, these data show that CYC residues are conserved in C4 and C16, are essential for Ku binding, and that the phenyl ring of the tyrosine residue forms the crux of this interaction.
Deletion of Both C4 and C16 Causes Attenuation of Virus Virulence
Previously, the virulence of a virus lacking C4 (vΔC4) was tested in two murine models of infection (Ember et al., 2012). In the i.n. model, the virus replicates in the lungs before spreading to other organs (Alcamí and Smith, 1992, Tscharke and Smith, 1999), whereas, in the intradermal (i.d.) model, infection of the ear pinna causes a localized lesion (Tscharke et al., 2002, Tscharke and Smith, 1999). Mice inoculated i.d. with vΔC4 had no change in lesion size compared to control viruses. In contrast, i.n. infection with vΔC4 induced less weight loss and milder signs of illness compared to controls (Ember et al., 2012). Similarly, a virus lacking C16 (vΔC16) had reduced virulence in the i.n. model but not in the i.d. model (Fahy et al., 2008). Both C4 and C16 are non-essential for virus replication in vitro (Ember et al., 2012, Fahy et al., 2008). To test if the loss of both genes altered virulence, a recombinant virus lacking both genes (vΔC16,ΔC4) was constructed using vΔC16 as the parental virus (Figure S3). C4 was then inserted back into vΔC16,ΔC4 as a revertant control (vΔC16-Rev).
Plaques formed by vΔC16,ΔC4 were significantly smaller than those formed by viruses lacking only C16 (Figure S4A). To test if this was due to reduced virus production or reduced spread, virus replication was examined after infection at low (0.01) or high (10) MOI. The production of intracellular mature virus (IMV) (Figures S4B and S4D) and extracellular enveloped virus (EEV) (Figures S4C and S4E) by vΔC16,ΔC4 were unaltered from control viruses, showing that the reduction in plaque size was not due to reduced replication but might be due to the effects of C4 on antiviral innate immune responses.
Skin lesions caused by i.d. infection with vΔC16,ΔC4 were significantly smaller from days 6 to 15 p.i. and healed sooner than those caused by viruses lacking only C16 (Figure 5A). This result indicates that C4 and C16 share redundancy in this model because the single deletion viruses had WT virulence. This may be due to the suppression of DNA-PK-mediated DNA sensing because this is the only known shared function. Following i.n. infection, animals lost weight after 3 days p.i. irrespective of the virus. However, mice infected with vΔC16,ΔC4 lost less weight from 5 to 11 days p.i. (Figure 5B), showed fewer signs of illness from day 7 onward (Figure 5C), and recovered faster compared to mice infected with viruses lacking only C16 (vΔC16 and vΔC16-Rev). This indicates that C4 functions in the absence of C16 and, therefore, does not require C16 for co-operativity. Measurements of viral titers revealed no difference at 2 days p.i., slightly lower titers at 5 days p.i., and significantly lower virus titers at 8 days p.i. in mice infected with vΔC16ΔC4 compared to control viruses (Figure 5D). This indicates better virus clearance at later time points. These data are consistent with C4 inhibiting the antiviral response in vivo.
Figure 5.

Absence of Both C16 and C4 Leads to Attenuation of Virus Virulence In Vivo
(A) Intradermal infection. C57BL/6 mice (n = 5) were infected i.d. with 104 PFU in both ears of the indicated viruses. Lesion sizes were measured daily with a micrometer.
(B–D) Intranasal infection. BALB/c mice (n = 5) were infected intranasally with 1 × 105 PFU per mouse of the indicated viruses and their weights and signs of illness were measured daily. (B) Weights. Data are expressed as a percentage of the mean weight of the same group of animals on day 0 ± SD. (C) Signs of illness. The mean ± SD score of each group of animals is shown. (D) The viral titers in lungs were determined by plaque assay. The horizontal bars indicate days where the lesion size, weight loss, or signs of illness induced by vΔC16/ΔC4 were statistically different (p < 0.05) from both vΔC16 and vΔC16/C4-Rev. The figure legend shown in (A) applies to (B) and (C). Each experiment was performed twice and one representative experiment is shown.
C4 Inhibits the Recruitment and Activation of Immune Cells by Suppressing Cytokine Production in an Intranasal Model of Infection
To understand the basis for the virulence phenotype observed in the i.n. model, cells from bronchoalveolar lavage (BAL) fluids and lungs of infected mice were extracted and analyzed by flow cytometry. Gating strategies are displayed in Figure S5. In the BAL fluid at day 5 p.i., vΔC16,ΔC4 gave a significantly higher number of macrophages compared to vΔC16 and vΔC16-Rev (Figures 6A and 6B). At day 8 p.i., neutrophil levels had significantly decreased, indicating that the inflammatory response in these mice had begun to subside. In the lungs, a greater number of natural killer (NK) cells were found at 2 and 5 days p.i. compared to control viruses (Figure 6C). To study the adaptive immune response, lung homogenates were stained for CD19 to assess the numbers of B cells and CD3, CD4, CD8, and CD69 to assess the recruitment and activation of T cells. No significant difference in the number of B cells was observed (Figure 6D); however, the number and activation status of CD4+ and CD8+ T cells was increased at 2 and 5 days p.i. compared to control viruses (Figures 6E–6H).
Figure 6.

Deletion of Both C16 and C4 Inhibits Recruitment of Immune Cells and Reduces Production of IL-6 and CXCL10 In Vivo
Groups of five BALB/c mice were infected intranasally with 1 × 105 PFU per mouse of the indicated viruses and at the indicated times p.i.
(A–H) Mice were sacrificed and cells were extracted from (A and B) BAL fluid and (C–H) lungs, counted, and analyzed by flow cytometry. (A) Macrophages, (B) neutrophils, (C) NK cells, and (D) B cells were identified and quantified by fluorescence-activated cell sorting (FACS). (E–H) The number and activation status (CD69+) of CD4+ and CD8+ T cells were analyzed by FACS.
(I and J) At the indicated times p.i., mice were killed and BAL fluid was extracted, and levels of (I) IL-6 and (J) CXCL10 were measured by ELISA (n = 5). Data are represented as means of cell counts ± SD. ∗p < 0.05 and ∗∗p < 0.01 for vΔC16/ΔC4, compared with both vΔC16 and vΔC16/C4-Rev. Each experiment was performed twice and one representative experiment is shown.
The mechanism underlying the effects of C4 on virulence and immune cell recruitment in vivo was then studied by measuring the levels of cytokines and chemokines in BAL fluids after i.n. infection with vΔC16,ΔC4 versus vΔC16 and vΔC16-Rev. Infection induced the production of IL-6 and CXCL-10, which both increased with time (Figures 6I and 6J). The deletion of C4 from vΔC16 resulted in enhanced secretion of these cytokines, particularly for IL-6, and is consistent with the impact of C4 on production of these inflammatory molecules in vitro. This was significant at 48 and 72 hr p.i. for IL-6 (p < 0.0001) (Figure 6I) and 48 hr for CXCL10 (p < 0.01) (Figure 6J).
Deletion of C4 from WT WR VACV also Results in Increased Recruitment of Innate Immune Cells and Activation of T Cells
The fact that deleting C4 from a WT VACV WR strain (that expresses C16) reduces virulence (Ember et al., 2012) shows that C4 contains non-redundant function(s) that contribute to virulence. C4 inhibits nuclear factor κB (NF-κB) signaling by an unknown mechanism (Ember et al., 2012), and this is a potential explanation, or C4 may have some other function(s). To study the effects of these non-redundant function(s) of C4 on the cellular inflammatory response, cells from BAL fluids and lungs of mice infected with vΔC4 and control viruses were analyzed by flow cytometry. The total number of viable cells in BAL fluid was significantly increased at 5 days p.i. with infection of vΔC4 compared to WT (Ember et al., 2012), and here, we examined whether this was due to any particular lymphoid subsets.
More macrophages and neutrophils in the BAL fluid and NK cells in the lungs were detected as early as 2 days p.i. (Figures 7A–7C). Greater increases in numbers for macrophages and neutrophils were seen at 5 days p.i. However, by day 8, neutrophil levels had decreased, indicating that the inflammatory response had begun to subside. To study the adaptive immune response, lung homogenates were stained for CD19 to assess the numbers of B cells and CD3, CD4, CD8, and CD69 to assess the recruitment and activation of T cells. No significant difference in the number of B cells was observed (Figure 7D). Although there were no statistically significant differences in the total number of CD4+ and CD8+ T cells, a measurement of their activation status indicated that more of these cell types displayed an activated phenotype (CD69+) by 5 days p.i. in the lungs of vΔC4-infected mice compared to control infections (Figures 7E–7H). Taken together, these data demonstrate that there are more innate immune cells and activated T cells in the lungs of mice infected with viruses lacking only C4, despite the presence of C16.
Figure 7.

Deletion of C4 from Wild-Type Western Reserve VACV Increases Recruitment of Innate Immune Cells and Activation of T Cells
BALB/c mice were infected intranasally with 5 × 103 PFU per mouse of the indicated viruses and at the indicated times p.i.; groups (n = 5) were sacrificed and cells were extracted from (A and B) BAL fluid and (C–H) lungs, counted, and analyzed by flow cytometry.
(A) Macrophages, (B) neutrophils, (C) NK cells, and (D) B cells were identified and quantified by FACS.
(E–H) The number and activation status (CD69+) of CD4+ and CD8+ T cells were analyzed by FACS.
Data are represented as means of cell counts ± SD. ∗p < 0.05 and ∗∗p < 0.01 for vΔC4, compared with both vC4 and vC4-Rev. Each experiment was performed twice and one representative experiment is shown.
Discussion
During infection, cytoplasmic DNA activates innate immune signaling that restricts virus replication and stimulates adaptive immunity. VACV produces cytoplasmic viral factories containing large amounts of virus DNA, and thus, evading recognition by cytosolic PRRs is beneficial to VACV. Here, we report that VACV protein C4 acts as another inhibitor of DNA-PK, emphasizing the biological role of DNA-PK as a DNA sensor. An interaction between C4 and Ku occurs at endogenous levels and blocks the binding of Ku to DNA to prevent the activation of IRF3 and the production of pro-inflammatory molecules. The mutagenesis of C4 showed that the C-terminal domain was needed for binding Ku and residues C174, Y175, and C176 are critical for this. Understanding the structural basis of this interaction will be valuable in the future.
C4 is related to another VACV protein, C16, and the C-terminal domain of C16 also inhibits DNA-PK-mediated DNA sensing via the same mechanism and to a similar level as C4. Moreover, mutagenesis of C16 at the CYC residues equivalent to those in C4 abrogated the C16-Ku interaction, indicating that C4 and C16 bind to Ku in a similar manner. A summary of the C4 and C16 functional domains is shown in Figure S6.
In vivo, C4 and C16 are non-redundant in that deletion of either caused reduced virulence after i.n. infection (Ember et al., 2012, Fahy et al., 2008), and a double deletion virus was attenuated further. In contrast, redundancy was observed in the i.d. model because deletion of either C16 or C4 alone did not affect virulence (Ember et al., 2012), but double deletion reduced virulence. This may be due to the common inhibition of DNA-PK, and the lack of redundancy in the i.n. model is likely due to other functions of these proteins (Figure S6). Indeed, C16 also induces a hypoxic response (Mazzon et al., 2013), reprogramming the central energy metabolism (Mazzon et al., 2015), and C4 inhibits NF-κB signaling (Ember et al., 2012), and other unknown functions may exist. The response to infection with vΔC16ΔC4 showed that C4 inhibits the recruitment of macrophages, neutrophils, and NK cells, and reduces the recruitment and activation of CD4+ and CD8+ T cells. C4 also inhibits the secretion of IL-6 and CXCL10 in vivo, and this may explain the changes in infiltrating leukocytes. Similar analyses with viruses expressing C4 and C16 with point mutations that abrogate each of their functions alone would be interesting. This was attempted for the C4 AAA mutant; however, expression of C4 AAA by the virus was too low to justify in vivo infection (data not shown).
It is remarkable that VACV proteins C4 and C16 perform the same function by the same mechanism. This might be explained by C4 and C16 having other functions that are enhanced by binding Ku, or redundancy might enable each protein to carry out other functions better. Alternatively, one protein alone might be insufficient to inhibit DNA-PK, particularly early in infection, given that DNA-PK is a very abundant complex. Both C4 and C16 are made early p.i. with nucleocytoplasmic localizations (Ember et al., 2012, Fahy et al., 2008); however, slight differences in expression levels and localization might exist. Having two proteins may also be beneficial should the host act to prevent either protein from functioning. Finally, because VACV has a broad host range, variations in Ku proteins in different hosts may require slightly different viral proteins to ensure functional binding. Interestingly, VACV was shown recently to also inhibit STING activation in a C16-independent manner by an alternative mechanism, acting at or downstream of cGAMP (Georgana et al., 2018).
The expression of multiple inhibitors of a single pathway is common in pathogen evasion. For example, >10 VACV proteins inhibit NF-κB activation (Bowie et al., 2000, Chen et al., 2008, Cooray et al., 2007, DiPerna et al., 2004, Ember et al., 2012, Gedey et al., 2006, Mansur et al., 2013, Myskiw et al., 2009, Schröder et al., 2008, Shisler and Jin, 2004, Stack et al., 2005), and these proteins are not redundant in vivo because, where tested, deletion of each one alone attenuates virulence (Bartlett et al., 2002, Benfield et al., 2013, Brandt and Jacobs, 2001, Chen et al., 2006, Ember et al., 2012, Harte et al., 2003, Mansur et al., 2013, Stack et al., 2005). C4 and C16 are also not the only example of VACV proteins that perform the same function via the same mechanism. The VACV enzymes D9 and D10 share ∼25% sequence identity and both decap host and viral mRNAs (Parrish et al., 2007, Parrish and Moss, 2007). This prevents an accumulation of dsRNAs to reduce the anti-viral response to this PAMP (Liu et al., 2015). Redundancy was found as WT virus and D9 or D10 catalytic site mutants retained virulence during i.n. infection, but a double mutant was attenuated (Liu et al., 2015). This is similar to viruses lacking C4, C16, or both proteins in the i.d. model.
cGAS has a central role in STING activation after DNA sensing, but it is unknown if most of the DNA sensors act independently or in cooperation with the cGAS-cGAMP pathway. This was addressed for IFI16 (Almine et al., 2017, Jønsson et al., 2017) and PQBP1 (Yoh et al., 2015) but remains to be tested for DNA-PK. Individual DNA sensors may share redundancy with each other but may be important for responding to specific types of DNA while avoiding self DNA. DNA-PK acts in a larger complex containing HEXIM1, NEAT1 long non-coding RNA, cGAS, and paraspeckle components (splicing factor, proline- and glutamine-rich [SFPQ]; MATRIN3; RNA binding protein 14 [RBM14]; paraspeckle component 1 [PSPC1]; and non-POU domain-containing octamer-binding protein [NONO]) (Morchikh et al., 2017). Upon stimulation of cells with ISD, DNA-PKcs was phosphorylated, paraspeckle proteins were released, STING was recruited, and IRF3 was activated, in a HEXIM1-dependent manner. The assembly of the complex, the precise function of each subunit, and whether they influence cGAS enzymatic activity are unknown. C4 and C16, being specific inhibitors, may be useful tools to investigate the role of DNA-PK.
DNA-PK can detect viruses other than VACV, including hepatitis B virus (Li et al., 2016), HSV-2 (Sui et al., 2017), and human T lymphotropic virus type 1 (Wang et al., 2017). Whether DNA-PK can sense other viruses and bacteria remains to be tested, and this testing should include nuclear DNA pathogens because DNA-PK is predominately nuclear. Other pathogens may have proteins to evade this receptor, yet VACV C4 and C16 are the only ones identified to date.
This paper describes the second DNA sensor inhibitor expressed by VACV, and, similar to the first, it targets DNA-PK to block DNA binding. Interestingly, VACV has evolved two proteins that inhibit DNA-PK; however, it is not known to encode proteins that directly inhibit any of the other reported DNA sensors. This emphasizes the importance of DNA-PK as a PRR. Learning more about the C4 and C16 interactions with Ku may allow for the development of small molecule inhibitors that mimic this interaction. This would be relevant to human autoimmune diseases associated with dysregulated DNA sensing, for example Aicardi-Goutières (Ahn and Barber, 2014). Our work on the viral inhibitors C4 and C16, along with similar studies, contributes to the understanding of the complex initial stages of DNA detection and the interplay between the DNA damage response and inflammation.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-DNA-PKcs | NeoMarkers | MS-423-P1 |
| Mouse anti-Ku70 | AbCam | ab3114 |
| Mouse anti-Ku80 | Santa Cruz | sc-1484 |
| Rabbit anti-FLAG | Sigma-Aldrich | F7425 |
| Mouse anti-HA | BioLegend | 16B12 |
| Rabbit anti-P-IRF3 | Abcam | ab76493 |
| Mouse α-tubulin | Merck Millipore | 05-829 |
| CD3 (clone 145-2C11) | ThermoFisher Scientific | 16-0031-81 |
| CD4 (GK1.5) | Biolegend | 100402 |
| CD8 (5H10-1) | Biolegend | 100802 |
| CD45R (RA-6B2) | ThermoFisher Scientific | MA1-70098 |
| NK1.1 (PK136) | ThermoFisher Scientific | 14-5941-81 |
| CD69 (H1.2F3) | Biolegend | 104502 |
| Ly6G (1A8) | Biolegend | 127602 |
| CD16/32 (2.4G2) | BD Biosciences | 553141 |
| F4/80 (BM8) | Biolegend | 123110 |
| Virus Strains | ||
| VACV strain Western Reserve: vC4-TAP | Maluquer de Motes et al., 2014 | N/A |
| VACV strain Western Reserve: vC6-TAP | Maluquer de Motes et al., 2014 | N/A |
| VACV strain Western Reserve: vΔC16 | Fahy et al., 2008 | N/A |
| VACV strain Western Reserve: vΔC16/ΔC4 | This paper | N/A |
| VACV strain Western Reserve: vΔC16/C4-Rev | This paper | N/A |
| VACV strain Western Reserve: vC4 | Ember et al., 2012 | N/A |
| VACV strain Western Reserve: vΔC4 | Ember et al., 2012 | N/A |
| VACV strain Western Reserve: vC4-Rev | Ember et al., 2012 | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| cOmplete Mini EDTA-free protease inhibitor cocktail | Roche | 11836170001 |
| Opti-MEM | GIBCO | 51985-026 |
| DMEM | GIBCO | 41966-029 |
| MEM | GIBCO | 31095-029 |
| Penicillin-Streptomycin | GIBCO | 15140-122 |
| MEM NEAAs | GIBCO | 11140050 |
| FBS | PAN-Biotech | P30-19375 |
| Critical Commercial Assays | ||
| HiSpeed Plasmid Maxi Kit | QIAGEN | 12663 |
| Lipofectamine 2000 | Life Technologies | 11668019 |
| Lipofectamine 3000 | Life Technologies | L3000015 |
| TransIT-LT1 | Mirus | MIR 2305 |
| Anti-FLAG M2 agarose gel beads | Sigma-Aldrich | A2220 |
| Strep-Tactin Superflow beads | IBA | 2-1206-002 |
| Streptavidin agarose gel beads | Thermo Scientific | 20357 |
| Mouse IL-6 DuoSet ELISA kit | R&D systems | DY406 |
| Mouse CXCL10 DuoSet ELISA kit | R&D systems | DY446 |
| Experimental Models: Cell Lines | ||
| BSC-1 | ATCC | CCL-26 |
| HEK293T | ATCC | CRL-11268 |
| RK-13 | ATCC | CCL-37 |
| HeLa | ATCC | CCL-2 |
| Xrcc5+/+/Tp53−/− MEFs | Prof. Paul Hasty | N/A |
| Prkdc−/− MEFs | Prof. Penelope Jeggo | N/A |
| HEK293 TRex | Life Technologies | R71007 |
| C4-TAP HEK293 TRex | This paper | N/A |
| C6-TAP HEK293 TRex | Maluquer de Motes et al., 2014 | N/A |
| Experimental Models: Organisms/Strains | ||
| BALB/c mice | Envigo | N/A |
| C57BL/6 mice | Envigo | N/A |
| Oligonucleotides | ||
| Primers for constructing pcDNA4/TO C4 HA, see Table S1. | This paper | N/A |
| Primers for constructing C4 mutant plasmids, see Table S1. | This paper | N/A |
| Primers for PCR analysis of recombinant VACV genomes, see Table S1. | This paper | N/A |
| Other | ||
| Sanger sequencing | Source Bioscience | https://www.sourcebioscience.com/ |
Contact for Reagents and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Geoffrey L. Smith (gls37@cam.ac.uk).
Experimental Model and Subject Details
Viruses
The recombinant VACV Western Reserve strain C6-TAP, C4-TAP, vC4, vΔC4, and vC4-Rev viruses were described (Maluquer de Motes et al., 2014) and (Ember et al., 2012). Recombinant viruses were constructed using a transient dominant selection method (Falkner and Moss, 1990) in the same manner as for construction of vΔC4 and vC4-Rev as described (Ember et al., 2012) but initiated by transfecting Z11ΔC4 into vΔC16-infected cells (to make vΔC16ΔC4) or Z11C4Rev into vΔC16ΔC4-infected cells (to make vΔC16-Rev). The viruses were genotyped using PCR and primers amplifying the flanking regions of the C4L and C16L genes.
Cell Lines
BSC-1 (ATCC CCL-26) and HEK293T (ATCC CRL-11268) cells were maintained in DMEM containing 10% fetal bovine serum (FBS) and penicillin-streptomycin (50 μg/ml; PS). RK-13 (ATCC CCL-37) cells were maintained in minimum essential medium (MEM), supplemented as above. HeLa (ATCC CCL-2) cells were cultured in MEM, supplemented above with the addition of 1% nonessential amino acids. MEFs (Xrcc5+/+/Tp53−/−, and Prkdc−/−) were grown in DMEM containing 15% FBS and PS. HEK293 TRex cells (Life Technologies) were grown in DMEM supplemented with 15% (v/v) FBS, PS, blasticidin (50 μ g/ml), and zeocin (100 μ g/ml).
Mice
Female BALB/c and C57BL/6 mice were handled as previously described (Alcamí and Smith, 1992, Williamson et al., 1990). This work was performed according to regulations of The Animals (Scientific Procedures) Act 1986. All procedures were carried out under the home office project licenses PPL 70/7116 and PPL 70/8524, as approved by the United Kingdom Home Office.
Method Details
Plasmids
Codon-optimized C4-TAP and C16-TAP plasmids were described previously (Ember et al., 2012; Peters et al., 2013). Site-directed mutagenesis of C4 and C16 was performed by amplifying from above plasmids and by using an overlapping PCR method as described in Higuchi et al. (1988) and oligonucleotide primers designed to contain the desired change and approximately 20 nucleotides 5′ and 3′ to the mutation site that were complementary to the template plasmid. PCR products for C4 truncation mutants were produced by amplification from the C4-TAP plasmid. All PCR products were cloned into the mammalian expression vector pcDNA4/TO (Invitrogen) with a C-terminal TAP tag. pcDNA4/TO was used in ELISAs and affinity purifications as EV. Plasmids used for construction of vΔC16ΔC4 and vΔC16-Rev were described previously (Ember et al., 2012).
Transfection of Cell Lines
HEK293T cells, HeLa cells, and MEFs were transfected with TransIT-LT1 (Mirus), Lipofectamine 3000 (Life Technologies), and Lipofectamine 2000 (Life Technologies), respectively, according to the manufacturer’s instructions.
Viruses
The recombinant VACV Western Reserve strain C6-TAP, C4-TAP, vC4, vΔC4, and vC4-Rev viruses were described (Maluquer de Motes et al., 2014, Ember et al., 2012). Recombinant viruses were constructed using a transient dominant selection method (Falkner and Moss, 1990) in the same manner as for construction of vΔC4 and vC4-Rev as described (Ember et al., 2012) but initiated by transfecting Z11ΔC4 into vΔC16-infected cells (to make vΔC16ΔC4) or Z11C4Rev into vΔC16ΔC4-infected cells (to make vΔC16-Rev). The viruses were genotyped using PCR and primers amplifying the flanking regions of the C4L and C16L genes.
Immunoprecipitation and Affinity Purification
HEK293T cells were seeded in 10-cm dishes and transfected the next day with pcDNA4/TO plasmids for 24 hr. Then, cells were washed once with PBS and lysed in immunoprecipitation (IP) lysis buffer (0.5% NP-40, PBS) supplemented with cOmplete Mini EDTA-free protease inhibitor cocktail (Roche). Insoluble material was collected by centrifugation at 21,000 × g for 15 min at 4°C and discarded. For anti-FLAG immunoprecipitations, lysates were incubated with FLAG M2 agarose beads (Sigma-Aldrich). For anti-streptavidin affinity purifications, lysates were incubated with streptavidin agarose (Thermo Scientific). For anti-Ku70 and anti-Ku80 immunoprecipitations, lysates were incubated with 2.5 μg of Ku70 antibody (Abcam, ab3114) or Ku80 antibody (Santa Cruz, sc-1484) and protein G-conjugated FastFlow Sepharose beads (GE Healthcare). Immunoprecipitations were incubated on a rotator overnight at 4°C and then washed four times with IP lysis buffer. Proteins were eluted by resuspension in Laemmli SDS-PAGE loading buffer and incubated at 95°C for 5 min.
Tandem Affinity Protein Purification
The C4-TAP HEK293 TRex cell line was induced with 2 μg/ml doxycycline overnight or uninduced, or HEK293T cells were infected with vC4-TAP or vC6-TAP at 2 PFU/cell overnight. Tandem affinity purification of C4-TAP and C6-TAP was carried out as described in Gloeckner et al. (2007) with FLAG M2 agarose beads (Sigma-Aldrich) and Strep-Tactin superflow beads (IBA). Protein complexes were separated by SDS-PAGE, and bands were visualized by silver staining.
DNA Affinity Purification
Double-stranded immunostimulatory DNA (sense sequence, TACAGATCTACTAGTGATCTATGACTGATCTGTACATGATCTACA) was biotinylated at its 3′ end and transfected into HEK293T cells at 7.5 μg/ml for 1 hr. Cells were lysed with virus overlay protein binding assay (VOPBA) buffer (100 mM Tris-Cl, pH 8, 0.2% Triton X-100, 2 mM MgCl2, 1 mM EDTA), followed by centrifugation at 600 × g for 3 min at 4°C. The supernatant was subjected to centrifugation at 21,000 × g for 10 min at 4°C, and the pellet was discarded to obtain the final crude cytoplasmic fraction. Anti-streptavidin agarose (Thermo Scientific) was used to purify biotinylated DNA and the associated proteins.
Immunoblotting
Proteins in cell lysates were separated by electrophoresis and transferred to a nitrocellulose Amersham Protran membrane (GE Healthcare). The membrane was blocked for 1 hr in 5% milk in PBS containing 0.1% Tween-20 at 23°C. Then, the membrane was incubated with primary antibody (Ab) diluted in blocking buffer at 4°C overnight. Antibodies used were from the following sources: DNA-PKcs (NeoMarkers, MS-423-P1, diluted 1:1000), Ku70 (AbCam, ab3114, diluted 1:1000), Ku80 (Santa Cruz, sc-1484, 1:500), FLAG (Sigma-Aldrich, F7425, diluted 1:5000), HA (Sigma-Aldrich, H6908, diluted 1:1000), P-IRF3 (Abcam, ab76493, diluted 1:1500), and α-tubulin (Merck Millipore, 05-829, diluted 1:10000). Membranes were probed and visualized with LI-COR Biosciences secondary Abs and the Li-Cor Odyssey infrared imaging system according to the manufacturer’s instructions. A quantification of protein bands was performed, where indicated, by using Odyssey software (LI-COR Biosciences).
Analysis of Plaque Size
The diameters of 30 plaques per virus were measured using Axiovision 4.6 on a Zeiss Axiovert 200 M microscope (Zeiss).
Analysis of Virus Growth Kinetics
Monolayers of BSC-1 cells were infected with either 10 or 0.01 PFU per cell with recombinant viruses for measurement of single-step or multi-step growth kinetics, respectively. After 1.5 hr of virus attachment and entry into cells, the infection medium was aspirated, the cells were washed with fresh medium, and then the flasks were returned to the incubator. At the indicated times post-infection, the medium was removed and cells were collected by centrifugation at 500 × g for 10 min. The supernatant was collected and titrated immediately on BSC-1 cells. The cells still attached to the flask were scraped into 3 ml of fresh medium and added to the pellet from the first centrifugation step. The cells were collected by centrifugation as above, resuspended into 500 μL infection medium and stored at −20°C until titration on BSC-1 cells.
Murine i.n. and i.d. Models of Infection
All viruses used for in vivo work were purified by sedimentation twice through a sucrose cushion prior to inoculation. For i.n. infection, groups of five female BALB/c mice (6–8 weeks old) were anaesthetized and inoculated with 1 × 105 PFU into both nostrils. Weight loss was measured and signs of illness were scored (hair ruffling, pneumonia, back arching, and decreased mobility) daily (Alcamí and Smith, 1992, Williamson et al., 1990). For the i.d. model of infection, groups of five female C57BL/6 mice (6–8 weeks old) were infected with 104 PFU in both ear pinna, and a micrometer was used to measure lesion diameter.
BAL fluids were harvested on the indicated days by flushing lungs with 1 ml of PBS containing 10 U/ml heparin and centrifuged at 500 × g. The pellet contained cells used for flow cytometry, and the supernatant was used for ELISA. The number of live cells in BAL fluids was quantified using a hemocytometer and trypan blue staining.
For extraction from lung, the lung tissue was cut into small pieces and incubated in 3 ml digestion buffer (RPMI-10 + 1 mg/ml collagenase + 30 μg/ml DNaseI), and then the lung homogenate was filtered through a cell strainer and 250 μl was collected for virus titration. The remainder of the lung homogenate was centrifuged at 500 × g to collect the cells. The cells were then resuspended in ACK buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM Na2EDTA, pH 7.4) and incubated at 23°C for 3 min. Nineteen milliliters of RPMI-10 buffer was added, and the cells were collected as before by centrifugation. The cells were then resuspended in 20% Percoll (Sigma) in RPMI-10 and were centrifuged at 1000 × g for 20 min. Finally, the cells were resuspended in 1 ml FACS buffer (0.1% [w/v] BSA, 0.1% [w/v] NaN3 in PBS, pH 7.4) and enumerated by trypan blue exclusion.
Cell Staining and Flow Cytometry
CD3 (clone 145-2C11), CD4 (GK1.5), CD8 (5H10-1), CD45R (RA-6B2), NK1.1 (PK136), CD69 (H1.2F3), Ly6G (1A8), and CD16/32 (2.4G2) antibodies were purchased from BD Biosciences, and F4/80 (BM8) antibody was purchased from BioLegend. These antibodies were purified or conjugated with PerCP/cy5.5, fluorescein isothiocyanate (FITC), APC/Cy7, APC, PE-Cy7, PE, or BV650. Relevant isotype controls were used as described elsewhere (Jacobs et al., 2006). Stained cells were measured by flow cytometry on a BD FACS CantoII flow cytometer, and these data were analyzed using Summit software (Beckman Coulter).
ELISA
CXCL10 and IL-6 levels in cell supernatants were measured with DuoSet ELISA kits (R&D Systems) in accordance with the manufacturer’s protocol.
Bioinformatic Analyses
Clustal Omega was used to perform sequence alignments, and these were manually curated using GeneDoc.
Quantification and Statistical Analysis
Data were analyzed by unpaired Student’s t tests by using GraphPad Prism 5 software. Quantification of immunoblot signal intensities was performed using a Li-Cor Odyssey infrared imaging system (LI-COR Biosciences) and Odyssey software (LI-COR Biosciences). Statistical details for individual experiments can be found in the figure legends. Experiments performed with cells in vitro were carried out at least three times, and in vivo mouse experiments were performed twice. One representative of each experiment is shown. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, n.s. = non-significant.
Acknowledgments
The authors thank Prof. Paul Hasty for supplying Xrcc5+/+/Tp53−/− MEFs and Prof. Penelope Jeggo for providing Prkdc−/− MEFs. We also thank Brian Ferguson for useful discussion regarding the DNA sensing ELISA assays. This work was funded by a grant from the Wellcome Trust. G.L.S. is a Wellcome Trust Principal Research Fellow, S.R.S. was supported by a Research Studentship from the Department of Pathology, University of Cambridge, and C.Y. was supported by a summer studentship from the Lister Institute.
Author Contributions
S.R.S., S.W.E., H.R., C.Y., C.A.L., M.M., and D.L.V. conducted experiments; G.L.S. and S.R.S. wrote the paper; G.L.S., R.P.S., and S.R.S. directed the research; and G.L.S., S.R.S., S.J.E., H.R., and D.L.V. analyzed the data.
Declaration of Interests
The authors declare no competing interests.
Published: November 13, 2018
Footnotes
Supplemental Information includes six figures and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.10.034.
Supplemental Information
References
- Ablasser A., Bauernfeind F., Hartmann G., Latz E., Fitzgerald K.A., Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009;10:1065–1072. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J., Barber G.N. Self-DNA, STING-dependent signaling and the origins of autoinflammatory disease. Curr. Opin. Immunol. 2014;31:121–126. doi: 10.1016/j.coi.2014.10.009. [DOI] [PubMed] [Google Scholar]
- Alcamí A., Smith G.L. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell. 1992;71:153–167. doi: 10.1016/0092-8674(92)90274-g. [DOI] [PubMed] [Google Scholar]
- Almine J.F., O’Hare C.A.J., Dunphy G., Haga I.R., Naik R.J., Atrih A., Connolly D.J., Taylor J., Kelsall I.R., Bowie A.G. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017;8:14392. doi: 10.1038/ncomms14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett N., Symons J.A., Tscharke D.C., Smith G.L. The vaccinia virus N1L protein is an intracellular homodimer that promotes virulence. J. Gen. Virol. 2002;83:1965–1976. doi: 10.1099/0022-1317-83-8-1965. [DOI] [PubMed] [Google Scholar]
- Benfield C.T.O., Ren H., Lucas S.J., Bahsoun B., Smith G.L. Vaccinia virus protein K7 is a virulence factor that alters the acute immune response to infection. J. Gen. Virol. 2013;94:1647–1657. doi: 10.1099/vir.0.052670-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie A.G., Unterholzner L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008;8:911–922. doi: 10.1038/nri2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie A., Kiss-Toth E., Symons J.A., Smith G.L., Dower S.K., O’Neill L.A. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc. Natl. Acad. Sci. USA. 2000;97:10162–10167. doi: 10.1073/pnas.160027697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt T.A., Jacobs B.L. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J. Virol. 2001;75:850–856. doi: 10.1128/JVI.75.2.850-856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker S.W., Bonham K.S., Zanoni I., Kagan J.C. Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller R.M., Smith G.L., Cremer K., Notkins A.L., Moss B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317:813–815. doi: 10.1038/317813a0. [DOI] [PubMed] [Google Scholar]
- Bürckstümmer T., Baumann C., Blüml S., Dixit E., Dürnberger G., Jahn H., Planyavsky M., Bilban M., Colinge J., Bennett K.L., Superti-Furga G. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 2009;10:266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- Chen R.A.J., Jacobs N., Smith G.L. Vaccinia virus strain Western Reserve protein B14 is an intracellular virulence factor. J. Gen. Virol. 2006;87:1451–1458. doi: 10.1099/vir.0.81736-0. [DOI] [PubMed] [Google Scholar]
- Chen R.A., Ryzhakov G., Cooray S., Randow F., Smith G.L. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 2008;4:e22. doi: 10.1371/journal.ppat.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu Y.H., Macmillan J.B., Chen Z.J. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell. 2009;138:576–591. doi: 10.1016/j.cell.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooray S., Bahar M.W., Abrescia N.G.A., McVey C.E., Bartlett N.W., Chen R.A.J., Stuart D.I., Grimes J.M., Smith G.L. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 2007;88:1656–1666. doi: 10.1099/vir.0.82772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPerna G., Stack J., Bowie A.G., Boyd A., Kotwal G., Zhang Z., Arvikar S., Latz E., Fitzgerald K.A., Marshall W.L. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by toll-like receptors. J. Biol. Chem. 2004;279:36570–36578. doi: 10.1074/jbc.M400567200. [DOI] [PubMed] [Google Scholar]
- Dombrowski Y., Peric M., Koglin S., Kammerbauer C., Göss C., Anz D., Simanski M., Gläser R., Harder J., Hornung V. Cytosolic DNA triggers inflammasome activation in keratinocytes in psoriatic lesions. Sci. Transl. Med. 2011;3:82ra38. doi: 10.1126/scitranslmed.3002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde N.C., Child S.J., Eickbush M.T., Kitzman J.O., Rogers K.S., Shendure J., Geballe A.P., Malik H.S. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell. 2012;150:831–841. doi: 10.1016/j.cell.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember S.W.J., Ren H., Ferguson B.J., Smith G.L. Vaccinia virus protein C4 inhibits NF-κB activation and promotes virus virulence. J. Gen. Virol. 2012;93:2098–2108. doi: 10.1099/vir.0.045070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy A.S., Clark R.H., Glyde E.F., Smith G.L. Vaccinia virus protein C16 acts intracellularly to modulate the host response and promote virulence. J. Gen. Virol. 2008;89:2377–2387. doi: 10.1099/vir.0.2008/004895-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkner F.G., Moss B. Transient dominant selection of recombinant vaccinia viruses. J. Virol. 1990;64:3108–3111. doi: 10.1128/jvi.64.6.3108-3111.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenner F., Henderson D.A., Arita I., Jezek Z., Ladnyi I.D. World Health Organization; 1988. Smallpox and Its Eradication. [Google Scholar]
- Ferguson B.J., Mansur D.S., Peters N.E., Ren H., Smith G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife. 2012;1:e00047. doi: 10.7554/eLife.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T., Yu J.-W., Datta P., Wu J., Alnemri E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedey R., Jin X.-L., Hinthong O., Shisler J.L. Poxviral regulation of the host NF-kappaB response: the vaccinia virus M2L protein inhibits induction of NF-kappaB activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J. Virol. 2006;80:8676–8685. doi: 10.1128/JVI.00935-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgana I., Sumner R.P., Towers G.J., Maluquer de Motes C. Virulent poxviruses inhibit DNA sensing by preventing STING activation. J. Virol. 2018;92 doi: 10.1128/JVI.02145-17. JVI.02145-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloeckner C.J., Boldt K., Schumacher A., Roepman R., Ueffing M. A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics. 2007;7:4228–4234. doi: 10.1002/pmic.200700038. [DOI] [PubMed] [Google Scholar]
- Gubser C., Hué S., Kellam P., Smith G.L. Poxvirus genomes: a phylogenetic analysis. J. Gen. Virol. 2004;85:105–117. doi: 10.1099/vir.0.19565-0. [DOI] [PubMed] [Google Scholar]
- Hanson D., Diven D.G. Molluscum contagiosum. Dermatol. Online J. 2003;9:2. [PubMed] [Google Scholar]
- Harte M.T., Haga I.R., Maloney G., Gray P., Reading P.C., Bartlett N.W., Smith G.L., Bowie A., O’Neill L.A.J. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J. Exp. Med. 2003;197:343–351. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi R., Krummel B., Saiki R.K. A general method of in vitro preparation and specific mutagenesis of DNA fragments: study of protein and DNA interactions. Nucleic Acids Res. 1988;16:7351–7367. doi: 10.1093/nar/16.15.7351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V., Ablasser A., Charrel-Dennis M., Bauernfeind F., Horvath G., Caffrey D.R., Latz E., Fitzgerald K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii K.J., Coban C., Kato H., Takahashi K., Torii Y., Takeshita F., Ludwig H., Sutter G., Suzuki K., Hemmi H. A Toll-like receptor-independent antiviral response induced by double-stranded B-form DNA. Nat. Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- Ishii K.J., Kawagoe T., Koyama S., Matsui K., Kumar H., Kawai T., Uematsu S., Takeuchi O., Takeshita F., Coban C., Akira S. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451:725–729. doi: 10.1038/nature06537. [DOI] [PubMed] [Google Scholar]
- Ishikawa H., Ma Z., Barber G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J., Reid T., Ruo L., Breitbach C.J., Rose S., Bloomston M., Cho M., Lim H.Y., Chung H.C., Kim C.W. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Isr. Med. Assoc. J. 2013;15:242. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs N., Chen R.A., Gubser C., Najarro P., Smith G.L. Intradermal immune response after infection with vaccinia virus. J. Gen. Virol. 2006;87:1157–1161. doi: 10.1099/vir.0.81556-0. [DOI] [PubMed] [Google Scholar]
- Jønsson K.L., Laustsen A., Krapp C., Skipper K.A., Thavachelvam K., Hotter D., Egedal J.H., Kjolby M., Mohammadi P., Prabakaran T. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 2017;8:14391. doi: 10.1038/ncomms14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T., Pazhoor S., Bao M., Zhang Z., Hanabuchi S., Facchinetti V., Bover L., Plumas J., Chaperot L., Qin J., Liu Y.J. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA. 2010;107:15181–15186. doi: 10.1073/pnas.1006539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn D.H., Thorne S.H. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- Kondo T., Kobayashi J., Saitoh T., Maruyama K., Ishii K.J., Barber G.N., Komatsu K., Akira S., Kawai T. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. USA. 2013;110:2969–2974. doi: 10.1073/pnas.1222694110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimasa A., Kumano S., Boubnov N.V., Story M.D., Tung C.S., Peterson S.R., Chen D.J. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol. Cell. Biol. 1999;19:3877–3884. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Wu Y., Zheng X., Cong J., Liu Y., Li J., Sun R., Tian Z.G., Wei H.M. Cytoplasm-translocated Ku70/80 complex sensing of HBV DNA induces hepatitis-associated chemokine secretion. Front. Immunol. 2016;7:569. doi: 10.3389/fimmu.2016.00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.W., Katsafanas G.C., Liu R., Wyatt L.S., Moss B. Poxvirus decapping enzymes enhance virulence by preventing the accumulation of dsRNA and the induction of innate antiviral responses. Cell Host Microbe. 2015;17:320–331. doi: 10.1016/j.chom.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke S., Paludan S.R. Molecular requirements for sensing of intracellular microbial nucleic acids by the innate immune system. Cytokine. 2017;98:4–14. doi: 10.1016/j.cyto.2016.10.003. [DOI] [PubMed] [Google Scholar]
- Mackett M., Smith G.L., Moss B. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proc. Natl. Acad. Sci. USA. 1982;79:7415–7419. doi: 10.1073/pnas.79.23.7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maluquer de Motes C., Schiffner T., Sumner R.P., Smith G.L. Vaccinia virus virulence factor N1 can be ubiquitylated on multiple lysine residues. J. Gen. Virol. 2014;95:2038–2049. doi: 10.1099/vir.0.065664-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansur D.S., Maluquer de Motes C., Unterholzner L., Sumner R.P., Ferguson B.J., Ren H., Strnadova P., Bowie A.G., Smith G.L. Poxvirus targeting of E3 ligase β-TrCP by molecular mimicry: a mechanism to inhibit NF-κB activation and promote immune evasion and virulence. PLoS Pathog. 2013;9:e1003183. doi: 10.1371/journal.ppat.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzon M., Peters N.E., Loenarz C., Krysztofinska E.M., Ember S.W.J., Ferguson B.J., Smith G.L. A mechanism for induction of a hypoxic response by vaccinia virus. Proc. Natl. Acad. Sci. USA. 2013;110:12444–12449. doi: 10.1073/pnas.1302140110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzon M., Castro C., Roberts L.D., Griffin J.L., Smith G.L. A role for vaccinia virus protein C16 in reprogramming cellular energy metabolism. J. Gen. Virol. 2015;96:395–407. doi: 10.1099/vir.0.069591-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morchikh M., Cribier A., Raffel R., Amraoui S., Cau J., Severac D., Dubois E., Schwartz O., Bennasser Y., Benkirane M. HEXIM1 and NEAT1 long non-coding RNA form a multi-subunit complex that regulates DNA-mediated innate immune response. Mol. Cell. 2017;67:387–399.e5. doi: 10.1016/j.molcel.2017.06.020. [DOI] [PubMed] [Google Scholar]
- Myskiw C., Arsenio J., van Bruggen R., Deschambault Y., Cao J. Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-kappaB, and IRF3 pathways. J. Virol. 2009;83:6757–6768. doi: 10.1128/JVI.02570-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicali D., Paoletti E. Construction of poxviruses as cloning vectors: insertion of the thymidine kinase gene from herpes simplex virus into the DNA of infectious vaccinia virus. Proc. Natl. Acad. Sci. USA. 1982;79:4927–4931. doi: 10.1073/pnas.79.16.4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicali D., Davis S.W., Weinberg R.L., Paoletti E. Construction of live vaccines by using genetically engineered poxviruses: biological activity of recombinant vaccinia virus expressing influenza virus hemagglutinin. Proc. Natl. Acad. Sci. USA. 1983;80:5364–5368. doi: 10.1073/pnas.80.17.5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannunzio N.R., Watanabe G., Lieber M.R. Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J. Biol. Chem. 2018;293:10512–10523. doi: 10.1074/jbc.TM117.000374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S., Moss B. Characterization of a second vaccinia virus mRNA-decapping enzyme conserved in poxviruses. J. Virol. 2007;81:12973–12978. doi: 10.1128/JVI.01668-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish S., Resch W., Moss B. Vaccinia virus D10 protein has mRNA decapping activity, providing a mechanism for control of host and viral gene expression. Proc. Natl. Acad. Sci. USA. 2007;104:2139–2144. doi: 10.1073/pnas.0611685104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters N.E., Ferguson B.J., Mazzon M., Fahy A.S., Krysztofinska E., Arribas-Bosacoma R., Pearl L.H., Ren H., Smith G.L. A mechanism for the inhibition of DNA-PK-mediated DNA sensing by a virus. PLoS Pathog. 2013;9:e1003649. doi: 10.1371/journal.ppat.1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A., Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Reynolds M.G., Cono J., Curns A., Holman R.C., Likos A., Regnery R., Treadwell T., Damon I. Human monkeypox. Lancet Infect. Dis. 2004;4:604–605. doi: 10.1016/S1473-3099(04)01139-9. discussion 605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts T.L., Idris A., Dunn J.A., Kelly G.M., Burnton C.M., Hodgson S., Hardy L.L., Garceau V., Sweet M.J., Ross I.L. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science. 2009;323:1057–1060. doi: 10.1126/science.1169841. [DOI] [PubMed] [Google Scholar]
- Schröder M., Baran M., Bowie A.G. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008;27:2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shisler J.L., Jin X.L. The vaccinia virus K1L gene product inhibits host NF-kappaB activation by preventing IkappaBalpha degradation. J. Virol. 2004;78:3553–3560. doi: 10.1128/JVI.78.7.3553-3560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G.L., Mackett M., Moss B. Infectious vaccinia virus recombinants that express hepatitis B virus surface antigen. Nature. 1983;302:490–495. doi: 10.1038/302490a0. [DOI] [PubMed] [Google Scholar]
- Smith G.L., Murphy B.R., Moss B. Construction and characterization of an infectious vaccinia virus recombinant that expresses the influenza hemagglutinin gene and induces resistance to influenza virus infection in hamsters. Proc. Natl. Acad. Sci. USA. 1983;80:7155–7159. doi: 10.1073/pnas.80.23.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G.L., Benfield C.T.O., Maluquer de Motes C., Mazzon M., Ember S.W.J., Ferguson B.J., Sumner R.P. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J. Gen. Virol. 2013;94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- Stack J., Haga I.R., Schröder M., Bartlett N.W., Maloney G., Reading P.C., Fitzgerald K.A., Smith G.L., Bowie A.G. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J. Exp. Med. 2005;201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson D.B., Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24:93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Sui H., Zhou M., Imamichi H., Jiao X., Sherman B.T., Lane H.C., Imamichi T. STING is an essential mediator of the Ku70-mediated production of IFN-λ1 in response to exogenous DNA. Sci. Signal. 2017;10 doi: 10.1126/scisignal.aah5054. eaah5054. [DOI] [PubMed] [Google Scholar]
- Sun L., Wu J., Du F., Chen X., Chen Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaoka A., Wang Z., Choi M.K., Yanai H., Negishi H., Ban T., Lu Y., Miyagishi M., Kodama T., Honda K. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Chen Z.J. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal. 2012;5:ra20. doi: 10.1126/scisignal.2002521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscharke D.C., Smith G.L. A model for vaccinia virus pathogenesis and immunity based on intradermal injection of mouse ear pinnae. J. Gen. Virol. 1999;80:2751–2755. doi: 10.1099/0022-1317-80-10-2751. [DOI] [PubMed] [Google Scholar]
- Tscharke D.C., Reading P.C., Smith G.L. Dermal infection with vaccinia virus reveals roles for virus proteins not seen using other inoculation routes. J. Gen. Virol. 2002;83:1977–1986. doi: 10.1099/0022-1317-83-8-1977. [DOI] [PubMed] [Google Scholar]
- Unterholzner L., Keating S.E., Baran M., Horan K.A., Jensen S.B., Sharma S., Sirois C.M., Jin T., Latz E., Xiao T.S. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Kang L., Song D., Liu L., Yang S., Ma L., Guo Z., Ding H., Wang H., Yang B. Ku70 senses HTLV-1 DNA and modulates HTLV-1 replication. J. Immunol. 2017;199:2475–2482. doi: 10.4049/jimmunol.1700111. [DOI] [PubMed] [Google Scholar]
- Williamson J.D., Reith R.W., Jeffrey L.J., Arrand J.R., Mackett M. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J. Gen. Virol. 1990;71:2761–2767. doi: 10.1099/0022-1317-71-11-2761. [DOI] [PubMed] [Google Scholar]
- Yang P., An H., Liu X., Wen M., Zheng Y., Rui Y., Cao X. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a β-catenin-dependent pathway. Nat. Immunol. 2010;11:487–494. doi: 10.1038/ni.1876. [DOI] [PubMed] [Google Scholar]
- Yoh S.M., Schneider M., Seifried J., Soonthornvacharin S., Akleh R.E., Olivieri K.C., De Jesus P.D., Ruan C., de Castro E., Ruiz P.A. PQBP1 is a proximal sensor of the cGAS-dependent innate response to HIV-1. Cell. 2015;161:1293–1305. doi: 10.1016/j.cell.2015.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Brann T.W., Zhou M., Yang J., Oguariri R.M., Lidie K.B., Imamichi H., Huang D.W., Lempicki R.A., Baseler M.W. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 2011;186:4541–4545. doi: 10.4049/jimmunol.1003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.