

Abstract
Patient: Male, 35
Final Diagnosis: Carney syndrome
Symptoms: Pain at the spine
Medication: —
Clinical Procedure: Retroperitoneal adrenalectomy
Specialty: Surgery
Objective:
Congenital defects/diseases
Background:
Carney complex (CNC) is a genetic disorder that presents as an adrenocorticotropic hormone (ACTH)-independent variant of endogenous Cushing syndrome. It was first reported in 1985 and was described as a form of multiple endocrine hyperplasia associated with mutations of the c-AMP-dependent protein kinase (PRKAR1A) gene that causes bilateral adrenal hyperplasia. We report a case of an incidentally found CNC in a 35-year-old male, and this case report focuses on the diagnostic scheme as well as the surgical treatment of this rare challenging condition.
Case Report:
A-35-year-old male presented with pathological thoracic spine fracture. The patient exhibited obesity, facial flushing, red-purplish streaks on the abdominal wall, multiple pigmented nevi of the trunk, and hypertension. Family history was positive for cardiac myxoma. Laboratory investigation showed ACTH-independent Cushing syndrome. Abdominal magnetic resonance imaging and computed tomography scan showed bilateral adrenal hyperplasia. The ensuing Liddle test revealed the characteristic paradox increase of 24-hours urine cortisol for CNC. After a bilateral retroperitoneoscopic adrenalectomy, histologic examination confirmed the presence of bilateral primary pigmented nodular adrenocortical disease (PPNAD). Genetic testing revealed a unique mutation of the responsible PRKAR1A gene.
Conclusions:
CNC presence was suspected due to the family history. Its characteristic pathologic manifestation called PPNAD, clinically presents as an ACTH-independent Cushing syndrome with paradoxical positive response of urinary glucocorticosteroid excretion after dexamethasone administration (Liddle’s test). Bilateral retroperitoneoscopic adrenalectomy constitutes an acceptable surgical option for PPNAD.
MeSH Keywords: Adrenal Cortex Neoplasms, Adrenalectomy, Carney Complex, Cushing Syndrome
Background
Carney complex (CNC) is a genetic disorder that presents as an adrenocorticotropic hormone (ACTH)-independent variant of endogenous Cushing syndrome [1]. It was first reported in 1985 and was described as a form of multiple endocrine hyperplasia associated with mutations of the c-AMP-dependent protein kinase (PRKAR1A) gene that causes bilateral adrenal hyperplasia [2,3].
We report a case of incidentally found CNC in a 35-year-old mal; this case report focuses on the diagnostic scheme as well as the surgical treatment of this rare challenging condition.
Case Report
A 35-year-old male was urgently admitted to our center due to a pathological thoracic spine fracture located at the T10 level, along with multiple herniations at various levels according to the chest and spine computed tomography (CT) and magnetic resonance imaging (MRI) studies (Figure 1). Technetium 99m scanning showed increased uptake at the T10-L1 level confirming the fracture location, whereas DEXA scanning of his left thigh (T score: −1.5, Z score: −1.4) and lumbar spine (T-score: −1.4, Z-score: −1.5) revealed the presence of osteopenia. No surgical intervention was needed for the spine fracture and the patient received a pain management scheme that included paracetamol and tramadol. After a 3 days analgesic course, the patient was referred for further workup.
Figure 1.
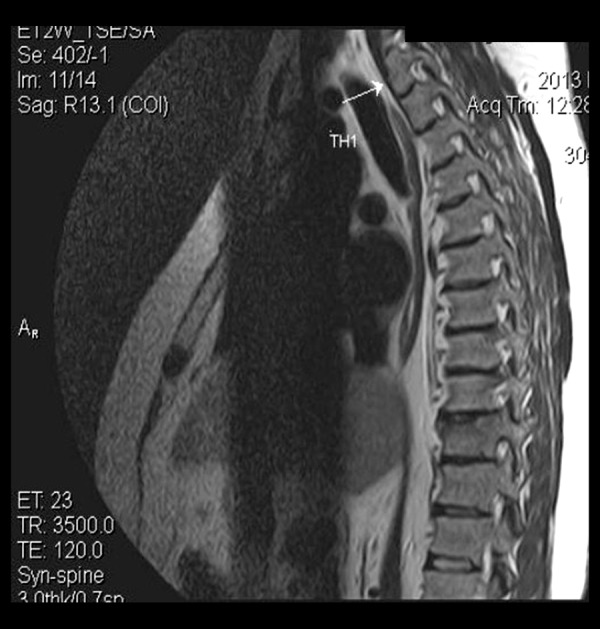
Thoracic spine magnetic resonance imaging: white arrow showing T1 vertebra.
The patient was obese (body mass index was 30.9 kg/m2) and he complained of progressive weakness and fatigue during the last 3 months. Physical examination revealed subtle facial flushing, red-purplish streaks on the lateral abdominal wall, and multiple pigmented nevi of the trunk. Hypertension was present (blood pressure was 160/105 mm Hg; heart rate was 100 beats per minute) and was treated with candesartan and hydrochlorothiazide. His past medical history included recurrent episodes of prostatitis and sterility, large body weight fluctuations (25 kg weight loss 4 years earlier and 40 kg weight gain thereafter), 2 urticaria episodes, fatty liver infiltration, and no major surgery in the past.
Family history revealed 2 aunts, 1 uncle and 2 cousins with history of cardiac myxoma (Figure 2). Laboratory investigation showed increased SGOT/SGPT/γGT values (63/90/111 IU/L respectively), hyperlipidemia (triglyceride level was 186 mg/dL, low-density lipid level was 210 mg/dL), normal alkaline phosphatase level at 98 IU/L, lactate dehydrogenase was 754 IU/L, creatine phosphokinase was 159 IU/L, normal calcium/parathyroid hormone values, normal thyroid function, normal aldosterone/renin/DHEA-S and mildly decreased luteinizing hormone/follicle-stimulating hormone/testosterone levels (1.7 mU/mL, 2.6 mU/ml, and 112 ng/dL respectively). On more elaborate endocrine testing, there was loss of serum cortisol diurnal rhythm (serum cortisol 24.13, 28.49, and 30.4 μg/dL at 8: 00, 16: 00, and 22: 00 hours, respectively) as well as increased 24-hour urine cortisol levels (167 μg/dL). Moreover, the absence of serum cortisol level suppression after low dose (1 mg) overnight dexamethasone test (serum cortisol: 44.05 μg/dL) established the presence of hypercortisolism. Proceeding to the differential diagnosis, there was low serum ACTH level (serum ACTH: 1 pg/mL) and absence of serum cortisol suppression after high dose (8 mg) dexamethasone test (serum cortisol: 29.94 μg/dL). Finally, the corticotropin-releasing hormone (CRH) test (CRH administration of 100 μg intravenously) revealed an unsuppressed serum cortisol value of 27.35 μg/dL that set the diagnosis of ACTH-independent Cushing syndrome.
Figure 2.

Family history of the patient.
Abdominal imaging studies with MRI and CT (Figure 3) showed nodular lesions in both adrenal glands, which were more prominent on the left gland and were consistent with the diagnosis of bilateral nodular adrenal hyperplasia.
Figure 3.
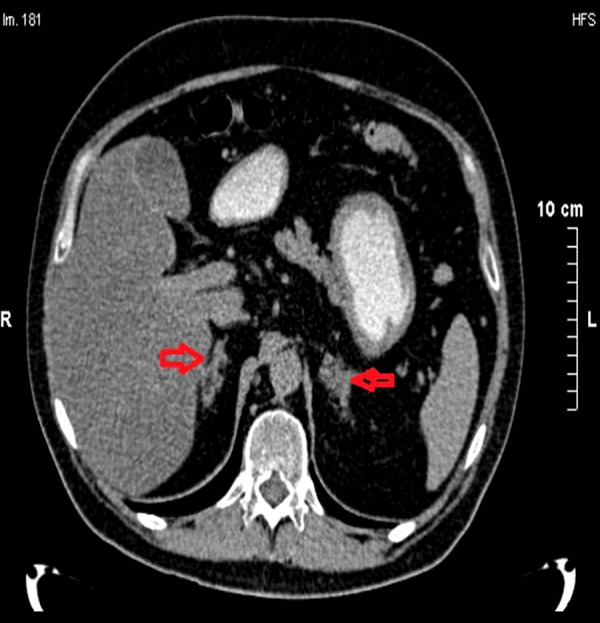
Abdominal computed tomography scan showing bilateral adrenal hyperplasia more prominent on the left side (right and left red arrows).
With the clinical suspicion of CNC, the patient underwent the Liddle test that revealed the characteristic paradox increase of 24-hour urine cortisol (Table 1). Cardiac triplex, pituitary MRI and scrotal and thyroid ultrasound revealed no pathologic findings. A bilateral retroperitoneoscopic adrenalectomy was subsequently performed and the patient had an uneventful postoperative course. The histology report confirmed the presence of bilateral adrenal hyperplasia due to primary pigmented nodular adrenocortical disease (PPNAD) (Figures 4, 5). Genetic testing and counseling revealed the presence of the c.487–488 delAC mutation in the PRKAR1A gene which had not been observed previously, and which lead to the genetic testing of his 30-year-old brother, resulting in the same diagnosis.
Table 1.
Liddle test: administration of Dexamethasone 0.5 mg every 6 hours for 2 days and then 2.0 mg every 6 hours for 2 days. Measurement of 24-hour urinary free cortisol. Pituitary tumor: fall; adrenal tumor: no response.
| UFC (mg/24-hour) | V (mL) | Days |
|---|---|---|
| 293 | 2450 | 1 |
| 167.5 | 2500 | 2 |
| 323.4 | 3000 | 3 |
| 650 | 3700 | 4 |
| 847.4 | 3800 | 5 |
| 909.5 | 3300 | 6 |
| 907.1 | 3100 | 7 |
UFC – urine free cortisol; V – volume.
Figure 4.
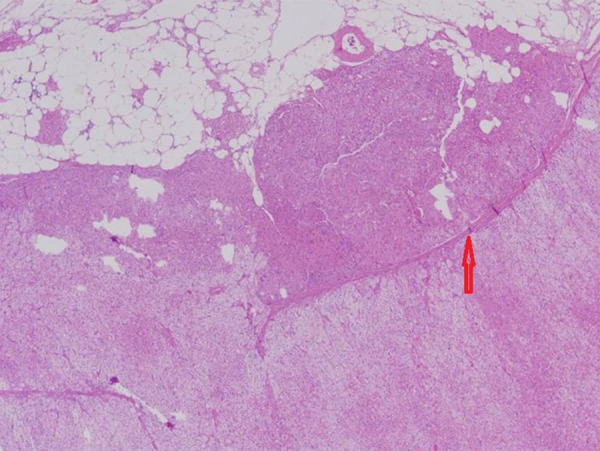
Hematoxylin and eosin stain: Projection of nodule in the periadrenal fat with atrophic internodular cortex (red arrow).
Figure 5.
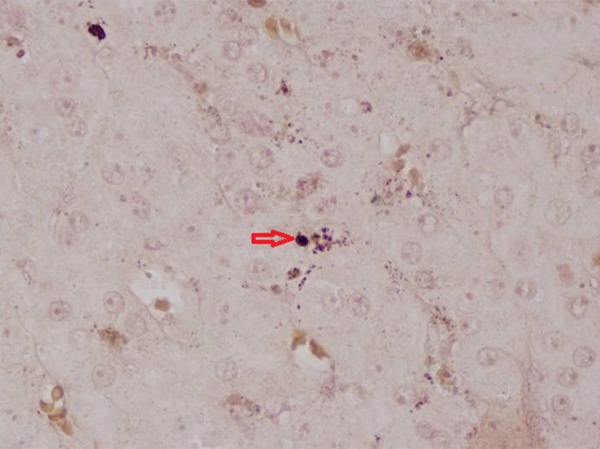
Positive Montana-Maison stain for melanin (red arrow) suggesting the presence of primary pigmented nodular adrenocortical disease.
Discussion
CNC was first reported in 1985 and was described as a form of multiple endocrine hyperplasia that involve 2 or more endocrine glands, including PPNAD, GH- and prolactin producing pituitary adenomas, testicular neoplasms, thyroid adenoma, and ovarian cysts [1]. It is a genetically heterogeneous, autosomal dominant disease, associated with mutations of the regulatory subunit type 1A of the cAMP-dependent protein kinase (PRKAR1A) gene found in nearly 60% of individuals with CNC [2].
The majority of CNC patients presents in the second or third decade of life. Spotty skin pigmentations, including blue nevi and café-au-lait spots, comprise the most common manifestation of the disease, although these signs are not invariably present. Cardiac myxomas might occur at a younger age, whereas myxomas in other sites, such as the eyelids, nipples, and external ear canals, have been also reported. Identification of cardiac myxomas in family history of our case was crucial since it set the suspicion of the presence of a genetic syndrome, highlighting the importance of this particular aspect of patient’s history inquiry.
Cushing syndrome is the most common endocrine manifestation and is caused by PPNAD, which typically consists of multiple, small, pigmented nodules and atrophy between them. CNC presents as an ACTH-independent Cushing syndrome with low plasma ACTH value, absence of serum cortisol suppression after CRH test, and absence of serum cortisol level suppression after low and high dose dexamethasone testing. Interestingly enough, the Liddle test, which helps identify PPNAD patients from those having Cushing syndrome caused by other primary adrenal disorders, showed the characteristic paradox increase of 24-hour urine cortisol level. Classic criterion for a positive response consistent with Cushing syndrome requires a >50% fall in 17 hydroxysteroid urine excretion on day 6, whereas an increase in urinary free cortisol excretion of 100% or more on day 6 of the Liddle test identifies only patients with PPNAD [3].
Even though laparoscopic adrenalectomy is the gold standard, the posterior retroperitoneoscopic method has become one of the standard approaches in minimally invasive adrenal surgery affording minimal pain and short convalescence to the patient [4,5]. The indication of bilateral adrenalectomy is best accomplished by the retroperitoneal approach because the procedure is fast and requires no further repositioning of the patient.
In our case, histologic sections revealed the presence of PPNAD with positive Fontana-Maison stain for melanin. Genetic testing confirmed the novel mutation c.487-488delAC in the PRKAR1A gene. This mutation causes a frameshift starting with codon threonine 163, changing the AA with cysteine residue, thus creating a premature stop codon at position 6 of the new reading frame. This mutation was not observed in 6500 individuals of European and African American ancestry [6], and to our knowledge has not been previously reported.
Conclusions
Family history set the suspicion of CNC presence on clinical grounds, thus highlighting the importance of patient’s history. PPNAD is the characteristic manifestation of CNC associated with ACTH-independent Cushing syndrome with paradoxical positive response of urinary glucocorticosteroid excretion after dexamethasone administration (Liddle test). Bilateral retroperitoneoscopic adrenalectomy constitutes an acceptable surgical option for PPNAD.
Footnotes
Cconflicts of interest
None.
References:
- 1.Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: Diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab. 2001;86:4041–46. doi: 10.1210/jcem.86.9.7903. [DOI] [PubMed] [Google Scholar]
- 2.Rothenbuhler A, Stratakis CA. Clinical and molecular genetics of Carney complex. Best Pract Res Clin Endocrinol Metab. 2010;24:389–99. doi: 10.1016/j.beem.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Stratakis CA, Sarlis N, Kirschner LS, et al. Paradoxical response to dexamethasone in the diagnosis of primary pigmented nodular adrenocortical disease. Ann Intern Med. 1999;131(8):585–91. doi: 10.7326/0003-4819-131-8-199910190-00006. [DOI] [PubMed] [Google Scholar]
- 4.Kiriakopoulos A, Petralias A, Linos D. Posterior retroperitoneoscopic versus laparoscopic adrenalectomy in sporadic and MENIIA pheochromocytomas. Surg Endosc. 2015;29(8):2164–70. doi: 10.1007/s00464-014-3912-0. [DOI] [PubMed] [Google Scholar]
- 5.Walz MK, Alesina PF, Wenger FA, et al. Posterior retroperitoneoscopic adrenalectomy: Results of 560 procedures in 520 patients. Surgery. 2006;140:943–48. doi: 10.1016/j.surg.2006.07.039. [DOI] [PubMed] [Google Scholar]
- 6.Horvath A, Bertherat J, Groussin L, et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-alpha of protein kinase A (PRKAR1A): An update. Hum Mutat. 2010;31:369–79. doi: 10.1002/humu.21178. [DOI] [PMC free article] [PubMed] [Google Scholar]