

Abstract
Background
Individuals with phenylketonuria (PKU) have a risk of cognitive impairment and inflammation. Many follow a low-phenylalanine (low-Phe) diet devoid of animal protein in combination with medical foods (MFs).
Objective
To assess lipid metabolism in participants with PKU consuming amino acid MFs (AA-MFs) or glycomacropeptide MFs (GMP-MFs), we conducted fatty acid and metabolomics analyses.
Methods
We used subsets of fasting plasma and urine samples from our randomized crossover trial in which participants with early-treated classical and variant (milder) PKU consumed a low-Phe diet combined with AA-MFs or GMP-MFs for 3 wk each. Fatty acid profiles of red blood cell (RBC) membranes were determined for 25 adults (aged 18–49 y) with PKU and 143 control participants. Metabolomics analyses of plasma and urine samples were conducted by Metabolon for 9–10 adolescent and adult participants with PKU and for 15 control participants.
Results
RBC fatty acid profiles were not significantly different with AA-MFs or GMP-MFs. PKU participants showed higher total n–6:n–3 (ω-6:ω-3) fatty acids (mean ± SD percentages of total fatty acids: AA-MF = 5.45% ± 1.07%; controls = 4.33%; P < 0.001) and lower docosahexaenoic acid (DHA; AA-MF = 3.21% ± 0.98%; controls = 3.70% ± 1.01%; P = 0.02) and eicosapentaenoic acid (AA-MF = 0.33% ± 0.12%; controls = 0.60% ± 0.43%; P < 0.001) in RBCs than did control participants. Despite higher carnitine intake from AA-MFs than GMP-MFs (mean ± SE intake: AA-MFs = 58.6 ± 5.3 mg/d; GMP-MFs = 0.3 ± 0.01 mg/d; P < 0.001), plasma concentrations of carnitine were similar and not different from those in the control group (AA-MF compared with GMP-MF, P = 0.73). AA-MFs resulted in higher urinary excretion of trimethylamine N-oxide (TMAO), which is synthesized by bacteria from carnitine, compared with GMP-MFs (mean ± SE scaled intensity—TMAO: AA-MFs = 1.2 ± 0.1, GMP-MFs = 0.9 ± 0.1; P = 0.005). Plasma deoxycarnitine was lower in PKU participants than in control participants, suggesting reduced carnitine biosynthesis in PKU (AA-MF = 0.9 ± 0.1; GMP-MF = 1.0 ± 0.1; controls = 1.3 ± 0.1; AA-MF compared with controls, P = 0.01; GMP-MF compared with controls, P = 0.04).
Conclusions
Supplementation with DHA is needed in PKU. Carnitine supplementation of AA-MFs shows reduced bioavailability due, in part, to bacterial degradation to TMAO, whereas the bioavailability of carnitine is greater with prebiotic GMP-MFs. This trial was registered at www.clinicaltrials.gov as NCT01428258.
Keywords: 3-hydroxy 3-methylglutarate, acylcarnitines, dihomo-γ-linolenic acid, deoxycarnitine, docosahexaenoic acid, eicosapentaenoic acid, gamma-linolenic acid, inflammation, trimethylamine, trimethylamine N-oxide
Introduction
Phenylketonuria (PKU; Online Mendelian Inheritance in Man 261600) is an inherited disorder of Phe metabolism characterized by a loss of function of hepatic phenylalanine hydroxylase (PAH; EC 1.14.16.1), thereby limiting the hydroxylation of phenylalanine to tyrosine (1). Diagnosis and initiation of a low-Phe diet in infancy are required to prevent severe cognitive impairment caused by accumulation of Phe in the brain (2). The mechanism of the neurotoxicity of elevated Phe is unknown and may involve oxidative stress and inflammation in the brain (3, 4). A strict low-Phe diet eliminates all animal protein and consists of a controlled amount of natural protein from plant sources to provide minimum Phe requirements and relies on the consumption of either elemental amino acid medical foods (AA-MFs) or glycomacropeptide medical foods (GMP-MFs) to provide the majority of dietary nitrogen and variable amounts of calories, carbohydrate, fat, minerals, and micronutrients (5, 6). Individuals with PKU have low intakes of long-chain (LC)–PUFAs, cholesterol, and carnitine due to the elimination of animal-based, protein-rich foods from their diets, which are the main dietary sources of LC-PUFAs, cholesterol, and carnitine. Instead, they obtain dietary fat from medical foods or whole foods, such as avocados, vegetable and tropical oils, and animal fats (i.e., bacon fat, lard, butter), that are used for cooking and baking. The FA content and composition of current medical foods, particularly the LC-PUFA content, vary greatly; many medical foods contain minimal or no fat.
Alterations in lipid intake leading to oxidative stress and inflammation observed in individuals with PKU managed with AA-MFs (7) are especially relevant to the brain, which has a high lipid content (3). A greater potential for oxidative stress and inflammation in PKU may reflect higher intakes of n–6 relative to n–3 PUFAs and changes in the gut microbiota (8–10). Reduced intake of LC-PUFAs is of concern because these FAs are found preformed only in animal foods and there is evidence of reduced concentrations of EPA (20:5n–3) and DHA (22:6n–3) in plasma and RBCs of individuals with treated PKU (11–14) that has been associated with cognition (15–18). There is also evidence in children with PKU of reduced free carnitine and acylcarnitines (activated FAs conjugated to carnitine) that may affect transport of FAs into the mitochondria for β-oxidation (14). Moreover, the concentration of cholesterol in plasma is noted to be lower in children with PKU with good metabolic control who consume minimal dietary cholesterol (11, 19).
Studies addressing lipid metabolism in PKU have largely focused on the essential FA and LC-PUFA status of children with PKU, not adults. Carnitine and cholesterol metabolism in PKU is poorly understood and has not been studied in adolescents and adults. To our knowledge, the impact on lipid metabolism of a low-Phe diet in combination with GMP-MFs compared with AA-MFs has not been reported and is of significance given the increased utilization of GMP-MFs to manage PKU. Moreover, GMP shows prebiotic properties associated with reduced inflammation (20, 21) and increases the bioavailability of Tyr compared with AA-MFs (22). Our objective was to assess lipid metabolism by using FA and metabolomics analyses in adults and adolescents with PKU who completed our randomized, controlled crossover study and consumed a low-Phe diet in combination with AA-MFs and GMP-MFs (23).
Methods
Study design and protocol
We determined the FA profiles of RBC membranes (n = 25 adults) and completed metabolomics analysis of plasma and urine (n = 9–10 adolescents and adults) from subsets of participants with early-treated PKU who completed our randomized, controlled crossover trial in which each participant consumed, for 3 wk each, their usual low-Phe diet combined with a mean intake of 0.74–0.76 g protein equivalent (PE) ⋅ kg−1 ⋅ d−1 from AA-MFs or GMP-MFs (23) (Table 1). Intake of medical foods, composed of primarily amino acids, are referred to as PEs. The treatments were separated by a 3-wk washout period with AA-MFs. The study protocol included baseline (day 1) and final (day 22) study visits for both AA-MF and GMP-MF treatments, at which fasting blood samples and 3-d food records were obtained (23). Food records were analyzed with Food Processor SQL (version 10.12.0; ESHA) to estimate the nutrient content of medical foods and natural foods as previously reported (23).
TABLE 1.
Participant characteristics1
| Study design | |||
|---|---|---|---|
| FA profile of RBC | Plasma | Urine | |
| membranes | metabolomics | metabolomics | |
| Variable | (n = 25) | (n = 10) | (n = 9) |
| Sex (F/M), n | 15/10 | 6/4 | 5/4 |
| Age group, n | |||
| Adults (≥18 y) | 25 | 8 | 6 |
| Adolescents (15–17 y) | 0 | 2 | 3 |
| Age, y | 29 ± 7 | 28 ± 10 | 25 ± 9 |
| Genotype, n | |||
| Classical PKU | 18 | 5 | 4 |
| Variant PKU | 7 | 5 | 5 |
| Sapropterin dihydrochloride use, n | 3 | 1 | 2 |
| BMI, kg/m2 | 26.2 ± 4.7 | 27.3 ± 5.8 | 25.8 ± 3.9 |
| Plasma Phe, µmol/L | 703 ± 295 | 635 ± 288 | 722 ± 257 |
| Plasma glucose, mg/dL | 84 ± 8 | 86 ± 9 | 87 ± 9 |
1Values are means ± SDs unless otherwise indicated. Information on participant characteristics shown here was obtained during visit 1 of the previously reported clinical trial (23). PKU, phenylketonuria.
To evaluate lipid metabolism in PKU, 3 study designs were used and included the following:
FA profiles of RBC membranes in 25 adult participants were analyzed. We removed 5 participants aged <18 y from the analysis due to limitations in age-matched control samples.
Metabolomics analysis of fasting plasma samples obtained from 10 participants with PKU who consumed AA-MFs and GMP-MFs for 3 wk each and from 15 age- and sex-matched control participants provided by Metabolon were evaluated.
Metabolomics analysis of urine samples obtained from 24-h urine collections in 9 participants with PKU who consumed AA-MFs and GMP-MFs were evaluated. Details of the design of this study have been previously reported (4).
Participants completed the study protocol at either the Waisman Center or at Boston Children's Hospital (23). The University of Wisconsin-Madison Health Sciences Institutional Review Board approved the study protocol. All of the participants provided written informed consent. The trial is registered at www.clinicaltrials.gov as NCT01428258. Participants consumed their preferred AA-MFs, resulting in the use of 15 different AA-MFs as previously reported (23). The GMP-MFs were donated by Cambrooke Therapeutics and contained Glytactin, a proprietary formulation of ∼70% GMP (cGMP-20; Arla Foods Ingredients) and ∼30% supplemental amino acids (Arg, His, Leu, Trp, Tyr).
Analytical methods
The total lipid FA profile of RBC membranes was determined on fasting samples at the Kennedy Krieger Institute (Baltimore, Maryland) by using capillary GC/MS (24). Briefly, 4 mL venous blood collected in EDTA-coated tubes was obtained in the Clinical Research Units at the University of Wisconsin Hospital and Clinics and Boston Children's Hospital. Plasma and RBCs were separated by centrifugation at 1700 × g for 20 min at 4°C, and the plasma removed. The buffy coat was discarded, and the RBCs were washed twice by resuspension in cold 0.01-M PBS, pH 7.4, followed by centrifugation at 4°C. The packed cells were stored under nitrogen at −70°C until analysis. The total lipid FAs were derivatized to pentafluorobenzyl bromide FA esters of chain lengths C10:0 to C30:0 and separated and quantified by capillary GC/MS (24). RBC FAs are presented as percentages of a total of 55 FAs measured. Data for PKU participants represent mean percentages of total FAs in RBC membranes on the basis of 3 determinations of fasting blood samples in 25 adult participants with PKU who consumed AA-MFs. RBC FA profiles of participants with PKU were compared with adult reference control participants that were also analyzed at the Kennedy Krieger Institute (mean ± SD age: 49.5 ± 17.0 y; range: 19–82 y; n = 143).
Metabolon, Inc. (Morrisville, NC), conducted the nontargeted metabolomics analysis on plasma and urine samples (n = 9–10). The data obtained for urine samples were normalized for osmolality before statistical analysis. Plasma and urine samples were processed and stored frozen at −70°C until analyzed. Compounds were identified by comparison to Metabolon's library of authenticated standards or recurrent unknown entities. The analysis identified significantly different concentrations of 185 and 214 known biochemicals in plasma of PKU participants (total of 766 compounds identified) with the ingestion of AA-MFs and GMP-MFs, respectively, compared with 15 healthy age- and sex-matched control participants. The profile of acylcarnitines in plasma determined with metabolomics analysis is shown in Supplemental Table 1.
Statistical analysis
Statistical analyses for the dietary intake and RBC FA percentage composition were performed in SAS version 9.4 (SAS Institute, Inc.). Assumptions of normality and equal variance were tested. Log transformations or nonparametric tests were used when data were skewed. RBC FA percentage composition data were analyzed by using PROC UNIVARIATE to compare participant mean ± SD percentages of composition against a control population. Dietary intake data were analyzed by using PROC MIXED. Analyses for dietary intake were analyzed by using ANOVA with effects for treatment (AA-MFs or GMP-MFs), genotype, and treatment by genotype interactions.
Statistical analyses for the metabolomics were conducted in Array Studio (Omicsoft, NC) version 7.2 and R (R Foundation for Statistical Computing) version 3.02. Biochemicals in the Metabolon analyses for plasma and urine samples were rescaled to set the median to 1, based on all samples. Statistical analyses were conducted to detect differences in fold-change (i.e., scaled intensity). To detect differences in the metabolomics of the plasma and urine samples, paired t tests were used to compare differences between dietary treatments. Welch's 2-sample t tests were used to detect differences between a dietary treatment or a PKU genotype against a control population. P values <0.05 were considered significant.
Results
Participants
The characteristics of all participants are summarized in Table 1. Participants were categorized with classical PKU on the basis of having a PAH genotype, an inadequate response to sapropterin dihydrochloride resulting in a severe PKU phenotype, or both. Sapropterin dihydrochloride is a synthetic form of the tetrahydrobiopterin cofactor for PAH (BioMarin Pharmaceutical, Inc.), which increases Phe tolerance in PKU patients with milder or variant forms of PKU (2). Participants were categorized with variant PKU on the basis of having a mild PAH genotype, a positive response to sapropterin dihydrochloride resulting in a mild PKU phenotype, or both. Participants taking sapropterin dihydrochloride maintained a consistent dosage throughout the study.
Intake profile of the diets
The low-Phe diets in combination with AA-MFs and GMP-MFs were generally constant with similar total intakes of energy, protein, carbohydrate, fat, Phe, and PE from medical foods (median: 53–59 g PEs/d), as shown in Supplemental Table 2 (25). Although total fat intake was not different between treatments, fat intake from medical foods was significantly greater with GMP-MFs than with AA-MFs. Notably, 22 of 25 adult participants consumed a Glytactin GMP-MF that provided a meaningful source of fat (>1 g fat/10 g PEs from medical foods), whereas only 8 of 25 adult participants consumed an AA-MF that provided a meaningful source of fat during the AA-MF treatment. Furthermore, 18 of 25 participants used a GMP-MF that was supplemented with DHA, whereas only 6 of 25 adult participants used an AA-MF that was supplemented with DHA. One of 25 adult participants used an AA-MF supplemented with both DHA and EPA. The GMP-MF products used by our participants in this study were not supplemented with EPA.
Erythrocyte membrane and plasma FA profiles
The profile of FAs in RBC membranes provides a reliable index of long-term intake of dietary FAs over the past several months (26). Thus, it is not surprising that the FA profile of RBC membranes was not different with the consumption of GMP-MFs compared with AA-MFs for 3 wk (data not shown). A comparison of the fasting RBC FA profile in 25 adult participants with PKU who consumed AA-MFs, based on measurements at 3 study visits compared with a control population, is shown in Table 2.
TABLE 2.
RBC FA profile in adult participants with PKU consuming a low-Phe diet in combination with AA-MFs compared with a control population1
| RBC FA percentage comparisons | ||||
|---|---|---|---|---|
| RBC FAs | PKU,2 % | Controls,3 % | P | RBC FAs in participants with PKU,4 µg/mL |
| n–9 FAs | ||||
| Oleic (18:1n–9) | 10.48 ± 1.06 | 11.31 ± 0.83 | <0.001 | 136.65 (125.4–157.8) |
| Mead (20:3n–9) | 0.03 ± 0.01 | 0.04 ± 0.01 | 0.51 | 0.43 (0.31–0.57) |
| Eicosenoic (20:1n–9) | 0.21 ± 0.09 | 0.20 ± 0.04 | 0.68 | 3.03 (2.29–3.87) |
| Nervonic (24:1n–9) | 4.64 ± 1.04 | 3.75 ± 0.52 | <0.01 | 59.24 (50.99–69.22) |
| Total n–9 FAs | 15.75 ± 1.47 | 15.59 ± 1.67 | 0.60 | 206.27 (187.40–240.10) |
| n–6 FAs | ||||
| α-Linoleic (18:2n–6)5 | 9.60 ± 1.24 | 9.19 ± 1.46 | 0.12 | 119.54 (114.9–141.4) |
| γ-Linolenic (18:3n–6) | 0.05 ± 0.02 | 0.03 ± 0.01 | 0.01 | 0.51 (0.38–0.74) |
| Dihomo-γ-linolenic (20:3n–6) | 1.77 ± 0.38 | 1.27 ± 0.29 | <0.001 | 21.91 (20.11–24.95) |
| Arachidonic (20:4n–6) | 12.10 ± 1.62 | 12.04 ± 1.27 | 0.85 | 153.78 (146.5–179.6) |
| Adrenic (22:4n–6) | 3.30 ± 0.60 | 2.46 ± 0.58 | <0.001 | 45.05 (40.35–48.88) |
| Docosapentaenoic (22:5n–6) | 0.59 ± 0.15 | 0.55 ± 0.19 | 0.39 | 7.69 (6.53–8.80) |
| Tetracosadienoic acid (24:2n–6) | 0.93 ± 0.23 | 0.55 ± 0.12 | <0.001 | 12.17 (10.93–14.13) |
| Total n–6 FAs | 29.22 ± 1.97 | 26.62 ± 2.03 | <0.001 | 388.94 (365.60–422.40) |
| n–3 FAs | ||||
| α-Linolenic (18:3n–3)5 | 0.13 ± 0.04 | 0.11 ± 0.03 | 0.003 | 1.67 (1.45–2.03) |
| EPA (20:5n–3) | 0.33 ± 0.12 | 0.60 ± 0.43 | <0.001 | 4.07 (3.40–4.90) |
| Docosapentaenoic (22:5n–3) | 1.87± 0.41 | 1.81 ± 0.36 | 0.47 | 24.64 (20.22–29.38) |
| DHA (22:6n–3) | 3.21 ± 0.98 | 3.70 ± 1.01 | 0.02 | 38.06 (33.11–50.45) |
| Total n–3 FAs | 5.55 ± 0.93 | 6.14 ± 1.50 | <0.01 | 72.38 (64.88–83.29) |
| SFAs | ||||
| Palmitic (16:0) | 19.94 ± 1.23 | 19.62 ± 1.08 | 0.20 | 266.37 (246.50–287.96) |
| Dimethyl acetyl (16:0) | 1.70 ± 0.28 | 1.56 ± 0.22 | 0.02 | 22.90 (19.97–24.32) |
| Stearic (18:0) | 13.90 ± 1.13 | 15.60 ± 1.03 | <0.001 | 185.40 (172.30–196.40) |
| Dimethyl acetyl (18:0) | 2.87± 0.44 | 2.91 ± 0.36 | 0.66 | 39.00 (33.49–40.92) |
| Behenic (22:0)6 | 1.56 ± 0.22 | 1.71 ± 0.19 | <0.01 | 20.55 (18.57–22.26) |
| Lignoceric (24:0)6 | 4.85 ± 0.66 | 5.07 ± 0.46 | 0.12 | 64.53 (57.65–70.76) |
| Total SFAs | 42.01 ± 2.14 | 43.78 ± 1.51 | <0.001 | 557.10 (509.70–600.60) |
| Total n–7 and n–5 FAs | 1.31 ± 0.25 | 1.60 ± 0.17 | <0.001 | 17.23 (14.86–19.87) |
| Total trans FAs | 0.60 ± 0.25 | 0.89 ± 0.29 | <0.001 | 7.47 (5.59–8.71) |
| Ratios | ||||
| n–6 to n–3 | 5.45 ± 1.07 | 4.33 | <0.001 | 5.44 (4.65–6.12) |
| n–3 to n–6 | 0.19 ± 0.04 | 0.23 | <0.001 | 0.19 (0.16–0.22) |
| 20:3n–9 to 20:4n–6 | 0.01 ± 0.01 | <0.01 | 0.61 | 0.0025 (0.002–0.003) |
| 20:4n–6 to 22:6n–3 | 4.10 ± 1.24 | 3.50 ± 1.14 | 0.02 | 4.13 (3.35–4.80) |
| 20:4n–6 to 20:3n–6 | 7.14 ± 1.72 | 9.45 | <0.001 | 7.16 (6.09–8.76) |
1Values are means ± SDs unless otherwise indicated. AA-MF, amino acid medical food; PKU, phenylketonuria.
2Values are expressed as percentages of total FAs, based on fasting blood samples at 3 study visits in adult participants with PKU who consumed AA-MFs (n = 25).
3Population mean reference values were based on male and female adult participants aged 19–82 y from the Kennedy Krieger Institute (n = 143).
4Values are medians (25th–75th percentiles) in adult participants with PKU who consumed AA-MFs (n = 25).
5Outliers were removed (n = 24).
6Means are based on 2 determinations due to the baseline effect of treatment by sequence interaction (n = 25).
The majority of fat is provided by vegetable oils within natural foods in the PKU diet because the fat content of the medical foods is generally low. Consistent with the absence of animal-derived products in the PKU diet, concentrations of oleic acid (18:1n–9), total SFAs, and trans FAs in RBCs were lower in PKU participants than in control participants. For the n–6 PUFAs in RBCs, PKU participants had higher concentrations of α-linoleic acid (18:2n–6), γ-linolenic acid (18:3n–6), and dihomo-γ-linolenic acid (20:3n–6), but similar concentrations of arachidonic acid (20:4n–6) and 4,7,10,13,16-docosapentaenoic acid (22:5n–6) compared with control participants.
For the n–3 PUFAs in RBCs, participants with PKU had significantly higher α-linolenic acid (18:3n–3) and no change for 7,10,13,16,19-docosapentaenoic acid (22:5n–3), but significantly lower EPA and DHA than did control participants. Metabolomics analysis showed higher plasma concentrations of stearidonic acid (18:4n–3), a precursor for EPA and DHA synthesized from 18:3n–3, in PKU participants compared with control participants (plasma stearidonic acid—scaled intensity means ± SEMs: control = 0.88 ± 0.20; GMP-MF = 1.88 ± 0.38; AA-MF = 1.95 ± 0.35; controls compared with PKU, P = 0.003; controls compared with AA-MF, P = 0.009; controls compared with GMP-MF, P = 0.02). Concentrations of DHA in PKU participants were 84% of control levels; 20 of 25 participants had DHA concentrations >1 SD below the mean control level with a low probability of adequacy. Interestingly, 4 of the 5 participants with DHA concentrations at or above the mean control level consumed AA-MFs that contained supplemental DHA according to the manufacturer's ingredient list, whereas the majority of the 20 participants with DHA concentrations >1 SD below the mean control level consumed AA-MFs without supplemental DHA. Concentrations of EPA in PKU participants were 56% of those in control participants; 24 of 25 participants had EPA concentrations 1 SD below the mean control level with a low probability of adequacy. Taken together, it appears that supplementation of medical foods with DHA is needed to ensure adequate intake in adults with PKU.
The sum of all n–3 FAs in RBCs was reduced in participants with PKU compared with control participants, whereas the sum of all n–6 FAs in RBCs increased compared with control participants. This resulted in a significantly higher ratio of n–6 to n–3 FAs based on percentage composition in participants with PKU. There were increased plasma concentrations of 13-hydroxyoctadecadienoic acid and 9-hydroxyoctadecadienoic acid, which are oxidized derivatives of 18:2n–6 (9), in participants with PKU who consumed AA-MFs compared with control participants (plasma 13-hydroxyoctadecadienoic acid and 9-hydroxyoctadecadienoic acid—scaled intensity means ± SEMs: control = 0.88 ± 0.17; AA-MF = 1.43 ± 0.23; GMP-MF = 1.12 ± 0.20; 1.63-fold of change increase in participants with PKU consuming AA-MFs compared with control participants, P < 0.04; GMP-MFs compared with control participants, P = 0.31). At the same time, a significantly lower ratio of 20:4n–6 to 20:3n–6 in RBCs was observed in participants with PKU compared with control participants, which may indicate a compensatory anti-inflammatory response (8). Consistent with adequate intakes of the essential FAs, the ratio of 20:3n–9 to 20:4n–6 in RBCs, an index of essential FA deficiency, was similar in PKU and control participants (27).
Metabolomics evidence relating supplementation of medical foods with carnitine
We found no significant differences in plasma carnitine concentrations, despite higher supplementation of AA-MFs with carnitine compared with GMP-MFs (Figure 1). The supplementation of AA-MFs with l-carnitine (∼5 μmol ⋅ kg−1 ⋅ d−1) provides an intake similar to typical carnitine intake from omnivorous Western diets of 2–10 μmol ⋅ kg−1 ⋅ d−1 (28). However, this is much less than typical carnitine supplementation of 100 mg l-carnitine ⋅ kg−1 ⋅ d−1 used in the treatment of other inborn errors of metabolism (29). Consistent with higher carnitine intake from AA-MFs, we observed a nonsignificant increase in the urinary excretion of carnitine and deoxycarnitine, the last intermediate in endogenous carnitine synthesis from Lys and Met (30), and 4-OH phenacetyl-carnitine (31), and a significant increase in the urinary excretion of trimethylamine N-oxide (TMAO), a product of bacterial metabolism of carnitine and choline (28, 32, 33), with AA-MFs compared with GMP-MFs (Figure 1 and Figure 2). Carnitine and choline are metabolized to trimethylamine by intestinal bacteria, which is further oxidized by the host to TMAO. In the current study, despite significantly higher choline intake from GMP-MFs compared with AA-MFs, urinary excretion of TMAO was significantly lower with GMP-MFs, suggesting that it was derived from carnitine and not choline degradation. Participants with PKU who consumed either AA-MFs or GMP-MFs showed 70% lower concentrations of deoxycarnitine in plasma compared with control levels, suggesting reduced endogenous carnitine synthesis. Plasma Phe based on metabolomics analysis was not significantly different with the ingestion of GMP-MFs than with AA-MFs, suggesting that observed changes in carnitine metabolism are independent of Phe concentrations. Overall, plasma concentrations of carnitine, acetylcarnitine (highest concentration and most functionally important acylcarnitine ester) (34), and >25 specific acylcarnitine species were not different with ingestion of AA-MFs or GMP-MFs than in control participants (Supplemental Table 1).
FIGURE 1.

Metabolomics of carnitine metabolism in participants with phenylketonuria with the use of AA-MFs and GMP-MFs. Values for dietary intakes of carnitine (A) and choline (B) are means ± SEMs and are based on 3-d food records; n = 30/treatment group. Values are scaled intensity means ± SEMs for plasma deoxycarnitine (C), carnitine (D), and choline (E); PKU, n = 10/treatment group; control group, n = 15. Values are scaled intensity means ± SEMs for urinary deoxycarnitine (F), carnitine (G), and TMAO (H); n = 9/treatment group. Labeled means without a common letter differ, P < 0.05. AA-MF, amino acid medical foods; GMP-MF, glycomacropeptide medical foods; PKU, phenylketonuria; TMAO, trimethylamine N-oxide.
FIGURE 2.
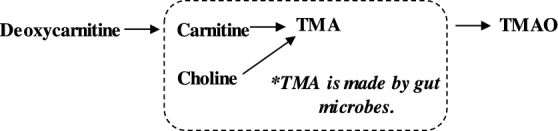
Carnitine synthesis and degradation pathway. TMA, trimethylamine; TMAO, Trimethylamine N-oxide.
Metabolomics evidence of alterations in cholesterol metabolism in variant compared with classical PKU
Because the PKU diet does not contain animal foods, which are the main dietary source of cholesterol, cholesterol consumption was minimal in participants with PKU, with <30% of typical cholesterol intakes of 240–350 mg cholesterol/d observed in the US population (Figure 3) (35). Participants with classical PKU had significantly lower plasma cholesterol concentrations compared with both variant PKU and control participants, whereas cholesterol concentrations in variant PKU were not significantly different from control participants despite much lower cholesterol intake. The higher plasma cholesterol concentrations in participants with variant PKU may reflect higher endogenous cholesterol synthesis, given significantly higher concentrations of the isoprenoid intermediate 3-hydroxy-3-methylglutarate in variant PKU compared with classical PKU and control participants. Steroids and bile acids, which are produced from cholesterol, were similar in participants with variant or classical PKU compared with control participants, suggesting that cholesterol intake in PKU is sufficient to support these downstream pathways. Interestingly, plasma concentrations of the phytosterols, β-sitosterol and campesterol, were higher in participants with variant than with classical PKU (scaled intensity means ± SEMs—β-sitosterol: variant PKU, 1.44 ± 0.23; classical PKU, 0.55 ± 0.08; P = 0.0002; campesterol: variant PKU, 1.69 ± 0.25; classical PKU, 0.70 ± 0.075; P = 0.0004). Plasma Phe concentrations and downstream metabolites of Phe based on metabolomics analyses were increased severalfold in PKU participants compared with control participants, but not significantly different in participants with variant compared with classical PKU. The type of medical food consumed did not affect cholesterol variables.
FIGURE 3.

Metabolomics of cholesterol metabolism in participants with PKU using AA-MFs and GMP-MFs. Intake of dietary cholesterol (A) and the metabolomics of cholesterol metabolism (B, C) are shown. Values are scaled intensity means ± SEMs for metabolites in plasma (classical PKU, n = 5; variant PKU, n = 5; controls, n = 15). Values for dietary intake are means ± SEMs. Total dietary cholesterol intakes are based on 3-d food records (classical PKU, n = 20; variant PKU, n = 10). Labeled means without a common letter differ, P < 0.05. AA-MF, amino acid medical foods; GMP-MF, glycomacropeptide medical foods; PKU, phenylketonuria.
Discussion
Given the absence of animal protein and its associated lipid components in a low-Phe diet and the reliance on medical foods used to manage PKU, there are concerns about FA, carnitine, and cholesterol metabolism. With the use of metabolomics and RBC FA profiles, this is the first study, to our knowledge, to assess how a low-Phe diet in combination with AA-MFs and Glytactin GMP-MFs affect variables of lipid metabolism, specifically essential FAs, carnitine, and cholesterol. Our findings extend the understanding of the impact of a low-Phe diet in children with PKU to adults with classical and variant PKU who consume a low-Phe diet in combination with medical foods.
Several indexes of RBC FA composition in participants with PKU with long-term intake of a low-Phe diet in combination with AA-MFs suggest a proinflammatory state (4, 7–9) associated with lipid metabolism. We observed the following in adults with PKU compared with control participants: 1) a higher ratio of n–6 to n–3 FAs; 2) increased plasma concentrations of oxidized derivatives of 18:2n–6 (13-hydroxyoctadecadienoic acid and 9-hydroxyoctadecadienoic acid); and 3) a lower ratio of 20:4n–6 to 20:3n–6 in RBCs, consistent with compensation for inflammation (8). Consistent with increased plasma concentrations of inflammatory cytokines (4) and altered RBC FA composition in our participants with PKU, Deon et al. (7) observed greater inflammation and lipid damage in PKU patients on the basis of a positive correlation of elevated plasma IL-1β with urinary isoprostanes.
Our data are in agreement with reports by Moseley et al. (13) for 25 adults with PKU, in that concentrations of LC-PUFAs, EPA, and DHA in adult participants with PKU remain lower than in control participants. This is surprising because RBC concentrations of α-linolenic acid and plasma stearidonic acid are higher and both are dietary precursors for EPA and DHA (Table 2) (13). Although these significantly lower concentrations of EPA and DHA do not indicate clinical deficiency (14), the data suggest diminished efficiency in the conversion of α-linolenic acid to EPA and DHA and the need to monitor EPA status in individuals with PKU and to consider supplemental DHA, given the importance of LC-PUFAs in cognition (15–18). More evidence with regard to the efficacy of supplemental EPA to improve EPA status in PKU is needed (36). Moreover, to reduce the potential for inflammation and oxidative stress, it appears prudent to emphasize vegetable oils in the PKU diet with a high proportion of n–3 linolenic acid (precursor to EPA and DHA with anti-inflammatory activities), such as canola and flaxseed oils, relative to n–6 linoleic acid (precursor to arachidonic acid and proinflammatory lipid mediators), which is present in corn and safflower oils (37).
Our data suggest lower bioavailability of carnitine from AA-MFs as reflected by the significantly higher urinary excretion of TMAO compared with GMP-MFs. Intestinal bacteria degrade poorly absorbed carnitine to trimethylamine, which is absorbed and converted to TMAO in the liver and then excreted in urine (28, 29, 32) (Figure 2). Higher plasma concentrations of TMAO have been correlated with the incidence of cardiovascular disease (32). Importantly, TMAO is considered to be a proatherosclerotic metabolite because it modulates cholesterol and sterol metabolism at multiple anatomic sites and reduces in vivo reverse cholesterol transport in mice (38). Koeth et al. (38) showed increased plasma and urinary TMAO with the ingestion of a 250-mg stable isotope–labeled l-carnitine supplement in 30 omnivorous participants, and when challenged with antibiotics, the production of TMAO in plasma and urine was suppressed after ingestion of the l-carnitine supplement. Therefore, the degradation of l-carnitine to TMAO is dependent on the intestinal microbiota and is inducible with the ingestion of carnitine (38).
Despite higher, but not excessive, intake of carnitine from AA-MFs, plasma concentrations of carnitine were similar when participants consumed either AA-MFs or GMP-MFs and were not different from control participants. This suggests adequate carnitine status in adolescents and adults, given the equilibrium of carnitine concentrations in plasma with skeletal muscle carnitine content, the primary carnitine reservoir (34). The ingestion of AA-MFs as compared with GMP-MFs was associated with changes in microbiome-derived metabolites of Tyr and lower bioavailability of Tyr from AA-MFs, suggesting differences in the composition of the intestinal microbiota that reduces the bioavailability of both Tyr and carnitine from AA-MFs compared with GMP-MFs (22). This observation supports greater degradation of carnitine by bacteria and subsequent excretion of TMAO with ingestion of AA-MFs. Thus, our results are consistent with lower bioavailability of supplemental l-carnitine (14–18%) compared with higher bioavailability of dietary carnitine from food sources, such as red meat, which is estimated at 54–87% (28, 34).
Studies report reduced plasma carnitine concentrations in healthy children following strict vegetarian diets (39) and in children with PKU (14) compared with age-matched control participants, which suggests that carnitine may be conditionally essential for growth in children. For children with PKU, carnitine supplementation of AA-MFs does not affect carnitine and acylcarnitine status (14, 40, 41). Taken together, our data suggest that reduced bioavailability of l-carnitine added to AA-MFs accounts for the inability to correct reduced carnitine concentrations in PKU, and that changes in the intestinal microbiota with the ingestion of GMP-MFs improve the bioavailability of carnitine. These observations raise questions about whether carnitine is conditionally essential in children with PKU and the best approach to improve the bioavailability of supplemental carnitine with consideration of changes in the intestinal microbiota due to AA-MFs (10, 22, 29).
Endogenous synthesis of carnitine from Lys and Met appears to be sufficient to support carnitine status in adults following strict vegan diets (39), although a sufficient intake of Lys and Met provided by either AA-MFs or GMP-MFs (23, 42) is needed to support endogenous carnitine synthesis. Participants with PKU in the current study who consumed either AA-MFs or GMP-MFs showed significantly reduced plasma concentrations of deoxycarnitine, the last intermediate in endogenous carnitine synthesis, which may suggest reduced endogenous synthesis of carnitine (30). Indeed, phenylacetate, a metabolite of Phe found in serum and urine of patients with PKU and shown to be 2-fold higher in the current study in participants with PKU than in control participants and not different with intake of AA-MFs and GMP-MFs, was noted to inhibit endogenous carnitine synthesis (31). In conclusion, these data provide support for the concept that the Phe metabolite phenylacetate inhibits endogenous carnitine synthesis.
The literature shows variable effects of PKU on plasma total cholesterol concentrations in children with treated PKU (11, 14, 19) and no difference in plasma total cholesterol in 25 adults with treated PKU (genotype not identified) compared with control participants (13). By using sensitive metabolomics analysis, we observed significantly lower concentrations of plasma total cholesterol in participants with classical PKU compared with both those with variant PKU and control participants. Consistent with our finding in adults, lower plasma cholesterol concentrations were also observed in children with classical PKU compared with hyperphenylalaninemia (a variant form of PKU) (19). Participants with variant PKU showed higher concentrations of the isoprenoid intermediate 3-hydroxy-3-methylglutarate, which may indicate greater endogenous cholesterol synthesis. Increased plasma Phe and its metabolites have been shown to inhibit cholesterol biosynthesis, and these inhibitory compounds were somewhat lower in participants with variant PKU who showed higher plasma concentrations of cholesterol (19). Intake of phytosterols noted to increase endogenous cholesterol synthesis (43) may also account for the higher plasma cholesterol concentration and apparent increase in endogenous cholesterol synthesis with variant PKU.
Strengths of our study include a crossover design and use of a control population for comparisons against our participants with PKU for the metabolomics analyses in plasma. Limitations of our study include the use of 3-wk dietary treatments for the assessment of RBC FA profiles, a small sample size for metabolomics analyses (n = 9–10), and the inability to estimate dietary intake of specific FAs.
In conclusion, this is the first intervention trial, to our knowledge, to assess how a low-Phe diet in combination with AA-MFs and Glytactin GMP-MFs affects metabolism of essential FAs, carnitine, and cholesterol in adults and adolescents with PKU. Our participants with PKU showed lower concentrations of DHA and EPA, and a higher ratio of total n–6 to n–3 FAs than did control participants. This FA profile suggests the need to supplement medical foods with DHA and the potential for oxidation and inflammation with relevance to altered neuronal lipid metabolism and cognition in PKU (3, 7, 18). Individuals with PKU may benefit from a diet that incorporates vegetable oils with a higher proportion of n–3 FAs, such as canola and flaxseed oils. Furthermore, despite higher l-carnitine supplementation in AA-MFs than in prebiotic GMP-MFs, we showed reduced bioavailability of carnitine from AA-MFs due, in part, to degradation of carnitine to proatherosclerotic TMAO by intestinal bacteria. This evidence questions the efficacy and safety of routine supplementation of medical foods with l-carnitine to improve carnitine status in individuals with PKU. More research is needed to determine the optimal FA supplementation scheme of medical foods that specifically addresses the needs of adults and children with PKU.
Supplementary Material
Acknowledgments
The authors’ responsibilities were as follows—BMS, DMN, FR, and HLL: designed the research and provided study oversight; BMS, NN, KB, SGM, and FR: conducted the research; BMS, NN, KB, and SGM: analyzed the data; BMS, DMN, and KB: wrote the manuscript; BMS and DMN: had primary responsibility for the final content; and all authors: read and approved the final manuscript.
Notes
Supported by Department of Health and Human Services grants R01 FD003711 from the FDA Office of Orphan Products Development (to DMN) and P30-HD-03352 and by the Clinical and Translational Science Award program, through the NIH National Center for Advancing Translational Sciences (grant UL1TR000427). Cambrooke Therapeutics, Inc., donated the glycomacropeptide medical foods used in this study and in conjunction with Arla Foods Ingredients provided funding for the metabolomics analyses but were not involved in the design or conduct of the study or in the collection, analysis, or interpretation of the data.
Supplemental Tables 1 and 2 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/jn/.
Abbreviations used:
- AA-MF
amino acid medical food
- GMP-MF
glycomacropeptide medical food
- LC
long-chain
- PAH
phenylalanine hydroxylase
- PE
protein equivalent
- PKU
phenylketonuria
- TMAO
trimethylamine N-oxide.
References
- 1. Flydal MI, Martinez A. Phenylalanine hydroxylase: function, structure, and regulation. IUBMB Life 2013;65:341–9. [DOI] [PubMed] [Google Scholar]
- 2. Vockley J, Andersson HC, Antshel KM, Braverman NE, Burton BK, Frazier DM, Mitchell J, Smith WE, Thompson BH, Berry SA et al. Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med 2014;16:188–200. [DOI] [PubMed] [Google Scholar]
- 3. Okano Y, Nagasaka H. Optimal serum phenylalanine for adult patients with phenylketonuria. Mol Genet Metab 2013;110:424–30. [DOI] [PubMed] [Google Scholar]
- 4. Stroup BM, Sawin EA, Murali SG, Binkley N, Hansen KE, Ney DM. Amino acid medical foods provide a high dietary acid load and increase urinary excretion of renal net acid, calcium, and magnesium compared with glycomacropeptide medical foods in phenylketonuria. J Nutr Metab 2017;2017:1–12, Article ID 1909101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Singh RH, Rohr F, Frazier D, Cunningham A, Mofidi S, Ogata B, Splett PL, Moseley K, Huntington K, Acosta PB et al. Recommendations for the nutrition management of phenylalanine hydroxylase deficiency. Genet Med 2014;16:121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Van Calcar SC, Ney DM. Food products made with glycomacropeptide, a low-phenylalanine whey protein, provide a new alternative to amino acid-based medical foods for nutrition management of phenylketonuria. J Acad Nutr Diet 2012;112:1201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deon M, Sitta A, Faverzani JL, Guerreiro GB, Donida B, Marchetti DP, Mescka CP, Ribas GS, Coitinho AS, Wajner M et al. Urinary biomarkers of oxidative stress and plasmatic inflammatory profile in phenylketonuric treated patients. Int J Dev Neurosci 2015;47:259–65. [DOI] [PubMed] [Google Scholar]
- 8. Sergeant S, Rahbar E, Chilton FH. Gamma-linolenic acid, dihommo-gamma linolenic, eicosanoids and inflammatory processes. Eur J Pharmacol 2016;785:77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vangaveti VN, Jansen H, Kennedy RL, Malabu UH. Hydroxyoctadecadienoic acids: oxidised derivatives of linoleic acid and their role in inflammation associated with metabolic syndrome and cancer. Eur J Pharmacol 2016;785:70–6. [DOI] [PubMed] [Google Scholar]
- 10. Pinheiro de Oliveira F, Mendes RH, Dobbler PT, Mai V, Pylro VS, Waugh SG, Vairo F, Refosco LF, Roesch LFW, Schwartz IVD. Phenylketonuria and gut microbiota: a controlled study based on next generation sequencing. PloS One 2016;11:e0157513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agostoni C, Scaglioni S, Bonvissuto M, Bruzzese MG, Giovannini M, Riva E. Biochemical effects of supplemented long-chain polyunsaturated fatty acids in hyperphenylalaninemia. Prostaglandins Leukot Essent Fatty Acids 2001;64:111–5. [DOI] [PubMed] [Google Scholar]
- 12. Demirdas S, van Spronsen FJ, Hollak CE, van der Lee JH, Bisschop PH, Vaz FM, Ter Horst NM, Rubio-Gozalbo ME, Bosch AM. Micronutrients, essential fatty acids and bone health in phenylketonuria. Ann Nutr Metab 2017;70:111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moseley K, Koch R, Moser AB. Lipid status and long-chain polyunsaturated fatty acid concentrations in adults and adolescents with phenylketonuria on phenylalanine-restricted diet. J Inherit Metab Dis 2002;25:56–64. [DOI] [PubMed] [Google Scholar]
- 14. Mutze U, Beblo S, Kortz L, Matthies C, Koletzko B, Bruegel M, Rohde C, Thiery J, Kiess W, Ceglarek U. Metabolomics of dietary fatty acid restriction in patients with phenylketonuria. PLoS One 2012;7:e43021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yi SH, Kable JA, Evatt ML, Singh RH. A cross-sectional study of docosahexaenoic acid status and cognitive outcomes in females of reproductive age with phenylketonuria. J Inherit Metab Dis 2011;34:455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ramakrishnan U, Gonzalez-Casanova I, Schnaas L, DiGirolamo A, Quezada AD, Pallo BC, Hao W, Neufeld LM, Rivera JA, Stein AD et al. Prenatal supplementation with DHA improves attention at 5 y of age: a randomized controlled trial. Am J Clin Nutr 2016;104:1075–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Helland IB, Smith L, Saarem K, Saugstad OD, Drevon CA. Maternal supplementation with very-long-chain n-3 fatty acids during pregnancy and lactation augments children's IQ at 4 years of age. Pediatrics 2003;111:e39–44. [DOI] [PubMed] [Google Scholar]
- 18. Janssen CI, Kiliaan AJ. Long-chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: the influence of LCPUFA on neural development, aging, and neurodegeneration. Prog Lipid Res 2014;53:1–17. [DOI] [PubMed] [Google Scholar]
- 19. Colome C, Artuch R, Lambruschini N, Cambra FJ, Campistol J, Vilaseca M. Is there a relationship between plasma phenylalanine and cholesterol in phenylketonuric patients under dietary treatment? Clin Biochem 2001;34:373–6. [DOI] [PubMed] [Google Scholar]
- 20. Sawin EA, De Wolfe TJ, Aktas B, Stroup BM, Murali SG, Steele JL, Ney DM. Glycomacropeptide is a prebiotic that reduces Desulfovibrio bacteria, increases cecal short-chain fatty acids, and is anti-inflammatory in mice. Am J Physiol Gastrointest Liver Physiol 2015;309:G590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ntemiri A, Chonchuir FN, O'Callaghan TF, Stanton C, Ross RP, O'Toole PW. Glycomacropeptide sustains microbiota diversity and promotes specific taxa in an artificial colon model of elderly gut microbiota. J Agric Food Chem 2017;65:1836–46. [DOI] [PubMed] [Google Scholar]
- 22. Ney DM, Murali SG, Stroup BM, Nair N, Sawin EA, Rohr F, Levy HL. Metabolomic changes demonstrate reduced bioavailability of tyrosine and altered metabolism of tryptophan via the kynurenine pathway with ingestion of medical foods in phenylketonuria. Mol Genet Metab 2017;121:96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ney DM, Stroup BM, Clayton MK, Murali SG, Rice GM, Rohr F, Levy HL. Glycomacropeptide for nutritional management of phenylketonuria: a randomized, controlled, crossover trial. Am J Clin Nutr 2016;104:334–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lagerstedt SA, Hinrichs DR, Batt SM, Magera MJ, Rinaldo P, McConnell JP. Quantitative determination of plasma C8-C26 total fatty acids for the biochemical diagnosis of nutritional and metabolic disorders. Mol Genet Metab 2001;73:38–45. [DOI] [PubMed] [Google Scholar]
- 25. Stroup BM, Ney DM, Murali SG, Rohr F, Gleason S, van Calcar SC, Harvey HL. Metabolomic insights into the nutritional status of adults and adolescents with phenylketonuria consuming a low-phenylalanine diet in combination with amino acid and glycomacropeptide medical foods, In Revision. J Nutr Metab. In press, December 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sun Q, Ma J, Campos H, Hankinson SE, Hu FB. Comparison between plasma and erythrocyte fatty acid content as biomarkers of fatty acid intake in US women. Am J Clin Nutr 2007;86:74–81. [DOI] [PubMed] [Google Scholar]
- 27. Gramlich L, Meddings L, Alberda C, Wichansawakun S, Robbins S, Driscoll D, Bistrian B. Essential fatty acid deficiency in 2015: the impact of novel intravenous lipid emulsions. JPEN J Parenter Enteral Nutr 2015;39:61S–6S. [DOI] [PubMed] [Google Scholar]
- 28. Rebouche CJ, Chenard CA. Metabolic fate of dietary carnitine in human adults: identification and quantification of urinary and fecal metabolites. J Nutr 1991;121:539–46. [DOI] [PubMed] [Google Scholar]
- 29. Miller MJ, Bostwick BL, Kennedy AD, Donti TR, Sun Q, Sutton VR, Elsea SH. Chronic oral L-carnitine supplementation drives marked plasma TMAO elevations in patients with organic acidemias despite dietary meat restrictions. JIMD Rep 2016;30:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vaz FM, Wanders RJ. Carnitine biosynthesis in mammals. Biochem J 2002;361:417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fischer GM, Nemeti B, Farkas V, Debreceni B, Laszlo A, Schaffer Z, Somogyi C, Sandor A. Metabolism of carnitine in phenylacetic acid-treated rats and in patients with phenylketonuria. Biochim Biophys Acta 2000;1501:200–10. [DOI] [PubMed] [Google Scholar]
- 32. Ussher JR, Lopaschuk GD, Arduini A. Gut microbiota metabolism of L-carnitine and cardiovascular risk. Atherosclerosis 2013;231:456–61. [DOI] [PubMed] [Google Scholar]
- 33. Ufnal M, Zadlo A, Ostaszewski R. TMAO: a small molecule of great expectations. Nutrition 2015;31:1317–23. [DOI] [PubMed] [Google Scholar]
- 34. Rebouche CJ. Kinetics, pharmacokinetics, and regulation of L-carnitine and acetyl-L-carnitine metabolism. Ann NY Acad Sci 2004;1033:30–41. [DOI] [PubMed] [Google Scholar]
- 35. U.S. Department of Health and Human Services and U.S. Department of Agriculture. 2015 - 2020 Dietary Guidelines for Americans. 8th Edition 2015. [DOI] [PubMed] [Google Scholar]
- 36. Lohner S, Fekete K, Decsi T. Lower n-3 long-chain polyunsaturated fatty acid values in patients with phenylketonuria: a systematic review and meta-analysis. Nutr Res 2013;33:513–20. [DOI] [PubMed] [Google Scholar]
- 37. Longo AB, Ward WE. PUFAs, bone mineral density, and fragility fracture: findings from human studies. Adv Nutr 2016;7:299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013;19:576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lombard KA, Olson AL, Nelson SE, Rebouche CJ. Carnitine status of lactoovovegetarians and strict vegetarian adults and children. Am J Clin Nutr 1989;50:301–6. [DOI] [PubMed] [Google Scholar]
- 40. Weigel C, Kiener C, Meier N, Schmid P, Rauh M, Rascher W, Knerr I. Carnitine status in early-treated children, adolescents and young adults with phenylketonuria on low phenylalanine diets. Ann Nutr Metab 2008;53:91–5. [DOI] [PubMed] [Google Scholar]
- 41. Schulpis KH, Nounopoulos C, Scarpalezou A, Bouloukos A, Missiou-Tsagarakis S. Serum carnitine level in phenylketonuric children under dietary control in Greece. Acta Paediatr Scand 1990;79:930–4. [DOI] [PubMed] [Google Scholar]
- 42. Stroup BM, Murali SG, Nair N, Sawin EA, Rohr F, Levy HL, Ney DM. Effects of a low-phenylalanine diet combined with amino acid medical foods or glycomacropeptide medical foods on dietary intake of amino acids and neuropsychological function in subjects with phenylketonuria. Data in Brief 2017;13:377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ostlund RE., Jr. Phytosterols and cholesterol metabolism. Curr Opin Lipidol 2004;15:37–41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.