

Abstract
Background
Klebsiella pneumoniae is an opportunistic pathogen and many strains are multidrug resistant. KPC is one of the most problematic resistance mechanisms, as it confers resistance to most β-lactams, including carbapenems. A promising platform technology for treating infections caused by MDR pathogens is the nucleic acid-like synthetic oligomers that silence bacterial gene expression by an antisense mechanism.
Objectives
To test a peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) in a mouse model of K. pneumoniae infection.
Methods
PPMOs were designed to target various essential genes of K. pneumoniae and screened in vitro against a panel of diverse strains. The most potent PPMOs were further tested for their bactericidal effects in broth cultures and in established biofilms. Finally, a PPMO was used to treat mice infected with a KPC-expressing strain.
Results
The most potent PPMOs targeted acpP, rpmB and ftsZ and had MIC75s of 0.5, 4 and 4 μM, respectively. AcpP PPMOs were bactericidal at 1–2 × MIC and reduced viable cells and biofilm mass in established biofilms. In a mouse pneumonia model, therapeutic intranasal treatment with ∼30 mg/kg AcpP PPMO improved survival by 89% and reduced bacterial burden in the lung by ∼3 logs. Survival was proportional to the dose of AcpP PPMO. Delaying treatment by 2, 8 or 24 h post-infection improved survival compared with control groups treated with PBS or scrambled sequence (Scr) PPMOs.
Conclusions
PPMOs have the potential to be effective therapeutic agents against KPC-expressing, MDR K. pneumoniae.
Introduction
Antibiotic resistance is a global threat.1 The percentage of isolates with MDR is increasing for many bacterial pathogens.2,3 In the USA, 2 million persons annually become severely infected with antibiotic-resistant bacteria and 23 000 of them die. In Europe, antibiotic-resistant bacteria kill at least 25 000 annually.
The main strategies for keeping ahead in the race against resistance have been to discover new antibiotics, chemically modify old ones so that they are not recognized as substrates for resistance mechanisms, or combine antibiotics with inhibitors of resistance.4–7 However, these strategies have not kept pace with the spread of resistance. Discovery of novel antibiotics has waned, effective modifications have become less frequent, inhibitors of resistance are limited to a few β-lactamases, and economic factors in the pharmaceutical industry dried up the developmental pipeline. As a result, a novel antibiotic scaffold effective against Gram-negative pathogens has not been approved in over 30 years.4,8
Some of the most problematic pathogens are a group of Gram-negative bacteria that includes Klebsiella pneumoniae.9,10K. pneumoniae is carried asymptomatically in the intestines and nasopharynx by a highly variable percentage of the population.11 It is an opportunistic pathogen that can infect many tissues, but is mostly associated with urinary tract infections, pneumonia, sepsis, skin infections and meningitis. Like other organisms in this group, K. pneumoniae has acquired an impressive array of antibiotic resistance mechanisms against all classes of antibiotics and the frequency of MDR isolates is growing.2,10
An alternative strategy for discovering and developing new antimicrobials is to synthesize nucleic acid mimetics that bind to specific bacterial RNA and inhibit growth. Phosphorodiamidate morpholino oligomers (PPMOs) are synthetic nucleotide analogues that prevent translation of a specific gene by selectively binding mRNA in an antisense manner.12,13 The structure differs from DNA by a morpholino ring that replaces the deoxyribose ring, and a charge-neutral phosphorodiamidate linker instead of the phosphodiester linker.14 An arginine-rich peptide is conjugated to one or the other end of the PPMO to enable penetration through the Gram-negative outer membrane.15,16
Currently, we have designed, synthesized and tested PPMOs targeted to a variety of essential genes in K. pneumoniae. We show here a proof-of-concept that a PPMO targeted to acpP (encoding an acyl carrier protein that is essential for lipid biosynthesis) protected mice in a lethal model of pneumonia. To our knowledge, this is the first time a gene-specific therapeutic agent has been shown to be effective against K. pneumoniae in vivo.
Materials and methods
Reagents
All PPMOs were synthesized and purified at Sarepta Therapeutics (Corvallis, Oregon, USA) as described previously.17 The PPMO nucleobase and amino acid sequences are shown in Table S1 (available as Supplementary data at JAC Online). Crystal violet was from Acros Chemical Co. (Geel, Belgium).
Bacterial strains, culture cells and growth conditions
K. pneumoniae OR1–OR20 are clinical isolates that were provided by Karim E. Morey (Oregon Public Health Laboratory, Portland, OR, USA). K. pneumoniae BAA-2146 and human A549 lung epithelial cells were obtained from ATCC (Manassas, VA, USA). K. pneumoniae NDM1-A, -B, -C and -D were provided by Susan M. Poutanen (Mount Sinai Hospital, Toronto, ON, Canada). K. pneumoniae 3190, 3290, 3426 and 3427 were provided by Carl Urban (New York Queens Hospital, NY, USA). K. pneumoniae NIH-1 and NIH-2 were provided by Karen Frank (NIH, Bethesda, MD, USA). All other strains were provided by BEI Resources (Manassas, VA, USA). Liquid cultures were grown in Mueller–Hinton II (MHII) (cation adjusted) broth. LB agar was used for growth on solid medium. Bacteria for in vivo studies were prepared from a log-phase culture. The culture was cooled on ice and the cells washed twice with ice-cold PBS by centrifugation (3 × 103 g). The washed cells were resuspended in PBS/10% glycerol to about 1 × 1011 cfu/mL and stored frozen at −75°C.
MIC and MBC
MICs were measured by the CLSI microdilution method.18 MBCs were measured by diluting and plating on LB agar samples from wells without visible growth after 20 h. The Petri dishes were incubated overnight at 37°C and the colonies were counted.
Cytotoxicity
Human lung epithelial cells (A549) were cultured with or without 10 μM PPMO as described in Figure S1. After 48 h, viable cells were measured by trypan blue exclusion.19
Biofilm
Biofilms were grown for 24 h and measured as described previously,20 except the crystal violet was solubilized in 95% ethanol and absorbance read at 595 nm. Viable cells in the biofilm were solubilized from a parallel microtitre plate by adding 100 μL of 0.1% Triton X-100. Preliminary tests found that Triton X-100 resulted in the solubilization of the maximal number of cells with no loss of viability. Liquid cultures from two additional, equivalent microtitre plates were removed at 24 h and wells washed with H2O and refilled with 100 μL of LB broth without or with various concentrations of PPMO. The plates were again incubated for an additional 24 h and then processed as described above.
Confocal microscopy
K. pneumoniae strain OR5 biofilm was grown and treated with one 24 h dose of PPMO in minimum biofilm eradication concentration (MBEC) plates, as described.21 Cut pegs were stained with Live/Dead BacLight Bacterial Viability Kit stain (Thermo Fisher Scientific) (3 μL of SYTO 9:3 μL of propidium iodide:994 μL of 150 mM NaCl) for 15 min and rinsed with 150 mM NaCl. Pegs were then imaged on a spinning-disc confocal microscope as described.21 Images were volumetrically rendered in Imaris software (Bitplane, Concord, MA, USA).
Ethics
All animal procedures were approved by the Oregon State University (approval number 4709) or University of Texas Southwestern Medical Center (approval number APN-2016-101626) Institutional Animal Care and Use Committees and comply with all local, state and federal laws.
Mouse survival experiments
Female BALB/c mice, aged 6–8 weeks (The Jackson Laboratory, Sacramento, CA, USA) were randomly assigned to treatment groups and then treated with 3 mg cyclophosphamide in 100 μL of PBS by intraperitoneal injection 1 and 4 days prior to infection. Mice were anaesthetized with isoflurane and infected intranasally with ∼3.0 × 107 cfu of K. pneumoniae NR15410 in 25 μL of PBS. Intranasal treatment with various doses of PPMO in 25 μL PBS (as indicated in figure legends) was initiated 0, 8, 24 or 48 h post infection and then repeated twice at 24 h intervals (so each mouse received a total of three doses of PPMO). Body temperature was measured in the ear using an infrared thermometer (Braun Thermoscan Pro 4000, Kaz USA, Marlborough, MA, USA).
Mouse lung burden
Six- to eight-week-old female BALB/c mice (The Jackson Laboratory, Bar Harbor, ME, USA) were treated with 3 mg cyclophosphamide in 100 μL of PBS by intraperitoneal injection 1 and 4 days prior to infection. Mice were anaesthetized with isoflurane and infected intranasally with 2.7–2.9 × 107 cfu of K. pneumoniae NR15410 (in 25 μL PBS). At 0 and 6 h post-infection, mice were treated intranasally with either 300 μg of PPMO in 25 μL PBS or 25 μL PBS. Mice were euthanized 24 h post-infection and whole lungs were collected, weighed, homogenized for 10 seconds in 1 mL of PBS and serially diluted and plated for cfu enumeration.
Statistical analysis
Data were analysed with GraphPad Prism 6 software (La Jolla, CA, USA). Treatment group cfu and mass means were compared by t-test (unpaired, two-tailed). All cfu data were transformed to log values prior to analysis. Survival was analysed by log-rank (Mantel–Cox) test.
Results
Optimization of PPMOs
Initially, 13 PPMOs were designed to target eight presumably essential genes, based on the effectiveness of PPMOs in other genera of bacteria (Figure 1).21,22 To reduce the possibility of off-target effects, PPMOs were positioned at unique 11-base sequences near the translation start codon and/or ribosome-binding site of the target gene. The 11-base length had been previously optimized to inhibit bacterial and not eukaryotic gene expression.23,24 The design algorithm is available on-line.25
Figure 1.

MIC values for various PPMOs in a panel of strains. NDM-1, New Delhi MBL; MEMR, meropenem resistant; FEPR, cefepime resistant; TZPR, piperacillin/tazobactam resistant; CIPR, ciprofloxacin resistant; PMBR, polymyxin B resistant. MIC75 is the minimum concentration required to inhibit growth of at least 75% of the strains tested. Colour code: red, 0.5–4 µM; blue, 8–16 µM; grey,>16 µM. This figure appears in colour in the online version of JAC and in black and white in the printed version of JAC.
Additionally, two control PPMOs with a scrambled sequence of bases (Scr) were also synthesized. All of the PPMOs had the same peptide [(RXR)4XB] attached to the 5′-end, except three (0802, 0017 and 1344) that used R6G. (RXR)4XB conjugates had been found previously to penetrate K. pneumoniae,26 but R6G conjugates had been tested only in Pseudomonas aeruginosa21 and not in Klebsiella. Three of the PPMOs targeted different positions on acpP: one (0276) spanning the start codon and two (0076 and 0802) starting three bases downstream of the start codon.
The MICs of all PPMOs were measured using a panel of 40 strains of K. pneumoniae. The results show that the 5′-(RXR)4XB-AcpP PPMO (0276) was the most potent with an MIC75 of 0.5 μM (Figure 1). This was followed closely by two other PPMOs targeted to acpP (0076 and 0802) with MIC75s of 1.0 μM and three PPMOs targeted to rpmB and ftsZ with MIC75s of 4 μM. The Scr PPMOs did not inhibit growth. Growth curves of K. pneumoniae NR 15410 with various concentrations of 0276 and 0802, and their control Scr PPMOs, indicated that growth was completely inhibited at or above the MIC values of 0276 or 0802, and the growth rate below the MIC was decreased compared with untreated cultures (Figure S2). The Scr PPMOs had no effect on growth.
To further optimize the PPMOs for use with K. pneumoniae, a set of AcpP PPMOs were synthesized with different peptides composed of arginine (R) and tyrosine (Y), glycine (G) or phenylalanine (F) instead of 6-aminohexanoic acid (X). The MIC of each PPMO was measured with the panel of strains. The results show that these changes made no difference in potency (Table 1).
Table 1.
Pairs of PPMOs comparing 5′ versus 3′ conjugation and various peptides
| PPMO no. | Target | Sequence (5′→3′) | Peptide sequence | End | MIC75 (μM) |
|---|---|---|---|---|---|
| 0076 | acpP +6 to +16 | CTTCGATAGTG | (RXR)4XB | 5′ | 1 |
| 0948 | acpP +6 to +16 | CTTCGATAGTG | (RXR)4XB | 3′ | 1 |
| 0802 | acpP +6 to +16 | CTTCGATAGTG | R6G | 5′ | 1 |
| 0155 | acpP +6 to +16 | CTTCGATAGTG | R6G | 3′ | 2 |
| 0276 | acpP −4 to +6 | TGCTCATACTC | (RXR)4XB | 5′ | 0.5 |
| 0398 | acpP −4 to +6 | TGCTCATACTC | (RXR)4XB | 3′ | 1 |
| 0016 | acpP −4 to +6 | TGCTCATACTC | R6G | 5′ | 2 |
| 0399 | acpP −4 to +6 | TGCTCATACTC | R6G | 3′ | 2 |
| 0605 | acpP +6 to +16 | CTTCGATAGTG | (RYR)4XB | 5′ | 1 |
| 0099 | acpP +6 to +16 | CTTCGATAGTG | (RYR)4XB | 3′ | 1 |
| 0606 | acpP +6 to +16 | CTTCGATAGTG | (RGR)4XB | 5′ | 1 |
| 0141 | acpP +6 to +16 | CTTCGATAGTG | (RGR)4XB | 3′ | 1 |
| 0621 | acpP +6 to +16 | CTTCGATAGTG | (RFR)4XB | 5′ | 1 |
| 0205 | acpP +6 to +16 | CTTCGATAGTG | (RFR)4XB | 3′ | 1 |
| 1073 | Scr | TCTCAGATGGT | (RXR)4XB | 5′ | >32 |
| 1344 | Scr | TCTCAGATGGT | R6G | 5′ | >32 |
We assessed another feature of the PPMOs, which is attaching the peptide to either the 5′ or 3′ end of the PPMO. Seven PPMOs (0948, 0155, 0398, 0399, 0099, 0141 and 0205) were synthesized with the same base sequence and peptides as 0076, 0802, 0276, 0016, 0605, 0606 and 0621, respectively, except the peptides were attached to the 3′ end. The PPMOs were tested by measuring the MIC in our panel of 40 strains. The results show that two of the 3′ conjugates were 2-fold less potent than their 5′ equivalents, but there was no difference in any of the other pairs (Table 1).
Minimal bactericidal concentration (MBC)
The MBCs of 0276 and 0802 were measured in two strains from our panel of K. pneumoniae: NR15410 and NIH-1. The results show that both PPMOs were bactericidal at 2 × MIC for both strains (Figure 2). Also, 0276 was bactericidal at 1 × MIC for both strains, whereas 0802 was only bactericidal at 1 × MIC for NR15410. Scr PPMOs had no effect on viability at any concentration tested.
Figure 2.

Minimal bactericidal concentrations (MBCs). Samples from cultures without visible growth in the MIC assay were diluted and spread onto LB agar Petri dishes. After overnight growth on the Petri dishes, colonies were enumerated. 2×, 1× and 0.5× refer to multiples of the MIC value of each AcpP PPMO. Error bars indicate standard deviation, n = 3 for AcpP PPMOs and n = 1 for Scr PPMOs.
Cytotoxicity
Although 11-base PPMOs do not affect the gene expression of eukaryotic cells, the sequences of both AcpP, FtsZ, RpmB and Scr PPMOs were nevertheless tested for cytotoxicity in cultures of A549 human lung epithelial cells. The results show that there was no reduction of growth or viability (Figure S2).
Testing of PPMOs on biofilm
PPMOs were tested for their ability to reduce established biofilms and viable cells within biofilms. Biofilms were established in microtitre plates by growing cultures of K. pneumoniae OR5 for 24 h. Following the establishment of biofilms, the spent medium was removed and replaced with fresh broth plus 8, 16 or 32 μM AcpP-PPMO (0276 or 0802) and grown for another 24 h. Control cultures included either no PPMO or 8, 16, or 32 μM Scr PPMO (1073 or 1344). The biofilm mass and viable cells in the biofilms were then measured. The results show that 32 μM PPMO 0276 or 0802 reduced the established biofilm mass by 32% and 22%, respectively, compared with the 24 h biofilm (Figure 3a). Furthermore, treatment with 32 μM PPMO 0276 or 0802 resulted in a 68% and 60% reduction in biofilm mass, respectively, compared with the 48 h culture without PPMO. Lower concentrations of the AcpP PPMOs resulted in a proportionally lower reduction in biofilm mass, but still significantly less biofilm than the untreated culture. The Scr PPMOs did not reduce biofilm at any concentration tested compared with the untreated culture.
Figure 3.

Biofilm penetration. Biofilms were established (for 24 h) and then treated with (a, b) 8, 16, or 32 µM AcpP PPMO (0276 or 0802) or Scr PPMO (1073 or 1344) in growth medium. After 48 h, (a) biofilm mass and (b) viable cells within the biofilm were measured. (a) N = 33 (no PPMO, 24 h, red bars), N = 36 (no PPMO, 48 h, blue bars), N = 12 for all others. (b) N = 6 (0276 and Scr 1073) and N = 4 (0802 and Scr 1344) for cfu. Error bars indicate means and standard deviations. Asterisks indicate highly significant difference (P < 0.0001) compared with no PPMO at 48 h; ## indicates significant difference (P < 0.05) compared with no PPMO at 24 h. (c, d, e, f) Confocal images of fluorescently stained K. pneumoniae cells within biofilm. Live cells are stained green and dead cells are stained red. (c) No PPMO; (d) 16 µM Scr PPMO (1344); (e) 16 µM AcpP PPMO (0802); and (f) 2 µM AcpP PPMO (0802). Upper images are perpendicular to surface, and lower images are parallel to surface. Bars in (c) to (f) represent 20 μm.
Treatment of established biofilms with the AcpP PPMOs also reduced the number of viable cells in the biofilm. Treatment with 8, 16, or 32 μM AcpP PPMO 0276 resulted in significantly fewer viable cells in the biofilm compared with the untreated culture [5.6 × 107, 3.8 × 107 or 2.0 × 107 cfu/mL, respectively, compared with 1.9 × 108 cfu/mL in the untreated culture at 48 h (Figure 3b)]. Similar reductions in viable cells were observed with the AcpP PPMO 0802 (Figure 3b). Neither Scr PPMO had any effect on viability of cells within the biofilm (Figure 3b).
PPMO penetration of established biofilms was also measured by confocal microscopy. Biofilms were established in cultures of K. pneumoniae OR5 for 24 h without PPMO. After establishing the biofilms, the growth medium was removed and replaced with fresh medium with or without PPMO and incubated for another 24 h. The biofilms were stained with fluorescent dyes to distinguish between live and dead cells (live cells stain green, dead cells stain red). The results show that treatment with either 16 μM or 2 μM AcpP PPMO decreased viable cells by 96% (pixels = 153) and 70% (pixels = 1111), respectively, compared with the untreated biofilm (pixels = 3750). AcpP PPMO treatment also increased the ratio of dead:live cells in 16 μM (3.5:1) or 2 μM (1.3:1) biofilms as compared with untreated (1:1) and 16 μM Scr controls (0.3:1) (Figure 3c–f). These results, taken together with those shown in Figure 3(a and b), support the conclusion that PPMO effectively penetrated Klebsiella biofilm.
Testing of AcpP PPMO in vivo
The efficacy of the R6G-AcpP PPMO (0802) was evaluated in vivo using a mouse pneumonia model. Mice were infected intranasally with the MDR clinical isolate K. pneumoniae NR15410 and then treated intranasally once daily for 3 days with various amounts of PPMO from 12.5 μg to 600 μg (∼0.6 to 30 mg/kg). Control groups were infected and treated similarly with either PBS or an Scr PPMO. The results show dose-dependent responses. Survival was 89%, 62% and 33% for mice treated with 600 μg, 200 μg or 67 μg (∼30, 10 and 3.3 mg/kg) of R6G-AcpP PPMO, respectively (Figure 4a). This was significantly greater (P < 0.002) than mice treated with either PBS or the Scr PPMO. All mice treated with lower amounts of the AcpP PPMO, PBS or 600 μg (∼30 mg/kg) of Scr PPMO died by day 5 post-infection. Mice treated with a lower dose (200 μg, ∼10 mg/kg) of Scr PPMO also died by day 5 (data not shown), strongly suggesting that deaths were not caused by toxicity of the Scr PPMO. Body temperature of surviving mice began to recover between 3 and 5 days post-infection, whereas it continued to decrease after day 2 or 3 in all mice that ultimately did not survive (Figure 4b).
Figure 4.
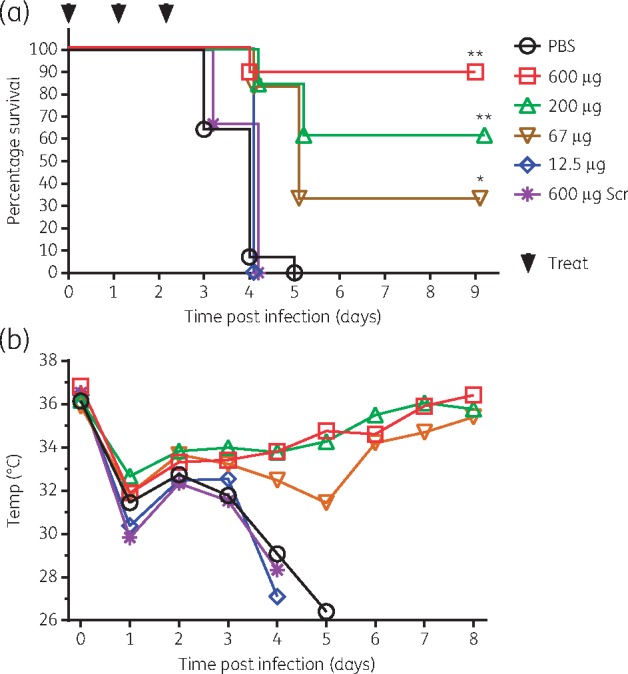
In vivo infectious challenge and treatment with R6G-AcpP PPMO (0802). Mice were infected with K. pneumoniae NR15410 and treated at 0, 24 and 48 h post-infection with 600 µg (n = 9), 200 µg (n = 13), 67 µg (n = 6) or 12.5 µg (n = 3) of AcpP-PPMO, 600 µg of Scr PPMO (Scr, 1344) (n = 9) or PBS (n = 14). (a) Survival; (b) body temperature. For Kaplan–Meier survival curves, **P < 0.0001, *P = 0.0016 by log-rank (Mantel–Cox) test. This figure appears in colour in the online version of JAC and in black and white in the printed version of JAC.
The R6G-AcpP PPMO (0802) was also tested therapeutically by administering it post-infection. Groups of mice were infected as described above and then treated with the first dose of 600 μg (∼30 mg/kg) of PPMO at 0 h, 8 h, 24 h or 48 h post-infection. Each group was treated twice more at 24 h intervals. When treatment was not delayed, 71% of the mice survived (Figure 5a). Delaying treatment for 8 h resulted in 33% survival, which is significantly greater than the PBS control group. There were no survivors in the PBS-treated group or when initial treatment was delayed for 24 or 48 h. However, median survival time in the 8 h (5.0 days) and 24 h (4.0 days) delayed-treatment groups was significantly (P < 0.01) greater than that in the PBS-treated group (3.0 days). Body temperature decreased less in mice treated initially at 0 or 8 h compared with groups treated initially at 24 or 48 h, or with PBS (Figure 5b).
Figure 5.
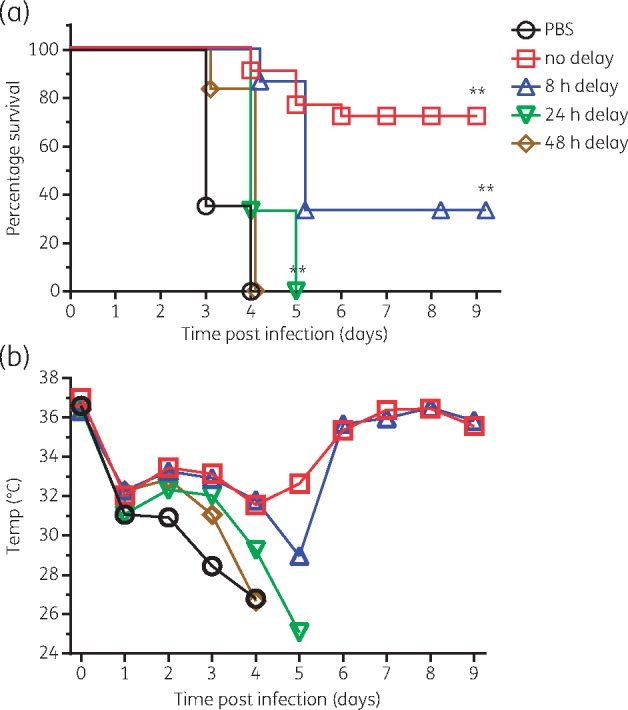
Therapeutic (delayed) treatment. Groups of mice were infected intranasally with K. pneumoniae NR15410. Initial treatment with ∼30 mg/kg R6G-AcpP PPMO (0802) was given at 0, 8, 24 or 48 h post-infection and then again at 24 h intervals for 3 days. (a) Survival and (b) body temperature were monitored. For Kaplan–Meier survival curves, **P < 0.0001 by log-rank (Mantel–Cox) test. N = 18 (PBS), 21 (no delay), 15 (8 h and 24 h delay), and 12 (48 h delay). This figure appears in colour in the online version of JAC and in black and white in the printed version of JAC.
In other experiments, the bacterial burden in the lungs of infected mice was measured. Groups of mice were infected intranasally and then treated intranasally at 0 and 6 h post infection with 300 μg (∼15 mg/kg) of R6G-AcpP PPMO (0802), R6G-Scr PPMO (1344) or PBS. After 24 h post-infection, lungs were removed, homogenized and spread on Petri dishes to determine viable bacteria. The results showed a highly significant (P < 0.0001), 3-log reduction in viable bacteria from the lungs of mice treated with AcpP PPMO 0802 compared with the mice treated with PBS (Figure 6). No statistically significant (P > 0.05) reduction was found in the group treated with Scr PPMO (1344) compared with the group treated with PBS.
Figure 6.

Bacterial lung burden. Mice were infected and treated at 0 and 6 h post-infection. Lungs were removed at 24 h post-infection, homogenized and then spread on LB agar Petri dishes, which were incubated for 18 h at 37°C. Bacterial colonies on Petri dishes were enumerated. An asterisk indicates a statistically significant difference (P < 0.0001).
Discussion
Our results show that PPMOs targeted to essential genes in K. pneumoniae inhibited growth in pure cultures of a panel of 40 diverse strains. The MIC75 values ranged from 0.5 μM to >16 μM. The MBC of two PPMOs targeted to acpP were either 1 or 2 × MIC, depending on the strain used for testing. These results are similar to previous reports that showed in vitro growth inhibition of K. pneumoniae using peptide-conjugated peptide nucleic acids targeted to various essential genes.27,28 It appears that the AcpP PPMOs are at least 20 times more potent than the peptide-conjugated peptide nucleic acids targeted to gyrA, ompA28 or rpoD.27 These differences in potency may be attributed to differences in the target gene, the membrane-penetrating peptides, the strains or methods used to measure MIC, or the position of the oligomers relative to the start codons or ribosome-binding sites. acpP appears to be a superior target across numerous pathogens. One possible explanation for this finding is that small decreases in acpP expression have a greater impact on growth rate than similar decreases in expression of other known essential genes.29 Importantly, PPMOs retained their activity in MDR strains of Klebsiella, which is similar to our experiences with other Gram-negative pathogens.
An unexpected result was the ability of the PPMOs to reduce both biofilm mass and viability of K. pneumoniae within established biofilms. Biofilms protect embedded bacteria by reducing the effectiveness of antibiotics, bacteriophage and the immune system.30 Although the exact mechanism(s) of biofilm protection against antibiotics is controversial, it seems dependent on the bacterium and the structures of the biofilm and antibiotic and not necessarily related to diffusion limitations linked to molecular weight.30–32 Our results are consistent with the hypothesis that Klebsiella biofilm does not pose a diffusion limitation, since the molecular weight of PPMOs is about 10–20 times that of most common antibiotics. Although the mechanism by which PPMOs reduce biofilm mass and gain access to kill the bacteria is unknown, our results from treating the established biofilm with PPMO in PBS are consistent with the PPMO penetrating the biofilm to gain access to the pathogen. Our results are inconsistent with the PPMO acting only on the planktonic cells. In addition, our results are inconsistent with the membrane-penetrating peptide portion of the PPMO simply solubilizing the biofilm, since an Scr PPMO with the same peptide attached did not reduce biofilm mass. These data suggest that PPMOs penetrate Klebsiella biofilms, kill cells within the biofilm and reduce biofilm mass. We have demonstrated a similar ability of PPMOs in biofilms of Pseudomonas.21
The Scr PPMO 1344 showed a slight, but statistically insignificant, reduction of bacterial lung burden. However, this was not sufficient to increase survival. No effect of the Scr PPMO was found in any of the in vitro assays. A previous report also found a positive effect of an Scr PPMO in vivo, but not in vitro.16 This trend seems to point to a non-sequence-specific effect. The cause of this effect is unknown, but we speculate that it might result from an interaction of the PPMO with the innate immune system. Alternatively, it is conceivable that it is related to changes in vasculature, possibly mediated through nitric oxide.
While previous reports have demonstrated the effectiveness of antisense antimicrobial oligomers in pure culture and in tissue culture,27,28,33 we believe that this is the first vetted report of the effectiveness of an antisense oligomer against K. pneumoniae in an animal model. Here we have shown that an AcpP PPMO improved survival and reduced morbidity in a dose-dependent manner and in a dosage range that is clinically achievable (3–30 mg/kg/dose once per day). The PPMO was well-tolerated in vivo and was effective therapeutically. Because the amount of each dose that actually reaches the site of infection in the lung may vary slightly with each intranasal administration, it is possible that the results underestimate the potency and efficacy of the AcpP PPMO.
These results open up a new possibility for future studies that will validate acyl carrier protein as the target, establish the pharmacokinetics and pharmacodynamics, and test the tolerability and toxicity in vivo. PPMOs have the potential for clinical applications against MDR Klebsiella.
Supplementary Material
Acknowledgements
We thank Karim Morey (Oregon Public Health Laboratory, Portland, OR), Susan Poutanen (Mount Sinai Hospital, Toronto, ON, Canada), Karen Frank (NIH, Bethesda, MD), and Carl Urban (New York Queens Hospital) for providing strains. We thank Stacey M. Bailey (formerly with Sarepta Therapeutics) for synthesizing and purifying the PPMOs. We acknowledge the UT Southwestern Live Cell Imagining Facility for use of their microscopes. We acknowledge the Texas Advanced Computing Center (TACC; http://www.tacc.utexas.edu) at The University of Texas at Austin for providing storage resources for microscopic imaging that contributed to the research results reported within this paper. Some of the in vitro results were presented in an abstract/poster (523) at the 2016 Interscience Conference on Antimicrobial Agents and Chemotherapy, Boston, MA, USA.
Funding
This project was supported by grants from the US National Institutes of Health grant R21/R33 AI111753 (D. E. G. and B. L. G.).
Transparency declarations
B. L. G. and D. E. G. are consultants to Sarepta Therapeutics and inventors on numerous patents and patent applications involving PPMOs. D. E. G. has received research support from Sarepta Therapeutics. Oregon State University and the University of Texas Southwestern Medical Center have licensed PPMO technologies to Sarepta Therapeutics. All other authors: none to declare.
Author contributions
B. L. G. and D. E. G. planned experiments, supervised the work and interpreted the results. L. L., F. M. and B. L. G. performed MIC assays and in vivo survival studies. F. M., E. S. and C. P. performed the biofilm experiments. C. P. performed confocal microscopy. C. R. S. and S. M. D. performed MIC and MBC assays, and planned, performed and analysed in vivo lung burden experiments. B. L. G. wrote the manuscript with significant contributions from D. E. G. and C. R. S. All authors helped edit the manuscript.
Supplementary data
Table S1 and Figures S1 and S2 available as Supplementary data at JAC Online.
References
- 1. Watkins RR, Bonomo RA.. Overview: global and local impact of antibiotic resistance. Infect Dis Clin North Am 2016; 30: 313–22. [DOI] [PubMed] [Google Scholar]
- 2. WHO. Antimicrobial Resistance. Global Report on Surveillance 2014; 257. http://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf.
- 3. Weiner LM, Webb AK, Limbago B. et al. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011-2014. Infect Control Hosp Epidemiol 2016; 37: 1288–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Butler MS, Blaskovich MA, Cooper MA.. Antibiotics in the clinical pipeline in 2013. J Antibiot 2013; 66: 571–91. [DOI] [PubMed] [Google Scholar]
- 5. Butler MS, Blaskovich MA, Owen JG. et al. Old dogs and new tricks in antimicrobial discovery. Curr Opin Microbiol 2016; 33: 25–34. [DOI] [PubMed] [Google Scholar]
- 6. Fischbach MA, Walsh CT.. Antibiotics for emerging pathogens. Science 2009; 325: 1089–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drawz SM, Papp-Wallace KM, Bonomo RA.. New β-lactamase inhibitors: a therapeutic renaissance in an MDR world. Antimicrob Agents Chemother 2014; 58: 1835–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boucher HW, Talbot GH, Benjamin DK Jr. et al. 10×'20 progress–development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America . Clin Infect Dis 2013; 56: 1685–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grundmann H, Glasner C, Albiger B. et al. Occurrence of carbapenemase-producing Klebsiella pneumoniae and Escherichia coli in the European survey of carbapenemase-producing Enterobacteriaceae (EuSCAPE): a prospective, multinational study. Lancet Infect Dis 2017; 17: 153–63. [DOI] [PubMed] [Google Scholar]
- 10. Navon-Venezia S, Kondratyeva K, Carattoli A.. Klebsiella pneumoniae: a major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol Rev 2017; 41: 252–75. [DOI] [PubMed] [Google Scholar]
- 11. Podschun R, Ullmann U.. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clinical Microbiol Rev 1998; 11: 589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stein D, Foster E, Huang SB. et al. A specificity comparison of four antisense types: morpholino, 2'-O-methyl RNA, DNA, and phosphorothioate DNA. Antisense Nucleic Acid Drug Dev 1997; 7: 151–7. [DOI] [PubMed] [Google Scholar]
- 13. Summerton J, Stein D, Huang SB. et al. Morpholino and phosphorothioate antisense oligomers compared in cell-free and in-cell systems. Antisense Nucleic Acid Drug Dev 1997; 7: 63–70. [DOI] [PubMed] [Google Scholar]
- 14. Sully EK, Geller BL.. Antisense antimicrobial therapeutics. Curr Opin Microbiol 2016; 33: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mellbye BL, Puckett SE, Tilley LD. et al. Variations in amino acid composition of antisense peptide-phosphorodiamidate morpholino oligomer affect potency against Escherichia coli in vitro and in vivo. Antimicrob Agents Chemother 2009; 53: 525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Greenberg DE, Marshall-Batty KR, Brinster LR. et al. Antisense phosphorodiamidate morpholino oligomers targeted to an essential gene inhibit Burkholderia cepacia complex. J Infect Dis 2010; 201: 1822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tilley LD, Hine OS, Kellogg JA. et al. Gene-specific effects of antisense phosphorodiamidate morpholino oligomer-peptide conjugates on Escherichia coli and Salmonella enterica serovar Typhimurium in pure culture and in tissue culture. Antimicrob Agents Chemother 2006; 50: 2789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; M7-A7; Broth Microdilution Method: CLSI, Wayne, PA, USA, 2006. [Google Scholar]
- 19. Strober W. Trypan blue exclusion test of cell viability Current Protocols in Immunology. 2001; 21: 3B:A.3B.1-A.3B.2. [DOI] [PubMed] [Google Scholar]
- 20. O’Toole GA. Microtiter dish biofilm formation assay. J Vis Exp 2011: pii=2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Howard JJ, Sturge CR, Moustafa DA. et al. Inhibition of Pseudomonas aeruginosa by peptide-conjugated phosphorodiamidate morpholino oligomers. Antimicrob Agents Chemother 2017; 61: e01938-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Geller BL, Marshall-Batty K, Schnell FJ. et al. Gene-silencing antisense oligomers inhibit Acinetobacter growth in vitro and in vivo. J Infect Dis 2013; 208: 1553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deere J, Iversen P, Geller BL.. Antisense phosphorodiamidate morpholino oligomer length and target position effects on gene-specific inhibition in Escherichia coli. Antimicrob Agents Chemother 2005; 49: 249–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Summerton JE. Morpholino, siRNA, and S-DNA compared: impact of structure and mechanism of action on off-target effects and sequence specificity. Curr Top Med Chem 2007; 7: 651–60. [DOI] [PubMed] [Google Scholar]
- 25. Daly SM, Sturge CR, Felder-Scott CF. et al. MCR-1 inhibition with peptide-conjugated phosphorodiamidate morpholino oligomers restores sensitivity to polymyxin in Escherichia coli. MBio 2017; 8: e01315-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sully EK, Geller BL, Li L. et al. Peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO) restores carbapenem susceptibility to NDM-1-positive pathogens in vitro and in vivo. J Antimicrob Chemother 2017; 72: 782–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bai H, You Y, Yan H. et al. Antisense inhibition of gene expression and growth in gram-negative bacteria by cell-penetrating peptide conjugates of peptide nucleic acids targeted to rpoD gene. Biomaterials 2012; 33: 659–67. [DOI] [PubMed] [Google Scholar]
- 28. Kurupati P, Tan KS, Kumarasinghe G. et al. Inhibition of gene expression and growth by antisense peptide nucleic acids in a multiresistant β-lactamase-producing Klebsiella pneumoniae strain. Antimicrob Agents Chemother 2007; 51: 805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goh S, Boberek JM, Nakashima N. et al. Concurrent growth rate and transcript analyses reveal essential gene stringency in Escherichia coli. PLoS One 2009; 4: e6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anderl JN, Franklin MJ, Stewart PS.. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob Agents Chemother 2000; 44: 1818–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Singh R, Sahore S, Kaur P. et al. Penetration barrier contributes to bacterial biofilm-associated resistance against only select antibiotics, and exhibits genus-, strain- and antibiotic-specific differences. Pathog Dis 2016; 74: pii=ftw056. [DOI] [PubMed] [Google Scholar]
- 32. Zahller J, Stewart PS.. Transmission electron microscopic study of antibiotic action on Klebsiella pneumoniae biofilm. Antimicrob Agents Chemother 2002; 46: 2679–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mondhe M, Chessher A, Goh S. et al. Species-selective killing of bacteria by antimicrobial peptide-PNAs. PLoS One 2014; 9: e89082. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.