

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is caused by mutations in either PKD1 or PKD2. It is one of the most common heritable human diseases with eventual development of renal failure; however, effective treatment is lacking. While inhibition of mechanistic target of rapamycin (mTOR) effectively slows cyst expansions in animal models, results from clinical studies are controversial, prompting further mechanistic studies of mTOR-based therapy. Here, we aim to establish autophagy, a downstream pathway of mTOR, as a new therapeutic target for PKD. We generated zebrafish mutants for pkd1 and noted cystic kidney and mTOR activation in pkd1a mutants, suggesting a conserved ADPKD model. Further assessment of the mutants revealed impaired autophagic flux, which was conserved in kidney epithelial cells derived from both Pkd1-null mice and ADPKD patients. We found that inhibition of autophagy by knocking down the core autophagy protein Atg5 promotes cystogenesis, while activation of autophagy using a specific inducer Beclin-1 peptide ameliorates cysts in the pkd1a model. Treatment with compound autophagy activators, including mTOR-dependent rapamycin as well as mTOR-independent carbamazepine and minoxidil, markedly attenuated cyst formation and restored kidney function. Finally, we showed that combination treatment with low doses of rapamycin and carbamazepine was able to attenuate cyst formation as effectively as a single treatment with a high dose of rapamycin alone. In summary, our results suggested a modifying effect of autophagy on ADPKD, established autophagy activation as a novel therapy for ADPKD, and presented zebrafish as an efficient vertebrate model for developing PKD therapeutic strategies.
Introduction
Polycystic kidney diseases (PKD) are a group of diseases that can be categorized into autosomal dominant PKD (ADPKD), autosomal recessive PKD (ARPKD) and other syndromic forms such as Meckel syndrome (1). ADPKD is caused by mutations in either PKD1 or PKD2. It is one of the leading life-threatening genetic diseases, affecting approximately 0.2% of the population regardless of sex, age, race, or ethnicity. Patients with PKD often develop renal failure and so require dialysis or a kidney transplant (2). The vasopressin receptor antagonist, tolvaptan, is approved recently for treatment of ADPKD in Japan, Canada and Europe, underscoring the feasibility of discovering novel therapeutic strategies via mechanistic studies of pathologic signalling pathways.
mTOR inhibition has been pursued as a candidate therapy for PKD. mTOR is a key component of two protein complexes: mTORC1 and mTORC2. While mTORC1 regulates protein translation via phosphorylation of 4E-BP, cell growth via phosphorylation of S6K and autophagy via phosphorylation of ULK1 and ATG13, mTORC2 controls cytoskeletal organization via phosphorylation of Akt (3–7). Mounting evidence suggests that the mTOR pathway plays an important role in cystogenesis. First, activation of the mTOR pathway contributes to PKD development. It has been shown that patients and animal models with mutations in TSC1 and TSC2, two upstream inhibitors of mTORC1, develop cysts. Second, activation of the mTOR pathway, including phosphorylation of mTOR and S6K, has been found in cells lining cysts in PKD patients and animal models (8–12). Third, inhibition of mTOR via rapamycin and its analogues, which bind to FKBP12 to inhibit mTOR activity, effectively attenuates cystic phenotypes in animal models (8,13–17). Unfortunately, these mTOR inhibitors failed to show the protective benefits in clinical trials, likely due to low target doses because human patients are less tolerant to rapamycin’s renal toxicity and non-renal side effects (18,19). While kidney-targeted delivery of folate-conjugated rapamycin may overcome the non-renal adverse effects, it may not help reduce the renal toxicity, given that the mTOR pathway is involved in multiple cellular functions (20).
Autophagy is a crucial downstream signalling branch of mTOR. It is a multi-step process, including autophagosome formation, in which a double-membrane structure of reticular origin sequesters cytoplasmic components, and autophagosome fusion with the lysosome to form the autolysosome, in which engulfed cargo is broken down by lysosome-derived acid hydrolases. Autophagy ensures that cells survive starvation and stress by degrading long-lived proteins and organelles for recycling. It is also responsible for the clearance of mutated/misfolded proteins (21–24). Deficiency of autophagy has been demonstrated to be a contributing factor for neurodegenerative disease development, cancer initiation and cardiac diseases; and induction of autophagy has shown therapeutic benefits for these conditions (25–28). Recent studies have preliminarily implicated autophagy in PKD (29). Cy/Cy Han:SPRD rat and congenital polycystic kidney (cpk) mouse exhibit increased expression of autophagosome marker LC3II, with the increase in cpk mouse likely resulting from a decreased autophagosome-lysosome fusion; Pkd1 knockout mouse embryonic fibroblasts fail to activate LC3II expression in response to glucose deprivation unlike wild type cells (30,31). However, the precise role of autophagy in the pathogenesis of PKD and whether this signalling branch can be leveraged for therapeutic benefits remains largely unknown.
The zebrafish has proven to be a relevant model to study PKD. The embryonic zebrafish kidney has conserved nephron segment organization and shares the same inductive events, signalling cascades, differentiation and morphogenesis process as the metanephric kidney in mammals (32,33). Mutations in genes associated with human PKD lead to cystogenesis in zebrafish (34–37), and stable zebrafish mutants can be efficiently generated via transcription activator-like effector nucleases (TALEN) technology (38–40). In addition, disease progression can be easily monitored owing to the translucent nature of zebrafish embryos, while the small body size enables drug treatment in a 24-well plate, presenting an excellent platform for small-molecule screening to identify compounds for potential therapies for PKD.
Here, we report the generation of a zebrafish ADPKD model by TALEN technology. Having confirmed the defective autophagy regulation, we demonstrated a beneficial effect of autophagy activation on cystogenesis. In addition, we found that rapamycin can be used at lower doses to achieve drug efficacy when applied together with carbamazepine, a US Food and Drug Administration (FDA)-approved drug and an mTOR-independent autophagy activator, suggesting a combinatorial strategy for ADPKD therapeutics.
Results
pkd1 deficient zebrafish embryos develop fully penetrant pronephric cysts
Mutations in PKD1 account for 85% of ADPKD cases; we therefore decided to generate a corresponding zebrafish model. Two orthologues of mammalian PKD1, pkd1a and pkd1b, have been identified in zebrafish and are expressed in multiple tissues, including head, trunk and lymph vessel (41,42). Their expression was also detected in the pronephric kidney (Supplementary Material, Fig. S1A and B). We generated stable pkd1a and pkd1b mutants using TALEN technology (38–40). Exon 7 of pkd1a and exon 9 of pkd1b were targeted, which correspond to exon 17 of mouse Pkd1 where PKD-causative mutations have been reported (43). TALEN pair for pkd1a was targeted to a SphI restriction enzyme cutting site (Fig. 1A). By screening TALEN-injected fish for loss of the SphI site, followed by sequencing, we identified a number of mutated alleles (Supplementary Material, Fig. S2A and data not shown). Two truncation alleles (M1 and M2) that cause a coding frameshift of the pkd1a gene were maintained for our study. The M1 allele has 4-bp deletion and results in a premature stop codon after coding 127 frameshifted amino acids; the M2 allele has a 10-bp deletion and encounters a stop codon after coding 127 frameshifted amino acids (Fig. 1A). Consistent with the notion that mRNAs containing premature stop codons are often degraded by nonsense-mediated mRNA decay, we observed reduction of pkd1a transcripts (Supplementary Material, Fig. S2B) (44). Because the two alleles gave rise to the same phenotypes, we only presented data from the M1 allele.
Figure 1.

Homozygous pkd1 TALEN mutants develop kidney cysts. (A) Schematic diagram of exon-intron structure and recognition sequences (underlined) by TALEN. Because the targeted genomic locus is repaired via a non-homologous recombination mechanism, truncation mutants are typically generated via TALEN owing to the shifted reading frame. Shown are two deletion alleles (M1 and M2) that cause a coding frameshift and premature stop codon (*). Deletions are indicated by dashes. Shown below the DNA sequence is the corresponding amino acid sequence. (B) The glomerulus-neck region of the pronephron was dilated in pkd1a-/- embryos (asterisks) when compared with wild-type siblings (pkd1a+/+). Shown is HE staining on JB-4 sections of day 3 embryos. (C) The pronephric tubule was dilated in pkd1a-/- embryos. Frozen sections of day 5 embryos were immunostained using atypical PKC (red) and Na+/K+ ATPase (α6F, green) antibodies. Blue: DAPI. Scale bar: 20 μm. (D) The pkd1a-/- embryos developed edema at 5 dpf. (E,F) Fluid excretion function of the kidney was impaired in the pkd1a-/- embryos. Using rhodamine-dextran (10 kD) as a fluid tracer, the fluorescence dye was injected into 2.5-day-old embryos and inspected at the cloaca (dashed circle) of embryos (E). Individual embryos were genotyped after the experiment. Quantification of pronephric fluid flow is shown in (F). (G) Cardiac function, as assessed by measuring the shortening fraction at 2.5 dpf, was normal in the pkd1a-/- embryos when compared with pkd1a+/+ fish. (H) pkd1a-/-;pkd1b-/- embryos formed pronephric cysts earlier than pkd1a-/- embryos. Cyst formation in day 2 and day 3 embryos was quantified by HE staining. Data are presented as means ± s.d. from three independent experiments. Eight to ten embryos per genotype were examined in each experiment. *: P < 0.05. **: P < 0.01. NS: not statistically significant (P > 0.05) (F-H). nc: notochord.
Homozygous pkd1a embryos developed pronephric cysts starting at 2 days post fertilization (dpf) at a low frequency, reaching 93% penetrance at 3 dpf, visualized as dilation of the glomerulus-neck region of the nephron in hematoxylin-eosin (HE) sections (Fig. 1B and H). The pronephric tubules were also progressively dilated in the mutants, with dilation consistently detected at 5 dpf by double immunostaining with anti-Na+/K+-ATPase (labelling the basolateral surface of the pronephric tubules) and anti-atypical PKC (recognizing the apical side of the pronephric tubules) (Fig. 1C). Notably, the apicobasolateral polarity of the tubular epithelial cells was not grossly altered in the mutants (Fig. 1C). All mutants exhibited edema at 5 dpf, which progressed to all tissues, resulting in death around 13 dpf (Fig. 1D and data not shown). To assess whether kidney function was affected, we examined fluid excretion from the kidney using a fluorescent dye injection assay (35,45). While rhodamine-dextran (10 kD) was observed at the cloaca of 95% of wild type embryos at 2.5 dpf, it was detected in only 45% of mutants (Fig. 1E and F). The kidney defect seems not secondary to cardiac edema because cardiac function, as reflected by shortening fraction, was normal at 2.5 dpf in the mutants but declined at 4 dpf (Fig. 1G and data not shown).
A previously identified pkd1ahu5855 mutant exhibited defects in lymphatic development (42). Consistently, we observed normal lymphatic precursor sprouting at 2.5 dpf and reduced lymphatic vessel morphogenesis at 4 dpf in our pkd1a-/- embryos by introducing the Tg(fil1:GFP) allele into the mutants (Supplementary Material, Fig. S2C and data not shown) (42). The onset of the lymphatic phenotype suggests that renal cysts are not secondary to the lymphatic defects but edema might be. In addition, the mutant embryos showed normal kidney pattern formation when pax2a was used as a marker (data not shown), excluding abnormal development as a source of cystogenesis. The pkd1a-/-embryos also displayed shorter jaw defects at 5 dpf as seen in pkd1a morphants (Supplementary Material, Fig. S2D and E) (41). Heterozygous pkd1a mutants were phenotypically normal and fertile for at least 12 months of age.
TALEN pair for pkd1b was targeted to an MfeI site in exon 9. Similarly, two truncation alleles of pkd1b were obtained (Supplementary Material, Fig. S3A and B). Homozygous pkd1b embryos, either the M1 or the M2 allele, did not show any visually noticeable phenotypes, and the data presented here were form the M2 allele (Supplementary Material, Fig. S3C). Quantitative PCR analysis showed a significant increase of pkd1a transcripts in the pkd1b-/-embryos, suggesting genetic compensation by pkd1a (Supplementary Material, Fig. S3D). Conversely, a significant increase of pkd1b transcripts was found in the pkd1a-/-embryos (Supplementary Material, Fig. S2B). To determine whether pkd1a and pkd1b have redundant functions, we generated pkd1a/b double mutants. In contrast to the 11% of pronephric cysts observed in pkd1a-/-embryos at 2 dpf, cystic dilation was found in 70% of pkd1a-/-;pkd1b-/-embryos, and at 3 dpf, cystogenesis was seen in all double mutants (Fig. 1H). Of note, the pkd1a-/-;pkd1b-/-embryos did not exhibit dorsal curvature and hydrocephalus, as reported in the pkd1a/b double morphants (41). This is unlikely due to the pkd1a or pkd1b mutants being hypomorphic because alternative splicing events resulting in skipping of the targeted exons were not detected (Supplementary Material, Fig. S4A and B). Taken together, our data reveal that pkd1a is the major isoform of PKD1 in zebrafish kidney, the depletion of which results in pronephric cysts. By contrast, pkd1b plays a redundant role, the depletion of which facilitates pkd1a-governed cystogenesis.
Pkd1 deficiency results in dysregulation of autophagy
We assessed the mTOR pathway in the pkd1a model and found increased phosphorylation of mTOR and S6 ribosomal proteins using whole embryo lysates (Fig. 2A and B). Additionally, phospho-S6 protein staining was specifically enhanced in the pronephric tubules of the mutants at 5 dpf (Fig. 2C). Activation of the mTOR pathway in the pkd1a mutants suggested a conserved mechanism of PKD pathogenesis from zebrafish to mammals.
Figure 2.

pkd1a-/- mutants show aberrant mTOR activation and dysfunctional autophagy. (A,B) Phospho-mTOR and -S6 proteins were increased in pkd1a-/- embryos. Embryo lysates from day 5 animals were analysed by Western blotting (A). The intensity of phospho-mTOR and -S6 bands was quantified and presented as means ± s.d. (B) (C) Phospho-S6 was specifically induced in the pronephric tubules of the pkd1a-/- embryos. Frozen sections of day 5 animals were immunostained using phospho-S6 ribosomal protein (red) and Na+/K+ ATPase (α6F, green) antibodies. Blue: DAPI. Scale bar: 20 μm. (D,E) LC3II levels in the pkd1a-/- embryos were not increased as effectively as those in siblings in response to BafA1 treatment. Embryo lysates from day 5 fish treated with vehicle or 167 nM BafA1 for 16 h were subjected to Western blot analysis. Actin was used as a loading control. The gel is representative of three independent experiments (D). The intensity of LC3II bands was quantified and presented as means ± s.d. (E). (F) Ubiquitin (Ub)-conjugated proteins were moderately increased in detergent-insoluble fractions of pkd1a-/- embryo lysates. The pkd1a-/- fish and siblings at 5 dpf were subjected to cell fractionation, and the resulting total lysates (T), soluble fractions (S), and insoluble fractions (P) analysed by Western blot using an Ub antibody. The blot is representative of three independent experiments. (G,H) Autophagosomes (arrows) in the pronephric cells of 5 dpf fish were evaluated by electron microscopy in the presence of BafA1. Shown are representative images (G) and quantification of autophagosome numbers per cell (H). A total of 100 cells from three embryos per group were scored. Scale bar: 1 μm. **: P < 0.01. NS: not statistically significant (P > 0.05).
Given that autophagy has been demonstrated to mediate mTOR functions in diseases such as neurodegenerative disorders (46–49), we measured autophagy marker LC3II, an autophagosome-associated, active form of LC3 that is converted from LC3I (50). The basal level of LC3II was not significantly altered in the pkd1a mutants at 5 dpf, but treatment with bafilomycin A1 (BafA1), a lysosomal inhibitor that prevents fusion of autophagosome with the lysosome, failed to increase LC3II levels in the mutants as efficiently as in the wild-type siblings, suggesting defects in autophagic flux (Fig. 2D and E) (50,51). Concurrently, the number of autophagosomes in the pronephric tubules of the mutants did not increase to the same extent as that of their siblings after BafA1 treatment (Fig. 2G and H). We then assessed whether the autophagic clearance of misfolded or aggregated proteins was affected because such proteins have been shown to accumulate in cells deficient of the autophagy pathway, including kidney epithelial cells (52–54). Protein aggregates are commonly tagged by ubiquitin, with those in the detergent insoluble fraction usually recognized and degraded by the autophagy-lysosome pathway and those in the soluble fraction usually eliminated by the proteasome system (28,52,55–57). We found more ubiquitinated protein aggregates in the pellets of the pkd1a mutants than in the wild type siblings (Fig. 2F), supporting the notion of impaired autophagy.
To better understand autophagy modulation during disease progression, we performed a time-course study. In addition to the insufficient autophagic flux at 5 dpf, when cystogenesis already reached full penetrance and edema developed, we chose to also assess autophagy at 2 dpf when pronephric cysts began to form in a small percentage of embryos and 8 dpf when severe edema was present in all tissues. At 2 dpf, LC3II proteins in the pkd1a-/- and wild type siblings were not significantly different either at the steady state or after BafA1 treatment. At 8 dpf, the mutants had significantly lower steady state LC3II levels and inadequate response to BafA1 treatment when compared with controls, suggesting a reduced autophagosome formation (Supplementary Material, Fig. S5A and B). In addition to LC3II, we examined p62/SQSTM1, which brings ubiquitinated substrates to the autophagic machinery by acting as a linker between LC3II and ubiquitin-tagged proteins, and therefore serves as a marker for autophagic degradation (23,58,59). Steady state levels of this protein were enhanced in the mutants, moderately at 2 dpf and significantly at 5 dpf and 8 dpf. Upon treatment with BafA1, p62 was similarly up-regulated at 2 dpf, but less efficiently increased at 5 dpf and 8 dpf in the mutants when compared with controls (Supplementary Material, Fig. S5C and D). Taken together, autophagy appeared to be dynamically impaired during disease progression in the zebrafish pkd1a-/- model, from nearly normal at 2 dpf to decreased autophagic flow at 5 dpf and to reduced autophagosome formation at 8 dpf.
Mammalian PKD1-null kidney epithelial cells exhibit multiple autophagic defects
To validate our findings in the zebrafish model, we used kidney epithelial cells isolated from E14.5 Pkd1del2/del2 (Pkd1-/-) and wild type (Pkd1+/+) mouse embryos. Pkd1-/-kidneys are probably only mildly cystic at this stage (60). Pkd1-/- cells displayed a significant increase in the LC3II protein level (Fig. 3A and B). This increase was unlikely attributable to autophagy activation, because the cells did not show decreased levels of p62 protein (data not shown), and did not accumulate LC3II as effectively as wild type cells in response to BafA1 treatment (Fig. 3A and B). Consistently, the Pkd1-/- cells contained more LC3-positive puncta in the absence of BafA1 and less LC3-positive puncta in the presence of BafA1 when compared with wild type cells (Supplementary Material, Fig. S6A and B). These observations suggest that an insufficient autophagic flux rather than enhanced autophagy underlies basal levels of LC3II in these Pkd1-deficient cells.
Figure 3.

Autophagy is dysregulated in mammalian PKD1-null kidney epithelial cells. (A,B) Kidney epithelial cells derived from Pkd1-null mouse embryos (Pkd1-/-) had higher basal LC3II levels but failed to significantly further increase LC3II expression after BafA1 treatment, compared with cells obtained from wild-type (wt) mouse embryos. (C,D) 9-12 cells isolated from ADPKD patients (PKD1-/-) had a lower LC3II expression before and after BafA1 treatment, compared with RCTE cells extracted from normal renal cortical tubular epithelia (wt). Cells were grown on plates for 24 h, incubated with 167 nM BafA1 or vehicle for 2 h, and then collected for LC3II analysis by Western blot (A,C) and normalization by Actin expression (B,D). (E,F) Human PKD1-/- cells contained fewer autophagosomes and autolysosomes, compared with wt cells. Cells were transfected with pBABE-puro mCherry-EGFP-LC3B plasmid and collected 24 h later for confocal microscopy imaging. Yellow puncta indicate autophagosomes and red dots indicate autolysosomes (E). Numbers of autophagosomes and autolysosomes per cell are quantified (F). (G,H) Human PKD1-/- cells accumulated more aggresomes, compared with wt cells. Cells were incubated with MG132 for 13 h to induce protein aggregation (left panels) and then returned to the normal growth condition for 11 h to allow clearance of the aggregates (right panels). Aggresomes (red) were detected using the ProteoStat Aggresome Detection Kit (G) and quantified (H). (I) Ub-conjugated proteins were accumulated in the detergent-insoluble fraction of human PKD1-/- cells. Cells were collected in the absence (lanes 1 and 2) or presence (lanes 3 and 4) of MG132 for 13 h, or 11 h after removal of MG132 (lanes 5 and 6). Insoluble fractions of cell lysates were subjected to Western blot analysis using an Ub antibody. (J) Ub-conjugated proteins were accumulated in detergent-insoluble fractions of mouse Pkd1-/- cells. The gel is representative of six (A,C) or three (I,J) independent experiments. Data are presented as means ± s.d. from six independent experiments (B,D) or from approximately 50-100 cells per group (F,H). *: P < 0.05. **: P < 0.01. NS: not statistically significant (P > 0.05). Blue: DAPI (E,G).
We further analysed the autophagy process using cyst-lining epithelial cells derived from an ADPKD patient (9–12; PKD1-/-) and cells isolated from normal human renal cortical tubule epithelium (RCTE; PKD1+/+) (61–63). LC3II levels were drastically reduced at steady state and poorly increased after BafA1 treatment in the PKD1-/- cells (Fig. 3C and D). To quantitatively study defects in autophagic progression, we counted numbers of the autophagosomes and autolysosomes via transfection of the pBABE-puro mCherry-EGFP-LC3B plasmid into the cells. In the autophagosome, the combination of both mCherry and EGFP shows yellow, while in the autolysosome, EGFP fluorescence is quenched by the acidic environment and thus only red is seen (64). We found that the number of both autophagosomes and autolysosomes in PKD1-/- cells was significantly decreased, and moreover, the number of autophagosomes failed to be up-regulated in response to BafA1 treatment when compared to controls (Fig. 3E and F), suggesting an inadequate autophagosome formation and diminished autophagosome-lysosome fusion. While human PKD1-/-cells shared the same deficiency in the autophagic flow with the mouse Pkd1-/- cells, they displayed an additional defect in the formation of autophagosomes. This difference could be attributed to several possibilities, such as species difference, subtle differences in cell type and SV40 expression though the mouse and human cells were both collecting duct origin and immortalized by SV40 large T antigen, or different stages of disease when either mouse or human cells were isolated, with the former from mouse embryos being mildly cystic and the latter from end-stage ADPKD patients (60–63). Given that the zebrafish pkd1a mutants exhibited an insufficient autophagic flux at 5 dpf and inadequate autophagosome formation at 8 dpf, we favour the hypothesis that disease severity underlies the severity of autophagy dysfunction.
To confirm the role of PKD1 in autophagy regulation, the PKD1-null cells were examined for clearance of protein aggregates. We first treated cells with MG132, a protease inhibitor, to induce aggresome formation. Using ProteoStat dye, a red fluorescent molecule that specifically binds to aggregated proteins within aggresomes, similar red puncta were detected at perinuclear sites of both the PKD1-/-and PKD1+/+ cells (Fig. 3G and H). When MG132 was withdrawn to allow degradation of the aggregates, red puncta were effectively eliminated from the wild-type cells, but they remained in the PKD1-/- cells (Fig. 3G and H). To test whether autophagy impairment accounts for the weakened ability to remove protein aggregates, we assayed ubiquitinated protein aggregates in the detergent-insoluble fraction. PKD1-/- cells accumulated more aggregates in the absence of any stress, and moreover, less efficiently broke down MG132-induced protein aggregation when compared with controls (Fig. 3I). Mouse Pkd1-/-cells showed a similar deficiency in the clearance of protein aggregates (Fig. 3J and data not shown). Combining our data from zebrafish and mammalian cells, we concluded that PKD1 deficiency results in multiple types of autophagy dysfunction.
Defective autophagy promotes cystogenesis in pkd1a-/- embryos
To pinpoint the role of autophagy in the pathogenesis of PKD, we examined cyst formation in embryos deficient for the core autophagy protein Atg5 (autophagy-related gene 5). Injection of atg5 morpholino (MO) resulted in multiple phenotypes, including loss of brain tissues, cardiac edema and twisted body as previously described (Fig. 4A) (65,66). Among these, cardiac defects and body curvature were most prominent, occurring in over 90% of injected embryos. These morphants exhibited cystic distention at 2 dpf, and severe cardiac edema prevented evaluation at later developmental stages (Fig. 4A’). Because cardiac edema occurs as early as day 1, it could well be the primary source of cystic distention. To prevent this complication, MO amount was titrated to eliminate apparent and early cardiac defects. We found that 1 ng of MO caused approximately 10% of morphants to show mild phenotypes, i.e., body curvature at 2 dpf and small pronephric cysts at 5 dpf (Fig. 4B and B’), suggesting that dysfunctional autophagy is less likely a causative event for overt cystogenesis.
Figure 4.

Knockdown of Atg5 promotes cyst formation in pkd1a-/- embryos. (A,A’) Injection of 4 ng of atg5 MO into one-cell staged embryos resulted in multiple defects, including loss of brain tissues, cardiac edema, and the body curvature (A, arrowhead). Cystic distension of the pronephric tubules (268/285) was observed in enlarged images (A’, arrow). Shown are lateral (A) and dorsal (A’) views of a day 2 embryo. (B,B’) Injection of 1 ng of atg5 MO led to body curvature (B, arrowhead) and small pronephric cysts (B’, arrow) in 19 out of 206 injected embryos. Shown is an embryo at 5 dpf. (C) Injection of 1 ng of atg5 MO increased the number of pkd1a-/- embryos with kidney cysts. MO was injected into embryos derived from heterozygous pkd1a inter-crosses. At 2 dpf, morphants displaying curly body phenotypes were discarded and normal-looking morphants were fixed in 4% formaldehyde, with the posterior tail collected for genotyping. Cyst formation was assessed by HE staining on JB-4 sections. Data are presented as means ± s.d. from three independent experiments. Ten to fifteen embryos per genotype were analysed in each experiment. *: P < 0.05.
We then asked whether Atg5 knockdown facilitates cystogenesis in the pkd1a-/- embryos. To be able to detect the acceleration of cyst formation, we injected the low-dose of MO into offspring of pkd1a+/- inter-crosses and collected normal looking embryos (not those with a curly body) at 2 dpf, a time point when only a small percentage of pkd1a-/- embryos developed pronephric cysts. We found that MO injection significantly increased the percentage of pkd1a-/- embryos with cysts from 10% to 48%. Meanwhile, wild type embryos injected with the MO did not display any pronephric cysts (Fig. 4C). Thus, Atg5 knockdown promotes pkd1a depletion-induced cystogenesis.
Induction of autophagy suppresses cyst formation and restores kidney function in pkd1a-/- embryos
The cyst-promoting effect of autophagy deficiency prompted us to hypothesize that autophagy induction might have a therapeutic benefit for PKD. To test this hypothesis, we first chose to activate autophagy using a specific autophagy inducer. A short peptide of the core autophagy protein Beclin 1 (amino acids 267-284) has been shown to specifically activate autophagy via binding to the autophagy inhibitor GAPR-1, and becomes cell-permeable by conjugation to HIV Tat protein transduction domain (67). As expected, the Tat-Beclin 1 treatment significantly increased LC3II levels in both the PKD1+/+ and PKD1-/-cells and effectively cleared accumulation of protein aggregates in the PKD1-/- cells (data not shown). Importantly, incubation with the Tat-Beclin 1 peptide enhanced LC3II levels in the wild type and pkd1a-/- embryos (Fig. 5A and B). To be able to test attenuation of cyst formation, embryos were treated with the Tat-Beclin 1 peptide for 16 h and collected at 3 dpf, a time point when >90% of pkd1a mutants displayed pronephric cysts. We found that administration of the Tat-Beclin1 peptide reduced cyst formation in the pkd1a mutants from 94% to 25% (Fig. 5C). Treatment with the peptide also restored kidney function, as reflected by an increase of fluorescent dye excretion from the pronephric tubule of mutant embryos from 58% to 95% (Fig. 5D). These results suggested the induction of autophagy as a potential therapeutic intervention for cystogenesis.
Figure 5.
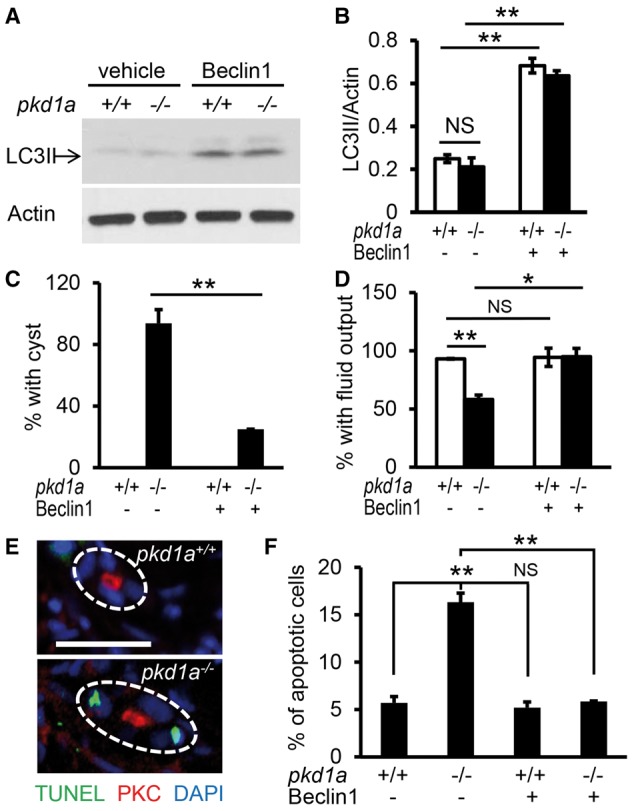
Autophagy induction by the Tat-Beclin 1 peptide suppresses cystogenesis and rescues kidney function in pkd1a-/- embryos. (A,B) Tat-Beclin 1 peptide induced autophagy in zebrafish embryos. Tat-Beclin-1 peptide (100 nM) was incubated with embryos at 2.5 dpf for 16 h, and Western blot analysis was performed using whole embryo lysates (A). Quantification of LC3II expression was presented as means ± s.d. from three independent experiments (B). (C) Tat-Beclin 1 peptide inhibited pronephric cyst formation in the pkd1a-/- embryos. Embryos derived from heterozygous pkd1a inter-crosses were incubated with the peptide (100 nM) at 2.5 dpf for 16 h, and then subjected to JB-4 embedding and HE staining. The percentage of embryos with kidney cysts was scored. (D) The Tat-Beclin 1 peptide rescued kidney excretion function in the pkd1a-/- embryos. Embryos at 2.5 dpf were treated with the peptide for 16 h, and then injected with rhodamine-dextran dye for assessment of kidney fluid flow. Individual embryos were genotyped at the end of the experiment. Data are presented as means ± s.d. from three independent experiments, 8-15 embryos per group per time were examined (C,D). (E) pkd1a mutants exhibited increased apoptosis in the pronephric kidney. Apoptotic cell death (green) was analysed by TUNEL assay on frozen sections of day 3 embryos, followed by immunostaining using atypical PKC (red) antibody to mark pronephric tubules. Blue: DAPI. Scale bar: 20 μm. (F) The Tat-Beclin 1 peptide decreased apoptotic cell death in the pkd1a-/- embryos. Embryos at 2.5 dpf were treated with the peptide for 16 h, and then processed for apoptosis analysis by the TUNEL assay. Quantification of apoptosis was expressed as the percentage of TUNEL-positive cells in total kidney epithelial cells examined, 80-100 cells from 4 embryos per group were scored. *: P < 0.05. **: P < 0.01. NS: not statistically significant (P > 0.05).
PKD patients and most models of PKD are characterized by an increased rate of apoptosis in the kidney epithelial cells (8,68–70). This feature was observed in the pkd1a model, as evidenced by more TUNEL-positive kidney epithelial cells in the mutants than in the wild type siblings (Fig. 5E and F). Because extensive crosstalk occurs between autophagy and apoptosis, we explored whether apoptosis regulation underlies the beneficial effect of the Tat-Beclin 1 peptide (71,72). Treatment with the peptide reduced numbers of TUNEL-positive pronephric epithelial cells in pkd1a-/- embryos to the level comparable to those in wild type siblings (Fig. 5F), suggesting that the Tat-Beclin 1 peptide ameliorates pronephric cysts, at least in part, through inhibition of apoptosis in the pkd1a model .
Both mTOR-dependent and mTOR-independent small molecule activators of autophagy exert therapeutic benefits in pkd1a-/- embryos
We then tested several known autophagy activators in PKD therapy. Treatment with rapamycin, the best-characterized mTOR-dependent autophagy inducer, inhibited mTOR signalling and activated autophagy in fish embryos, as demonstrated by a decrease of phospho-ribosomal S6 and an increase of LC3II expression, respectively (Fig. 6A and B). Importantly, rapamycin treatment was able to reduce cyst formation in the pkd1a mutants from 93% to 38% and rescue dye excretion function of the kidney from 58% to 94% (Fig. 6C and D), indicating that the therapeutic benefit of rapamycin is conserved in zebrafish, and autophagy activation could be a contributing factor. We went on to assess the effect of mTOR-independent up-regulation of autophagy in cystogenesis. We elected to treat embryos with carbamazepine and minoxidil, two FDA-approved drugs. Carbamazepine is currently used as an anticonvulsant and stimulates autophagy by inhibiting inositol monophosphatase (IMPase), which leads to lower intracellular inositol levels and suppression of the phosphoinositol cycle, while minoxidil, a K + ATP channel opener, is clinically prescribed as an antihypertensive agent and activates autophagy by decreasing L-type Ca2+ channel currents, which initiates a cyclical mTOR-independent pathway, involving Ca2+–calpain–Gsα-cAMP–IP3 signalling (73,74). In fish embryos, these two compounds activate autophagy without affecting the mTOR pathway, as manifested by higher LC3II and normal phospho-ribosomal S6 levels (Fig. 6A and B). Similar to rapamycin treatment, we found that incubation with either of these two compounds suppressed cyst formation, and rescued kidney function in the mutants (Fig. 6C and D). Moreover, we assayed autophagy-mediated clearance of protein aggregation using the PKD1-/- cells. Rapamycin, carbamazepine and minoxidil modulated mTOR or autophagy signalling in the same manner as they did in the embryos (data not shown). They all promoted the removal of MG132-induced protein aggregation in the PKD1-/- cells (Fig. 6E). Together, our results suggest that autophagy activation in general, either mTOR dependently or independently, exerts therapeutic benefits on PKD.
Figure 6.

Autophagy activators in the mTOR-dependent or mTOR-independent pathways both ameliorate cystic phenotypes in pkd1a-/- embryos. (A,B) Rapamycin, carbamazepine, and minoxidil all enhanced LC3II expression while differentially affecting phospho-S6 levels. Zebrafish embryos were treated with DMSO (D), rapamycin (R, 400 nM), carbamazepine (C, 20 μM), or minoxidil (M, 400 nM) at 4 dpf for 16 h, and Western blot analysis was performed using whole embryo lysates. The gel is representative of three independent experiments (A), and LC3II levels were normalized by actin expression and presented as means ± s.d. (B). (C) Autophagy activators suppressed cystogenesis in the pkd1a-/- embryos. Embryos derived from heterozygous pkd1a inter-crosses were incubated with vehicle (D) or autophagy activators (R: 400 nM,; C: 20 μM; or M: 400 nM) at 2.5 dpf for 16 h. Percentage of embryos with kidney cysts was analysed by HE staining of JB-4 sections. Note: rapamycin, but not other drugs, led to cyst formation in 14% of wild type embryos. (D) Autophagy activators preserved kidney excretion function in the pkd1a-/- embryos. Embryos at 2.5 dpf were treated with vehicle (D) or autophagy activators (R, C, or M) for 16 h, and kidney fluid flow was analysed via dye injection. (E) Autophagy activators promoted the clearance of ubiquitinated protein aggregates. Human PKD1-/- cells were incubated with MG132 and 13 h later MG132 was removed and replaced with vehicle (D) or autophagy activators (R, C, or M) for 11 h. Insoluble fractions of the cell lysates were subjected to Western blot analysis using an Ub antibody. Shown is a representative blot of three independent experiments. (F) Low doses of rapamycin significantly alleviated cyst formation in the pkd1a-/- embryos when combined with low doses of carbamazepine. Embryos at 2.5 dpf were treated with 40 nM rapamycin (R), 2 μM carbamazepine (C), or 40 nM rapamycin plus 2 µM carbamazepine (R + C). Data are presented as means ± s.d. from three independent experiments, and 7 to24 embryos per group were scored in each experiment (C,D,F). *: P < 0.05. **: P < 0.01. NS: not statistically significant (P > 0.05).
Since mTOR-independent autophagy inducer carbamazepine or minoxidil also diminished cystogenesis, we speculated that the combination of lower doses of rapamycin with, for example, carbamazepine might have an additive or synergistic effect. A ten-fold reduction of rapamycin concentration to 40 nM had marginal efficacy, as 85% of the pkd1a mutants still displayed kidney cysts (Fig. 6F). This dose did not cause any abnormality in the pkd1a+/+ embryos, compared with 14% of wild type embryos showing cystic dilation after incubation with 400 nM of rapamycin (Fig. 6C and data not shown). Further reducing rapamycin to 4 nM resulted in no beneficial effect, nor side effects (data not shown). A ten-fold reduction of carbamazepine to 2 µM also exhibited marginal efficacy (Fig. 6F). However, a combination of 40 nM of rapamycin and 2 µM of carbamazepine suppressed cystogenesis to a level comparable to that of single use of 400 nM of rapamycin (42% vs 38%) (Fig. 6C and F). These data suggest a new direction in PKD therapy by combining low doses of mTOR-dependent and mTOR-independent autophagy activators to potentially reduce drug toxicity.
Discussion
pkd1 embryonic zebrafish model is amenable for functional studies of ADPKD and small-molecule screening
Using TALEN technology, we reported the generation of stable pkd1 zebrafish mutants. We were able to detect kidney cysts in 11% of pkd1a-/- and 70% of pkd1a-/-;pkd1b-/- mutants at 2 dpf via HE staining, with near fully penetrant phenotypes for both genotypes at 3 dpf. The frequency is higher than that of morpholino-mediated depletion, whereby 10–15% of pkd1a or pkd1a/1b morphants formed cystic dilations by visual observation at 2 dpf (41). Besides stable mutation vs transient depletion, different detection methods also might contribute to the difference in penetrance. For example, in our experience, microscopic inspection of pronephric dilation at 2 dpf was not reliable. The mutants and morphants shared additional phenotypes, such as shorter jaw and edema. However, the mutants did not recapitulate body curvature and hydrocephalus that were reported in the morphants. Lacking these phenotypes is less likely due to maternal expression of pkd1 in the mutants or TALEN targeting sites different from morpholinos because both translation-blocking and splice-donor blocking morpholinos caused the same abnormalities. In fact, recent studies have noted a poor correlation between morpholino-induced and mutant phenotypes (75–77). For instance, pkd2 zebrafish mutants fail to recapitulate cystic and hydrocephalus phenotypes of pkd2 morphants; pkd1ahu5855 mutants do not display body curvature phenotype as pkd1a morphants do (34,37,41,42,45). Several possibilities have been proposed to explain the discrepancy between mutants and morphants (77–79). Specifically for the pkd1 mutants, we did not detect a hypomorphic mutant allele due to exon skipping or genetic compensation by pkd2 (data not shown). Other potential explanations, including synthesis of a functional C-terminal domain using an alternative ATG downstream truncation site, genetic compensation by genes not belonging to pkd family, or off-target effects of morpholinos, need to be further investigated.
Given efficient approaches of TALEN, CRISPR/Cas9 and knock-in for mutant generation in zebrafish, this study highlights the fish model as a practicable platform for functional analysis of various mutations identified in human PKD1 (39,80–85). Embryonic zebrafish models are particularly attractive for drug screening because their small body size consumes much less drug and ensures low maintenance costs (86). Powerful genetics and higher housing capacity also render zebrafish embryos ideal for genetic modifier screens. Meanwhile, there are limitations associated with zebrafish embryonic models. For example, quantifiable methods of measuring cyst size and number in zebrafish embryos have yet to be developed; methods allowing kidney-specific analysis of molecular/cellular changes and physiological assessment need to be further established. Moreover, PKD is a progressive disease, with symptoms primarily occurring later in adult life (87). It is conceivable that the embryonic model might only recapitulate certain perspectives of PKD pathogenesis. Therefore, we do consider it essential to substantiate results obtained from embryonic fish models in either adult zebrafish or mammalian PKD models.
Autophagy defects are important pathological events during the pathogenesis of PKD
PKD is characterized by increased rates of both proliferation and apoptosis in epithelial cells lining the cyst (88,89). Here, we revealed autophagy dysfunction as an additional pathological event in PKD. Using the zebrafish pkd1 mutants, we detected sequential dynamic changes of autophagy during disease progression: nearly normal at 2 dpf, when cystogenesis is about to initiate, insufficient autophagic flux at 5 dpf, after cystogenesis had reached full penetrance, and eventually suppressed autophagosome formation at 8 dpf when severe edema was present in all tissues. This time-dependent autophagy modulation was partially reflected in mammalian PKD null kidney epithelial cells. While cells obtained from Pkd1 knockout mouse embryos with probably mild cysts manifest diminished autophagic flux, cells derived from late-stage ADPKD patient exhibit impairment in autophagosome formation (60,61). Together, our data suggest dynamic changes in autophagy during the development and progression of PKD, which require validation in the kidneys from slowly progressing PKD models that more closely mimic the human disease.
The dynamic modulation of autophagy implies that dysfunctional autophagy is secondary to PKD-associated events, such as mTOR activation and/or defective cilia signalling. While we and others did not detect alterations in the cilium length and number (data not shown) (41), changes in cilia-related signalling cannot be ruled out. In support of this hypothesis, there is no evidence indicating direct interaction of PC1 with components in the autophagy cascade. On the other hand, results from knocking down the core autophagy protein Atg5, under the condition that prevents earlier and severe cardiac edema, suggest that dysfunctional autophagy is not a causative event for cystogenesis, but rather a modifying mid-step, representing a failure in maintaining homeostasis or repairing damages. Strengthening this notion, no renal cysts have been reported in Atg5 or Atg7 KO mice up to 9 months (53,54). Our discovery advocates autophagy as an important area for future research to understand the pathogenesis of PKD. As has been recognized in other diseases, relationship among autophagy, proliferation, apoptosis and cilia signalling should be further elucidated (71,72,90,91).
Up-regulation of autophagy as a novel therapeutic strategy for PKD
Treatment with the specific autophagy inducer Beclin 1 peptide provides proof-of-principle evidence to support that activation of autophagy is of therapeutic benefit in PKD. This observation echoes the protective function of autophagy activation during acute kidney injury and podocyte aging (53,54,92–96). Then, small molecular activators of autophagy, including rapamycin, carbamazepine or minoxidil, all partially ameliorate kidney cysts and preserve kidney function. The phenomenon that cysts remained in some of the mutants following autophagy induction supports our conclusion that autophagy modulation is a modifying event for cystogenesis. The phenomenon could also be explained by the non-isogenic genetic background and lack of quantifiable methods of measuring cyst size and number in zebrafish embryos.
In addition to demonstrating autophagy activation as a novel therapeutic strategy for PKD, our data offer a potential explanation for the mechanisms underlying several other beneficial compounds for PKD. For instance, the AMPK activator metformin, HSP90 inhibitor STA-2842, and histone deacetylases (HDAC) inhibitor trichostatin A, have all been shown to slow renal cystogenesis in animal models, and have also been shown in separate studies as activators of autophagy in cultured cells (86,97–100). However, to firmly establish that these compounds execute their therapeutic effects through autophagy activation, evaluation of drug efficacy in autophagy-deficient PKD models will be indispensable. One beneficial effect of autophagy activation might be ascribed to the removal of aggregate-forming proteins. Future identification of aggregate-forming proteins and understanding of their roles in PKD pathogenesis will provide more insights into the molecular nature of PKD pathogenesis.
Future in vivo experiments are warranted to determine which autophagy activators exert better therapeutic effects than rapamycin while causing less renal and non-renal toxicity (18,19). Here, our studies suggest that lower doses of rapamycin, when combined with mTOR-independent autophagy inducer carbamazepine, can achieve efficacy comparable to that of using higher doses of rapamycin alone. This combination strategy may thus reduce the side effects associated with rapamycin, and be safer for long-term clinical use. Notably, carbamazepine, minoxidil and metformin are FDA-approved drugs that are used to treat seizures, hypertension and type 2 diabetes, respectively; HSP90 inhibitors and HDAC inhibitors are currently under clinical development for cancer therapy (101,102). Therefore, our present studies in zebrafish highlight the promising future of drug repurposing strategies in ADPKD therapy, which may also be beneficial for other PKD forms.
Materials and methods
Zebrafish husbandry
WIK fish were maintained under standard laboratory conditions. The animal experiments were approved by the Mayo Clinic College of Medicine Institutional Animal Care and Use Committee.
Generation of zebrafish pkd1a and pkd1b mutants by TALEN
TALENs were constructed using the Golden Gate TALEN assembly protocol and library (Addgene) (38). Fifteen pg of TALEN mRNAs were injected into wild type embryos at the one-cell stage. Sixteen embryos from each injection group were used for genomic DNA isolation to evaluate the efficiency of the TALENs by PCR-RFLP (restriction fragment length polymorphism). Embryos injected with efficient TALENs were raised to adulthood and outcrossed. Individual F1 adults were genotyped to identify germ line mutations, and F1carriers were then sequenced to search for frame-shift mutations. Two distinct frame-shift deletion alleles for each gene were selected. Selected F1 founders were further outcrossed to generate F2 fish in an effort to reduce potential off-target effects. All experiments performed in this study used F2 fish for breeding.
Cell culture
Mouse kidney epithelial cells were isolated from Pkd1+/+;H2kb-tsA58+ and Pkd1-/-;H2kb-tsA58+ mice at embryonic day (E)14.5 by interbreeding Pkd1+/del2 and transgenic H2kb-tsA58 mice (Immortomouse, Charles River) (60). Kidneys were placed in the cold dissection solution (10 mM glucose, 5 mM glycine, 1 mM alanine and 15 mM HEPES in Hanks buffered saline solution), transferred and resuspended in the digestion solution (0.1% collagenase D (Roche) and 0.125% hyaluronidase (Sigma-Aldrich) in Advanced DMEM/F12 (Invitrogen)), disrupted using 22 gauge needles and 1mL syringes, incubated at 37 °C for 30 min, and pulse vortexed once during incubation. Cells were pelleted by centrifugation at 1,000 g for 5 min, resuspended in Advanced DMEM/F12 containing 20% FBS, penicillin/streptomycin, glutamine, 0.1% plasmocin (Invivogen) and 10ng/mL recombinant mouse interferon gamma (Millipore), plated on collagen I coated 25 cm2 flasks, and incubated at 33 °C to induce transgene expression. The cells were plated at low density to allow clonal expansions, and collecting duct epithelial cells were obtained by screening for cell morphology and expression of E-cadherin and Aquaporin-2. Human cell lines were previously derived from normal renal cortical tubular epithelia (PKD1+/+) and ADPKD cyst-lining epithelia (PKD1–/–) (61,62). Human cell lines were transfected with the pBABE-puro mCherry-EGFP-LC3B plasmid (Addgene) on coverslips using Lipofectamine 2000 (Life Technologies) according to the manufacturer’s protocol. Twenty-four hours after transfection, cells were fixed with 4% formaldehyde for 10 min and mounted with Vectashield mounting medium (Vector Laboratories) for confocal microscopy imaging. Aggresome detection was performed using the ProteoStat Aggresome Detection Kit (Enzo Life Sciences, Inc.), following the manufacturer’s protocol.
Reagents
The following reagents were used: translation-blocking MO of atg5 (Gene Tools, LLC, 4 ng or 1 ng) (65), Tat-Beclin 1 peptide (AnaSpec, 100 nM), rapamycin (LC Laboratories, 400 nM or 40 nM), carbamazepine (Tocris, 20 µM or 2 µM), minoxidil (Sigma-Aldrich, 400 nM), bafilomycin A1 (Sigma-Aldrich, 167 nM) and MG-132 (Sigma-Aldrich, 5 µM).
Histological and ultrastructural analysis
Embryos for histological analysis were individually fixed in 4% formaldehyde at 2 dpf or 3 dpf, with the tail region collected for genotyping. On the next day, wild-type and mutant embryos were embedded in JB-4 resin (Polysciences, Inc.) and sectioned at 4 µm thickness. The sections were then stained with HE, dried and mounted with Permount (Fisher Scientific) (103). Embryos for ultrastructural analysis were fixed in Trump fixative (4% paraformaldehyde, 1% glutaraldehyde) at 5 dpf. The remaining procedure was performed according to standard methods by the Electron Microscopy Core Facility at Mayo Clinic in Rochester, Minnesota.
Immunofluorescence
Immunofluorescence labelling was performed on cryosectioned materials (104). Embryos were fixed in 4% formaldehyde overnight and embedded in 1.2% agarose/5% sucrose. The agarose blocks were then saturated in 30% sucrose overnight, transferred into a tissue freezing medium (Electron Microscopy Sciences), and frozen on a freeze platform. Then 10-µm sections were incubated with primary antibodies against phospho-S6 ribosomal protein (Cell Signaling Technology, 1:200) and α6F (Developmental Studies Hybridoma Bank, 1:25), followed by Alexa Fluor conjugated secondary antibodies (Life Technologies). Images were acquired using an Axioplan II Zeiss microscope equipped with ApoTome.TdT-UTP nick end labelling (TUNEL) assay was performed on frozen-sectioned day 3 embryos using In Situ Cell Death Detection Kit, following the manufacturer’s protocol (Roche).
Western blotting
Zebrafish embryos or cultured cells were homogenized in RIPA lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 2 mM EDTA, 1 mM phenylmethylsulphonyl fluoride and protease inhibitors) containing stainless steel beads using the Bullet Blender tissue homogenizer (Next Advance, Inc). Western blotting was performed according to standard protocols. The following antibodies were used: anti-mTOR, anti-phospho-mTOR (Ser2481), anti-S6 ribosomal protein, anti-phospho-S6 ribosomal protein (Ser235/236), anti-P62 (Cell Signaling Technology), anti-actin (Sigma-Aldrich), anti-LC3 (Novus Biologicals) and anti-ubiquitin (Thermo Scientific). For analysis requiring cell fractionation, embryos or cultured cells were homogenized in lysis buffer (100 mM HEPES, 1% Triton X-100, 300 mM NaCl, 1 mM phenylmethylsulphonyl fluoride and protease inhibitors), pre-cleared at 2,000 g, and then centrifuged at 12,000 g to separate extracts into soluble (S) and insoluble pellet (P) fractions. The isolated P fraction was further lysed by adding additional 0.5% SDS (52). 5% of the total lysate (T), 5% of the S fraction, and 20% of the P fraction were resolved on 4% to 20% gels.
Fluorescent dye injection
The pronephric fluid output assay was performed as previously described (35,45). Briefly, 2.5 dpf embryos that were anaesthetized were injected with 1% rhodamine-dextran (10 kD, Molecular Probes) via the common cardinal vein, and were then examined using a Zeiss Axioplan microscope equipped with a Nikon camera.
Measurement of shortening fraction (SF) of the ventricular chambers
SF was measured as previously described (105). Briefly, zebrafish embryos at 2.5 dpf were anaesthetized and positioned on a microscope slide with a thin layer of 3% methyl cellulose (Sigma-Aldrich). Videos of beating hearts from a lateral view were recorded using a Zeiss Axioplan microscope equipped with a Nikon camera and used for maximum diastole and maximum systole measurements. SF= (maximum diastole—maximum systole)/maximum diastole ×100.
Statistical analysis
All data were expressed as mean ± s.d. Comparison between two groups was performed by a two-tailed t test. A P value <0.05 was considered statistically significant.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We thank Bingquan Huang of the Mayo Clinic Electron Microscopy Core Facility for expert transmission electron microscopy assistance, Caroline Sussman for critical reading of the manuscript, and Diana Escobar, Katharina Hopp and Vladimir Gainullin for kindly providing us mouse and human cell lines.
Conflict of Interest statement. None declared.
Funding
This work was partially supported by the Mayo Translational Polycystic Kidney Disease Center (MTPC) Pilot and Feasibility grant (NIDDK DK90728) to X.L.
References
- 1. Harris P.C., Torres V.E. (2009) Polycystic kidney disease. Annu. Rev. Med., 60, 321–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Torres V.E., Harris P.C. (2014) Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J. Am. Soc. Nephrol., 25, 18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kim Y.C., Guan K.L. (2015) mTOR: a pharmacologic target for autophagy regulation. J. Clin. Invest., 125, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Laplante M., Sabatini D.M. (2012) mTOR signaling in growth control and disease. Cell, 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Laplante M., Sabatini D.M. (2009) mTOR signaling at a glance. J. Cell Sci., 122, 3589–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Torres V.E., Boletta A., Chapman A., Gattone V., Pei Y., Qian Q., Wallace D.P., Weimbs T., Wuthrich R.P. (2010) Prospects for mTOR inhibitor use in patients with polycystic kidney disease and hamartomatous diseases. Clin. J. Am. Soc. Nephrol., 5, 1312–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huber T.B., Walz G., Kuehn E.W. (2011) mTOR and rapamycin in the kidney: signaling and therapeutic implications beyond immunosuppression. Kidney Int., 79, 502–511. [DOI] [PubMed] [Google Scholar]
- 8. Shillingford J.M., Murcia N.S., Larson C.H., Low S.H., Hedgepeth R., Brown N., Flask C.A., Novick A.C., Goldfarb D.A., Kramer-Zucker A., et al. (2006) The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl Acad. Sci. U S A, 103, 5466–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brook-Carter P.T., Peral B., Ward C.J., Thompson P., Hughes J., Maheshwar M.M., Nellist M., Gamble V., Harris P.C., Sampson J.R. (1994) Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease–a contiguous gene syndrome. Nat. Genet., 8, 328–332. [DOI] [PubMed] [Google Scholar]
- 10. Zhou J., Brugarolas J., Parada L.F. (2009) Loss of Tsc1, but not Pten, in renal tubular cells causes polycystic kidney disease by activating mTORC1. Hum. Mol. Genet., 18, 4428–4441. [DOI] [PubMed] [Google Scholar]
- 11. Bonnet C.S., Aldred M., von Ruhland C., Harris R., Sandford R., Cheadle J.P. (2009) Defects in cell polarity underlie TSC and ADPKD-associated cystogenesis. Hum. Mol. Genet., 18, 2166–2176. [DOI] [PubMed] [Google Scholar]
- 12. Fischer D.C., Jacoby U., Pape L., Ward C.J., Kuwertz-Broeking E., Renken C., Nizze H., Querfeld U., Rudolph B., Mueller-Wiefel D.E., et al. (2009) Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol. Dial. Transplant, 24, 1819–1827. [DOI] [PubMed] [Google Scholar]
- 13. Wahl P.R., Serra A.L., Le Hir M., Molle K.D., Hall M.N., Wuthrich R.P. (2006) Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transplant, 21, 598–604. [DOI] [PubMed] [Google Scholar]
- 14. Wu M., Wahl P.R., Le Hir M., Wackerle-Men Y., Wuthrich R.P., Serra A.L. (2007) Everolimus retards cyst growth and preserves kidney function in a rodent model for polycystic kidney disease. Kidney Blood Press. Res., 30, 253–259. [DOI] [PubMed] [Google Scholar]
- 15. Zafar I., Belibi F.A., He Z., Edelstein C.L. (2009) Long-term rapamycin therapy in the Han:SPRD rat model of polycystic kidney disease (PKD). Nephrol. Dial. Transplant, 24, 2349–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tao Y., Kim J., Schrier R.W., Edelstein C.L. (2005) Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J. Am. Soc. Nephrol., 16, 46–51. [DOI] [PubMed] [Google Scholar]
- 17. Shillingford J.M., Piontek K.B., Germino G.G., Weimbs T. (2010) Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol., 21, 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Walz G., Budde K., Mannaa M., Nurnberger J., Wanner C., Sommerer C., Kunzendorf U., Banas B., Horl W.H., Obermuller N., et al. (2010) Everolimus in patients with autosomal dominant polycystic kidney disease. N. Engl J. Med., 363, 830–840. [DOI] [PubMed] [Google Scholar]
- 19. Serra A.L., Poster D., Kistler A.D., Krauer F., Raina S., Young J., Rentsch K.M., Spanaus K.S., Senn O., Kristanto P., et al. (2010) Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N. Engl J. Med., 363, 820–829. [DOI] [PubMed] [Google Scholar]
- 20. Shillingford J.M., Leamon C.P., Vlahov I.R., Weimbs T. (2012) Folate-conjugated rapamycin slows progression of polycystic kidney disease. J. Am. Soc. Nephrol., 23, 1674–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rubinsztein D.C., Codogno P., Levine B. (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov., 11, 709–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Green D.R., Levine B. (2014) To be or not to be? How selective autophagy and cell death govern cell fate. Cell, 157, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mizushima N., Komatsu M. (2011) Autophagy: renovation of cells and tissues. Cell, 147, 728–741. [DOI] [PubMed] [Google Scholar]
- 24. Yang Z., Klionsky D.J. (2010) Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol., 22, 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. White E. (2015) The role for autophagy in cancer. J. Clin. Invest., 125, 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang X., Overholtzer M., Thompson C.B. (2015) Autophagy in cellular metabolism and cancer. J. Clin. Invest., 125, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lavandero S., Chiong M., Rothermel B.A., Hill J.A. (2015) Autophagy in cardiovascular biology. J. Clin. Invest., 125, 55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frake R.A., Ricketts T., Menzies F.M., Rubinsztein D.C. (2015) Autophagy and neurodegeneration. J. Clin. Invest., 125, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ravichandran K., Edelstein C.L. (2014) Polycystic kidney disease: a case of suppressed autophagy?. Semin. Nephrol., 34, 27–33. [DOI] [PubMed] [Google Scholar]
- 30. Rowe I., Chiaravalli M., Mannella V., Ulisse V., Quilici G., Pema M., Song X.W., Xu H., Mari S., Qian F., et al. (2013) Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med., 19, 488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Belibi F., Zafar I., Ravichandran K., Segvic A.B., Jani A., Ljubanovic D.G., Edelstein C.L. (2011) Hypoxia-inducible factor-1alpha (HIF-1alpha) and autophagy in polycystic kidney disease (PKD). Am. J. Physiol. Renal Physiol., 300, F1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Drummond I.A., Majumdar A., Hentschel H., Elger M., Solnica-Krezel L., Schier A.F., Neuhauss S.C., Stemple D.L., Zwartkruis F., Rangini Z.. et al. (1998) Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development, 125, 4655–4667. [DOI] [PubMed] [Google Scholar]
- 33. Wingert R.A., Selleck R., Yu J., Song H.D., Chen Z., Song A., Zhou Y., Thisse B., Thisse C., McMahon A.P., et al. (2007) The cdx genes and retinoic acid control the positioning and segmentation of the zebrafish pronephros. PLoS Genet., 3, 1922–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sun Z., Amsterdam A., Pazour G.J., Cole D.G., Miller M.S., Hopkins N. (2004) A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development, 131, 4085–4093. [DOI] [PubMed] [Google Scholar]
- 35. Kramer-Zucker A.G., Olale F., Haycraft C.J., Yoder B.K., Schier A.F., Drummond I.A. (2005) Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer's vesicle is required for normal organogenesis. Development, 132, 1907–1921. [DOI] [PubMed] [Google Scholar]
- 36. Zhao C., Malicki J. (2007) Genetic defects of pronephric cilia in zebrafish. Mech. Dev., 124, 605–616. [DOI] [PubMed] [Google Scholar]
- 37. Schottenfeld J., Sullivan-Brown J., Burdine R.D. (2007) Zebrafish curly up encodes a Pkd2 ortholog that restricts left-side-specific expression of southpaw. Development, 134, 1605–1615. [DOI] [PubMed] [Google Scholar]
- 38. Cermak T., Doyle E.L., Christian M., Wang L., Zhang Y., Schmidt C., Baller J.A., Somia N.V., Bogdanove A.J., Voytas D.F. (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res., 39, e82.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Huang P., Xiao A., Zhou M., Zhu Z., Lin S., Zhang B. (2011) Heritable gene targeting in zebrafish using customized TALENs. Nat. Biotechnol., 29, 699–700. [DOI] [PubMed] [Google Scholar]
- 40. Miller J.C., Tan S., Qiao G., Barlow K.A., Wang J., Xia D.F., Meng X., Paschon D.E., Leung E., Hinkley S.J., et al. (2011) A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol., 29, 143–148. [DOI] [PubMed] [Google Scholar]
- 41. Mangos S., Lam P.Y., Zhao A., Liu Y., Mudumana S., Vasilyev A., Liu A., Drummond I.A. (2010) The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis. Model Mech., 3, 354–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coxam B., Sabine A., Bower N.I., Smith K.A., Pichol-Thievend C., Skoczylas R., Astin J.W., Frampton E., Jaquet M., Crosier P.S., et al. (2014) Pkd1 regulates lymphatic vascular morphogenesis during development. Cell Rep., 7, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Boulter C., Mulroy S., Webb S., Fleming S., Brindle K., Sandford R. (2001) Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc. Natl Acad. Sci. U S A, 98, 12174–12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chang Y.F., Imam J.S., Wilkinson M.F. (2007) The nonsense-mediated decay RNA surveillance pathway. Annu. Rev. Biochem., 76, 51–74. [DOI] [PubMed] [Google Scholar]
- 45. Obara T., Mangos S., Liu Y., Zhao J., Wiessner S., Kramer-Zucker A.G., Olale F., Schier A.F., Drummond I.A. (2006) Polycystin-2 immunolocalization and function in zebrafish. J. Am. Soc. Nephrol., 17, 2706–2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ravikumar B., Duden R., Rubinsztein D.C. (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet., 11, 1107–1117. [DOI] [PubMed] [Google Scholar]
- 47. Ravikumar B., Vacher C., Berger Z., Davies J.E., Luo S., Oroz L.G., Scaravilli F., Easton D.F., Duden R., O'Kane C.J., et al. (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet., 36, 585–595. [DOI] [PubMed] [Google Scholar]
- 48. Berger Z., Ravikumar B., Menzies F.M., Oroz L.G., Underwood B.R., Pangalos M.N., Schmitt I., Wullner U., Evert B.O., O'Kane C.J., et al. (2006) Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum. Mol. Genet., 15, 433–442. [DOI] [PubMed] [Google Scholar]
- 49. Wang T., Lao U., Edgar B.A. (2009) TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J. Cell Biol., 186, 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mizushima N., Yoshimori T., Levine B. (2010) Methods in mammalian autophagy research. Cell, 140, 313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J.A., et al. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy, 8, 445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lu K., Psakhye I., Jentsch S. (2014) Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell, 158, 549–563. [DOI] [PubMed] [Google Scholar]
- 53. Kimura T., Takabatake Y., Takahashi A., Kaimori J.Y., Matsui I., Namba T., Kitamura H., Niimura F., Matsusaka T., Soga T., et al. (2011) Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J. Am. Soc. Nephrol., 22, 902–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jiang M., Wei Q., Dong G., Komatsu M., Su Y., Dong Z. (2012) Autophagy in proximal tubules protects against acute kidney injury. Kidney Int., 82, 1271–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Qin Z.H., Wang Y., Kegel K.B., Kazantsev A., Apostol B.L., Thompson L.M., Yoder J., Aronin N., DiFiglia M. (2003) Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet., 12, 3231–3244. [DOI] [PubMed] [Google Scholar]
- 56. Kirkin V., Lamark T., Sou Y.S., Bjorkoy G., Nunn J.L., Bruun J.A., Shvets E., McEwan D.G., Clausen T.H., Wild P., et al. (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell, 33, 505–516. [DOI] [PubMed] [Google Scholar]
- 57. Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y.S., Ueno I., Sakamoto A., Tong K.I.. et al. (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol., 12, 213–223. [DOI] [PubMed] [Google Scholar]
- 58. Bjorkoy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol., 171, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bartlett B.J., Isakson P., Lewerenz J., Sanchez H., Kotzebue R.W., Cumming R.C., Harris G.L., Nezis I.P., Schubert D.R., Simonsen A., et al. (2011) p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy, 7, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Muto S., Aiba A., Saito Y., Nakao K., Nakamura K., Tomita K., Kitamura T., Kurabayashi M., Nagai R., Higashihara E., et al. (2002) Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum. Mol. Genet., 11, 1731–1742. [DOI] [PubMed] [Google Scholar]
- 61. Loghman-Adham M., Nauli S.M., Soto C.E., Kariuki B., Zhou J. (2003) Immortalized epithelial cells from human autosomal dominant polycystic kidney cysts. Am. J. Physiol. Renal Physiol., 285, F397–F412. [DOI] [PubMed] [Google Scholar]
- 62. Nauli S.M., Rossetti S., Kolb R.J., Alenghat F.J., Consugar M.B., Harris P.C., Ingber D.E., Loghman-Adham M., Zhou J. (2006) Loss of polycystin-1 in human cyst-lining epithelia leads to ciliary dysfunction. J. Am. Soc. Nephrol., 17, 1015–1025. [DOI] [PubMed] [Google Scholar]
- 63. Gainullin V.G., Hopp K., Ward C.J., Hommerding C.J., Harris P.C. (2015) Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J. Clin. Invest., 125, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cortes C.J., Miranda H.C., Frankowski H., Batlevi Y., Young J.E., Le A., Ivanov N., Sopher B.L., Carromeu C., Muotri A.R., et al. (2014) Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci., 17, 1180–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lee E., Koo Y., Ng A., Wei Y., Luby-Phelps K., Juraszek A., Xavier R.J., Cleaver O., Levine B., Amatruda J.F. (2014) Autophagy is essential for cardiac morphogenesis during vertebrate development. Autophagy. 10, 572–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hu Z., Zhang J., Zhang Q. (2011) Expression pattern and functions of autophagy-related gene atg5 in zebrafish organogenesis. Autophagy. 7, 1514–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Shoji-Kawata S., Sumpter R., Leveno M., Campbell G.R., Zou Z., Kinch L., Wilkins A.D., Sun Q., Pallauf K., MacDuff D., et al. (2013) Identification of a candidate therapeutic autophagy-inducing peptide. Nature, 494, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Woo D. (1995) Apoptosis and loss of renal tissue in polycystic kidney diseases. N. Engl J. Med., 333, 18–25. [DOI] [PubMed] [Google Scholar]
- 69. Lager D.J., Qian Q., Bengal R.J., Ishibashi M., Torres V.E. (2001) The pck rat: a new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int., 59, 126–136. [DOI] [PubMed] [Google Scholar]
- 70. Nauli S.M., Alenghat F.J., Luo Y., Williams E., Vassilev P., Li X., Elia A.E., Lu W., Brown E.M., Quinn S.J., et al. (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet., 33, 129–137. [DOI] [PubMed] [Google Scholar]
- 71. Maiuri M.C., Zalckvar E., Kimchi A., Kroemer G. (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol., 8, 741–752. [DOI] [PubMed] [Google Scholar]
- 72. Rubinstein A.D., Kimchi A. (2012) Life in the balance - a mechanistic view of the crosstalk between autophagy and apoptosis. J. Cell Sci., 125, 5259–5268. [DOI] [PubMed] [Google Scholar]
- 73. Williams A., Sarkar S., Cuddon P., Ttofi E.K., Saiki S., Siddiqi F.H., Jahreiss L., Fleming A., Pask D., Goldsmith P., et al. (2008) Novel targets for Huntington's disease in an mTOR-independent autophagy pathway. Nat. Chem. Biol., 4, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sarkar S., Ravikumar B., Floto R.A., Rubinsztein D.C. (2009) Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ., 16, 46–56. [DOI] [PubMed] [Google Scholar]
- 75. Law S.H., Sargent T.D. (2014) The serine-threonine protein kinase PAK4 is dispensable in zebrafish: identification of a morpholino-generated pseudophenotype. PLoS One, 9, e100268.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Schulte-Merker S., Stainier D.Y. (2014) Out with the old, in with the new: reassessing morpholino knockdowns in light of genome editing technology. Development, 141, 3103–3104. [DOI] [PubMed] [Google Scholar]
- 77. Kok F.O., Shin M., Ni C.W., Gupta A., Grosse A.S., van Impel A., Kirchmaier B.C., Peterson-Maduro J., Kourkoulis G., Male I., et al. (2015) Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish. Dev. Cell, 32, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rossi A., Kontarakis Z., Gerri C., Nolte H., Holper S., Kruger M., Stainier D.Y. (2015) Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature, 524, 230–233. [DOI] [PubMed] [Google Scholar]
- 79. Stainier D.Y., Kontarakis Z., Rossi A. (2015) Making sense of anti-sense data. Dev. Cell, 32, 7–8. [DOI] [PubMed] [Google Scholar]
- 80. Chang N., Sun C., Gao L., Zhu D., Xu X., Zhu X., Xiong J.W., Xi J.J. (2013) Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res., 23, 465–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sander J.D., Cade L., Khayter C., Reyon D., Peterson R.T., Joung J.K., Yeh J.R. (2011) Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat. Biotechnol., 29, 697–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R., Joung J.K. (2013) Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol., 31, 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zu Y., Tong X., Wang Z., Liu D., Pan R., Li Z., Hu Y., Luo Z., Huang P., Wu Q., et al. (2013) TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat. Methods, 10, 329–331. [DOI] [PubMed] [Google Scholar]
- 84. Bedell V.M., Wang Y., Campbell J.M., Poshusta T.L., Starker C.G., Krug R.G. 2nd, Tan W., Penheiter S.G., Ma A.C., et al. (2012) In vivo genome editing using a high-efficiency TALEN system. Nature, 491, 114–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Auer T.O., Duroure K., De Cian A., Concordet J.P., Del Bene F. (2014) Highly efficient CRISPR/Cas9-mediated knock-in in zebrafish by homology-independent DNA repair. Genome Res., 24, 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cao Y., Semanchik N., Lee S.H., Somlo S., Barbano P.E., Coifman R., Sun Z. (2009) Chemical modifier screen identifies HDAC inhibitors as suppressors of PKD models. Proc. Natl Acad. Sci. U S A., 106, 21819–21824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Igarashi P., Somlo S. (2002) Genetics and pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol., 13, 2384–2398. [DOI] [PubMed] [Google Scholar]
- 88. Torres V.E., Harris P.C. (2009) Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int., 76, 149–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Harris P.C., Torres V.E. (2014) Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Invest., 124, 2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Cianfanelli V., Fuoco C., Lorente M., Salazar M., Quondamatteo F., Gherardini P.F., De Zio D., Nazio F., Antonioli M., D'Orazio M., et al. (2015) AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol., 17, 20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Delgado M.E., Dyck L., Laussmann M.A., Rehm M. (2014) Modulation of apoptosis sensitivity through the interplay with autophagic and proteasomal degradation pathways. Cell Death Dis., 5, e1011.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Liu S., Hartleben B., Kretz O., Wiech T., Igarashi P., Mizushima N., Walz G., Huber T.B. (2012) Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy. 8, 826–837. [DOI] [PubMed] [Google Scholar]
- 93. Livingston M.J., Dong Z. (2014) Autophagy in acute kidney injury. Semin. Nephrol., 34, 17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell, 15, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hartleben B., Godel M., Meyer-Schwesinger C., Liu S., Ulrich T., Kobler S., Wiech T., Grahammer F., Arnold S.J., Lindenmeyer M.T., et al. (2010) Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin. Invest., 120, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Huber T.B., Edelstein C.L., Hartleben B., Inoki K., Jiang M., Koya D., Kume S., Lieberthal W., Pallet N., Quiroga A., et al. (2012) Emerging role of autophagy in kidney function, diseases and aging. Autophagy, 8, 1009–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Takiar V., Nishio S., Seo-Mayer P., King J.D. Jr., Li H., Zhang L., Karihaloo A., Hallows K.R., Somlo S., Caplan M.J. (2011) Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl Acad. Sci. U S A, 108, 2462–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Xie Z., Lau K., Eby B., Lozano P., He C., Pennington B., Li H., Rathi S., Dong Y., Tian R., et al. (2011) Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes, 60, 1770–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Seeger-Nukpezah T., Proia D.A., Egleston B.L., Nikonova A.S., Kent T., Cai K.Q., Hensley H.H., Ying W., Chimmanamada D., Serebriiskii I.G., et al. (2013) Inhibiting the HSP90 chaperone slows cyst growth in a mouse model of autosomal dominant polycystic kidney disease. Proc. Natl Acad. Sci. U S A, 110, 12786–12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Cao D.J., Wang Z.V., Battiprolu P.K., Jiang N., Morales C.R., Kong Y., Rothermel B.A., Gillette T.G., Hill J.A. (2011) Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl Acad. Sci. U S A, 108, 4123–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Proia D.A., Bates R.C. (2014) Ganetespib and HSP90: translating preclinical hypotheses into clinical promise. Cancer Res., 74, 1294–1300. [DOI] [PubMed] [Google Scholar]
- 102. West A.C., Johnstone R.W. (2014) New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest., 124, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Sullivan-Brown J., Bisher M.E., Burdine R.D. (2011) Embedding, serial sectioning and staining of zebrafish embryos using JB-4 resin. Nat. Protoc., 6, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Macdonald R. (1999) Molecular Methods in Developmental Biology: Xenopus and Zebrafish. In Guille Matt. (ed.) 1st edn.Humana Press. [Google Scholar]
- 105. Huang W., Zhang R., Xu X. (2009) Myofibrillogenesis in the developing zebrafish heart: A functional study of tnnt2. Dev. Biol., 331, 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.