

Abstract
Atmospheric autoxidation of volatile organic compounds (VOC) leads to prompt formation of highly oxidized multifunctional compounds (HOM) that have been found crucial in forming ambient secondary organic aerosol (SOA). As a radical chain reaction mediated by oxidized peroxy (RO2) and alkoxy (RO) radical intermediates, the formation pathways can be intercepted by suitable reaction partners, preventing the production of the highest oxidized reaction products, and thus the formation of the most condensable material. Commonly, NO is expected to have a detrimental effect on RO2 chemistry, and thus on autoxidation, whereas the influence of NO2 is mostly neglected. Here it is shown by dedicated flow tube experiments, how high concentration of NO2 suppresses cyclohexene ozonolysis initiated autoxidation chain reaction. Importantly, the addition of NO2 ceases covalently bound dimer production, indicating their production involving acylperoxy radical (RC(O)OO•) intermediates. In related experiments NO was also shown to strongly suppress the highly oxidized product formation, but due to possibility for chain propagating reactions (as with RO2 and HO2 too), the suppression is not as absolute as with NO2. Furthermore, it is shown how NOx reactions with oxidized peroxy radicals lead into indistinguishable product compositions, complicating mass spectral assignments in any RO2 + NOx system. The present work was conducted with atmospheric pressure chemical ionization mass spectrometry (CIMS) as the detection method for the highly oxidized end-products and peroxy radical intermediates, under ambient conditions and at short few second reaction times. Specifically, the insight was gained by addition of a large amount of NO2 (and NO) to the oxidation system, upon which acylperoxy radicals reacted in RC(O)O2 + NO2 → RC(O)O2NO2 reaction to form peroxyacylnitrates, consequently shutting down the oxidation sequence.
Keywords: Autoxidation, Highly oxidized multifunctional compounds, Highly oxygenated molecules, HOM, acylperoxy radicals, dimers, nitrogen oxides, peroxyacylnitrate
Introduction
Autoxidation of volatile organic compounds (VOCs) is a rapid process by which volatile, gas-phase hydrocarbon precursors rapidly evolve into very low volatile end-products capable of acting as in situ atmospheric aerosol embryos.1−5 It is a pseudounimolecular chain of reactions the efficiency of which relies on facile hydrogen abstraction isomerization reactions of the intermediate peroxy radicals (RO2; see Figure 1). The chain reaction begins with a single oxidant attack forming a carbon-centered radical which rapidly adds an O2 molecule producing a peroxy radical. If the RO2 structure is right, that is, if the radical has a loosely bound H atom in the carbon backbone that is about 5 to 8 atoms away from the oxygen atom containing the radical center, the peroxy radical can isomerize by an internal hydrogen abstraction reaction (i.e., H-shift) leading to another carbon-centered radical, and another prompt O2 addition.3,6−8 This forms a hydroperoxyalkylperoxy radical (commonly denoted as OOQOOH) that is potentially able to undergo a second internal isomerization reaction and an O2 addition. This chain of reactions then repeats until a suitable reaction partner comes along (bimolecular termination), or if through transfer of the radical site the molecule reaches a structure which is prone to decomposition (unimolecular termination). As the oxidation chain reaction advances, the further H-shifts generally become easier due to addition of oxygen- bearing, electron-withdrawing substituents that loosen the H-binding to the nearest carbon atoms: hence, the term autoxidation–autocatalytic oxidation. Autoxidation is pseudounimolecular in a sense that oxygen addition reactions to carbon-centered radicals are very rapid under ambient atmospheric conditions. Thus, the unimolecular hydrogen shift isomerization reactions of the peroxy radicals constitute the bottlenecks of the oxidation chain.
Figure 1.

First steps of gas-phase cyclohexene oxidation illustrating the formation of primary acylperoxy radicals (RC(O)O2•; in red). Shown are the OH and O3 initiated oxidation pathways. NO3 radical and Cl-atom initiated oxidation would likely proceed analogous to OH initiation. Note that the dialdehyde structure is formed in both cases but only the O3 reaction directly leads to RC(O)O2 in pseudounimolecular steps after a single-oxidant attack.
The oxidation chain is mediated by peroxy radical intermediates and has been observed to complete even in a subsecond time scale.5 This oxidation progression has been found especially efficient in ozonolysis of endocyclic alkenes, which often generate peroxy radicals with an aldehyde functionality as the primary oxidation product. Previously, an aldehydic 1,4 H-shift rate has been determined at 0.5 s–1 for a methacrolein derived RO2,9 and larger H-shifts with less strained transition states are likely to be even faster than this. Thus, the looseness of the aldehydic carbon–hydrogen (C(O)—H) bond provides for a facile first isomerization step, overcoming the bottleneck of the ensuing chain of reactions.2 The subsequent autoxidation sequence of a VOC leads to a prompt formation of highly oxidized products that have been found crucial in forming ambient secondary organic aerosol (SOA),1 even contribute to atmospheric new particle formation.10 Because of the prevalence of aldehydic functionality in the primary atmospheric oxidation products (see, e.g., refs (3, 11, and 12)), the formation of acyl radicals (i.e., radicals with a radical site located at a terminal carbonyl carbon atom; RC(O)•) are currently assumed ubiquitous in autoxidation pathways.
Oxygen addition to acyl radicals produces a special type of a peroxy radical, an acylperoxy radical (RC(O)OO•; RC(O)O2). Common peroxy radicals (RO2) are rather unreactive free radicals and are primarily consumed in reactions with other peroxy radicals (RO2 and HO2) and with nitrogen oxides (NOx = NO and NO2) but also in internal isomerization reactions as in autoxidation. These bimolecular RO2 reactions lead into oxidation chain termination according to reactions 1–5, but also to oxidation chain propagation by reactions 6–8. The termination reactions are composed of hydroperoxide (ROOH) formation with HO2 in reaction 1, organic peroxide (ROOR) formation with RO2 in reaction 2, accompanied by carbonyl and alcohol coproducts from reaction 3, organic nitrate formation with NO in reaction 4, and peroxynitrate formation with NO2 in reaction 5. The propagating channels 6 and 7 in reactions with RO2 and NO have generally high branching factors and lead to very reactive alkoxy radical (RO) generation. Acylperoxy radicals exhibit exceptionally fast rates in all these bimolecular reactions12−21 and also show an unusual chain branching behavior in reaction with HO2, resulting in high yields of oxy radical intermediates (RO and OH) and O3 production in reactions 8 and 9, respectively.22,23 Crucially, the NO2reaction 5 is the only reaction that according to current knowledge exclusively leads to oxidation chain termination without a possibility for propagating channels.
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
As opposed to general primary, secondary, and tertiary RO2 radical reactions with NO2 forming unstable peroxynitrates (RO2NO2) in reaction 5 (i.e., usual gas-phase RO2NO2 lifetimes are around 0.1 to 1 s11,12,24−27), the acylperoxy radicals frequently react faster and form considerably more stable peroxyacylnitrate (RC(O)OONO2; PAN) species, which are known air pollutants and constituents of the photochemical smog and in the atmosphere the main compounds responsible for long-range transport of NOx28,29 (see Figure 1 for a schematic of acylperoxy radical formation). Previously, acylperoxy radical combination reactions 2 have been suggested to lead to diacylperoxides,3,6,30−32 constituting one of the pathways proposed to account for gas-phase organic dimer formation (i.e., products detected with more carbon atoms than the apparent parent VOC, but not necessarily twice the amount). These dimers have been implied experimentally as especially important for atmospheric new particle formation due to their very low vapor pressures.9,33,34 A very recent theoretical work inspecting saturation vapor pressures of modeled highly oxidized product structures found them as the most likely species capable of condensing even onto the smallest of the atmospheric nanoparticles.35 Several other pathways to gas-phase dimers have been proposed, which include, for example, reactions of stabilized Criegee intermediates (sCI) with certain oxygenated VOCs36 and with RO2.37
Currently the involvement of acylperoxy radicals has only been implied based on theoretical considerations of the reactions propagating autoxidation. Only Berndt et al.5 have investigated the moderate addition of NO2 (around 1011 cm–3) to the cyclohexene autoxidation reaction mixture in short reaction time-scale experiments. They reported a single apparent peroxyacylnitrate compound (C6H9O8NO2) and derived a rate coefficient for its formation of 1.6 ± 0.5 × 10–12 cm–3 molecule–1 s–1, without delineating mechanistic insight. Here it is shown by a simple experimental arrangement how NO2 intercepts the oxidation process and even ceases the oxidized dimer formation completely. This further implies the crucial role of acylperoxy radicals as the mediators of the autoxidation chain, and their nature as the source of the observed highly oxidized dimer compounds. The essence of this work is the realization that whereas RO2 unimolecular reactions and bimolecular reactions with the common coreagents HO2, RO2, and NO can all propagate the oxidation sequence, the NO2 reaction exclusively leads to termination of the oxidation progression, and thus prevents the HOM and subsequent particle formation processes.
Results and Discussion
Cyclohexene ozonolysis initiated autoxidation experiments were performed in Quartz flow tube reactors under ambient atmospheric conditions with various concentrations of cyclohexene, ozone, and NOx and at a 2–20 s reaction time. The setup was similar as used in our previous investigations,3,6 and more details of the investigations and the setup are given in the Supporting Information (SI).
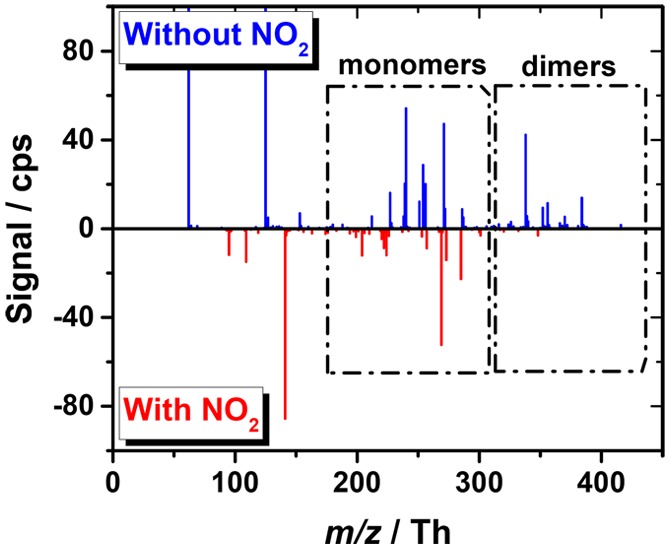
The addition of a large amount of NO2 into the flow tube gas mixture interferes with the highly oxidized product formation by cyclohexene autoxidation, and most significantly, ceases the dimer formation (Figure 2). Here, the dimer species constitute all the products observed with more carbon atoms than the parent cyclohexene, that is, a C7 compound would be considered a dimer in this case. This change in the oxidized product distribution is proposed to result mainly from NO2 reacting with the acylperoxy radical pool forming peroxyacylnitrates (PAN) according to reaction 5, as the common primary (R-COO•), secondary (RC(OO•)R), and tertiary (R3COO•) peroxy radicals do not generally result in stable reaction products with NO2 and also have generally slower reaction rates. As shown in Figure 1 (and in Scheme S1), sequential cyclohexene oxidation inevitably leads to acylperoxy radicals, and offers an ideal system to infer this mechanistic insight.
Figure 2.
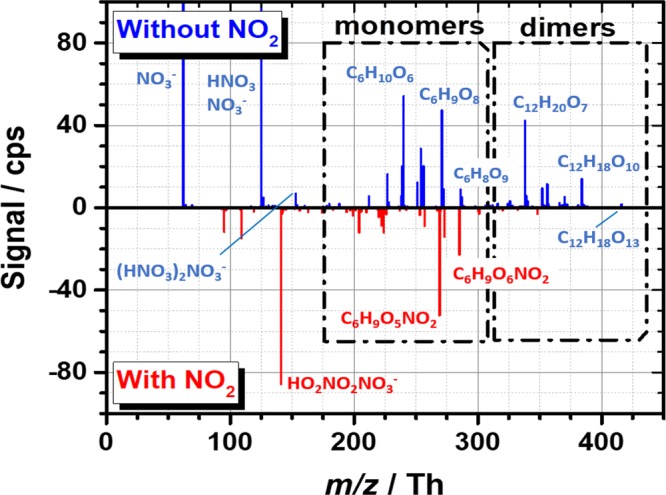
An example of NO3– chemical ionization mass spectra illustrating the suppression of highly oxidized dimer product formation by NO2. In the upper panel a spectrum measured in absence of NO2 (blue) and in the lower panel with a 100 ppb NO2 addition (red) at a 5.9 s reaction time, are shown.
The most prominent mass spectral peaks observed in the experiments with NOx addition have been collected in Table 1. The peaks observed without NOx addition are given in Table S1 of the Supporting Information. Majority of the products were detected as clusters with NO3–, excluding a few dicarboxylic acids such as glutaric (C5H8O4) and adipic (C6H10O4) acids, which were also detected as deprotonated product ions (i.e., C5H7O4– and C6H9O4–, see the SI Table S1). By glancing at Table 1 it becomes immediately evident that NO and NO2 reactions with oxidized peroxy radicals lead into similar product compositions and exemplifies the ambiguity of simply assigning measured product compositions to certain chemical compounds. The peroxyacylnitrate (and potentially some peroxynitrate) compounds observed here could be confused with organonitrates, which are expected to have a significantly different subsequent chemistry; organonitrates formed in reaction 4 are generally much more stable compounds and undergo deposition in the atmosphere, whereas the similar composition bearing peroxynitrates (RO2NO2) obtained from reaction 5(11,12,24−27,38) are expected to thermally decompose in a relatively short time frame. In the case of the more stable peroxyacylnitrates (RC(O)O2NO2), the delayed release of acylperoxy and NO2 participants constitutes the main long-range transport of NOx in the atmosphere with the decomposition rate (and thus the distance) heavily influenced by the ambient temperature.26,27,39,40
Table 1. Most Prominent Mass Peaks Measured with NO and NO2 Addition to the Gas Mixturea.
| composition | mass/Th | NO2 additionb | NO additionb,c |
|---|---|---|---|
| C5H8O4d | e194.0306 | x | |
| C5H8O5 | 210.0250 | x | x |
| C6H8O5 | 222.0255 | x | x |
| C5H9O6 | 227.0283 | x | x |
| C6H8O6 | 238.0205 | x | x |
| C6H9O6 | 239.0283 | x | x |
| C6H10O6 | 240.0361 | x | x |
| C5H9O4NO2f | 241.0314 | x | x |
| C6H9O4NO2 | 253.0314 | x | |
| C6H8O7 | 254.0154 | x | x |
| C5H9O5NO2 | 257.0263 | x | x |
| C6H9O5NO2 | 269.0263 | x | x |
| C6H8O8 | 270.0103 | x | |
| C6H8O9 | 271.0181 | x | |
| C5H9O6NO2 | 273.0212 | x | x |
| C6H9O6NO2 | 285.0212 | x | x |
| C6H8O9 | 286.0052 | x | x |
| C6H9O9 | 287.0130 | x | x |
| C6H9O7NO2 | 301.0161 | x | x |
| C6H9O8NO2 | 317.0110 | x | x |
| C6H10O5NO3NO2 | 332.0219 | xg | |
| C6H9O9NO2 | 333.0059 | xg | x |
| C6H10O6NO3NO2 | 348.0168 | xg | xg |
The peaks measured without NOx addition are given in the Supporting Information (Table S1).
[NO2] addition was about 400 ppb; [NO] addition was about 200 ppb.
Note that the rapid increase of NO2 in the system potentially enables acylperoxynitrate formation, too.
Observed product composition in the spectrum.
Observed product exact mass in the spectrum (including the mass of NO3– of 61.9884 Th).
Nitrogen-containing products have been marked with italic font and the identical compositions observed in both NO and NO2 addition experiments have been additionally marked with bold font.
Significantly smaller intensity, but nevertheless present. All masses are given in Thomson units; 1 Th = u/e, where e is elementary charge and u is the atomic mass unit.
Whereas NO2 addition only suppresses the oxidized product formation, NO exerts a more complicated influence. At a low addition level, NO aids the C6H8O8 HOM product formation (see Figure S3) by reactive alkoxy radical (RO) formation in reaction 7(41) and illustrates the oxidation enhancing influence of generating highly reactive RO radicals (also possible in reactions 6 and 8(42−44)). These reactive intermediates enable certain bond breaking reactions43 (e.g., Figure 1) and internal isomerizations42 and thus can propagate the oxidation chain reaction, as discussed previously by Kurtén et al.44 As the NO reaction can occur multiple times during the oxidation sequence under the present experimental conditions, a later NO reaction can still terminate the oxidation chain by organonitrate formation in reaction 4. Commonly, this is a minor pathway of this type of reaction,12 but the yield has been observed to increase with the size and functionalization of the R group,45 although certain oxidized RO2 have shown lower yields.46 Note that RO can also react with NO and form the corresponding nitrite (RONO) compounds but are not expected to do so due to their very short lifetimes.47 If present, however, also they could not be distinguished from other nitrogen bearing compounds due to their perfect mass spectral overlap.
In stark contrast to NO’s potentially enhancing influence, the NO2 reaction does not have any propagating channels, and thus only results in suppression of the oxidation (Figure 2). NO2 also competes for OH radicals with cyclohexene in this system due to its high concentration and fast reaction at atmospheric pressure (i.e., k298 K(NO2 + OH) = 1.1 × 10–1148 and k298 K(cyclohexene + OH) = 6.4 × 10–11,49 both in cm3 molecule–1 s–1) and thereby also suppresses the secondary chemistry in the system. The OH + NO2 reaction produces gas-phase nitric acid (HNO3) to the sample gas flow, which is known to influence the individual detection sensitivities of HOM compounds in NO3– CIMS.50 Specifically, additional HNO3 has been shown to favor the highest oxidized species due to their ability to form stronger HOM*NO3– clusters,50,51 and should thus lead to overestimation of the highest oxidized products. However, the opposite is observed here, as the highest oxidized compounds (e.g., C6H9O8 and C6H8O9) are strongly depleted in the obtained spectrum after NO2 was introduced into the flow.
The complexity of the oxidation progression, even in such a symmetric and simple molecule such as cyclohexene, becomes quickly evident by the subtle changes observed in the product distribution as a function of the initial radical concentration, reaction time, and the amount of NOx. Both the individual product signal heights and the extent of oxidation were influenced, and a shift from RO2 radical dominated product distribution at low loadings to closed-shell dominant products at higher loadings was observed (see Figure S5 for changes as a function [RO2] and [NO2]). For example, a moderate NO2 addition (about 100 ppb) was needed under high [RO2] conditions and at a 6 s reaction time for the nitrate peaks to outcompete the pure C, H, O containing HOM product peaks, whereas the highest oxidized closed-shell, monomer species (C6H8O9*NO3– at 286.0052 Th) was almost lost already at a significantly lower NO2 addition. At relatively low [NO2] and [RO2], the oxidation advanced apparently largely unhindered and the highest-oxidized peroxyacylnitrate compounds were observed at 317.0110 Th (C6H9O8NO2*NO3–) and 333.0059 Th (C6H9O9NO2*NO3–), together with the most prominent highly oxidized radical at 271.0181 Th (C6H9O8*NO3–). Under low [RO2] and high [NO2], the oxidation was terminated in the early part of the chain and resulted in significantly less oxidized PAN products. These features aptly illustrate the dynamic nature of the autoxidation progression in which the NOx addition can occur in many instances of the oxidation chain and thus have a significantly different outcome on the specific product formation. To map out the whole product dependencies on the full parameter space (i.e., over concentrations of reagents, oxidants and reaction conditions) is a challenging task and not resolvable with the current mass spectrometric technique lacking structural information but potentially necessary to overcome in the most detailed treatments concerning e.g., specific pollutant mitigation strategies and VOC combustion optimization. The changes observed in the product distribution in this work also potentially imply that the peroxy-H-shift-dynamics even in the cyclohexene system could be yet more complicated than currently thought (as noted recently for isoprene52) and that acylperoxy radicals may be abundant in every ladder of the oxidation progression. Previously Berndt et al.5 showed that the RO2 distribution in cyclohexene ozonolysis is practically fixed already at a 1.5 s reaction time, illustrating the very rapid interconversion of the intermediate peroxy radicals.
The amount of NO2 required to influence the oxidation sequence will always remain intimately coupled to the structure of the RO2 being oxidized. Currently the data on RC(O)O2 + NO2 (and RO2 + NO2) reaction rate coefficients are scarce, and the rates of the large cyclohexene derived RO2’s can only be estimated at best. In the simplest form, the RC(O)O2 + NO2 is in competition with the unimolecular initiation of the radical chain reaction, and thus the probability for NO2 prematurely terminating the sequence is a simple comparison of these two rates (see Figure 3). Typical NO2 concentrations encountered in the ambient atmosphere usually fall between <0.01 ppb in remote background regions to about few tens to hundreds of ppb found in polluted urban environments with relatively high variability due to localized sources and sinks.53 If the NO2 reaction rate is estimated by the previously reported prototypical CH3C(O)O2 and C2H5O2 reaction rate coefficients with NO2 (k298K(CH3C(O)O2 + NO2) = 1.5 × 10–11 cm3 molecule–1 s–1 and k298K(C2H5O2 + NO2) = 8.8 × 10–12 cm3 molecule–1 s–1),54 then around 0.5 ppb of NO2 in the case of ethylperoxy radical and 0.27 ppb in the case of acylperoxy can compete with autoxidation rates that are in the order of 0.1 s–1, which is an approximate limit at which autoxidation becomes competitive even under moderately polluted environments.2 If compared to the only available experimental determination by Berndt et al.5 (i.e., k = 1.6 × 10–12 cm3 molecule–1 s–1), the needed NO2 amount is abut 2.5 ppb. If, however, the rates for the large functionalized RO2 are much faster (i.e., k(RO2 + NO2) close to collision rate of 10–10 cm3 molecule–1 s–1), then only about 0.04 ppb NO2 is needed to effectively compete against autoxidation initiation.
Figure 3.
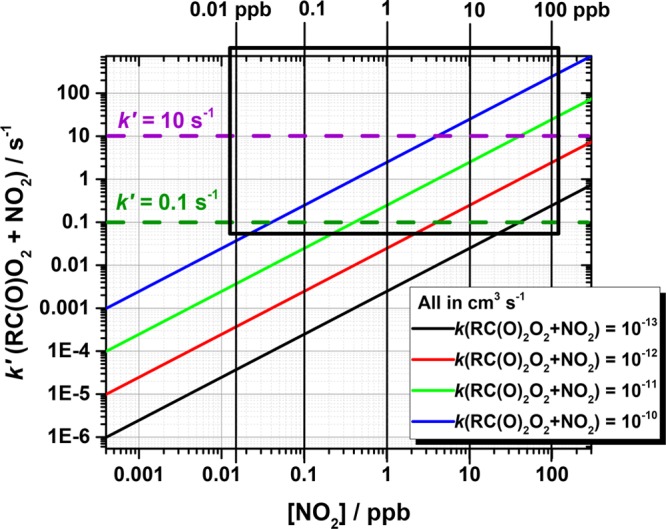
Bimolecular RC(O)O2 + NO2 reaction rate (in s–1) as a function of NO2 concentration in comparison to H-shift isomerization rates. At a rate of 0.1 s–1 (green dashed horizontal line) autoxidation is expected to compete, whereas at 10 s–1 rate (purple dashed horizontal line) the NO2 reaction will likely dominate. The most common ambient NO2 concentration range has been marked with a hollow black box.
In addition to RO2’s reactions with NOx, the cross reactions of peroxy radicals become more important with increasing [RO2] and reaction time, and thus have implications on organic peroxide dimer formation (2) and radical pooling (6) in the system. All the experiments were performed in 2–20 s reaction time, during which a shift from unimolecularly dominated chemistry to bimolecularly dominated regime was observed, evident from, for example, significantly enhanced dimer production. Acylperoxy radicals have anomalously fast recombination rate coefficients but also cross combination reactions with other common peroxy radicals (on the order of 10–11 cm3 molecule–1 s–112−21), which increases their contribution over most other RO2 in the reaction mixture. Previously the cross- and recombination rate coefficients of RO2 have been reported to span over 6 orders of magnitude and increase in the order: tertiary < secondary < primary < acyl (see, e.g., refs (13) and (55)) with the presence of electron-withdrawing oxygenated functional groups close to the reaction center, such as these oxidized RO2 certainly have, been observed to increase the RO2 + RO2 reaction rates. Several recent experimental studies1,3−6,10,41,45 have indicated reaction 2 as the likely source of the observed gas-phase dimer compounds. The current observation of oxidation suppression by NO2 with concurrent disappearance of dimers (all the dimer compounds observed without NOx addition decreased by over 95% when sufficient NO2 (or NO) was introduced into the flow) strongly implies acylperoxy radicals as they key intermediates in the oxidation chain, and likely dimerizing according to reaction 2.
The organic dimer formation by acylperoxy radicals in reaction 2 is in competition with other RC(O)OO• loss processes, mainly with the fast-unimolecular reactions of acyl and acylperoxy radicals. Before the formation of acylperoxy radicals occurs, CO-loss from acyl radicals can propagate the radical chain, concurrently converting acyl radicals into alkyl radicals (i.e., (RC(O)• → R• + CO) and accounting for certain products observed with less carbon atoms than the parent VOC.3,9,56 Perhaps more importantly, ultrafast hydrogen shift isomerization reactions of the RO2 can interconvert peroxy and hydroperoxide functionalities (see Figure 4) and has been reported to favor peroxyacids (i.e., R–C(O)OOH) over hydroperoxides,57,58 with peroxyacids previously identified in the gas-phase cyclohexene oxidation by Iyer et al.59 From the current results it is evident that the RC(O)OO• + NO2 reactions are fast enough to compete with the ultrafast H-shifts even to overcome them at moderately high [NO2], making interception of acylperoxy chemistry possible. Alternatively, the equilibrium between hydroperoxide and peroxyacid functionalities is more even in these highly oxidized RO2 than previously assumed based on theoretical calculations, making the interception easier. Furthermore, it should be emphasized that for smaller more abundant atmospheric RO2s, reaction 6 is far more likely than the dimerization reaction 2, and in the case of RC(O)OO• results in acyloxy radicals (RC(O)O•) and subsequent CO2 loss.43 However, currently the information on the branching factors of acylperoxy radicals with other peroxy radicals (or with themselves) contributing to reaction 2 versus reaction 6 is scarce,12−21 mainly due to the immense difficulty in studying radical–radical reaction product channels in required detail. If the dimer species observed in the current work would be due to other RO2s than RC(O)OO•, they would likely be formed in the postacylperoxy chemistry, as the apparent interception of RC(O)OO• by NO2 ceases the organic dimer formation. On the other hand, it is possible that only acylperoxy radicals have fast enough reactions 2 to contribute significantly to dimer formation in these short reaction time experiments. Regardless of the ultimate reason, these observations imply the special importance of acylperoxy radicals in directing autoxidation phenomena.
Figure 4.

Competing reaction steps in acylperoxy radical formation and subsequent chemistry. As mentioned in the text, post acylperoxy radical intermediates such as P1, an acyloxy radical, could also account for the dimer formation, although oxy radical lifetimes do not generally allow for significant bimolecular reactions except with O2. Color coding in the figure: Green arrows show the autoxidation pathway leading to molecular growth, black arrows show inhibition of autoxidation and molecular fragmentation, red arrows show the reactions contributing to PAN formation, and blue arrows show the rapid interconversion of isomers. Acylperoxy radicals have been marked with red color, and the H atoms undergoing H-shifts have been explicitly indicated.
Possibilities for misinterpreting the importance of acylperoxy radicals in the current system could result from (i) other oxidized RO2s reacting rapidly with NO2 forming stable nitrates within the reaction time, (ii) certain carbon-centered radicals living long enough to react with NO2 forming nitro compounds (R-NO2, for example, due to longer lifetime of R enabled by equilibrium of the type R + O2 ↔ RO2), (iii) stabilized Criegee intermediate (sCI) reactions, sCI + NO2, scavenging the prestages of the peroxy radicals before the formation of highly oxidized species occurs, or (iv) that all O3 is scavenged by NO2 to form NO3, and that NO3 is not able to initiate HOM formation from cyclohexene. However, (i) only RC(O)O2 reactions are known to lead to stable enough products with NO2, (ii) strong resonances able to shift the equilibrium to room temperature are most likely absent from the intermediate RO2, (iii) cyclohexene ozonolysis does not produce a stabilized CI60 to react with NO2, and finally, (iv) only a small fraction of O3 will be consumed under these short reaction times due to the slow NO2 + O3 → NO3 + O2 reaction,61 and with such a high [NO2] most of the NO3 will be rapidly converted into N2O5.62 Nevertheless, two apparent minor compounds with two nitrogen atoms attached were detected in the high concentration experiments (see Table 1 and Figure S5), which could have potentially formed in NO3 initiated, and NOx terminated, cyclohexene oxidation. However, these compositions could also result from product molecules clustering with the reagent ion dimer (i.e., HNO3NO3–), and although there is currently no possibility to discriminate between either origin the charging by reagent dimer seems far more likely under the current experimental conditions (see the SI for more discussion).
By using mass spectrometric detection methods, it is not generally possible to obtain branching fractions to isomeric products due to their perfect overlap in the mass spectra. Furthermore, the detection sensitivity of different oxidized states is known to differ using chemical ionization as a probing method,50,63 increasing uncertainty in using a single calibration factor for all the detected species. By applying this procedure, however, concentrations ranging from fractions of ppt to some tens of ppt have been reported previously for individual HOMs in cyclohexene autoxidation with the total measured HOM ranging from below one ppt to hundreds of ppt.3,5 Yet, it remains unclear how many different acylperoxy radicals there are in the gas mixture, what are their concentrations, and how big a portion of these result in dimer compounds observed in the current CIMS spectra. Nevertheless, it is possible to lean on previous theoretical and experimental foundation on RO2 + NO2 reactions,12,24,25,38 and on previous knowledge on cyclohexene autoxidation,1,3,5,41,59 to imply the mechanistic details presented in the current work. Cyclohexene ozonolysis initiated autoxidation is a close to an ideal autoxidation system that proceeds largely unhindered due to close-to-optimal structures of the formed intermediates,3 and has a correspondingly high HOM molar yield of 4.8% (average of three determinations with NO3– CIMS technique1,3,5). Yet even in this highly efficient autoxidation progression, NO2 is able to interfere and cuts down the oxidation sequence, preventing formation of the least volatile compounds that would be the most potent in forming ambient particulate matter.1,10,35 Just alone substituting the potential peroxy- or carboxylic acid terminal groups into nitrates or peroxynitrates, as is the case here, constitutes a considerable increase in the resulting product volatility,64,65 and corresponding reduction in SOA formation potential. The current finding further suggests that the common procedure of conducting chamber experiments, in which unnaturally high NO2 concentration is first equilibrated in a chamber environment and then photolyzed to obtain a uniform O3 concentration (or a suitable NO to NO2 ratio, or both), poorly reflects the conditions of the true atmosphere. Such an experimental design has the prospective to unintendedly suppress acylperoxy chemistry and thereby also the low-volatile product formation, consequently leading to underestimation of the SOA forming potential of a VOC. Undoubtedly more molecular-level detailed work is required to understand the intricacies of acylperoxy radicals in directing autoxidation pathways.
Conclusions
Prevention of gas-phase highly oxidized product formation by NO2 was illustrated in the cyclohexene ozonolysis system, supporting the involvement of acylperoxy radicals as the key intermediates in the autoxidation chain reaction. By addition of NO2 to the reacting gas mixture, the oxidation pathways were influenced, and the highest-oxidized product signal levels were observed to plummet. Most notably, the dimer formation ceased, which implied that the acylperoxy radicals derived from cyclohexene reacted fast enough with NO2 and slowly enough unimolecularly that the interception of the autoxidation progression became possible. Furthermore, the NO and NO2 reactions with the same oxidized RO2 were observed to lead to indistinguishable product compositions. These observations have consequences on our understanding of the atmospheric oxidation phenomena and exemplify the importance of NO2 reactions preventing the formation of in situ ambient aerosol precursors. The current finding thus also demonstrates the significance of separating NOx measurement data into respective NO and NO2 values as the outcome of a certain VOC oxidation will differ significantly from NO to NO2 dominated environments, that is, in moving away from primary emission sources. Even more so, this becomes important as the oxidized products are likely to have significantly different chemical nature, yet are not distinguishable from their mass spectral signals, which is the current primary method for ambient gas-phase aerosol precursor compound detection.
Acknowledgments
The author is grateful for the funding from Academy of Finland project 299574.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsearthspacechem.8b00123.
Methods. Influence of charging probabilities and transmission on measured concentrations. Oxidation pathways. Suppression kinetics. NO’s enhancing influence. Table S1: Mass peaks measured without NOx addition. Table S2: Competition of RC(O)O2 + NO2 and H-shift reactions. Scheme S1: Further acylperoxy chemistry. Figure S1: Experimental setup. Figure S2: Potential PAN structures. Figure S3: C6H8O8 product formation. Figure S4: Nitrate signals plummeting. Figure S5: NOx addition at various stages during autoxidation (PDF)
The author declares no competing financial interest.
Supplementary Material
References
- Ehn M.; Thornton J. A.; Kleist E.; Sipilä M.; Junninen H.; Pullinen I.; Springer M.; Rubach F.; Tillmann R.; Lee B.; Lopez-Hilfiker F.; Andres S.; Acir I.-H.; Rissanen M.; Jokinen T.; Schobesberger S.; Kangasluoma J.; Kontkanen J.; Nieminen T.; Kurtén T.; Nielsen L. B.; Jørgensen S.; Kjaergaard H. G.; Canagaratna M.; Dal Maso M.; Berndt T.; Petäjä T.; Wahner A.; Kerminen V.-M.; Kulmala M.; Worsnop D. R.; Wildt J.; Mentel T. F. A large source of low-volatility secondary organic aerosol. Nature 2014, 506, 476–479. 10.1038/nature13032. [DOI] [PubMed] [Google Scholar]
- Crounse J. D.; Nielsen L. B.; Jørgensen S.; Kjaergaard H. G.; Wennberg P. O. Autoxidation of Organic Compounds in the Atmosphere. J. Phys. Chem. Lett. 2013, 4, 3513–3520. 10.1021/jz4019207. [DOI] [Google Scholar]
- Rissanen M. P.; Kurtén T.; Sipilä M.; Thornton J. A.; Kangasluoma J.; Sarnela N.; Junninen H.; Jørgensen S.; Schallhart S.; Kajos M. K.; Taipale R.; Springer M.; Mentel T. F.; Ruuskanen T.; Petäjä T.; Worsnop D. R.; Kjaergaard H. G.; Ehn M. The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene. J. Am. Chem. Soc. 2014, 136, 15596–15606. 10.1021/ja507146s. [DOI] [PubMed] [Google Scholar]
- Jokinen T.; Sipila M.; Richters S.; Kerminen V.-M.; Paasonen P.; Stratmann F.; Worsnop D. R.; Kulmala M.; Ehn M.; Herrmann H.; Berndt T. Rapid Autoxidation Forms Highly Oxidized RO2 Radicals in the Atmosphere. Angew. Chem., Int. Ed. 2014, 53, 14596–14600. 10.1002/anie.201408566. [DOI] [PubMed] [Google Scholar]
- Berndt T.; Richters S.; Kaethner R.; Voigtländer J.; Stratmann F.; Sipilä M.; Kulmala M.; Herrmann H. Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO2 Radicals and Their Reactions with NO, NO2, SO2, and Other RO2 Radicals. J. Phys. Chem. A 2015, 119, 10336–10348. 10.1021/acs.jpca.5b07295. [DOI] [PubMed] [Google Scholar]
- Rissanen M. P.; Kurtén T.; Sipila M.; Thornton J. A.; Kausiala O.; Garmash O.; Kjaergaard H. G.; Petäjä T.; Worsnop D. R.; Ehn M.; Kulmala M. Effects of Chemical Complexity on the Autoxidation Mechanisms of Endocyclic Alkene Ozonolysis Products: From Methylcyclohexenes toward Understanding α-Pinene. J. Phys. Chem. A 2015, 119, 4633–4650. 10.1021/jp510966g. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Popolan-Vaida D. M.; Chen B.; Moshammer K.; Mohamed S. Y.; Wang H.; Sioud S.; Raji M. A.; Kohse-Höinghaus K.; Hansen N.; Dagaut P.; Leone S. R.; Sarathy S. M. Unraveling the structure and chemical mechanisms of highly oxygenated intermediates in oxidation of organic compounds. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 13102–13107. 10.1073/pnas.1707564114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savee J. D.; Papajak E.; Rotavera B.; Huang H.; Eskola A. J.; Welz O.; Sheps L.; Taatjes C. A.; Zádor J.; Osborn D. L. Direct observation and kinetics of a hydroperoxyalkyl radical (QOOH). Science 2015, 347, 643–646. 10.1126/science.aaa1495. [DOI] [PubMed] [Google Scholar]
- Crounse J. D.; Knap H. C.; Ørnsø K. B.; Jørgensen S.; Paulot F.; Kjaergaard H. G.; Wennberg P. O. Atmospheric Fate of Methacrolein. 1. Peroxy Radical Isomerization Following Addition of OH and O2. J. Phys. Chem. A 2012, 116, 5756–5762. 10.1021/jp211560u. [DOI] [PubMed] [Google Scholar]
- Kirkby J.; Duplissy J.; Sengupta K.; Frege C.; Gordon H.; Williamson C.; Heinritzi M.; Simon M.; Yan C.; Almeida J.; Tröstl J.; Nieminen T.; Ortega I. K.; Wagner R.; Adamov A.; Amorim A.; Bernhammer A.-K.; Bianchi F.; Breitenlechner M.; Brilke S.; Chen X.; Craven J.; Dias A.; Ehrhart S.; Flagan R. C.; Franchin A.; Fuchs C.; Guida R.; Hakala J.; Hoyle C. R.; Jokinen T.; Junninen H.; Kangasluoma J.; Kim J.; Krapf M.; Kürten A.; Laaksonen A.; Lehtipalo K.; Makhmutov V.; Mathot S.; Molteni U.; Onnela A.; Peräkylä O.; Piel F.; Petäjä T.; Praplan A. P.; Pringle K.; Rap A.; Richards N. A. D.; Riipinen I.; Rissanen M. P.; Rondo L.; Sarnela N.; Schobesberger S.; Scott C. E.; Seinfeld J. H.; Mikko Sipilä; Steiner G.; Stozhkov Y.; Stratmann F.; Tomé A.; Virtanen A.; Vogel A. L.; Wagner A. C.; Wagner P. E.; Weingartner E.; Wimmer D.; Winkler P. M.; Ye P.; Zhang X.; Hansel A.; Dommen J.; Donahue N. M.; Worsnop D. R.; Baltensperger U.; Kulmala M.; Carslaw K. S.; Curtius J. Ion-induced nucleation of pure biogenic particles. Nature 2016, 533, 521–526. 10.1038/nature17953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson R.; Arey J. Atmospheric Degradation of Volatile Organic Compounds. Chem. Rev. 2003, 103, 4605–4638. 10.1021/cr0206420. [DOI] [PubMed] [Google Scholar]
- Orlando J. J.; Tyndall G. S. Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance. Chem. Soc. Rev. 2012, 41, 6294–6317. 10.1039/c2cs35166h. [DOI] [PubMed] [Google Scholar]
- Shallcross D. E.; Raventos-Duran M. T.; Bardwell M. W.; Bacak A.; Solman Z.; Percival C. J. A semi-empirical correlation for the rate coefficients for cross- and self-reactions of peroxy radicals in the gas-phase. Atmos. Environ. 2005, 39, 763–771. 10.1016/j.atmosenv.2004.09.072. [DOI] [Google Scholar]
- Villenave E.; Lesclaux R.; Seefeld S.; Stockwell W. R. Kinetics and atmospheric implications of peroxy radical cross reactions involving the CH3C(O)O2 radical. J. Geophys. Res. 1998, 103, 25273–25285. 10.1029/98JD00926. [DOI] [Google Scholar]
- Villenave E.; Lesclaux R. Kinetics of the cross reactions of CH3O2 and C2H5O2 radicals with selected peroxy radicals. J. Phys. Chem. 1996, 100, 14372–14382. 10.1021/jp960765m. [DOI] [Google Scholar]
- Moortgat G.; Veyret B.; Lesclaux R. Absorption-spectrum and kinetics of reactions of the acetylperoxy radical. J. Phys. Chem. 1989, 93, 2362. 10.1021/j100343a032. [DOI] [Google Scholar]
- Wallington T. J.; Dagaut P.; Kurylo M. J. Ultraviolet Absorption Cross Sections and Reaction Kinetics and Mechanisms for Peroxy Radicals in the Gas Phase. Chem. Rev. 1992, 92, 667–710. 10.1021/cr00012a008. [DOI] [Google Scholar]
- Bridier I.; Veyret B.; Lesclaux R.; Jenkin M. E. Flash Photolysis Study of the UV Spectrum and Kinetics of Reactions of the Acetonylperoxy Radical. J. Chem. Soc., Faraday Trans. 1993, 89, 2993–2997. 10.1039/ft9938902993. [DOI] [Google Scholar]
- Maricq M. M.; Szente J. J. Kinetics of the Reaction between Acetylperoxy and Ethylperoxy Radicals. J. Phys. Chem. A 2000, 104, 7239–7245. 10.1021/jp9930649. [DOI] [Google Scholar]
- Tyndall G. S.; Cox R. A.; Granier C.; Lesclaux R.; Moortgat G. K.; Pilling M. J.; Ravishankara A. R.; Wallington T. J. Atmospheric chemistry of small organic peroxy radicals. J. Geophys. Res. 2001, 106, 12157–12182. 10.1029/2000JD900746. [DOI] [Google Scholar]
- Maricq M. M.; Szente J. J. The CH3C(O)O2 Radical. Its UV Spectrum, Self-Reaction Kinetics, and Reaction with CH3O2. J. Phys. Chem. 1996, 100, 4507. 10.1021/jp9533234. [DOI] [Google Scholar]
- Gross C. B. M.; Dillon T. J.; Schuster G.; Lelieveld J.; Crowley J. N. Direct Kinetic Study of OH and O3 Formation in the Reaction of CH3C(O)O2 with HO2. J. Phys. Chem. A 2014, 118, 974–985. 10.1021/jp412380z. [DOI] [PubMed] [Google Scholar]
- Hasson A. S.; Kuwata K. T.; Arroyo M. C.; Petersen E. B. Theoretical studies of the reaction of hydroperoxy radicals (HO2) with ethyl peroxy (CH3CH2O2•), acetyl peroxy (CH3C(O)O2•) and acetonyl peroxy (CH3C(O)CH2O2• radicals. J. Photochem. Photobiol., A 2005, 176, 218–230. 10.1016/j.jphotochem.2005.08.012. [DOI] [Google Scholar]
- Zabel F. Unimolecular decomposition of peroxynitrates, Zeitschrift. Z. Phys. Chem. 1995, 188, 119–142. 10.1524/zpch.1995.188.Part_1_2.119. [DOI] [Google Scholar]
- Atkinson R. Atmospheric chemistry of VOCs and NOx. Atmos. Environ. 2000, 34, 2063–2101. 10.1016/S1352-2310(99)00460-4. [DOI] [Google Scholar]
- Zabel F.; Reimer A.; Becker K. H.; Fink E. H. Thermal decomposition of alkyl peroxynitrates. J. Phys. Chem. 1989, 93, 5500–5507. 10.1021/j100351a036. [DOI] [Google Scholar]
- Kirchner F.; Mayer–Figge A.; Zabel F.; Becker K. H. Thermal stability of peroxynitrates. Int. J. Chem. Kinet. 1999, 31, 127–144. . [DOI] [Google Scholar]
- Nielsen T.; Samuelsson U.; Grennfelt P.; Thomsen E. L. Peroxyacetyl nitrate in long-range transported polluted air. Nature 1981, 293, 553. 10.1038/293553a0. [DOI] [Google Scholar]
- Singh H. B. Reactive nitrogen in the troposphere. Environ. Sci. Technol. 1987, 21, 320. 10.1021/es00158a001. [DOI] [PubMed] [Google Scholar]
- Ziemann P. J. Evidence for Low-Volatility Diacyl Peroxides as a Nucleating Agent and Major Component of Aerosol Formed from Reactions of O3 with Cyclohexene and Homologous Compounds. J. Phys. Chem. A 2002, 106, 4390–4402. 10.1021/jp012925m. [DOI] [Google Scholar]
- McDowell C. A.; Sifniades S. Oxygen-18 tracer evidence for termination mechanism in photochemical oxidation of acetaldehyde. Can. J. Chem. 1963, 41, 300–307. 10.1139/v63-046. [DOI] [Google Scholar]
- McDowell C. A.; Sharples L. K. The Oxidation of Aldehydes in the Gaseous Phase Part I. The Kinetics of the Photochemical Oxidation of Acetaldehyde. Can. J. Chem. 1958, 36, 251–257. 10.1139/v58-034. [DOI] [Google Scholar]
- Tröstl J.; Chuang W. K.; Gordon H.; Heinritzi M.; Yan C.; Molteni U.; Ahlm L.; Frege C.; Bianchi F.; Wagner R.; Simon M.; Lehtipalo K.; Williamson C.; Craven J. S.; Duplissy J.; Adamov A.; Almeida J.; Bernhammer A.-K.; Breitenlechner M.; Brilke S.; Dias A.; Ehrhart S.; Flagan R. C.; Franchin A.; Fuchs C.; Guida R.; Gysel M.; Hansel A.; Hoyle C. R.; Jokinen T.; Junninen H.; Kangasluoma J.; Keskinen H.; Kim J.; Krapf M.; Kürten A.; Laaksonen A.; Lawler M.; Leiminger M.; Mathot S.; Möhler O.; Nieminen T.; Onnela A.; Petäjä T.; Piel F. M.; Miettinen P.; Rissanen M. P.; Rondo L.; Sarnela N.; Schobesberger S.; Sengupta K.; Sipilä M.; Smith J. N.; Steiner G.; Tomè A.; Virtanen A.; Wagner A. C.; Weingartner E.; Wimmer D.; Winkler P. M.; Ye P.; Carslaw K. S.; Curtius J.; Dommen J.; Kirkby J.; Kulmala M.; Riipinen I.; Worsnop D. R.; Donahue N. M.; Baltensperger U. The role of low-volatility organic compounds in initial particle growth in the atmosphere. Nature 2016, 533, 527–531. 10.1038/nature18271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr C.; Lopez-Hilfiker F. D.; Yli-Juuti T.; Heitto A.; Lutz A.; Hallquist M.; D’Ambro E. L.; Rissanen M. P.; Hao L.; Schobesberger S.; Kulmala M.; Mauldin R. L.; Makkonen U.; Sipilä M.; Petäjä T.; Thornton J. A. Ambient observations of dimers from terpene oxidation in the gas phase: Implications for new particle formation and growth. Geophys. Res. Lett. 2017, 44, 2958–2966. 10.1002/2017GL072718. [DOI] [Google Scholar]
- Kurtén T.; Tiusanen K.; Roldin P.; Rissanen M.; Luy J.-N.; Boy M.; Ehn M.; Donahue N. α-Pinene Autoxidation Products May Not Have Extremely Low Saturation Vapor Pressures Despite High O: C Ratios. J. Phys. Chem. A 2016, 120, 2569–2582. 10.1021/acs.jpca.6b02196. [DOI] [PubMed] [Google Scholar]
- Kristensen K.; Cui T.; Zhang H.; Gold A.; Glasius M.; Surratt J. D. Dimers in α-pinene secondary organic aerosol: Effect of hydroxyl radical, ozone, relative humidity and aerosol acidity. Atmos. Chem. Phys. 2014, 14, 4201–4218. 10.5194/acp-14-4201-2014. [DOI] [Google Scholar]
- Zhao Y.; Wingen L. M.; Perraud V.; Greaves J.; Finlayson-Pitts B. J. Role of the reaction of stabilized Criegee intermediates with peroxy radicals in particle formation and growth in air. Phys. Chem. Chem. Phys. 2015, 17, 12500–12514. 10.1039/C5CP01171J. [DOI] [PubMed] [Google Scholar]
- Atkinson R.; Aschmann S. M.; Carter W. P. L.; Winer A. M.; Pitts J. N. Alkyl Nitrate Formation from the NOx-Air Photooxidations of C2-C8 n-Alkanes. J. Phys. Chem. 1982, 86, 4563–4569. 10.1021/j100220a022. [DOI] [Google Scholar]
- Bridier I.; Caralp F.; Loirat H.; Lesclaux R.; Veyret B.; Becker K. H.; Reimer A.; Zabel F. Kinetic and theoretical studies of the reactions CH3C(O)O2 + NO2 + M ⇔ CH3C(O)O2NO2 + M between 248 and 393 K and between 30 and 760 Torr. J. Phys. Chem. 1991, 95, 3594–3600. 10.1021/j100162a031. [DOI] [Google Scholar]
- Fischer E. V.; Jacob D. J.; Yantosca R. M.; Sulprizio M. P.; Millet D. B.; Mao J.; Paulot F.; Singh H. B.; Roiger A.; Ries L.; Talbot R. W.; Dzepina K.; Pandey Deolal S. Atmospheric peroxyacetyl nitrate (PAN): a global budget and source attribution. Atmos. Chem. Phys. 2014, 14, 2679–2698. 10.5194/acp-14-2679-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentel T. F.; Springer M.; Ehn M.; Kleist E.; Pullinen I.; Kurtén T.; Rissanen M.; Wahner A.; Wildt J. Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships. Atmos. Chem. Phys. Discuss. 2015, 15, 2791–2851. 10.5194/acpd-15-2791-2015. [DOI] [Google Scholar]
- Vereecken L.; Peeters J. A structure-activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals. Phys. Chem. Chem. Phys. 2010, 12, 12608–12620. 10.1039/c0cp00387e. [DOI] [PubMed] [Google Scholar]
- Vereecken L.; Peeters J. Decomposition of substituted alkoxy radicals—part I: a generalized structure–activity relationship for reaction barrier heights. Phys. Chem. Chem. Phys. 2009, 11, 9062–9074. 10.1039/b909712k. [DOI] [PubMed] [Google Scholar]
- Kurtén T.; Rissanen M. P.; Mackeprang K.; Thronton J. A.; Jorgensen S.; Ehn M.; Kjaergaard H. Computational Study of Hydrogen Shifts and Ring-Opening Mechanisms in α-Pinene Ozonolysis Products. J. Phys. Chem. A 2015, 119, 11366–11375. 10.1021/acs.jpca.5b08948. [DOI] [PubMed] [Google Scholar]
- Jokinen T.; Berndt T.; Makkonen R.; Kerminen V.-M.; Junninen H.; Paasonen P.; Stratmann F.; Herrmann H.; Guenther A. B.; Worsnop D. R.; Kulmala M.; Ehn M.; Sipilä M. Production of extremely low volatile organic compounds from biognenic emissions: Measured yields and atmospheric implications. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 7123–7128. 10.1073/pnas.1423977112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien J. M.; Czuba E.; Hastie D. R.; Francisco J. S.; Shepson P. B. Determination of the Hydroxy Nitrate Yields from the Reaction of C2–C6Alkenes with OH in the Presence of NO. J. Phys. Chem. A 1998, 102, 8903–8908. 10.1021/jp982320z. [DOI] [Google Scholar]
- Orlando J. J.; Tyndall G. S.; Wallington T. J. The atmospheric chemistry of alkoxy radicals. Chem. Rev. 2003, 103, 4657–4689. 10.1021/cr020527p. [DOI] [PubMed] [Google Scholar]
- Mollner A. K.; Valluvadasan S.; Feng L.; Sprague M. K.; Okumura M.; Milligan D. B.; Bloss W. J.; Sander S. P.; Martien P. T.; Harley R. A.; McCoy A. B.; Carter W. P. L. Rate of Gas Phase Association of Hydroxyl Radical and Nitrogen Dioxide. Science 2010, 330, 646–649. 10.1126/science.1193030. [DOI] [PubMed] [Google Scholar]
- Aschmann S. M.; Arey J.; Atkinson R. Kinetics and Products of the Reactions of OH Radicals with Cyclohexene, 1-Methyl-1-cyclohexene, cis-Cyclooctene, and cis-Cyclodecene. J. Phys. Chem. A 2012, 116, 9507–9515. 10.1021/jp307217m. [DOI] [PubMed] [Google Scholar]
- Hyttinen N.; Kupiainen-Määttä O.; Rissanen M. P.; Muuronen M.; Ehn M.; Kurtén T. Modeling the Detection of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization. J. Phys. Chem. A 2015, 119, 6339–6345. 10.1021/acs.jpca.5b01818. [DOI] [PubMed] [Google Scholar]
- Hyttinen N.; Rissanen M. P.; Kurtén T. Computational Comparison of Acetate and Nitrate Chemical Ionization of Highly Oxidized Cyclohexene Ozonolysis Intermediates and Products. J. Phys. Chem. A 2017, 121, 2172–2179. 10.1021/acs.jpca.6b12654. [DOI] [PubMed] [Google Scholar]
- Teng A. P.; Crounse J. D.; Wennberg P. O. Isoprene peroxy dynamics. J. Am. Chem. Soc. 2017, 139, 5367–5377. 10.1021/jacs.6b12838. [DOI] [PubMed] [Google Scholar]
- Wayne R. P.Chemistry of Atmospheres, 3rd ed.; Oxford University Press, 2000. [Google Scholar]
- Atkinson R.; Baulch D. L.; Cox R. A.; Hampson R. F. Jr.; Kerr J. A.; Rossi M. J.; Troe J. Evaluated kinetic, photochemical and heterogeneous data for atmospheric chemistry: supplement V, IUPAC subcommittee on gas kinetic data evaluation for atmospheric chemistry. J. Phys. Chem. Ref. Data 1997, 26, 521–1011. 10.1063/1.556011. [DOI] [Google Scholar]
- Burkholder J. B.; Sander S. P.; Abbatt J. P. D.; Barker J. R.; Huie R. E.; Kolb C. E.; Kurylo M. J.; Orkin V. L.; Wilmouth D. M.; Wine P. H.. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies, Evaluation No. 18, JPL Publication 15-10, Jet Propulsion Laboratory, Pasadena, 2015; https://jpldataeval.jpl.nasa.gov/.
- Setokuchi O. Trajectory calculations of OH radical- and Cl atom-initiated reaction of glyoxal: atmospheric chemistry of the HC(O)CO radical. Phys. Chem. Chem. Phys. 2011, 13, 6296–6304. 10.1039/c0cp01942a. [DOI] [PubMed] [Google Scholar]
- Jørgensen S.; Knap H. C.; Otkjær R. V.; Jensen A. M.; Kjeldsen M. L. H.; Wennberg P. O.; Kjaergaard H. G. Rapid Hydrogen Shift Scrambling in Hydroperoxy-Substituted Organic Peroxy Radicals. J. Phys. Chem. A 2016, 120, 266–275. 10.1021/acs.jpca.5b06768. [DOI] [PubMed] [Google Scholar]
- Knap H. C.; Jørgensen S. Rapid Hydrogen Shift Reactions in Acyl Peroxy Radicals. J. Phys. Chem. A 2017, 121, 1470–1479. 10.1021/acs.jpca.6b12787. [DOI] [PubMed] [Google Scholar]
- Iyer S.; He X.; Hyttinen N.; Kurtén T.; Rissanen M. P. Computational and Experimental Investigation of the Detection of HO2 Radical and the Products of Its Reaction with Cyclohexene Ozonolysis Derived RO2 Radicals by an Iodide-Based Chemical Ionization Mass Spectrometer. J. Phys. Chem. A 2017, 121, 6778–6789. 10.1021/acs.jpca.7b01588. [DOI] [PubMed] [Google Scholar]
- Donahue N. M.; Drozd G. T.; Epstein S. A.; Presto A. A.; Kroll J. H. Adventures in Ozoneland: down the rabbit hole. Phys. Chem. Chem. Phys. 2011, 13, 10848–10857. 10.1039/c0cp02564j. [DOI] [PubMed] [Google Scholar]
- Cox R. A.; Coker G. B. Kinetics of the reaction of nitrogen dioxide with ozone. J. Atmos. Chem. 1983, 1, 53–63. 10.1007/BF00113979. [DOI] [Google Scholar]
- Hahn J.; Luther K.; Troe J. Experimental and Theoretical Study of the Temperature and Pressure Dependences of the Recombination Reactions O+NO2(+M)→NO3(+M) and NO2+NO3(+M)→ N2O5 (+M). Phys. Chem. Chem. Phys. 2000, 2, 5098–5104. 10.1039/b005756h. [DOI] [Google Scholar]
- Hyttinen N.; Otkjær R. V.; Iyer S.; Kjaergaard H. G.; Rissanen M. P.; Wennberg P. O.; Kurten T. Computational Comparison of Different Reagent Ions in the Chemical Ionization of Oxidized Multifunctional Compounds. J. Phys. Chem. A 2018, 122, 269–279. 10.1021/acs.jpca.7b10015. [DOI] [PubMed] [Google Scholar]
- Pankow J. F.; Asher W. E. SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds. Atmos. Chem. Phys. 2008, 8, 2773–2796. 10.5194/acp-8-2773-2008. [DOI] [Google Scholar]
- Compernolle S.; Ceulemans K.; Muller J.-F. EVAPORATION: a new vapour pressure estimation method for organic molecules including non-additivity and intramolecular interactions. Atmos. Chem. Phys. 2011, 11, 9431–9450. 10.5194/acp-11-9431-2011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.