

Abstract
Objective
To investigate the diagnostic challenges of congenital myasthenic syndromes (CMS) in adult neuromuscular practice.
Methods
We searched the Mayo Clinic database for patients with CMS diagnosed in adulthood in the neuromuscular clinic between 2000 and 2016. Clinical, laboratory, and electrodiagnostic data were reviewed.
Results
We identified 34 patients with CMS, 30 of whom had a molecular diagnosis (14 DOK7, 6 RAPSN, 2 LRP4, 2 COLQ, 2 slow-channel syndrome, 1 primary acetylcholine receptor deficiency, 1 AGRN, 1 GFPT1, and 1 SCN4A). Ophthalmoparesis was often mild and present in 13 patients. Predominant limb-girdle weakness occurred in 19 patients. Two patients had only ptosis. Age at onset ranged from birth to 39 years (median 5 years). The median time from onset to diagnosis was 26 years (range 4–56 years). Thirteen patients had affected family members. Fatigable weakness was present when examined. Creatine kinase was elevated in 4 of 23 patients (range 1.2–4.2 times the upper limit of normal). Repetitive nerve stimulation revealed a decrement in 30 patients. Thirty-two patients were previously misdiagnosed with seronegative myasthenia gravis (n = 16), muscle diseases (n = 15), weakness of undetermined cause (n = 8), and others (n = 4). Fifteen patients received immunotherapy or thymectomy without benefits. Fourteen of the 25 patients receiving pyridostigmine did not improve or worsen.
Conclusion
Misdiagnosis occurred in 94% of the adult patients with CMS and causes a median diagnostic delay of nearly 3 decades from symptom onset. Seronegative myasthenia gravis and muscle diseases were the 2 most common misdiagnoses, which led to treatment delay and unnecessary exposure to immunotherapy, thymectomy, or muscle biopsy.
Congenital myasthenic syndromes (CMS) encompass a group of underdiagnosed but treatable hereditary disorders of the neuromuscular junction1 that typically present with neonatal to early childhood onset with fatigable weakness affecting ocular, bulbar, and limb-girdle muscles. Some patients may have predominant limb-girdle weakness with sparing or subtle involvement of the oculobulbar muscles (limb-girdle subtype [LG-CMS]) or at a later age at onset.1 Mutations in at least 30 genes have been linked to CMS, but 40% of patients with CMS remain genetically undiagnosed.2,3 An abnormal decrement on low-frequency repetitive nerve stimulation (RNS) or an increased jitter on single-fiber EMG (SF-EMG) confirms the underlying neuromuscular transmission defect in CMS, but these electrodiagnostic findings are like those in myasthenia gravis (MG).1,4 A repetitive compound muscle action potential (CMAP) after a single nerve stimulus can be seen in endplate acetylcholine esterase deficiency or slow-channel CMS but not in MG.1 The distinction between CMS and seronegative MG is clinically and electrophysiologically challenging.1,4 Distinguishing CMS from seronegative MG can prevent patients from undergoing unnecessary immunotherapy and thymectomy and allow proper pharmacologic treatment.
The diagnosis of CMS in a pediatric population was often delayed to the mean age of 4 years, despite mean age at symptom onset being 1 year.5 Approximately 80% of patients in the pediatric cohort were misdiagnosed. Similar data for adult patients with CMS are lacking. Here we review the data of adult patients with CMS at our institution to ascertain the delay in diagnosis and to identify any feature that might have obviated the delay.
Methods
Patient selection
We searched the Mayo Clinic database for adult patients (≥18 years old) with a diagnosis of a CMS seen in neurology clinics at the Mayo Clinic (Rochester, MN) between January 1, 2000, and December 31, 2016. Patients were diagnosed with CMS if they had 1 or more of the following features: (1) mutations in known CMS genes; (2) ≥1 similarly affected family members; (3) repetitive CMAP without signs of organophosphate intoxication or excessive exposure to acetylcholinesterase inhibitors; (4) improvement with fluoxetine; or (5) positive response to 3,4-DAP in the absence of diagnostic findings (facilitation and/or voltage gated calcium channel antibody) of Lambert-Eaton myasthenic syndrome. We excluded patients who were diagnosed with CMS before 18 years of age. Clinical, laboratory, electrodiagnostic, radiologic, and pathologic information was extracted by retrospective chart review. We used the term classic CMS to indicate patients with ophthalmoparesis because extraocular muscle involvement is a common finding in CMS, although not always present (e.g., rapsyn-CMS).6 We defined LG-CMS as CMS with limb-girdle weakness and normal extraocular movements. The severity of weakness was graded on a Medical Research Council scale (5 = no weakness, 4+ = subtle, 4 = mild, 3 = moderate, 0–2 = severe).
Fatigability
Fatigability on clinical examination was defined as worsening ptosis after a 1-minute sustained upward gaze, the inability to maintain arm abduction at 90° for at least 1 minute or increased weakness of the arm abductor muscles after a 1-minute arm abduction, or difficulty performing 10 consecutive squats or worsening of hip flexor weakness after 10 consecutive squats.
Electrodiagnostic testing
Electrophysiologic studies were performed according to the standard methods used in the EMG laboratory at the Mayo Clinic. Most patients underwent 2-Hz repetitive stimulation of at least 2 of the following nerve-muscle pairs: facial nerve–nasalis, peroneal nerve–tibialis anterior, spinal accessory nerve–trapezius, and ulnar nerve–abductor digiti minimi. Rare patients also underwent repetitive stimulation of axillary, femoral, musculocutaneous, or radial nerves at the discretion of electromyographers. Decremental EMG response was defined as a >10% decrease in amplitude or area of the fourth CMAP compared to the first CMAP on 2-Hz RNS of selected motor nerves. Repetitive CMAP was defined as the presence of >1 CMAP evoked by a single nerve stimulus. When a muscle showed a reduced CMAP amplitude and a decremental response, facilitation was evaluated after a 10-second maximal volitional activity of that specific muscle. In the absence of decremental response, SF-EMG and a conditioning 10-Hz RNS for 5 minutes (brief interruption every 1.5 minutes to conduct 2-Hz RNS) were performed to search for neuromuscular transmission defects. If the CMAP amplitude declined with 10-Hz RNS over 5 minutes, the CMAP amplitude elicited by single stimuli was monitored for the next 10 minutes or until the CMAP amplitude returned to baseline.
Statistical analysis
Descriptive summaries are presented as frequencies and percentages for categorical variables and as median and ranges for continuous variables. The Fisher exact test was used to compare the frequencies of clinical and electrophysiologic features between patients with LG-CMS and patients with other CMS. The Mann-Whitney U test was used to compare continuous variables between these 2 groups. Values of p < 0.05 were considered significant.
Standard protocol approvals, registrations, and patient consents
The Mayo Clinic Investigational Review Board approved the study. The patients included in this study had given consent to review of their medical records for research purposes.
Data availability statement
No unpublished data related to this study are publicly available.
Results
Genotype and phenotype of undiagnosed adult patients with CMS at presentation
We identified 34 patients from 33 unrelated families. Thirty patients received a genetic diagnosis of a specific CMS, including 14 DOK7, 6 RAPSN, 2 LRP4 (in 2 sisters), 2 COLQ, 2 slow-channel (1 CHRNA1 and 1 CHRND), 1 primary acetylcholine receptor (AChR) deficiency (CHRNE), and single patients with mutations in AGRN, GFPT1, and SCN4A. Among the 30 patients with genetically characterized CMS, 26 underwent Sanger sequencing of individual genes on a research basis, and 4 underwent next-generation sequencing of CMS-related genes at a commercial laboratory. Listing the molecular variants for each patient is beyond the scope of this article, but most of them were previously reported by the authors.6–10 Mutations identified by next-generation sequencing in 4 patients (3 DOK7 and 1 RAPSN) were known pathogenic mutations in 2 and a combination of known pathogenic and novel, predicted pathogenic (frameshift) mutations in others. Among 4 patients without a molecular diagnosis, CMS was diagnosed on the basis of a positive family history (n = 1), presence of repetitive CMAP (n = 1), and responsiveness to 3,4-DAP therapy without diagnostic features of Lambert-Eaton myasthenic syndrome (n = 2). Thirteen patients reported other family members with similar symptoms. No patient had antibodies directed against the AChR or muscle-specific kinase (MuSK). No patient underwent LRP4 antibody testing.
Figure 1 shows the patients with a molecular diagnosis of CMS. Among 13 patients with classic CMS, extraocular muscle impairment was graded as mild in 10 patients, moderate in 2 patients (1 SCN4A and 1 primary AChR deficiency), and severe in 1 patient (genetically unknown). All but 2 patients with classic CMS also had ptosis, which was graded as mild in 10 patients and moderate in 1 patient. Among 19 patients with LG-CMS, 11 patients had no ptosis and 8 patients had ptosis (7 mild and 1 moderate). Two patients (1 DOK7 and 1 RAPSN) had only ptosis with no limb or extraocular muscle weakness. Facial or bulbar weakness was observed in 11 patients with classic CMS (8 mild, 2 moderate, and 1 severe weakness), 8 patients with LG-CMS with ptosis (5 mild and 3 moderate weakness), and 2 patients with LG-CMS without ptosis (2 subtle weakness). Fatigability was present in all 30 patients examined, including fatigable arm elevation (n = 26) with median arm elevation time of 21 seconds (range 5–60 seconds, data available in 18 patients), fatigable ptosis (n = 3), and difficulty with squatting (n = 3) with a median of 10 squats (range 4–10). Leg elevation time was recorded in 2 patients (15 and 20 seconds).
Figure 1. Molecular diagnosis in an adult CMS cohort with classic or LG phenotype or patients with isolated ptosis.
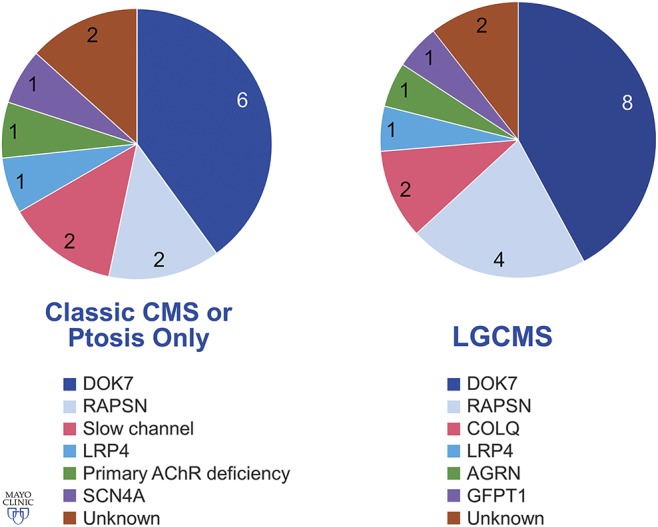
AChR = acetylcholine receptor; CMS = congenital myasthenic syndrome; LG = limb girdle. Used with permission of Mayo Foundation for Medical Education and Research. All rights reserved.
Diagnostic delay and misdiagnosis
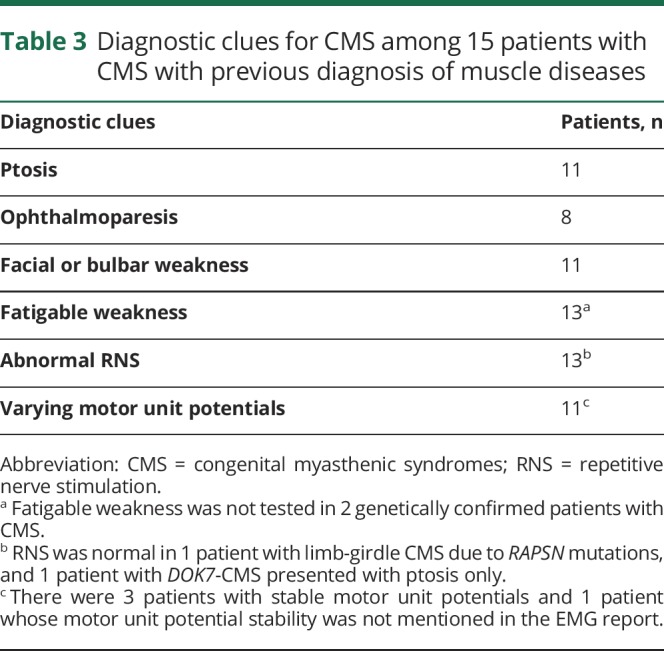
Table 1 shows median age at symptom onset, age at diagnosis, and time from symptom onset to diagnosis in each group of patients. Symptoms first manifested at or before 5 years of age in 28 patients (11 patients had onset at birth or during infancy) and between 6 and 10, 11 and 19, 20 and 29, and 30 and 39 years of age in 2, 2, 1, and 1 patient, respectively. The initial presenting symptoms included delayed crawling or sitting (n = 2); delayed walking (n = 7); slow runner, frequent falls, or inability to keep up with peers (n = 14); ptosis (n = 5); dyspnea (n = 3); dysphagia (n = 2); fatigue (n = 2); and difficulty with heavy lifting (n = 1). Parents reported decreased fetal movements in 1 patient. Two patients described >1 initial symptom. Among 19 female patients, 8 (2 DOK7, 2 RAPSN, 2 slow-channel, 1 AGRN, and 1 genetically undiagnosed CMS) manifested or reported significant worsening of weakness during pregnancy. The median time from symptom onset to diagnosis was 30.5 years for patients who manifested during the first year of age and 24.5 years for patients who presented after infancy (p = 0.659). In only 7 of 34 patients, including 2 sisters diagnosed at our institution with LRP4-CMS, the possibility of CMS was previously entertained because they had no antibodies against AChR and MuSK and on account of an affected sibling (LRP4-CMS sisters). Thirty-two patients were previously misdiagnosed with other disorders (figure 2). Eight patients had >1 provisional diagnosis. Table 2 highlights diagnostic clues in 16 patients with genetically proven CMS with provisional diagnosis of seronegative MG. Table 3 shows diagnostic clues in 15 patients (13 genetically proven and 2 genetically undiagnosed) with a provisional diagnosis of myopathy or muscular dystrophy.
Table 1.
Comparison between LG-CMS and classic CMS or CMS with isolated ptosis

Figure 2. Misdiagnosis in an adult CMS cohort with classic or LG phenotype or patients with isolated ptosis.

CMS = congenital myasthenic syndrome; LG = limb girdle. Used with permission of Mayo Foundation for Medical Education and Research. All rights reserved.
Table 2.
Diagnostic clues for CMS among 16 genetically diagnosed patients with CMS presenting with the diagnosis of seronegative MG

Table 3.
Diagnostic clues for CMS among 15 patients with CMS with previous diagnosis of muscle diseases
Laboratory and electrodiagnostic features
Creatine kinase (CK) level was elevated in 4 of 23 patients tested (2 LRP4, 1 DOK7, and 1 GFPT1). The median CK level in those with hyperCKemia was 2.85 times the upper limit of normal (range 1.2–4.2 times the upper limit of normal). Thirty of 33 patients who underwent electrodiagnostic studies at our institution had a decremental response on 2-Hz stimulation (29 patients with standard RNS and 1 SCN4A-CMS patient after a 5-minute conditioning stimulation train at 10 Hz). No patient had facilitation to suggest presynaptic neuromuscular transmission defect. Seven patients who had borderline decrement (10%–15% decrement) or decrement documented in only 1 nerve also underwent SF-EMG at the discretion of electromyographers, all of which were abnormal. Two patients with isolated ptosis did not have decremental response. Two patients with RAPSN-CMS with no decrement on RNS (1 with standard RNS and 1 after the prolonged 10-Hz RNS) had abnormal SF-EMGs. Three patients (2 slow-channel CMS and 1 with an unknown genetic defect) had repetitive CMAP. One patient with DOK7-CMS who had abnormal SF-EMG at another institution did not undergo electrodiagnostic studies at the Mayo Clinic. Needle EMG revealed small and varying motor unit potentials in 25 patients, while the others had normal or small motor unit potentials, most of which were varying. Fibrillation potentials were observed in only 2 patients (1 GFPT1 with tubular aggregates and 1 LRP4). Data on RNS performed earlier at other institutions were available in 14 patients, 11 of whom reportedly showed a decrement; of the 3 patients with normal RNS, 1 had abnormal SF-EMG and 2 had decremental response at our institution.
Muscle pathology
Twenty-four patients underwent a muscle biopsy during their evaluation. Among 17 muscle biopsies of genetically confirmed CMS that were either performed at our institution or performed elsewhere but reviewed by us, we revealed normal findings (6), nonspecific myopathic changes (1 DOK7 and 1 LRP4), and tubular aggregates (1 GFPT1). Three patients with myopathic changes on biopsy had hyperCKemia (range 1.71–4.2 times the upper limit of normal). Other myopathologic findings included type 1 fiber preponderance (n = 5), type 2 fiber atrophy (n = 2), and features suggestive of denervation atrophy (n = 3). Three of 4 patients with genetically undiagnosed CMS underwent muscle biopsy (1 was normal, 1 showed type 1 fiber atrophy, and 1 showed type 2 fiber atrophy).
Treatment
Fifteen patients with genetically confirmed CMS (previous diagnosis of seronegative MG [n = 13], polymyositis [n = 1], and myopathy not otherwise specified [n = 1]) had undergone immunosuppressant or immunomodulatory therapy (5 prednisone monotherapy, 5 prednisone and IV immunoglobulin [IVIG], 2 IVIG monotherapy, 1 prednisone and plasma exchange, 1 IVIG and plasma exchange, and 1 prednisone, IVIG, and plasma exchange) or thymectomy with or without immunosuppressive therapy (1 thymectomy alone and 2 thymectomy and immunosuppressive agents) before our evaluation. All except 3 patients reported no improvement from immunotherapy. Two patients with DOK7-CMS reported subjective improvement with prednisone, and 1 patient with slow-channel CMS reported subjective improvement with IVIG. Responses to nonimmunomodulatory therapy in all 34 patients are summarized in table 4. The degree of clinical improvement with nonimmunomodulatory therapy was available in some patients, and most were previously reported by the authors.6–10 Among 4 nonambulatory patients (table 1), we had follow-up data on 3 patients, including 2 patients (1 AGRN and 1 COLQ) who were able to walk independently after the introduction of appropriate treatment and 1 patient (DOK7) who became physically more active and experienced much less fatigability with therapy.
Table 4.
Clinical responses to nonimmunotherapeutic drugs in adult CMS cohort

Discussion
Nearly 90% of patients in our adult CMS cohort received a molecular diagnosis in our clinic compared to 60% in a combined cohort of adult and pediatric patients with CMS.2,3 DOK7 and RAPSN mutations were the most common causes of CMS in our cohort, accounting for ≈57% and 20% of the genetically identified cases, respectively. The proportion of DOK7-CMS in our adult CMS cohort is 6 times higher than previously reported in a cohort of combined adult and pediatric patients with CMS,11–14 while there is no significant difference in the proportion of RAPSN-CMS between our adult CMS cohort and a previously reported CMS cohort of combined adult and pediatric patients.11,14 Cases of LG-CMS are overrepresented in our adult CMS group (56%) compared to only 6.5% of the pediatric CMS cohort with the LG-CMS phenotype.5 Mutations in DOK7 and RAPSN are known to cause the LG-CMS phenotype,3,6,15 so our finding is not surprising. However, there may be an element of referral bias in the high proportion of patients with LG-CMS in our adult cohort. Such patients may constitute a greater diagnostic challenge and hence are more likely to be referred to a tertiary center for evaluation. A milder phenotype of DOK7-CMS and RAPSN-CMS could also explain the overrepresentation of these cases of CMS in our adult CMS cohort.
As table 1 shows, the median age at onset was 1.5 years in the classic CMS group and 5 years in the LG-CMS group (p < 0.05). While ≈80% of our patients had presented in early childhood (≤5 years old), only 32% of patients manifested within the first year of life. Patients with classic CMS developed symptoms at birth or had developmental delay more frequently than patients with LG-CMS (p < 0.05). In contrast to pediatric patients with CMS, our adult patients with CMS had milder weakness.5 Among all adult patients diagnosed in our neuromuscular clinic, only 10% were nonambulatory and 15% required feeding tube or respiratory support compared to 35% and 50% of pediatric patients,5 respectively.
Our study highlights the significant delay in the diagnosis journey of patients with CMS and should alert physicians to consider CMS as a concrete potential etiology of weakness in the adult neuromuscular patient population. The time lag to diagnosis in CMS is in keeping with previous studies, which have shown a delayed diagnosis even in children.4,5 Such a diagnostic delay is magnified in the adult neuromuscular clinical practice with a median time from onset to diagnosis in our adult CMS cohort of 29 years in classic CMS and 24 years in LG-CMS. Misdiagnosis occurred in 94% of our adult patients with CMS (figure 2) compared to the reported 80% in a pediatric CMS cohort.5 The most common provisional diagnoses in our cohort were seronegative MG (47%) and myopathies (44%) of various types, including muscular dystrophies. Conversely, in the pediatric CMS cohort, <10% of patients were misdiagnosed as having MG and nearly 60% as having congenital myopathies.5 The proportion of patients with CMS misdiagnosed with muscle diseases is similar between the classic and LG-CMS groups. This could be explained by the mildness of the ophthalmoparesis in most of our patients with the classic CMS phenotype. Misdiagnosis of muscle diseases led to unnecessary muscle biopsy and deprived patients of available effective treatment (table 4). Because all patients misdiagnosed with muscle diseases in our cohort had fatigable weakness when examined, bedside fatigability testing should be performed in all patients with proximal weakness despite the absence of ptosis or ophthalmoparesis. The presence of fatigable weakness should prompt the clinicians to consider a possibility of both acquired and hereditary neuromuscular junction disorders. HyperCKemia, not present in the majority of our adult patients with CMS but reported in some cases of CMS and ranging from slight to marked, can further misdirect the diagnosis of CMS.16,17 Needle EMG studies typically show short-duration and low-amplitude motor unit potentials in most patients, which can suggest a myopathy. Motor unit instability, suggestive of neuromuscular junction dysfunction, was not noted in all patients, perhaps because it was subtle or overlooked. Muscle biopsy from patients with CMS may reveal myopathic changes such as core- or minicore-like structures, tubular aggregates, autophagic vacuoles, or nonspecific myopathic changes, in addition to type 1 fiber preponderance or fiber type disproportion.5,18–21
In our cohort, 13 of 16 patients (81%) misdiagnosed with seronegative MG were unnecessarily exposed to immunotherapy, thymectomy, or both. The clinical history is crucial in distinguishing seronegative MG from CMS. In ≈80% of our patients with CMS, the symptoms appeared in early childhood. This is consistent with the international consensus guidance for the management of MG that recommends considering the diagnosis of CMS in all children diagnosed with seronegative MG.22 Indeed, sequencing of the CMS genes would help distinguish most CMS from juvenile autoimmune myasthenia.1,23 A positive family history favors CMS, but only 5 patients with provisional diagnosis of seronegative MG in our cohort had similarly affected family members. Clinical response to therapy can be informative for diagnosis. Some CMS such as DOK7 myasthenia, the slow-channel syndrome, and COLQ–CMS fail to respond to or are worsened by cholinergic medications, whereas AChR antibody–positive MG is improved with these drugs.7,24 However, refractoriness to cholinergic drugs is also a feature of MuSK antibody–positive MG25; therefore, worsening of the myasthenic symptoms in MuSK antibody–negative MG should also prompt the consideration of CMS before the initiation of immunotherapy and thymectomy. In our cohort, 3 patients with genetically confirmed CMS reported subjective improvement with immunotherapy, a finding that was previously reported.4 A repetitive CMAP can be a diagnostic clue to the slow-channel syndrome or endplate acetylcholinesterase deficiency.1,5 In our cohort, a repetitive CMAP was present in only 2 of 4 patients with these CMS.
Our study underscores the importance of considering the diagnosis of CMS in adults with myasthenic features to warrant appropriate treatment and to prevent exposure to unnecessary interventions. Bedside examination for fatigable weakness, 2-Hz RNS in patients evaluated for myopathies, and the inclusion of genes causative of CMS within myopathy/muscular dystrophy next-generation sequencing panels may overcome the diagnostic challenge of CMS and facilitate appropriate therapy. Prolonged RNS at 10 Hz for 5 minutes or SF-EMG should be performed to search for a defect of neuromuscular transmission in patients with a high index of suspicion for CMS who had normal RNS at 2 Hz. CMS genes mutational screening should be considered in patients with seronegative MG, especially if the patient has childhood symptom onset, positive family history, repetitive CMAP, or worsening or lack of beneficial effect with pyridostigmine. However, the absence of affected family members does not exclude CMS given that only a small proportion of patients have a positive family history.
Glossary
- AChR
acetylcholine receptor
- CK
creatine kinase
- CMAP
compound muscle action potential
- CMS
congenital myasthenic syndromes
- IVIG
intravenous immunoglobulin
- LG-CMS
limb-girdle congenital myasthenic syndromes
- MG
myasthenia gravis
- MuSK
muscle-specific kinase
- RNS
repetitive nerve stimulation
- SF-EMG
single-fiber EMG
Author contributions
Dr. Kao: acquisition, analysis, and interpretation of data and wrote the first draft of the manuscript. Dr. Milone, Dr. Selcen, Dr. Shen, and Dr. Engel: critical revision of the manuscript for important intellectual content. Dr. Liewluck: study concept and design, analysis and interpretation of data, statistical analysis, and study supervision.
Study funding
No targeted funding reported.
Disclosure
The authors report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
Publication history
Received by Neurology May 9, 2018. Accepted in final form July 27, 2018.
References
- 1.Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. Lancet Neurol 2015;14:461. [DOI] [PubMed] [Google Scholar]
- 2.Nicole S, Azuma Y, Bauche S, Eymard B, Lochmuller H, Slater C. Congenital myasthenic syndromes or inherited disorders of neuromuscular transmission: recent discoveries and open questions. J Neuromuscul Dis 2017;4:269–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Connor E, Topf A, Zahedi R, et al. Clinical and research strategies for limb-girdle congenital myasthenic syndromes. Ann NY Acad Sci 2018;1412:102–112. [DOI] [PubMed] [Google Scholar]
- 4.Garg N, Yiannikas C, Hardy TA, et al. . Late presentations of congenital myasthenic syndromes: how many do we miss? Muscle Nerve 2016;54:721–727. [DOI] [PubMed] [Google Scholar]
- 5.Kinali M, Beeson D, Pitt MC, et al. Congenital myasthenic syndromes in childhood: diagnostic and management challenges. J Neuroimmunol 2008;201–202:6–12. [DOI] [PubMed] [Google Scholar]
- 6.Milone M, Shen XM, Selcen D, et al. Myasthenic syndrome due to defects in rapsyn: clinical and molecular findings in 39 patients. Neurology 2009;73:228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liewluck T, Selcen D, Engel AG. Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and Dok-7 myasthenia. Muscle Nerve 2011;44:789–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Selcen D, Milone M, Shen XM, et al. Dok-7 myasthenia: phenotypic and molecular genetic studies in 16 patients. Ann Neurol 2008;64:71–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selcen D, Shen XM, Milone M, et al. GFPT1-myasthenia: clinical, structural, and electrophysiologic heterogeneity. Neurology 2013;81:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Selcen D, Ohkawara B, Shen XM, McEvoy K, Ohno K, Engel AG. Impaired synaptic development, maintenance, and neuromuscular transmission in LRP4-related myasthenia. JAMA Neurol 2015;72:889–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engel AG, Shen XM, Selcen D. The unfolding landscape of the congenital myasthenic syndromes. Ann NY Acad Sci 2018;1413:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palace J, Lashley D, Newsom-Davis J, et al. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain 2007;130:1507–1515. [DOI] [PubMed] [Google Scholar]
- 13.Barisic N, Chaouch A, Muller JS, Lochmuller H. Genetic heterogeneity and pathophysiological mechanisms in congenital myasthenic syndromes. Eur J Paediatr Neurol 2011;15:189–196. [DOI] [PubMed] [Google Scholar]
- 14.Natera-de Benito D, Topf A, Vilchez JJ, et al. Molecular characterization of congenital myasthenic syndromes in Spain. Neuromuscul Disord 2017;27:1087–1098. [DOI] [PubMed] [Google Scholar]
- 15.Natera-de Benito D, Bestue M, Vilchez JJ, et al. Long-term follow-up in patients with congenital myasthenic syndrome due to RAPSN mutations. Neuromuscul Disord 2016;26:153–159. [DOI] [PubMed] [Google Scholar]
- 16.Ohno K. Is the serum creatine kinase level elevated in congenital myasthenic syndrome?. J Neurol Neurosurg Psychiatry 2016;87:801. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez Cruz PM, Belaya K, Basiri K, et al. Clinical features of the myasthenic syndrome arising from mutations in GMPPB. J Neurol Neurosurg Psychiatry 2016;87:802–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein A, Pitt MC, McHugh JC, et al. DOK7 congenital myasthenic syndrome in childhood: early diagnostic clues in 23 children. Neuromuscul Disord 2013;23:883–891. [DOI] [PubMed] [Google Scholar]
- 19.Senderek J, Muller JS, Dusl M, et al. Hexosamine biosynthetic pathway mutations cause neuromuscular transmission defect. Am J Hum Genet 2011;88:162–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cossins J, Belaya K, Hicks D, et al. Congenital myasthenic syndromes due to mutations in ALG2 and ALG14. Brain 2013;136:944–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liewluck T, Shen XM, Milone M, Engel AG. Endplate structure and parameters of neuromuscular transmission in sporadic centronuclear myopathy associated with myasthenia. Neuromuscul Disord 2011;21:387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for management of myasthenia gravis: executive summary. Neurology 2016;87:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Della Marina A, Trippe H, Lutz S, Schara U. Juvenile myasthenia gravis: recommendations for diagnostic approaches and treatment. Neuropediatrics 2014;45:75–83. [DOI] [PubMed] [Google Scholar]
- 24.Witting N, Vissing J. Pharmacologic treatment of downstream of tyrosine kinase 7 congenital myasthenic syndrome. JAMA Neurol 2014;71:350–354. [DOI] [PubMed] [Google Scholar]
- 25.Morren J, Li Y. Myasthenia gravis with muscle-specific tyrosine kinase antibodies: a narrative review. Muscle Nerve 2018;58:344–358. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
No unpublished data related to this study are publicly available.