

Abstract
The induction of mitochondrial biogenesis could potentially alleviate mitochondrial and muscle disease. We show here that dimethyl fumarate (DMF) dose-dependently induces mitochondrial biogenesis and function dosed to cells in vitro, and also dosed in vivo to mice and humans. The induction of mitochondrial gene expression is more dependent on DMF's target Nrf2 than hydroxycarboxylic acid receptor 2 (HCAR2). Thus, DMF induces mitochondrial biogenesis primarily through its action on Nrf2, and is the first drug demonstrated to increase mitochondrial biogenesis with in vivo human dosing. This is the first demonstration that mitochondrial biogenesis is deficient in Multiple Sclerosis patients, which could have implications for MS pathophysiology and therapy. The observation that DMF stimulates mitochondrial biogenesis, gene expression and function suggests that it could be considered for mitochondrial disease therapy and/or therapy in muscle disease in which mitochondrial function is important.
Introduction
Inheritance of defects in mitochondrial genes causes mitochondrial disease (1); and at the current time there is no effective or approved therapy for mitochondrial disease. One therapeutic strategy for mitochondrial disease is to increase mitochondrial biogenesis, the idea being that a small defect in function might be ameliorated by increased mitochondrial mass or function overall (2).
The co-transcriptional regulation factor peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) is a well-known marker of mitochondrial biogenesis (3). PGC1α induces the expression of the transcription factors, nuclear respiration factor 1 (NRF1) (4). NRF1 was initially identified to regulate nuclear-encoded mitochondrial complex expression (5). However, it has more recently been observed to be involved in mitochondrial replication and even drive the expression of mitochondrially encoded genes (6,7). Together, PGC1α and NRF1 mediate the expression of mitochondrial transcription factor A (TFAM), a major regulator of mitochondrial replication and transcription (8,9). Also, expression of TFAM has been shown to be proportional to alterations in mtDNA copy number (10). Thus, TFAM and NRF1 are robust markers of mitochondrial proliferation.
Dimethyl fumarate (DMF) is known for its anti-inflammatory and cytoprotective properties (11,12). It is currently used to treat multiple sclerosis (MS) and psoriasis and is marketed under the name Tecfidera (13) and Fumaderm (14), respectively. DMF is known to stimulate the activity of the transcription factor, nuclear factor (erythroid-derived 2)-like 2 (Nrf2, also known as NFE2L2) and the G protein coupled receptor, hydroxycarboxylic acid receptor 2 (HCAR2) (15).
Nrf2 helps to maintain cellular redox homeostasis by regulating a number of genes involved in antioxidant protection including, but not limited to, glutathione (16,17), thioredoxin (18), heme oxygenase (HO1), and NAD(P)H dehydrogenase (NQO1) (19,20). It was previously discovered that monomethyl fumarate (MMF), a metabolite of DMF, mediates Nrf2 activation by modifying numerous cysteine (Cys) residues of the Kelch-like ECH-associated protein 1 (KEAP1). The modification of KEAP1 then drives the dissociation and translocation of Nrf2 into the nucleus, initiating the transcription of many phase II antioxidant enzymes that contain the antioxidant response element (ARE) promoter sequence (21–23). It is known that knocking out Nrf2 is detrimental to mitochondrial health, and activation of the Nrf2 pathway by DMF is thought to be beneficial to mitochondria by mitigating reactive oxygen species (ROS)-related damage (24).
In addition, Nrf2 is also thought to be involved in the induction of mitochondrial biogenesis. Specifically, Nrf2 is known to positively regulate NRF1 by binding to the four ARE promoter sequences of NRF1, leading to the activation of NRF1 mediated mitochondrial biogenesis pathway (25). In concurrence, a study by Shen et al. 2008 has shown that treatment of murine 3T3-L1 adipocytes with (R)-α-lipoic acid and acetyl-L-carnitine, known activators of Nrf2 induces mitochondrial proliferation and observed increased mtDNA, mitochondrial complex expression, oxygen consumption, and increased expressions of mitochondrial biogenesis biomarkers such as PGC1α, TFAM and NRF1 (26).
HCAR2 is involved in the regulation of anti-inflammatory activity and fat metabolism. DMF's major metabolite MMF is known to be a potent agonist of HCAR2 (27). The effects of DMF on HCAR2 remain largely unclear. However, DMF's protective effect in MS may include its metabolism to MMF that agonizes HCAR2 to cause anti-inflammatory activity in the mouse EAE model of MS (15).
As a consequence of screening drugs for effect on mitochondrial functions, a group of mitoactive drugs were identified including DMF (28). We studied the effects of DMF on mitochondria in human fibroblasts, C57BL/6 mice and human MS patients. We report a novel mitochondrial biogenesis effect of DMF; to increase mtDNA copy number and expression of mitochondria complexes in vitro and in vivo, in cells and mice and humans. Furthermore, we note a mitochondrial gene expression deficit in human MS patients, which could potentially be used as a biomarker of disease severity, and that there is a mitochondrial biogenesis defect in MS, which is rescued by DMF. Mechanistically by knockdown, we show that DMF's mitochondrial biogenesis effect is attributable to Nrf2 rather than HCAR2.
Currently there is no FDA-approved drug for mitochondrial disease, and a drug that increases mitochondrial biogenesis could have an impact for muscle disease as well. DMF is already approved for other indications, and because it increases mitochondrial mass and activity in vitro and in vivo, it could potentially have impact for diseases with mitochondrial deficiency.
Results
DMF increases mitochondrial DNA copy number, biogenesis marker and subunit expression, in human fibroblasts
Healthy human fibroblast cells were treated with 0.1% DMSO (vehicle), 3μM, 10μM or 30μM DMF for 48 h. Mitochondrial DNA (mtDNA) copy number was analyzed from total DNA isolates by measuring the ratio of mitochondrial to nuclear DNA (mtDNA/nDNA). While no changes were observed in the 3μM-dosed fibroblasts, 10μM and 30μM DMF treatment showed a 1.51 fold (P < 0.036, n = 3) and 1.75 fold (P < 0.0098, n = 3) increase in mtDNA copy number compared to vehicle control (Fig. 1A). Similarly, expression of mitochondrial biogenesis marker TFAM increased at 10μM and 30μM DMF when compared to vehicle control (Fig. 1B). In addition, we observed an increase in mitochondrial protein VDAC and mt-ND6 at 30μM DMF compared to vehicle control by western blot analysis (Supplementary Material, Fig. S1)
Figure 1.

DMF increases mtDNA copy number, mitochondrial biogenesis marker expression and mitochondrial complex expression in human fibroblasts. Human fibroblast cells were treated with 0.1% DMSO vehicle, 3µM, 10µM or 30µM DMF for 48 h. (A) qPCR analysis of mitochondrial DNA copy number over nuclear DNA copy number (MT-TL1/B2M). (B) qPCR analysis of TFAM normalized to β-Actin. (C) qPCR analysis of two subunits of complex 1-5. Bars represent averages ± standard deviations (n = 3, *P < 0.05, **P < 0.01,***P < 0.001). One way ANOVA with post-hoc dunnett test comparing all groups to vehicle, was used to measure statistical significance.
To elucidate whether increased mitochondrial proliferation affects the abundance of the ETC mitochondrial complexes, qPCR analysis was performed to quantify the mRNA expression of numerous complex subunits: complex 1 subunits MT-ND2 and MT-ND6, complex 2 subunits SDHA and SDHB, complex 3 subunits MT-CO1 and MT-CO2, complex 4 subunits MT-CYB and CYC1, and complex 5 subunits ATP5B and MT-ATP6 (Supplementary Material, Table S1). Consistent with the DMF-dependent increase in mtDNA copy number and TFAM expression, the subunit expression of the mitochondrial complexes also exhibited a dose-dependent response to DMF treatment. Compared to vehicle treated fibroblasts, average induction of all the complexes were significant when dosed with 10μM and 30μM of DMF at 1.42 fold (P < 5.7×10−6, n = 10) and 2.65 fold (P < 4.8×10−6, n = 10) increase (Fig. 1C). Interestingly, the expression of complex 5 subunits was less increased in response to DMF treatment compared to other complex subunits, suggesting that there may be a greater need for complexes 1–4 in maintaining a proton gradient necessary for ATP synthesis during mitochondrial biogenesis. Taken together, human fibroblast cells increased mitochondrial biogenesis measured by mtDNA copy number, mitochondrial proliferator marker TFAM expression and mitochondrial complex expression.
Dimethyl fumarate increases oxygen consumption rate in human fibroblasts
To elucidate the bioenergetic effects of DMF-dependent increases in mtDNA copy number and mitochondrial complex expression in human fibroblasts, oxygen consumption rate (OCR) was measured. Human fibroblasts were treated with 0.1% DMSO, 3μM, 10μM or 30μM DMF for 48 h, and oxygen consumption rate (OCR) was sequentially measured in the presence of oligomycin (complex 5 inhibitor), FCCP (mitochondrial uncoupler), and rotenone/antimycin A (Complex 1/3 inhibitor) (Fig. 2A). The basal OCR reading of fibroblasts treated with 10μM and 30μM DMF showed a relative OCR increase of 1.59 fold (P < 3.1×10−5, n = 8) and 1.66 fold (P < 5.7×10−5, n = 8) compared to vehicle control, respectively (Fig. 2B). The maximal OCR measured after FCCP injection was elevated after treatment of 3μM, 10μM and 30μM DMF with a relative OCR increase of 1.20 fold (P < 3.2×10−3, n = 8), 1.35 fold (P < 3.1×10−5, n = 8), and 1.47 fold (P < 9.1×10−5, n = 8) compared to vehicle control, respectively (Fig. 2C). OCR in the presence of oligomycin and rotenone/antimycin A were not significantly different between the DMF treatment groups and vehicle control (Fig. 2A). Taken together, human fibroblasts showed DMF dose-dependent induction of basal and maximal (FCCP-treated) OCR at the 48-h time point. Consistent with the idea that DMF increases mtDNA copy number and mitochondrial complex expression; the effects of DMF are nullified in the presence of oligomycin and rotenone/antimycin A, which inhibits the mitochondrial electron transport chain that is responsible for mitochondrial oxygen consumption.
Figure 2.

DMF increases basal and maximal mitochondrial oxygen consumption rates. (A) Oxygen consumption rates (OCR) of human fibroblasts treated with 0.1% DMSO vehicle, 3µM, 10µM or 30µM DMF for 48 h. (B) Relative basal oxygen consumption normalized to vehicle-treated fibroblasts, noted as segment B in Figure 2A. (C) Relative maximal oxygen consumption of mitochondrial uncoupled (FCCP) cells normalized to vehicle-treated fibroblasts, noted as segment C in Figure 2A. Bars represent averages ± standard deviations (n = 8, *P < 0.05, **P < 0.01, ***P < 0.001). One way ANOVA with post-hoc dunnett test comparing all groups to vehicle, was used to measure statistical significance.
Dimethyl fumarate increases mitochondrial DNA copy number and mitochondrial complex expression in mice
To understand whether the DMF-dependent mitochondrial biogenesis is only applicable to the human fibroblast cells, we examined the effects of DMF on wild type C57BL/6 mice. The mice were dosed with 10mg/kg DMF daily, and skeletal muscle, cerebellum, liver, and heart tissues were collected after two weeks of treatment. We utilized qPCR to analyze, mtDNA copy number as ratio of mt-Nd1 to Cftr and mitochondrial complex expression, in these tissues. Of the four tissues tested, skeletal muscle, cerebellum and liver tissues showed increase in mtDNA copy number by1.45 fold (P < 0.021, n = 6), 1.29 fold (P < 0.010, n = 6) and 1.34 fold (0.027, n = 6) respectively. Heart tissue showed no significant change in mtDNA copy number (Fig. 3A). While DMF seems to induce mitochondrial replication as indicated by mtDNA copy number, it does not seem to affect all tissues equally and suggests that tissue-specific regulation might contribute to this finding.
Figure 3.

DMF increases mtDNA copy number and mitochondrial complex expression in mouse skeletal muscle, cerebellum and liver. C57BL/6 mice were intraperitoneally injected daily with 10mg/kg of DMF for two weeks. (A) qPCR analysis of mitochondrial DNA copy number over nuclear DNA copy number (mt-Nd1/Cftr). (B) qPCR analysis of the mitochondrial complex subunits mt-Nd2, mt-Co1 and mt-Atp6 normalized to β-Actin. Bars represent averages ± standard deviations (n = 6, *P < 0.05, **P < 0.01, ***P < 0.001). Student’s t-test was used to measure statistical significance.
For the mitochondrial complex expression, skeletal muscle and cerebellar tissue showed significant induction. Both tissues showed an increase in mt-Co1 (complex 4) and mt-Atp6 (complex 5) expression. mt-Co1 and mt-Atp6 was increased 2.38 fold (P < 0.0029, n = 6) and 1.77 fold (P < 0.029, n = 6) respectively in skeletal muscle, and 1.92 fold (P < 4.5×10−4, n = 6) and 1.82 fold (P < 0.020, n = 6) respectively in cerebellar tissue. Additionally, skeletal muscle showed 4.68 fold (P < 0.0030, n = 6) increase in mt-Nd2 expression as a result of two weeks of DMF treatment (Fig. 3B). Taken together, C57BL/6 mice when dosed with 10mg/kg of DMF for two weeks show increased mtDNA copy number and mitochondrial complex expression in multiple tissues. Interestingly, DMF did not increase mtDNA copy in all tissues equally and the expression of some mitochondrial complexes was more induced than others.
Dimethyl fumarate increases mitochondrial DNA copy number and mitochondrial complex expression in MS patients
DMF is a FDA approved drug, currently being used to treat adult patients with relapsing form of MS. To confirm whether DMF is contributing to mitochondrial biogenesis when given to human patients, we used qPCR to study mitochondrial DNA copy number and mitochondrial complex subunit expression in peripheral blood lymphocytes isolated from 11 MS patients at baseline and those same patients 3 months after DMF treatment, and 10 controls. We see significant increase in mitochondrial DNA copy number by 71% (P < 0.0075, n = 11) in MS patients’ treated with DMF for 3 months compared to its own baseline (Fig. 4A). When compared to healthy control group, MS patient at baseline has decreased mtDNA copy number by 25% (P < 0.062, n = 11) and 3 month DMF treatment seems to rescue the defect by increasing the mitochondrial DNA copy number back to healthy control level (Fig. 4B).
Figure 4.

DMF increases mtDNA copy number and mitochondrial complex expression in MS patients. PBMCs were collected from whole blood of MS patients before and after 3months of DMF treatment and healthy individuals. (A) qPCR analysis of mitochondrial DNA copy number over nuclear DNA copy number (MT-TL1/B2M) in MS patients treated with DMF relative to its own baseline. (B) qPCR analysis of mitochondrial DNA copy number over nuclear DNA copy number (MT-TL1/B2M) in MS patients before and after treatment relative to healthy control group (C) qPCR analysis of mitochondrial complex subunits mt-ND6, mt-CYB, mt-CO2 and mt-ATP6 in MS patients treated with DMF relative to its own baseline. (D) qPCR analysis of average mitochondrial complex mRNA expression of mt-ND6, mt-CYB, mt-CO2 and mt-ATP6 in MS patients before and after treatment relative to healthy control group. Bars represent averages ± standard deviations (n = 11, *P < 0.05, **P < 0.01, ***P < 0.001). Paired t-test (A, C) and One way ANOVA with post-hoc tukey test comparing all groups to each other (B, D), was used to measure statistical significance.
Similarly, DMF treated MS patients’ shows significant increase in mitochondrial complex subunit expression of mt-ND6 (complex 1), mt-CYB (complex 3), mt-CO2 (complex 4) and mt-ATP6 (complex 5) by 3.13 fold (P < 0.0358, n = 12), 2.87 fold (P < 0.016, n = 12), 2.34 fold (P < 0.041, n = 12) and 3.74 fold (P < 0.014, n = 12) respectively, when normalized to its own baseline. (Fig. 4C). We also studied the expressions of mitochondrial genes: mt-ND6, mt-CYB, mt-CO2 and mt-ATP6 in healthy individuals and compared them to MS patients at baseline and 3 months DMF treatment. We see significant defect in average mitochondrial gene expression in MS patients, a 56% (P < 0.0018, n = 11) reduction compared to healthy individuals. Following DMF treatment, MS patients showed significant recovery of 88% in average mitochondrial gene expression (Fig. 4D).
DMF’s induction of mitochondrial proliferation is dependent on Nrf2 more than HCAR2
DMF is known to mediate antioxidant cellular defense by Nrf2 activation, and it suppresses inflammatory signaling by binding to and activating HCAR2 (11,15). To further understand the Nrf2 and HCAR2 dependent effects of DMF on mitochondrial biogenesis, we analyzed changes in mitochondrial proliferation in Nrf2 siRNA knockdown and HCAR2 siRNA knockdown fibroblasts.
The siNrf2 knockdown significantly reduced Nrf2 expression to 2.5% (P < 6.04×10−5, n = 3) of control siRNA treated cells (siCTL). Nrf2 knockdown significantly decreases expression of - NQO1, a downstream target and positive control for Nrf2 activation; TFAM and NRF1, mitochondrial proliferative marker and mt DNA copy number (Fig. 5A and B). DMF treatment of siCNT cells (siCNT + DMF) showed significant induction of Nrf2, NQO1, TFAM, NRF1 and mtDNA copy number compared to siCNT. DMF treatment of siNrf2 cells (siNrf2 + DMF) also significantly increases expression of Nrf2, NQO1 and mtDNA copy number compared to siNrf2 cells. However, the induction of mitochondrial proliferative marker by siNrf2 + DMF is significantly reduced as compared to siCTL + DMF. In addition, there was no significant induction of TFAM and NRF1 in siNrf2 + DMF. Thus, the results indicate that Nrf2 pathway plays a major role in DMF mediated induction of mitochondrial proliferation.
Figure 5.

Stimulation of mitochondrial proliferation and mitochondrial complex transcription, by DMF, is mediated by Nrf2. Human fibroblast cells were treated with control or Nrf2 siRNA for 48 h followed by 0.1% DMSO vehicle, 3µM, 10µM or 30µM DMF treatment for 48 h. (A) qPCR analysis of Nrf2, NQO1 and NRF1 normalized to β-Actin. (B) qPCR analysis of mitochondrial DNA copy number over nuclear DNA copy number (MT-TL/B2M). (C) qPCR analysis of MT-ND2 (complex 1), SDHB (complex 2), CYC1 (complex 3), MT-CO2 (complex 4), and ATP5B (complex 5) normalized to β-Actin. Bars represent averages ± standard deviations (n = 3, *P < 0.05, **P < 0.01, ***P < 0.001). One way ANOVA with post-hoc tukey test comparing all groups to each other, was used to measure statistical significance.
Quantitative PCR analysis was performed to assess the effect of the Nrf2 pathway on mitochondrial complex subunit expression after DMF treatment. With the exception of mitochondrial complex 4 expressions, siNrf2 + DMF cells showed general reduction in mitochondrial complex expression. In addition, siNrf2 basally reduces the expressions of the mitochondrial complexes when vehicle treated (Fig. 5C). It is apparent that Nrf2 plays a key role in basal and DMF-induced transcription of mitochondrial complex. It is however also important to note that inhibition of DMF induced mitochondrial complex expression range from slight inhibition in complex 4 indicated by MT-CO2 to large inhibition in complex 2 indicated by SDHB (Fig. 5C). Taken together, these results indicate dependence of DMF-mediated mitochondrial biogenesis effect on Nrf2 pathway measured by mtDNA copy number and mitochondrial complex expressions.
As mentioned earlier, DMF is also known to suppress inflammatory signal by binding to HCAR2 receptor (11,15). While HCAR2 knockdown significantly reduced HCAR2 expression, DMF treatment of siHCAR2 cells has no significant difference in inducibility of NRF1, mtDNA copy number and mitochondrial complex 2–5 subunit gene expression compared to siCTL + DMF (Supplementary Material, Fig. S2). These results indicate that HCAR2 is not involved in DMF mediated mitochondrial proliferation, unlike Nrf2. Interestingly; in HCAR2 knockdown cells, there was some effect of DMF treatment on inducibility of complex 1 subunit gene (Supplementary Material, Fig. S2C). This induction may be due to an alternative pathway discussed later.
Discussion
Need for mitochondrial disease therapy
Inherited mitochondrial defects cause serious and lethal disease, for which there is no FDA-approved therapy (2). Identifying pharmacological compounds that can safely and dose-dependently increase mtDNA copy number and mitochondrial complex expression has potential benefit for those with mitochondrial disease and multiple muscle disease (29,30). Many muscle diseases depend on mitochondrial function. Muscles contain a paracrystalline formation of mitochondria, whose function is closely tied to overall muscle function. These include not only the mitochondrial myopathies, but also several other muscle dystrophies, including Duchenne dystrophies (31) and ALS (32), which have increasing evidence of mitochondrial involvement. We show here that DMF, an FDA-approved compound, induces mitochondrial biogenesis in healthy human fibroblasts, mouse tissues and humans. DMF dosed systemically in mice clearly produced mitochondrial biogenesis and increased mitochondrial gene expression in muscle, which suggests the potential for ameliorating muscle diseases, which have some mitochondrial pathophysiology as their basis.
Nrf2 and mitochondrial biogenesis
The pharmacological basis of DMF's activity is thought to proceed through its targets Keap1/Nrf2 and the G-protein coupled receptor, HCAR2 (11,33). While Nrf2 is most commonly known as a major regulator of the antioxidant cellular defense (34), we show that Nrf2 is also necessary for basal mitochondrial maintenance and DMF-induced mitochondrial biogenesis. The transcription factor NRF1 is a key regulator of mitochondrial biogenesis (6–8) with involvements in mitochondrial replication (10) and mtDNA transcription (5,35). Our data show that basal NRF1 expression was reduced, and its induction by DMF was strikingly diminished in the Nrf2 knockdown line (Fig. 5A). This observation can be attributed to Nrf2 positive regulation of NRF1 expression by its four ARE motifs (25). Together, the data suggest that DMF activity in part depends on Nrf2-driven NRF1 expression regulating mitochondrial biogenesis. This idea is supported by previous reports showing that Nrf2 binds to the ARE sequence of the NRF1 promoter, inducing mitochondrial biogenesis in cardiomyocytes (25).
In addition to regulating markers of mitochondrial biogenesis, DMF was observed to regulate mitochondrial replication and transcriptions of mitochondrial complexes Nrf2- dependently. We observed a reduction in mtDNA copy number and its reduced inducibility by DMF as a consequence of Nrf2 knock down (Fig. 5B). Similarly, A previous study by Zhang et al. 2013 showed reduction of mtDNA copy number in the livers of Nrf2 knockout mice (36). The reduction of mtDNA copy as a result of Nrf2 knockdown can be a consequence of diminished mitochondrial proliferation. Additionally, knocking down Nrf2 reduced both basal expression and inducibility of mitochondrial complex subunits expressions by DMF (Fig. 5C). Similarly, treatments with Nrf2 activators (R)-α-lipoic acid and acetyl-L-carnitine can promote mitochondrial proliferation and function in adipocytes (26). Taken together, induction of mitochondrial replication and mitochondrial complex expressions by DMF is dependent on Nrf2 pathway.
Aside from promoters and binding, there is also the physiological question of why Nrf2 and mitochondrial biogenesis pathways are co-regulated in both positive and negative directions. Positively, one could imagine that increased fat content in the diet and agonism of the HCAR2 beta-hydroxybutyrate receptor could signal increased mitochondrial biogenesis in order to carry out more active oxidative phosphorylation on the more reduced fatty foodstuff, which in turn would presumably produce more ROS and thus require more Nrf2. In the negative direction, a suppression of Nrf2 results in a decreased antioxidant response, and cells may protect themselves by decreasing mitochondrial number, as mitochondria are a major contributor to ROS production.
DMF’s tissue-specific effects
Similar to the in vitro responses seen in fibroblast cells, two-week intraperitoneal DMF dosing of C57BL/6 mice showed increase in mtDNA copy number and mitochondrial complex expression in vivo (Fig. 3). Three of the four tissues tested: skeletal muscle (gastrocnemius), whole cerebellum, and whole liver, were observed to have increased mtDNA in DMF-treated mice as compared to vehicle-treated mice (Fig. 3A). We additionally observed induction of mitochondrial complexes in the skeletal muscle and cerebellum (Fig. 3B), effects in liver or heart tissues were more minor [not shown]. DMF's tissue-specific effects on mitochondrial gene expression could result from differential expression of its two known targets Nrf2 (37–40) and HCAR2 (41–43).
Contribution of HCAR2 to DMF's effects on mitochondrial complex I
Of the two DMF targets, Nrf2 and HCAR2, our results suggest that HCAR2 plays a smaller role in the mitochondrial biogenesis effect of DMF, except for some small effects on mitochondrial complex 1, or additive effects. When HCAR2 expression is knocked down, complex 1 subunit MT-ND2 became uninducible by DMF (Supplementary Material, Fig. S2). HCAR2 is also known as the NIACR1 (niacin receptor), and thus can detect the bioavailability of nicotinic acid (44), a precursor to nicotinic dinucleotide (NAD+) (45), the major redox substrate of mitochondrial complex 1 (1). Thus HCAR2 knockdown could reduce the signaling of DMF and other HCAR2 agonists such as niacin in a feedforward stimulation of complex 1. But besides complex 1, knockdown of HCAR2 did not affect DMF-dependent mitochondrial gene expression in a major way (Supplementary Material, Fig. S2). It is possible that there is some synergy among DMF's Nrf2 and HCAR2-dependent mitochondrial effects, with Nrf2 the main player but HCAR2 playing an additive role (Fig. 6).
Figure 6.
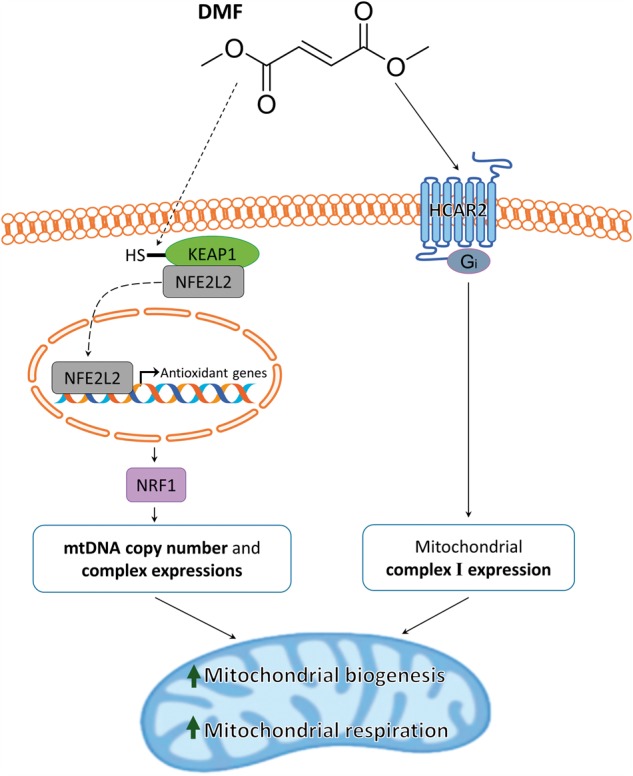
Mechanistic diagram of dimethyl fumarate-induced mitochondrial biogenesis.
Deficient mitochondrial biogenesis as potential mechanism of MS
Mitochondrial dysfunction is considered one of the several causes of axonal neuron degradation in MS (46). Ours is the first reported demonstration that mtDNA copy number and mitochondrial gene expression is decreased in MS blood lymphocytes relative to healthy controls (Fig. 4). This brings up the pathophysiological hypothesis that the neurodegenerative disease MS is the result of deficient mitochondrial biogenesis. There are several connections between MS and mitochondrial disease, especially Leber's hereditary Optic Neuropathy (LHON). It has been demonstrated that MS is both more common and more aggressive in patients bearing mitochondrial LHON mutations (47,48). Furthermore, the clinical severity of LHON mutations has been suggested to be controlled by mitochondrial biogenesis, i.e. LHON carriers that can carry out mitochondrial biogenesis are more protected (49). Putting the data from these disparate studies together, we could form the hypothesis that MS is a disease of defective mitochondrial biogenesis. Furthermore, that the increased severity of MS in patients bearing LHON mutations is the result of a defect in mitochondrial proliferation. This is a testable hypothesis. It also suggests that other agents that increase mitochondrial biogenesis may be of benefit in MS.
Deficient mitochondrial biogenesis as a potential biomarker of MS
Given the observation that mtDNA copy number and gene expression is deficient in blood samples from MS patients, and that mitochondrial biogenesis could be a cause of MS (as hypothesized above), suggests that mtDNA copy number and mitochondrial gene expression could be used as a biomarker of clinical severity of MS, disease form (progressive or relapsing-remitting), and response to treatment. The defect in mtDNA copy number was about 70% residual in MS patients (Fig. 4). The defect in mitochondrial gene expression by QRTPCR averaged about 45% residual, with each individual gene (mtND6, mtCYB, mtCO2, mtATP6) significantly decreased in MS vs CTL comparisons. Thus, blood-based evaluation of mitochondrial biogenesis could apparently be used as a biomarker of MS, and future studies could determine the linearity of the mitochondrial biogenesis effect related to clinical severity, age of onset and response to treatment.
Prospects for DMF as a drug for mitochondrial biogenesis and muscle disease
The findings reported here demonstrate novel mitochondrial proliferative pharmacological properties of the FDA-approved drug dimethyl fumarate, which appear to depend mainly on the Nrf2 pathway (Fig. 6). DMF increases mtDNA copy number (Figs 1A and 3A) and mitochondrial complex mRNA expression (Figs 1C and 3B) in human fibroblasts and WT mice, and rescues a mitochondrial deficit in MS patients (Fig. 4). In contrast to other potential mitoproliferative compounds not clinically available, DMF is an approved drug in US and Europe with demonstrated mitoproliferative activity. DMF is currently used to treat psoriasis, an autoimmune disease, and Multiple Sclerosis, a demyelinating disease (13). In the past, the drug pioglitazone, a thiazolidinedione used to treat diabetic patients, and bezafibrate (2) were shown to have mitochondrial proliferative effect. Pioglitazone was later shown to inhibit mitochondrial complex I (50). DMF on the other hand does not appear to have mitochondrial complex inhibition activity as indicated by induction of both basal and maximal respiration (Fig. 2) while simultaneously increasing mitochondrial DNA copy number and mitochondrial gene expression (Fig. 1). Thus, the FDA-approved drug DMF could be considered for diseases in which there is reduced mitochondrial number and function, including mitochondrial and muscle diseases.
Materials and Methods
Fibroblast cell culture and drug treatment
The healthy human fibroblast cell line AG09429 (Coriell Institute, Camden, NJ, USA) was maintained at 37 °C in a humidified atmosphere with 5% CO2. DMEM (Corning, Inc., Corning, NY, USA) supplemented with 10% fetal bovine serum (JR-Scientific, Woodland, CA, USA), 1x Penicillin-Streptomycin Solution (Corning, Inc., Corning, NY, USA) was used as growth media. Media was changed every two days.
The human fibroblasts were plated in a 12-well format at 0.1×106 cells per well. The cells were incubated with 0.1% DMSO as vehicle control or 3–30 µM of dimethyl fumarate (Sigma-Aldrich, St. Louis, MO, USA) dissolved in DMSO. Total RNA and DNA were extracted following a 48-h incubation period.
Patient consent and blood collection
The local Ethics Committee of the University Federico II of Naples, approved the study. Patients were recruited from the Multiple Sclerosis Center of the Federico II University of Naples. All patients gave written informed consent before any activity linked to the study was started. Healthy controls were recruited at the clinic through students and site personnel. The study included 12 MS patients and 11 healthy individuals. Samples were obtained on the day before and after 3 months of continuous DMF treatment. Whole blood was collected in EDTA containing Leucosep® tubes (Greiner bio-one, Frickenhausen, Germany) and frozen at −80 °C until analysis. PBMCs were isolated from 30 ml of EDTA anticoagulated whole blood for RNA extraction.
Nrf2 and HCAR2 siRNA knockdown in human fibroblast cells
Fibroblast cells were seeded in six-well plates at 0.2×106 cells per well and transfected with negative control siRNA (cat. 12935300, Thermo-Fisher, Waltham, MA, USA), pooled Nrf2 siRNA (cat. HSS107130, HSS181505, HSS181506, Thermo-Fisher, Waltham, MA, USA) or pooled HCAR2 siRNA (cat. L-006688-02-0005, Dharmacon, Lafayette, CO, USA) using Lipofectamine RNAiMAX following manufacturer’s instruction. After a 48-h incubation, subsequent drug treatment was conducted with the cells.
Mouse models, drug treatment, and dissection
C57/bl6 wild-type mice were housed in a vivarium maintained at 22–24 °C and 40–60% relative humidity with a 12-h light/12-h dark cycle. All experimental procedures were approved by the University of California Institutional Animal Care and Use Committee.
The stock dimethyl fumarate solution was made by dissolving 50mg/ml of DMF into DMSO. Prior to injection, 0.5 mg/ml of working DMF solution (1:100 dilution) was made by diluting the stock solution into phosphate-buffered saline with 5% Tween-20 and 5% polyethylene glycol (Sigma-Aldrich, St. Louis, MO, USA). The mice were injected intraperitoneally every day for 14 days with 10 mg/kg of DMF.
The mice were euthanized with CO2 followed by cervical dislocation and tissues were immediately removed then flash frozen with liquid nitrogen. Samples were stored in −80 °C until utilized for experiments.
DNA and RNA extraction
Total DNA was extracted from human fibroblast and mouse tissues using DNeasy plus mini kit and DNeasy blood & tissue kit (Qiagen, Valencia, CA, USA), respectively, following manufacturer’s instruction. DNA was quantified by a NanoDrop 2000c Spectrophotometer (Thermo Scientific, Waltham, MA, USA).
Total RNA was extracted from human fibroblast cells and PBMCs from whole blood using RNeasy plus mini kit (Qiagen, Valencia, CA) and RIboPure RNA Purification Kit (ThermoFisher Scientific, Waltham, MA, USA), respectively following manufacturer’s instruction. RNA quantity and quality were measured by a NanoDrop 2000c Spectrophotometer (Thermo Scientific, Waltham, MA, USA).
Quantitative RT-PCR
cDNA was synthesized from mRNA with iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, USA) per manufacturer’s instruction in a C1000 Touch Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA). A SensiFAST SYBR No-ROX Kit (Bioline, Taunton, MA, USA) was used to perform qPCR on the synthesized cDNA in a Roche Lightcycler 480 (Roche Diagnostics, Indianapolis, IN, USA). The second derivative of the amplification curve was used to determine the cycle threshold, and the data were analyzed by a delta delta CT calculation. Primer sets used in qPCR are listed in Supplementary Material, Table S1.
Measurement of oxygen consumption in DMF-treated fibroblasts by seahorse XF analyzer
Fibroblast cell lines were seeded at a density of 60,000 cells/well in 200µL of culture medium in a 24-well seahorse tissue culture plate (Seahorse Biosciences, Billerica, MA, USA). Following a 24-h incubation, the medium was replaced and incubated with 200 µl of 0.1% DMSO or 3, 10 and 30 µM dimethyl fumarate in 0.1% DMSO. Prior to reading the oxygen consumption, the medium was changed to unbuffered DMEM without phenol red (Corning, Inc., Corning, NY, USA), 10% fetal bovine serum (JR-Scientific, Woodland, CA, USA), 200mm glutamax, 100mM sodium pyruvate, 25mm glucose (Invitrogen, Waltham, MA, USA) and was adjusted to pH 7.4. Cells were pre-equilibrated for 20 min; oxygen consumption rate (OCR) and proton production rate (PPR) were recorded with the Seahorse XF-24 after sequential addition of oligomycin (1µg/ml), FCCP (10µM) and a combination of antimycin A (1µM) and rotenone (1µM) (Sigma-Aldrich, St. Louis, MO, USA). Total protein in each well was measured and protein concentration was used to normalize the readings.
Protein extraction and western blot analysis
Fibroblast cells were homogenized with 1x cell lysis buffer (Cell Signaling Technologies, Danvers, MA, USA) containing 1x Halt phosphatase and protease inhibitor cocktail (Thermo-Fisher, Waltham, MA, USA) and 1% PMSF (Sigma-Aldrich, St. Louis, MO, USA). 50µg of lysates were loaded per lane into 4–12% Bis–Tris gels (Invitrogen, Waltham, MA, USA). Electrophoresis was carried out according to the manufacturer’s recommendations. Following electrophoresis, the proteins were transferred to nitrocellulose membranes by the iBlot device (Invitrogen, Waltham, MA, USA) and blocked with an Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE, USA) for 1 h. Membranes were incubated overnight with the following primary antibodies in blocking buffer: 1:500 dilution mt-ND6 (SC-200667, Santa Cruz Biotech), 1:500 dilution HO1 (ab13248, Abcam), 1:1000 dilution VDAC (D73D12, Cell Signaling Technologies), 1:2000 dilution β-tubulin (T8328, Sigma-Aldrich) and 1:2000 dilution β-actin (A5441, Sigma-Aldrich). Subsequently, the membranes were incubated with a corresponding pair of IRDye 680CW and IRDye 800CW-coupled secondary antibodies (LI-COR Biosciences, Lincoln, NE, USA) at 1:20,000 dilution. Proteins were visualized with the Odyssey infrared imager and software (LI-COR Biosciences, Lincoln, NE, USA) according the manufacturer’s instruction.
Data analysis
Data analysis was carried out with GraphPad Prism 5.0 statistics software (GraphPad Software, La Jolla, CA, USA). A list of analysis includes student’s t-test, paired t-test, one-way ANOVA with post-hoc Dunnett test comparing all groups to vehicle or post-hoc Tukey test comparing all groups to each other.
Supplementary Material
Supplementary Material is available at HMG online.
Supplementary Material
Acknowledgements
We would like to thank Jennifer A. Gray for her generous contribution to this work as a professional editor.
Conflict of Interest statement. None declared.
Funding
RO1NS07777, RO1EY12245, PO1AG025532 and a grant from the Friedreich's ataxia Research Alliance (FARA) to G.A.C.
References
- 1. Schapira A.H. (2006) Mitochondrial disease. Lancet (London, England), 368, 70–82. [DOI] [PubMed] [Google Scholar]
- 2. Wenz T., Diaz F., Spiegelman B.M., Moraes C.T. (2008) Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab., 8, 249–256. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3. Puigserver P., Wu Z., Park C.W., Graves R., Wright M., Spiegelman B.M. (1998) A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell, 92, 829–839. [DOI] [PubMed] [Google Scholar]
- 4. Virbasius J.V., Scarpulla R.C. (1994) Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc. Natl Acad. Sci. U.S.A., 91, 1309–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gugneja S., Virbasius C.M., Scarpulla R.C. (1996) Nuclear respiratory factors 1 and 2 utilize similar glutamine-containing clusters of hydrophobic residues to activate transcription. Mol. Cell. Biol., 16, 5708–5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Evans M.J., Scarpulla R.C. (1990) NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev., 4, 1023–1034. [DOI] [PubMed] [Google Scholar]
- 7. Virbasius C.A., Virbasius J.V., Scarpulla R.C. (1993) NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev., 7, 2431–2445. [DOI] [PubMed] [Google Scholar]
- 8. Wu Z., Puigserver P., Andersson U., Zhang C., Adelmant G., Mootha V.. et al. (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell, 98, 115–124. [DOI] [PubMed] [Google Scholar]
- 9. Gleyzer N., Vercauteren K., Scarpulla R.C. (2005) Control of mitochondrial transcription specificity factors (TFB1M and TFB2M) by nuclear respiratory factors (NRF-1 and NRF-2) and PGC-1 family coactivators. Mol. Cell. Biol., 25, 1354–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ekstrand M.I., Falkenberg M., Rantanen A., Park C.B., Gaspari M., Hultenby K.. et al. (2004) Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet., 13, 935–944. [DOI] [PubMed] [Google Scholar]
- 11. Linker R.A., Lee D.H., Ryan S., van Dam A.M., Conrad R., Bista P.. et al. (2011) Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain, 134, 678–692. [DOI] [PubMed] [Google Scholar]
- 12. Scannevin R.H., Chollate S., Jung M.Y., Shackett M., Patel H., Bista P.. et al. (2012) Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther., 341, 274–284. [DOI] [PubMed] [Google Scholar]
- 13. Fox R.J., Miller D.H., Phillips J.T., Hutchinson M., Havrdova E., Kita M.. et al. (2012) Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N. Eng. J. Med., 367, 1087–1097. [DOI] [PubMed] [Google Scholar]
- 14. Mrowietz U., Altmeyer P., Bieber T., Rocken M., Schopf R.E., Sterry W. (2007) Treatment of psoriasis with fumaric acid esters (Fumaderm). J. Dtsch. Dermatol. Ges., 5, 716–717. [DOI] [PubMed] [Google Scholar]
- 15. Chen H., Assmann J.C., Krenz A., Rahman M., Grimm M., Karsten C.M.. et al. (2014) Hydroxycarboxylic acid receptor 2 mediates dimethyl fumarate's protective effect in EAE. J. Clin. Invest., 124, 2188–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wild A.C., Moinova H.R., Mulcahy R.T. (1999) Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem., 274, 33627–33636. [DOI] [PubMed] [Google Scholar]
- 17. Harvey C.J., Thimmulappa R.K., Singh A., Blake D.J., Ling G., Wakabayashi N.. et al. (2009) Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med., 46, 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kensler T.W., Wakabayashi N., Biswal S. (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Ann. Rev. Pharmacol. Toxicol., 47, 89–116. [DOI] [PubMed] [Google Scholar]
- 19. Calkins M.J., Johnson D.A., Townsend J.A., Vargas M.R., Dowell J.A., Williamson T.P.. et al. (2009) The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox Signal., 11, 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dinkova-Kostova A.T., Abramov A.Y. (2015) The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med., 88, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brennan M.S., Matos M.F., Li B., Hronowski X., Gao B., Juhasz P.. et al. (2015) Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PloS One, 10, e0120254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yamamoto T., Suzuki T., Kobayashi A., Wakabayashi J., Maher J., Motohashi H.. et al. (2008) Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol. Cell. Biol., 28, 2758–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nguyen T., Sherratt P.J., Nioi P., Yang C.S., Pickett C.B. (2005) Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem., 280, 32485–32492. [DOI] [PubMed] [Google Scholar]
- 24. Dinkova-Kostova A.T., Abramov A.Y. (2015) The emerging role of Nrf2 in mitochondrial function. Free Radic. Biol. Med., 88, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Piantadosi C.A., Carraway M.S., Babiker A., Suliman H.B. (2008) Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ. Res., 103, 1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen W., Liu K., Tian C., Yang L., Li X., Ren J.. et al. (2008) R-alpha-lipoic acid and acetyl-L-carnitine complementarily promote mitochondrial biogenesis in murine 3T3-L1 adipocytes. Diabetologia, 51, 165–174. [DOI] [PubMed] [Google Scholar]
- 27. Tang H., Lu J.Y., Zheng X., Yang Y., Reagan J.D. (2008) The psoriasis drug monomethylfumarate is a potent nicotinic acid receptor agonist. Biochem. Biophys. Res.Commun., 375, 562–565. [DOI] [PubMed] [Google Scholar]
- 28. Sahdeo S., Tomilov A., Komachi K., Iwahashi C., Datta S., Hughes O.. et al. (2014) High-throughput screening of FDA-approved drugs using oxygen biosensor plates reveals secondary mitofunctional effects. Mitochondrion, 17, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhat A.H., Dar K.B., Anees S., Zargar M.A., Masood A., Sofi M.A.. et al. (2015) Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother., 74, 101–110. [DOI] [PubMed] [Google Scholar]
- 30. Katsetos C.D., Koutzaki S., Melvin J.J. (2013) Mitochondrial dysfunction in neuromuscular disorders. Semin. Pediatr. Neurol., 20, 202–215. [DOI] [PubMed] [Google Scholar]
- 31. Percival J.M., Siegel M.P., Knowels G., Marcinek D.J. (2013) Defects in mitochondrial localization and ATP synthesis in the mdx mouse model of Duchenne muscular dystrophy are not alleviated by PDE5 inhibition. Hum. Mol. Genet., 22, 153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cozzolino M., Carrì M.T. (2012) Mitochondrial dysfunction in ALS. Prog. Neurobiol., 97, 54–66. [DOI] [PubMed] [Google Scholar]
- 33. Parodi B., Rossi S., Morando S., Cordano C., Bragoni A., Motta C.. et al. (2015) Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol., 130, 279–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ma Q. (2013) Role of nrf2 in oxidative stress and toxicity. Ann. Rev. Pharmacol.Toxicol., 53, 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fisher R.P., Clayton D.A. (1988) Purification and characterization of human mitochondrial transcription factor 1. Mol. Cell. Biol., 8, 3496–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang Y.K., Wu K.C., Klaassen C.D. (2013) Genetic activation of Nrf2 protects against fasting-induced oxidative stress in livers of mice. PloS One, 8, e59122.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cho H.Y., Reddy S.P., Yamamoto M., Kleeberger S.R. (2004) The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J., 18, 1258–1260. [DOI] [PubMed] [Google Scholar]
- 38. Li J., Stein T.D., Johnson J.A. (2004) Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol. Genomics, 18, 261–272. [DOI] [PubMed] [Google Scholar]
- 39. Lee D.H., Gold R., Linker R.A. (2012) Mechanisms of Oxidative Damage in Multiple Sclerosis and Neurodegenerative Diseases: Therapeutic Modulation via Fumaric Acid Esters. Int. J. Mol. Sci., 13, 11783–11803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Al-Sawaf O., Fragoulis A., Rosen C., Keimes N., Liehn E.A., Holzle F.. et al. (2014) Nrf2 augments skeletal muscle regeneration after ischaemia-reperfusion injury. J. Pathol., 234, 538–547. [DOI] [PubMed] [Google Scholar]
- 41. Li X., Millar J.S., Brownell N., Briand F., Rader D.J. (2010) Modulation of HDL metabolism by the niacin receptor GPR109A in mouse hepatocytes. Biochem. Pharmacol., 80, 1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rahman M., Muhammad S., Khan M.A., Chen H., Ridder D.A., Muller-Fielitz H.. et al. (2014) The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun., 5, 3944. [DOI] [PubMed] [Google Scholar]
- 43. Couturier A., Ringseis R., Most E., Eder K. (2014) Pharmacological doses of niacin stimulate the expression of genes involved in carnitine uptake and biosynthesis and improve the carnitine status of obese Zucker rats. BMC Pharmacol. Toxicol., 15, 37.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Soga T., Kamohara M., Takasaki J., Matsumoto S., Saito T., Ohishi T.. et al. (2003) Molecular identification of nicotinic acid receptor. Biochem. Biophys. Res. Commun., 303, 364–369. [DOI] [PubMed] [Google Scholar]
- 45. Hara N., Yamada K., Shibata T., Osago H., Hashimoto T., Tsuchiya M. (2007) Elevation of cellular NAD levels by nicotinic acid and involvement of nicotinic acid phosphoribosyltransferase in human cells. J. Biol. Chem., 282, 24574–24582. [DOI] [PubMed] [Google Scholar]
- 46. Mao P., Reddy P.H. (2010) Is multiple sclerosis a mitochondrial disease? Biochim. Biophys. Acta, 1802, 66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Harding A., Sweeney M., Miller D., Mumford C., Kellar-Wood H., Menard D.. et al. (1992) Occurrence of a multiple sclerosis-like illness in women who have a Leber's hereditary optic neuropathy mitochondrial DNA mutation. Brain, 115, 979–989. [DOI] [PubMed] [Google Scholar]
- 48. Pfeffer G., Burke A., Yu-Wai-Man P., Compston D.A.S., Chinnery P.F. (2013) Clinical features of MS associated with Leber hereditary optic neuropathy mtDNA mutations. Neurology, 81, 2073–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Giordano C., Iommarini L., Giordano L., Maresca A., Pisano A., Valentino M.L.. et al. (2014) Efficient mitochondrial biogenesis drives incomplete penetrance in Leber’s hereditary optic neuropathy. Brain, 137, 335–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Garcia-Ruiz I., Solis-Munoz P., Fernandez-Moreira D., Munoz-Yague T., Solis-Herruzo J.A. (2013) Pioglitazone leads to an inactivation and disassembly of complex I of the mitochondrial respiratory chain. BMC Biol., 11, 88.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.