

Abstract
Chlamydia secrete into host cells a diverse array of effector proteins, but progress in characterizing the spatiotemporal localization of these proteins has been hindered by a paucity of genetic approaches in Chlamydia and also by the challenge of studying these proteins within the live cellular environment. We adapted a split-green fluorescent protein (GFP) system for use in Chlamydia to label chlamydial effector proteins and track their localization in host cells under native environment. The efficacy of this system was demonstrated by detecting several known Chlamydia proteins including IncA, CT005 and CT694. We further used this approach to detect two chlamydial deubiquitinases (CT867 and CT868) within live cells during the infection. CT868 localized only to the inclusion membrane at early and late developmental stages. CT867 localized to the chlamydial inclusion membrane at an early developmental stage and was concomitantly localized to the host plasma membrane at a late stage during the infection. These data suggest that chlamydial deubiquitinase play important roles for chlamydial pathogenesis by targeting proteins at both the plasma membrane and the chlamydial inclusion membrane. The split-GFP technology was demonstrated to be a robust and efficient approach to identify the secretion and cellular localization of important chlamydial virulence factors.
Keywords: Chlamydia, effector, localization, deubiquitinase, split-GFP
Live imaging of chlamydial effectors using split-GFP.
INTRODUCTION
Chlamydia are obligate intracellular pathogens that cause a spectrum of human diseases, including sexually transmitted diseases, infectious blindness and respiratory tract infection (Gerbase et al.1998; Burton and Mabey 2009; Johnson et al.2014). Chlamydia species have a unique developmental cycle that occurs entirely within a membrane-bound intracellular vacuole called an inclusion (Moulder 1991). It has been shown that the temporal expression of chlamydial genes is tightly regulated throughout the developmental cycle and is important for controlling the developmental stages of the infection cycle (Shaw et al.2000; Nicholson et al.2003).
Bacterial pathogens have the ability to modulate host cellular functions by secretion of various virulence factors. It is thought that Chlamydia employ a number of effector proteins that modulate host cellular functions in order to create a favorable intracellular environment for their survival (Valdivia 2008; Aeberhard et al.2015; Mirrashidi et al.2015; Elwell, Mirrashidi and Engel 2016). More than 20 putative effector proteins have been detected in the host cytoplasmic compartment during the chlamydial developmental cycle (Kleba and Stephens 2008). Thus, identification of secreted effector proteins and their subcellular localization in vivo will be a significant step for understanding chlamydial pathogenesis. The conventional method for identifying the localization of chlamydial proteins requires antigen-specific antisera and subsequent immunostaining of samples at a single time point (Li et al.2008; Bauler and Hackstadt 2014; Weber et al.2015). However, immunostaining approaches require fixed cells during infection, and can result in false positive or false negative signals due to non-specific binding or poor sensitivity for low abundant proteins. Fluorescent proteins, such as green fluorescent protein (GFP), provide an alternative opportunity for tracking protein localization in living cells; however, fusion of bacterial effector proteins to a large 27 kDa GFP may perturb the folding and localization of tagged proteins in a native environment and also may block the secretion by bacterial type-III secretion systems due to the very stable fold of GFP (Akeda and Galan 2005).
Chlamydia encode two enzymes with deubiquitinating and deneddylating activies in vitro (Misaghi et al.2006), although the subcellular localization and their native functions are not well characterized. Ubiquitination is a crucial post-translational protein modification that plays a decisive role in regulating a variety of cellular processes in eukaryotes, including immune responses (Bhoj and Chen 2009), transcriptional regulation (Muratani and Tansey 2003), endocytic trafficking (Molfetta et al.2014) and the cell cycle (Nakayama and Nakayama 2006). Because the important functions of ubiquitination in eukaryotes, some pathogens have evolved strategies to exploit the host ubiquitin system in order to manipulate the host defense system or to obtain nutrients (Ashida, Kim and Sasakawa 2014; Huang and Brumell 2014). The presence of deubiquitinase enzymes in Chlamydia implies that this pathogen may employ these proteins to modulate host cellular processes by disruption of the host ubiquitin system. If the role of these proteins is interfacing with host ubiquitinated proteins, they should be localized with access to the cytosolic compartment of the infected host cell.
Based on the advance to transform chlamydiae (Wang et al.2011), we present a novel approach to label chlamydial effector proteins and track their localization within living host cells using split-GFP technology (Cabantous, Terwilliger and Waldo 2005). Using this approach, the localization of several chlamydial effector proteins was characterized to establish the method for use in Chlamydia research. The localization of two chlamydial deubiquitinase enzymes, CT867 and CT868, was characterized that provided new insights into the roles of these deubiquitinating enzymes in chlamydial pathogenesis. Our data indicate that split-GFP system is an efficient method for detecting the secretion and subcellular localization of chlamydial virulence factors during infection.
MATERIALS AND METHODS
Cell lines and bacterial strains
Hela and L929 cells were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum (FBS). McCoy (ATCC; Manassas, VA, USA) and A549-GFP1-10 cell lines (stably expressing the GFP1-10 protein, kindly provided by Dr Nadia Naffakh, Institute Pasteur Cells) were maintained in Dulbecco's Modified Eagle's medium (DMEM) medium (Invitrogen) supplemented with 10% FBS. All cell lines were grown at 37°C in an atmosphere containing 5% CO2. XL10-Gold ultracompetent cells were obtained from Agilent Technologies. Rabbit anti-Flag antibody (Cat No: 637301) was obtained from Biolegend. The anti-major outer membrane protein (MOMP) antibody was a stock from our laboratory and produced from mouse. HRP-conjugated Mouse anti-rabbit IgG (Cat No: SC-2357) and HRP-conjugated Goat anti-mouse IgG (Cat No: NB7539) antibodies were purchased from Santa Cruz Biotechnology and Novus Biologicals, respectively. Dam−/Dcm− Escherichia coli cells were obtained from New England Biolabs. Chlamydia trachomatis lymphogranuloma venereum serovar L2 (L2/434/Bu) EB organisms were purified from L929 cells as described previously (Abromaitis and Stephens 2009).
Construction of E.coli–Chlamydia shuttle vectors for expressing GFP11-fused effector proteins
To construct the constitutive expression vector, the pUC replication origin (ori) and β-lactamase (bla) gene were amplified from pGFP-SW2 (kindly provided by Dr Ian Clarke, University of Southampton) with the following primers: p1-F and p1-R (Table 1) that encoded BamHI and XhoI, respectively. The resulting PCR product was digested with BamHI and XhoI restriction endonycleases. The wild-type C. trachomatis DnaK promoter was synthesized by annealing primers pdnaK-F and pdnaK-R (Table 1). The annealed double-stranded dnaK promoter was ligated with purified ori-bla fragment, the resulting plasmid was named pdnaK. The sequence encoding Flag-GFP11 peptide was synthesized by annealing the primers GFP11-F and GFP11-R (Table 1), and the annealed product was inserted into the NotI and NheI restriction sites of pdnaK. The mKate2 gene containing ribosomal binding site (RBS) at the 5' end was amplified from plasmid pTRL-309a (kindly provided by Dr Scott Hefty, University of Kansas) with primers pmKate-F and pmKate-R. The PCR product was inserted into the pdnaK plasmid with NheI and SalI restriction sites, and then transformed into XL-10 strain. The resulting plasmid was named pdnaK-GFP11. To construct the shuttle plasmid, pdnaK-SW2-GFP11, pGFP-SW2 plasmids were digested with BamHI, and the chlamydial plasmid SW2 fragment was purified from DNA agarose gels. The plasmid pdanK-GFP11 was also digested with BamHI. The resulting product was ligated with digested SW2 fragment, and then transformed into the XL-10 strain. The resulting shuttle vector was confirmed by PCR and DNA sequencing.
Table 1.
Primers used in this study.
| Primers name | Sequencea |
|---|---|
| CT867-F | CGCCCATGGCAGAACCAATTCATAATCCT |
| CT867-R | ACCGGCGGCCGCCATCCGTAGTTGGCCAGCTCAAAGAT |
| CT867-F1 | GCGCTCGAGATGGAACCAATTCATAATCCT |
| CT867-R1 | GCGGGATCCATCCGTAGTTGGCCAGCTCAAAGAT |
| CT868-F | CGCCCATGGCATTGTCTCCCACCAACTCAACT |
| CT868-R | GGTAGCGGCCGCCGAAAAGAGCTTTTGCTTCAG |
| TmeA-F | GACCCATGGCAATGAGTATTCGACCTACT |
| TmeA-R | ACCGGCGGCCGCCGTCTAAGAAAACAGAAGAAGT |
| IncA-F | GCGCCATGGGTATGACAACGCCTACTCTAATCGT |
| IncA-R | GACGGCGGCCGCCGGAGCTTTTTGTAGAGGGT |
| CT005-F | GCCCCATGGCAATGACTCCAGTAACAC |
| CT005-R | GGTAGCGGCCGCCTTTACGAGAGGGTTTCTTCTTTTGA |
| CT733-F | GCGCCATGGCAATGTTAATAAACTTTACCTTTCGCA |
| CT733-R | GGTAGCGGCCGCCTAAATGGATACTAACGGTTCCA |
| GFP11-F | (Phos)GGCCGCATCTTCTGATTATAAAGATGATGACGATAAATCTTCTGGTAGAGATCATATGGTTTTGCATGAATATGTTAA TGCAGCAGGTATTACTTAAG |
| GFP11-R | (Phos)CTAGCTTAAGTAATACCTGCTGCATTAACATATTCATGCAAAACCATATGATCTCTACCAGAAGATTTATCGTCATCA TCTTTATAATCAGAAGATGC |
| P1-F | CGGGGATCCGGTTCTATAGTGTCACCTA |
| P1-R | CAGCTGCTCGAGGCCGGTCTCCCTA |
| P2-F | GCGGATCCATTCTTGACCGGTGGAGACGGTTTTCTTATAATGACACCGACTTATGGAAAATAGAGGTTCAT GGTCTCTGAAC |
| P2-R | GCGGGATCCTTTAGAGCTTGACGGGGAAAG |
| P3-F | AGAGCTAGCGTTGACCTGTGAAGTGAA |
| P3-R | GAGCCATGGGTATATCTCCTTCTTAAA |
| pdnaK-F | (Phos)TCGAGATTCTTGACCGGTGGAGACGGTTTTCTTATAGAGGTTCCATGGGTGGCGGCCGCAGCGCTAGCGGCGCCGGTA CCGTCGACG |
| pdnaK-R | (phos)GATCCGTCGACGGTACCGGCGCCGCTAGCGCTGCGGCCGCCACCCATGGAACCTCTATTTTCCATAAGTCGGTGTCAT TATAAGAAAACCGTCTCCACCGGTCAAGAATC |
| pmKate-F | AGGGCTAGCGAGAGGATTTCATGGTCTCTGAACTTATC |
| pmKate-R | CGCGTCGACTTAGCGGTGTCCAAGTT TAGA |
aThe restriction enzyme site was shown in bold type
To construct the plasmids for the constitutive expression of CT694 (TmeA), IncA and CT005, the respective genes were amplified with listed primers (Table 1) by using L2 genomic DNA as the template. The PCR products were digested with NcoI and NotI enzymes, and then inserted into pdnaK-SW2-GFP11 vector digested with the same restriction enzymes. To construct the CT733 expression vector with pdnaK-SW2-GFP11, the original NcoI sequence located between amino acid A372 and G374 of CT733 was mutated without changing amino acid coding sequence, the resulting CT733 gene construct was used as template to amplify CT733 gene with CT733-F and CT733-R. The PCR product was digested with NcoI and NotI, and then cloned into pdnaK-SW2-GFP11. The plasmids were verified by sequencing and restriction endonuclease digestion.
To construct the conditional split-GFP expression system, the full-length CT867, CT868 genes without the stop codons were amplified with their specific primers (Table 1) from C. trachomatis L2 DNA. The DNA sequence encoding GFP11 fusion peptide was synthesized by annealing the primers GFP11-F and GFP11-R that encoded NotI and NheI site, respectively. CT867 and GFP11 fragments were digested by NotI enzyme and ligated with T4 DNA ligase. The resulting fragment was further amplified using primers CT867-F and GFP11-R and named CT867-GFP11. The tetracycline-inducible expression vector containing GFP and mKate2 gene expression elements was amplified from pTRL2a-309 with primers P2-F and p2-R that both encoded BamHI site at 5' terminus. The PCR product was digested with BamHI enzyme and ligated with T4 DNA ligase, and then transformed into XL-10 strain. The resulting plasmid was used as template to amplify the tetracycline-inducible vector including mKate2 gene expression element with primers P3-F and P3-R that encoded NcoI and NheI site, respectively. The PCR product was further digested with enzymes NcoI and NheI, and then ligated with CT867-GFP11 that digested with same enzymes. The resulting plasmid was named pTet-CT867.
To construct the shuttle vector expressing GFP11 fusion protein, pGFP-SW2 plasmid was digested with BamHI, and the digested SW2 plasmid was ligated with the same enzyme digested pTet-CT867, and then transformed into XL-10 strain. To express GFP11-fused CT868 in Chlamydia, the plasmid pTet-SW2-CT867 was digested with NcoI and NotI, and the digested plasmid was purified by gel extraction and named pTet-SW2-GFP11. The amplified CT868 product was digested with NcoI and NotI, and then inserted into pTet-SW2-GFP11 vector. The resulting plasmid was transformed into XL-10 strain. All resulting shuttle vectors were confirmed by PCR and DNA sequencing.
The sequences of primers used are indicated in Table 1. The transformation of C. trachomatis was performed as described previously (Wang et al.2011).
Construction and transfection of mammalian expression plasmids
To construct mammalian expression vector CT867-EGFP, the full length CT867 gene was amplified from C. trachomatis L2 genomic DNA with the primers CT867-F1 and CT867-R1 (Table 1). The PCR product was digested with enzymes XhoI and BamHI, and then ligated with the same enzymes digested plasmid EGFP-N1, the resulting plasmid was then transformed into XL-10 competent cells. The insert CT867 was verified by sequencing. Transfection of CT867-EGFP vector was performed with lipofectmine 2000 as described by Invitrogen. The expression was confirmed by immunoblotting and immunofluorescence assay.
Identification of effector proteins localization with split-GFP using fluorescence microscopy
A549-GFP1-10 cells grown in 4-well chambered coverglasses were infected with C. trachomatis expressing various GFP11-fused proteins. For the cells infected with Chlamydia constitutively expressing fusion protein, the fluorescence was detected at various time points during the infection. For the conditional split-GFP expression system, the infected cells were induced with 50 ng/ml of anhydrotetracycline (ATc) for 6 h at 18 and 34 hpi. As a negative control, the infected cells were also treated with protein synthesis inhibitor chloramphenicol during the induction. To track the protein localization, the infected cells were rinsed and incubated with PBS buffer, and the chambers were secured and imaged live on a Zeiss Axiovert 200 inverted microscope fitted with Orca-R2 cooled digital CCD camera.
For direct split-GFP staining, Hela cells grown in 8-well chambered coverglass were infected with transformed C. trachomatis. After 30 hpi, the cells were fixed with 4% paraformaldehyde in PBS for 30 min at room temperature (RT). The cells were washed three times with PBS, and permeabilized with 0.1% Triton X-100 in PBS for 15 min at RT. After three washes, the cells were incubated with 0.1 mg/ml of purified His-GFP1-10 protein for 5 h at RT. The split-GFP fluorescence was detected by fluorescence microscopy.
RESULTS
Construction of an E.coli-Chlamydia shuttle vector with the capability of constitutively expressing effector proteins fused with the GFP11 peptide
The split-GFP methodology (Cabantous, Terwilliger and Waldo 2005) uses a small 15-amino acid peptide (GFP11), corresponding to the 11th β-strand domain of the GFP β-barrel, to tag proteins of interest. Their localization is detected in vivo by the self-assembly capacity of GFP11 with the first 10 β-strands of GFP (GFP1-10) resulting in the functional restoration the GFP chromophore. Thus, the cellular distribution of GFP11-tagged proteins can be detected by the expression of GFP110 in trans within cells or, alternatively, by staining fixed cells with purified GFP1-10 fragment (Cabantous, Terwilliger and Waldo 2005). The successful transformation of Chlamydia with plasmids (Wang et al.2011) provides the possibility of expressing GFP11-tagged chlamydial effector proteins during infection. Thus, this system should enable live detection of chlamydial proteins secreted to the cytoplasmic compartment, or exposed to the cytoplasm, of host cells expressing cytoplasmic GFP1-10 in trans. Proteins that are not secreted or secreted within organelles can be detected by staining fixed cells with the purified GFP1-10 fragment (Kaddoum et al.2010). In the present study, a split-GFP system was developed for Chlamydia by genetic fusion of the small GFP11 peptide to the C-terminus of chlamydial proteins. The chlamydial DnaK promoter was used to drive expression of the fused hybrid genes in an Escherichia coli–Chlamydia shuttle vector (Fig. 1A). The chlamydial plasmid pSW2 fragment was used to maintain the replication of shuttle vector in Chlamydia as described previously (Wang et al.2011). To facilitate the detection of chlamydial organisms in living cells, a variant of mKate2 chromophore optimized for fluorescence in Chlamydia (Wickstrum et al.2013) was co-expressed with GFP11-fused proteins by adding a RBS before the start codon of mKate2 sequence.
Figure 1.

Detection of localization of TmeA, IncA and CT005 with the split-GFP system during C. trachomatis infection. (A) The constitutive expression vector was constructed by ligating the chlamydial pSW2 fragment to a modified E. coli vector that contains an origin (ori) and beta-lactamase gene (bla). Constitutive expression of GFP11-fused effector proteins is driven by the chlamydial DnaK promoter. The restriction sites NcoI and NotI were used to insert genes encoding chlamydial effector proteins. (B) Chlamydia trachomatis transformants expressing control vector, IncA-GFP11, CT005-GFP11 or TmeA-GFP11 were used to infect A549-GFP1-10 cells at a multiplicity of infection (MOI) of 10. The localization of IncA, CT005 and TmeA were visualized at 24 hpi by live imaging, respectively. Scale bar, 32 μm.
To test whether the split-GFP technology was applicable to label and characterize the localization of chlamydial effector proteins, several well-characterized chlamydial proteins were tested with this system. TmeA is a chlamydial type-III secretion system effector protein that localizes to the host plasma membrane (Bullock, Hower and Fields 2012), whereas CT119 (IncA) is a protein that localizes to the inclusion membrane (Bannantine et al.1998). The plasmids constitutively expressing TmeA-GFP11 or IncA-GFP11 fusion proteins were transformed into Chlamydia, respectively, and the transformed strains were used to infect A549 (GFP1-10) cells that stably express the non-fluorescent complementary GFP1-10 fragment (Avilov et al.2012). Consistent with previous reports (Bannantine et al.1998; Bullock, Hower and Fields 2012), it was shown that TmeA localized to the plasma membrane, whereas IncA localized to the inclusion membrane during the course of infection (Fig. 1B). The localization of another effector protein, CT005, was also tested because of its unusual localization to the host nucleus as shown by transient over-expression of GFP-tagged cytoplasmic fragment of CT005 in Hep2 cells (Sisko et al.2006). Since the nuclear localization was performed by ectopic expression of carboxyl-terminal domain of CT005 in yeast cells, it is not known whether CT005 localizes to host nucleus under the native infection environment. Using the split-GFP approach, we found that CT005 only localized to the inclusion membrane (Fig. 1B), which is consistent with the localization of the whole CT005 when over expressed in infected cells (Sisko et al.2006). Notably, we also observed a development delay and the formation of aberrant-looking inclusions within cells infected by Chlamydia expressing GFP11-CT005. The reason causing the abnormal development of CT005-overexpression Chlamydia is not clear. It is possible that uncontrollable overexpression of CT005 during chlamydial growth may influence the chlamydial development.
Overall, our data demonstrate that the split-GFP system is a powerful method for identifying the expression and localization of secreted Chlamydia effector proteins and analysis is additionally enabled during the developmental course of infection by live-imaging fluorescence microscopy.
Staining of GFP11-fused effector proteins with purified GFP1-10
Our data indicated that the split-GFP approach was an effective tool to characterize the localization of chlamydial-secreted effector proteins within the native cellular environment. When testing uncharacterized proteins, a negative finding needs to be confirmed that the protein is produced in detectable amounts and, in the case of chlamydiae, its possible location may be sequestered from the cytosol within the inclusion vacuole or within EB. To demonstrate the utility of this application, Hela cells infected with Chlamydia expressing GFP11-fused chlamydial proteins were fixed and permeabilized, and then probed with purified recombinant GFP1-10. Consistent with the live-imaging results, it was shown that TmeA was detected at the plasma membrane, IncA was stained at both the inclusion membrane and the inclusion lumen, and CT005 was only observed at the inclusion membrane (Fig. 2 A–C). To test the breadth of split-GFP for detecting non-secreted chlamydial proteins, CT733, a protein of unknown function that has been shown to localize within the inclusion (Finco et al.2011), was inserted into the shuttle vector and its localization was tested by the split-GFP method by live imaging. Fluorescence of CT733 was not detectable when A549 GFP1-10 cells were infected with Chlamydia expressing CT733-GFP11 (data not shown). Using the complementary GFP1-10 indirect staining strategy, infected cells exhibited detectable fluorescence but only within the inclusion (Fig. 2D), suggesting that CT733 was not secreted into host cells during infection. These data indicate that the split-GFP approach was also applicable for the detection of either secreted or non-secreted proteins.
Figure 2.

GFP1-10 staining assay for chlamydial protein localization. Chlamydia trachomatis strains expressing IncA-GFP11, TmeA-GFP11, CT005-GFP11 or CT733-GFP11 were used to infect Hela cells with an MOI of 10. The cells were fixed at 24 hpi with 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100 as described in Experimental procedures. The localization of IncA (A), CT005 (B), TmeA (C) and CT733 (D) was detected by staining with purified GFP1-10 fragment. Scale bar, 16 μm.
Characterizing the localization of chlamydial deubiquitinase proteins with the conditional split-GFP expression system
Chlamydia selectively express and secrete effector proteins at different developmental stages to support their growth in a hostile host cell environment. A limitation may be raised that constitutive over-expression of GFP11-fused effector proteins in the split-GFP system may alter the characteristics of some protein's natural expression during chlamydial infection that may result in biologic toxicity and inhibition of chlamydial growth. It has been shown that a tetracycline-inducible promoter from E. coli can control gene expression in Chlamydia (Wickstrum et al.2013). To overcome temporal expression bias, a tetracycline-inducible promoter from pASK-IBA33plus was used to drive the expression of GFP11-fused effector genes, whereas expression of mKate2 gene was controlled by the constitutive chlamydial DnaK promoter in the E. coli–Chlamydia shuttle vector providing simultaneous detection of transfected organisms (Fig. 3A). The improved expression system should enable the characterization of the localization of secreted chlamydial effector proteins at experimentally defined development stages during infection.
Figure 3.

Localization of CT868 during C. trachomatis infection. (A). The tetracycline-inducible shuttle vector was constructed by ligating the chlamydial pSW2 fragment to a modified E. coli vector derived from pASK-IBA33plus as described in Experimental procedures. The mKate2 gene is constitutively expressed under control of the chlamydial DnaK promoter. Conditional expression of GFP11-fused effector proteins is driven by an inducible tetracycline promoter (pTet). The restriction sites NcoI and NotI were used to insert genes encoding chlamydial effector proteins. (B). Chlamydia trachomatis L2 transforming pCT868 plasmid was used to infect Hela cells, and then induced by 50 ng/ml of anhydrotetracycline (ATc) for 6 h at 24 hpi. The expression of CT868 was evaluated by immunoblotting using anti-Flag antibody (1:1000). Immunoblotting of MOMP using anti-MOMP antibody (1:1000) was used as a control. (C). A549-GFP1-10 cells were infected with CT868-GFP11 expression vector-transformed C. trachomatis, and induced with 50 ng/ml of ATc for 6 h, the localization of CT868 was visualized by live imaging at 24 and 40 hpi following induction. As control, cells were only infected without induction, or with induction in the presence of 100 μg/ml of chloramphenicol (Cm) during the induction. Scale bar, 27 μm.
Chlamydia encodes two proteins (CT867 and CT868) that have been shown to have deubiquitinating activity (Misaghi et al.2006). It is presumed that these proteins are secreted to the host cytosol; however, their secretion, localization and substrate targets during chlamydial infection have not been well characterized. We tested the hypothesis that these proteins are secreted to a host cytosolic compartment, and reasoned that the split-GFP system should be an effective approach for characterizing their localization by fusing the small 11 amino acid GFP peptide to each of the target proteins.
CT868 has been demonstrated to inhibit the NF-κB signaling pathway (Misaghi et al.2006), as well as to stabilize the apoptosis regulator Mcl-1 (Fischer et al.2017) and host glucose transporter 1 (GLUT1) (Wang, Hybiske and Stephens 2017) by preventing the ubiquitination-mediated degradation of these host proteins. Unfortunately the attempts to detect the localization of CT868 from these studies showed inconsistent results (Misaghi et al.2006; Fischer et al.2017). To test the efficiency of the established inducible system, we detected the expression of CT868 by western blot analysis using a Flag tag antibody, which probed a small Flag tag that fused between chlamydia effector protein and GFP11 peptide. We have shown that expression of CT868 within infected cells was significantly induced after 6 h incubation with 50 ng/ml ATc (Fig. 3B). To address the localization of this protein with the our approach, the expression of CT868-GFP11 fusion protein in Chlamydia was induced at early and late time points post-infection, and the localization of CT868 was detected by live imaging as early as 6 h after induction. It was shown that CT868 was strongly localized to inclusion membranes at both early and late stages of infection after induction (Fig. 3C). As control, CT868 was not detected within cells that were not induced or induced in the presence of 100 μg/ml of chloramphenicol during the infection (Fig. 3C). These data demonstrate that CT868 is an inclusion membrane-associated protein and likely participates in its functional activities for proteins co-localized at that site, which was consistent with the recent report (Fischer et al.2017).
Expression of GFP11-tagged CT867 in Chlamydia transformed with the tetracycline-inducible vector was induced after infection of A549 GFP1-10 cells, resulting in entire GFP fluorescence around the inclusion membrane at early time points post-infection (Fig. 4A). It has previously been shown in native chlamydiae that CT867 is strongly transcribed early and throughout the developmental cycle (Nicholson et al.2003), thus CT867 is likely localized to the inclusion membrane at early and middle developmental stages. GFP signal was also localized to the inclusion membrane at late time points following infection but additionally became localized to the host cell plasma membrane at a later stage (Fig. 4A). As control, the expression and localization of CT867 were not detected within cells that were only infected without induction, or with induction in the presence of 100 μg/ml of chloramphenicol during the infection (Fig. 4B).
Figure 4.

Localization of CT867 during C. trachomatis infection. A549-GFP1-10 cells were infected with pCT867-transformed C. trachomatis, and then induced with 50 ng/ml of ATc for 6 h; the localization of CT867 was visualized by live imaging at 24 and 40 hpi following induction (A). The infected cells without induction or with induction in the presence of 100 μg/ml of chloramphenicol were used as negative controls (B). Scale bar, 16 μm.
The detection of CT867 at the plasma membrane was unexpected. To confirm the host cell plasma membrane localization, CT867 was fused to the N-terminus of GFP and ectopically expressed in Hela cells. It was shown that CT867-fused to GFP localized to the plasma membrane (Fig. 5), which is consistent with the split-GFP data. These data suggest that CT867 is secreted into host cells and localizes to the plasma membrane during chlamydial infection. Significantly, this experimental strategy tested the ability for plasma membrane localization in the absence of chlamydial infection as CT867 localized to the plasma membrane without additional requirements of other chlamydial proteins after its secretion into the cytosol.
Figure 5.
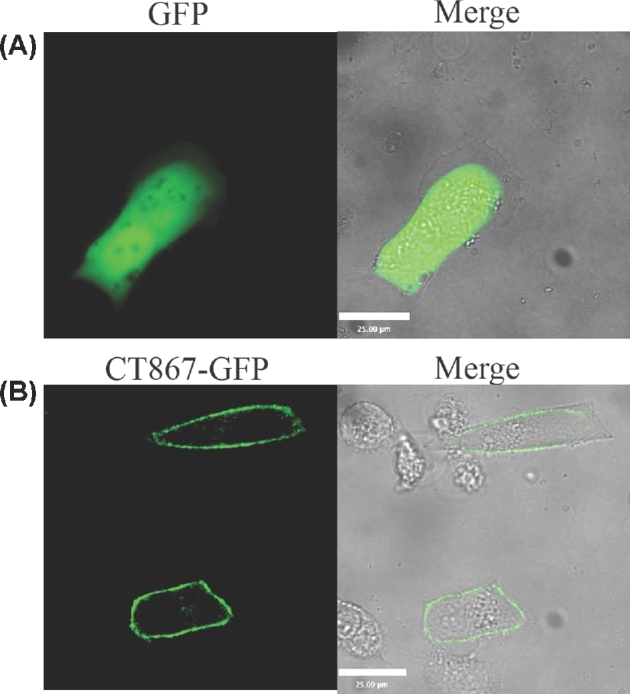
Host membrane localization of CT867. Hela cell monolayers were transfected for 24 h with plasmid EGFP-N1 (A), or the EGFP-CT867 expression plasmid (B). Images were captured directly by immunofluorescence. Scale bar, 25 μm.
DISCUSSIONS
A reliable and specific method to detect the localization of chlamydial effector proteins in the native cellular environment during infection was established using a split-GFP system. Putative chlamydial effector protein genes were genetically fused to the small GFP11 peptide-coding sequence and were transformed and expressed in Chlamydia. When the analyte-GFP11 fusion protein is secreted into cytosolic compartments and reaches the large parental fragment, GFP1-10, they combine by spontaneous association resulting in the formation of a bright fluorophore. We have shown using live fluorescence microscopy that the split-GFP system can successfully label and track the localization of chlamydial effector proteins in real time during the course of infection. This methodology enables a new experimental approach for identifying those unknown, but secreted virulence factors of Chlamydia. Additionally, it is expected that this method will be also useful for identifying the new virulence factors of other intracellular pathogens as previously shown for Salmonella (Van Engelenburg and Palmer 2010).
After demonstrating utility using chlamydial effector proteins of known location, we tested the hypothesis that two proteins whose functional attributes predict a cytosolic location are located in the cytosol. An important finding for this study is that CT867, an enzyme with deubiquitinating and deneddylating activities (Misaghi et al.2006), localizes to the cytosolic side of the inclusion membrane at early time points post-infection and at later time points additionally localizes to the inner leaflet of the host plasma membrane. This finding suggests that CT867 is an important virulence factor that likely deubiquitinates target host proteins localized at both the inclusion membrane and the host plasma membrane, significantly at different developmental stages during the course of infection.
Chamydia encode another deubiquitinating enzyme, CT868, with a demonstrated function of suppressing NF-κB by targeting IκBα ubiquitination (Le Negrate et al.2008). Although it was proposed that this effector protein was secreted into cells following a proteolytic cleavage event during the chlamydial infection, the proteolytic products were not detected by immunoblotting using an affinity-purified antibody against CT868 (Le Negrate et al.2008). In contrast, we found that CT868 was strongly localized to the inclusion membrane at early and late developmental stages during the course of infection using the split-GFP system. Although a proteolytic cleavage event could release at protein fragment, it would likely contain the carboxy-terminal GFP-11 peptide, thus this approach should be capable of tracking the cleaved peptide to a new location; however, there was no evidence that cleavage was observed. While it is nevertheless possible that localization of potential effector proteins may not be detectable in living cells if there is a cleavage event resulting in the loss of GFP11 peptide before or during protein translocation, an alternative strategy is to fuse the small GFP11 peptide to different domains of effector proteins.
The roles for ubiquitination include post-translational bypass of protein recycling (Thoms and Erdmann 2006), ubiquitination-mediated signaling for innate and adaptive immunity (Bhoj and Chen 2009), cell cycle progression (Nakayama and Nakayama 2006) and vacuolar sorting (Scheuring et al.2012). It has been demonstrated that bacterial pathogens have evolved diverse strategies to subvert or exploit host ubiquitin systems for their own benefit (Ashida, Kim and Sasakawa 2014). Therefore, the deubiquitination activity of CT868 and CT867, as well as their specific localization to membranes during chlamydial development suggests their important functions in chlamydial pathogenesis. Notably, we have reported recently that CT868 can stabilize the host glucose transporter GLUT1, while the latter plays an important role during the chlamydial growth (Wang, Hybiske and Stephens 2017). In addition, another recent study has demonstrated that CT868 localized in the inclusion membrane to interfere host defense by stabilizing apoptosis regulator Mcl-1 with its deubiquitination activity (Fischer et al.2017).
The secretion of different virulence factors is spatiotemporally controlled by bacterial pathogens during the infection cycle, especially for Chlamydia. A better understanding of secretion systems and their effectors’ translocation will rely on the assays to measure their activities in real time under a native environment. The present study relies on the reconstitution of fluorescence with a native effector protein fused to the small GFP11 peptide and GFP1-10 enabling its spatiotemporal characterization. Although we have demonstrated the use of this system for tracking native secreted proteins, this approach will have important application to the analysis of protein function by tracking modified proteins by expression of mutated genes either as a control for appropriate localization or as a means to characterize the requirements for protein secretion and cellular localization.
Acknowledgements
We would like to thank Dr Nadia Naffakh (Institute Pasteur) for providing stable cell line A549-GFP1-10. We would also like to thank Dr Ian N. Clarke (University of Southanpton Medical School) for providing plasmid pGFP::SW2, and also thank Dr Scott Hefty (University of Kansas) for providing plasmid pTRL2a-309 with tetracycline-inducible promoter.
Footnotes
Present address: Division of Infectious Diseases, Department of Medicine, Brigham and Women's Hospital and Harvard Medical School, 181 Longwood Ave, Boston, Ma 02115, USA
FUNDING
This research was supported by National Institutes of Health (NIH) award AI091851.
Conflict of interest. None declare.
REFERENCES
- Abromaitis S, Stephens RS. Attachment and entry of Chlamydia have distinct requirements for host protein disulfide isomerase. PLoS Pathog 2009;5:e1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeberhard L, Banhart S, Fischer M et al. . The proteome of the isolated Chlamydia trachomatis containing vacuole reveals a complex trafficking platform enriched for retromer components. PLoS Pathog 2015;11:e1004883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akeda Y, Galan JE. Chaperone release and unfolding of substrates in type III secretion. Nature 2005;437:911–5. [DOI] [PubMed] [Google Scholar]
- Ashida H, Kim M, Sasakawa C. Exploitation of the host ubiquitin system by human bacterial pathogens. Nat Rev Microbiol 2014;12:399–413. [DOI] [PubMed] [Google Scholar]
- Avilov SV, Moisy D, Munier S et al. . Replication-competent influenza A virus that encodes a split-green fluorescent protein-tagged PB2 polymerase subunit allows live-cell imaging of the virus life cycle. J Virol 2012;86:1433–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannantine JP, Stamm WE, Suchland RJ et al. . Chlamydia trachomatis IncA is localized to the inclusion membrane and is recognized by antisera from infected humans and primates. Infect Immun 1998;66:6017–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauler LD, Hackstadt T. Expression and targeting of secreted proteins from Chlamydia trachomatis. J Bacteriol 2014;196:1325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature 2009;458:430–7. [DOI] [PubMed] [Google Scholar]
- Bullock HD, Hower S, Fields KA. Domain analyses reveal that Chlamydia trachomatis CT694 protein belongs to the membrane-localized family of type III effector proteins. J Biol Chem 2012;287:28078–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton MJ, Mabey DC. The global burden of trachoma: a review. PLoS Neglect Trop D 2009;3:e460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol 2005;23:102–7. [DOI] [PubMed] [Google Scholar]
- Elwell C, Mirrashidi K, Engel J. Chlamydia cell biology and pathogenesis. Nat Rev Microbiol 2016;14:385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finco O, Frigimelica E, Buricchi F et al. . Approach to discover T- and B-cell antigens of intracellular pathogens applied to the design of Chlamydia trachomatis vaccines. P Natl Acad Sci USA 2011;108:9969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Harrison KS, Ramirez Y et al. . Chlamydia trachomatis-containing vacuole serves as deubiquitination platform to stabilize Mcl-1 and to interfere with host defense. Elife 2017;6:e21465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerbase AC, Rowley JT, Heymann DH et al. . Global prevalence and incidence estimates of selected curable STDs. Sex Transm Infect 1998;74:S12–6. [PubMed] [Google Scholar]
- Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol 2014;12:101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson NB, Hayes LD, Brown K et al. . CDC National Health Report: leading causes of morbidity and mortality and associated behavioral risk and protective factors–United States, 2005–2013. MMWR Suppl 2014;63:3–27. [PubMed] [Google Scholar]
- Kaddoum L, Magdeleine E, Waldo GS et al. . One-step split GFP staining for sensitive protein detection and localization in mammalian cells. Biotechniques 2010;49:727–28. [DOI] [PubMed] [Google Scholar]
- Kleba B, Stephens RS. Chlamydial effector proteins localized to the host cell cytoplasmic compartment. Infect Immun 2008;76:4842–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Negrate G, Krieg A, Faustin B et al. . ChlaDub1 of Chlamydia trachomatis suppresses NF-kappaB activation and inhibits IkappaBalpha ubiquitination and degradation. Cell Microbiol 2008;10:1879–92. [DOI] [PubMed] [Google Scholar]
- Li Z, Chen C, Chen D et al. . Characterization of fifty putative inclusion membrane proteins encoded in the Chlamydia trachomatis genome. Infect Immun 2008;76:2746–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirrashidi KM, Elwell CA, Verschueren E et al. . Global mapping of the Inc-human interactome reveals that retromer restricts Chlamydia infection. Cell Host Microbe 2015;18:109–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misaghi S, Balsara ZR, Catic A et al. . Chlamydia trachomatis-derived deubiquitinating enzymes in mammalian cells during infection. Mol Microbiol 2006;61:142–50. [DOI] [PubMed] [Google Scholar]
- Molfetta R, Quatrini L, Gasparrini F et al. . Regulation of fc receptor endocytic trafficking by ubiquitination. Front Immunol 2014;5:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder JW. Interaction of chlamydiae and host cells in vitro. Microbiol Rev 1991;55:143–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratani M, Tansey WP. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol 2003;4:192–201. [DOI] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 2006;6:369–81. [DOI] [PubMed] [Google Scholar]
- Nicholson TL, Olinger L, Chong K et al. . Global stage-specific gene regulation during the developmental cycle of Chlamydia trachomatis. J Bacteriol 2003;185:3179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuring D, Kunzl F, Viotti C et al. . Ubiquitin initiates sorting of Golgi and plasma membrane proteins into the vacuolar degradation pathway. BMC Plant Biol 2012;12:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw EI, Dooley CA, Fischer ER et al. . Three temporal classes of gene expression during the Chlamydia trachomatis developmental cycle. Mol Microbiol 2000;37:913–25. [DOI] [PubMed] [Google Scholar]
- Sisko JL, Spaeth K, Kumar Y et al. . Multifunctional analysis of Chlamydia-specific genes in a yeast expression system. Mol Microbiol 2006;60:51–66. [DOI] [PubMed] [Google Scholar]
- Thoms S, Erdmann R. Peroxisomal matrix protein receptor ubiquitination and recycling. Biochim Biophys Acta 2006;1763:1620–8. [DOI] [PubMed] [Google Scholar]
- Valdivia RH. Chlamydia effector proteinsnew insights into chlamydial cellular microbiology. Curr Opin Microbiol 2008;11:53–59. [DOI] [PubMed] [Google Scholar]
- Van Engelenburg SB, Palmer AE. Imaging type-III secretion reveals dynamics and spatial segregation of Salmonella effectors. Nat Methods 2010;7:325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hybiske K, Stephens RS. Orchestration of the mammalian host cell glucose transporter proteins-1 and 3 by Chlamydia contributes to intracellular growth and infectivity. Pathog Dis 2017;75:ftx108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kahane S, Cutcliffe LT et al. . Development of a transformation system for Chlamydia trachomatis: restoration of glycogen biosynthesis by acquisition of a plasmid shuttle vector. PLoS Pathog 2011;7:e1002258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber MM, Bauler LD, Lam J et al. . Expression and localization of predicted inclusion membrane proteins in Chlamydia trachomatis. Infect Immun 2015;83:4710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstrum J, Sammons LR, Restivo KN et al. . Conditional gene expression in Chlamydia trachomatis using the tet system. PLoS One 2013;8:e76743. [DOI] [PMC free article] [PubMed] [Google Scholar]