

Abstract
Background
Cisplatin is a potent chemotherapeutic drug whose nephrotoxic effect is a major complication and a dose-limiting factor for antitumoral therapy. There is much evidence that inflammation contributes to the pathogenesis of cisplatin-induced nephrotoxicity. We found that cilastatin, a renal dehydropeptidase-I inhibitor, has protective effects in vitro and in vivo against cisplatin-induced renal damage by inhibiting apoptosis and oxidation. Here, we investigated the potential use of cilastatin to protect against cisplatin-induced kidney injury and inflammation in rats.
Methods
Male Wistar rats were divided into four groups: control, cilastatin-control, cisplatin and cilastatin-cisplatin. Nephrotoxicity was assessed 5 days after administration of cisplatin based on blood urea nitrogen, creatinine, glomerular filtration rate (GFR), kidney injury molecule (KIM)-1 and renal morphology. Inflammation was measured using the electrophoretic mobility shift assay, immunohistochemical studies and evaluation of inflammatory mediators.
Results
Compared with the control rats, cisplatin-administered rats were affected by significant proximal tubule damage, decreased GFR, increased production of inflammatory mediators and elevations in urea, creatinine and tissue KIM-1 levels. Cilastatin prevented these changes in renal function and ameliorated histological damage in cisplatin-administered animals. Cilastatin also reduced pro-inflammatory cytokine levels, activation of nuclear factor-κB and CD68-positive cell concentrations.
Conclusions
Cilastatin reduces cisplatin-induced nephrotoxicity, which is associated with decreased inflammation in vivo. Although the exact role of decreased inflammation in nephroprotection has not been fully elucidated, treatment with cilastatin could be a novel strategy for the prevention of cisplatin-induced acute kidney injury.
Keywords: acute kidney injury, cilastatin, cisplatin, inflammation, nephroprotection
INTRODUCTION
Cisplatin (cis-diamminedichloroplatinum II) is a potent antineoplastic agent that is widely used to treat various types of tumors [1, 2]. However, its clinical application is limited by the development of time- and dose-dependent nephrotoxicity, which affects about one-third of patients receiving high-dose cisplatin [3, 4].
Although the mechanisms underlying cisplatin-induced nephrotoxicity are not fully understood, apoptosis, oxidative stress and inflammation are reported to be involved in the development and amplification of kidney injury [1, 5, 6]. Recent studies highlight the role of inflammation in this process, with activation of tumor necrosis factor α (TNFα) and nuclear factor-κB (NF-κB) considered to play a decisive role by inducing a variety of signaling pathways, leading to pro-inflammatory cytokine and chemokine production and cell infiltration, which exacerbates kidney injury [7–11]. In fact, inhibition or prevention of the activity of these molecules is associated with improvement of cisplatin-induced kidney injury [11–13]. Therefore, the search for new molecules that modulate renal inflammation is an important research area that may lead to therapies to prevent cisplatin-induced kidney injury.
Cilastatin is a specific inhibitor of renal dehydropeptidase-I (DHP-I), which is located on epithelial brush-border cholesterol lipid rafts [14]. It was initially designed to inhibit hydrolysis and uptake of the antibiotic imipenem, thus enabling it to be less toxic and more easily eliminated through urine [15, 16]. However, older clinical studies support the protective role of cilastatin (imipenem–cilastatin) against cyclosporin (CsA)-induced nephrotoxicity [17–19], and recent experimental evidence from our group indicates that cilastatin protects proximal tubular epithelial cells from toxic apoptotic and oxidative damage induced by cisplatin both in vitro and in vivo, with no reduction in the effect of cisplatin on cancer cells [20–23].
The aim of this study was to evaluate the effects of cilastatin on cisplatin-induced renal inflammation and assess the protective effects of cilastatin against cisplatin-induced nephrotoxicity in a rat model. Our findings could have important implications for the prevention of cisplatin-induced nephrotoxicity and for the treatment of other inflammatory kidney diseases.
MATERIALS AND METHODS
Animals and drug treatment
Male Wistar rats (Charles River, Wilmington, MA, USA) were housed in a humidity- and temperature-controlled environment with a 12-h light–dark cycle and allowed free access to food and water. The animals were weighed at the beginning of the experiment and immediately before being euthanized. All the procedures were approved by the Ethics Committee on Animal Experimentation from the Hospital Gregorio Marañón, Madrid, Spain, and animals were cared for in accordance with Directive 2010/63/EU and Spanish Royal Decree 53/2013 on the protection of animals used for experimentation and other scientific purposes.
At the start of the experiments, the rats were 7 weeks of age and weighed 250–270 g. Animals were divided into four groups: untreated control rats (n = 7); cilastatin-treated rats (n = 7); cisplatin-injected rats (n = 8); and cilastatin-treated, cisplatin-injected rats (n = 8). Cisplatin 5 mg/kg (Pharmacia Nostrum S.A., Barcelona, Spain) or its vehicle (0.9% saline serum) was administered by a single intraperitoneal injection to all rats in the same manner and volume (1 mL/100 g). Cilastatin 75 mg/kg (Merck Sharp and Dohme S.A., Madrid, Spain) was injected intraperitoneally twice daily (0.25 mL/100 g) from the first day cisplatin was administered until the end of the experiment. The first dose was started immediately before administration of cisplatin. Cilastatin was substituted by vehicle (0.9% saline solution) in the other groups.
The dose and administration period of cisplatin were selected based on the proven effectiveness of the drug in inducing nephrotoxicity [12, 21, 24, 25]. The dose of cilastatin was selected based on previous studies by our group [21–23] and considering earlier evaluations which showed that imipenem/cilastatin reduced CsA-induced nephrotoxicity [26, 27].
On the fifth day after cisplatin or saline injection, the animals were euthanized and samples collected (see Supplementary data for further details).
Analysis of renal function
Serum and urine levels of creatinine and blood urea nitrogen (BUN) were measured automatically in a Dimension RxL autoanalyzer (Dade-Behring, Siemens, Eschborn, Germany). The glomerular filtration rate (GFR) was calculated, as was the creatinine clearance rate. The kidney:body weight ratio was expressed as a percentage.
Histology
Paraffin-embedded renal sections (4-µm thick) were stained with periodic acid–Schiff (PAS). Renal tubular damage (see Supplementary data) in PAS sections was scored using a previously described semiquantitative scale [21, 28].
Analysis of transcription factor activity
NF-κB activity was evaluated by binding 70 µg of protein extracts from renal tissue with an oligonucleotide consensus sequence labeled with γ-[32P]ATP, and the complexes formed were analyzed using electrophoretic mobility shift assay (EMSA). Nuclear protein extraction and EMSA were performed as previously described [29].
Western blot analysis
Western blot analysis was performed as previously described [20, 30]. The antibodies used are detailed in Supplementary data.
Immunohistochemistry
Immunohistochemistry was carried out as previously described [21, 28]. The antibodies used are detailed in Supplementary data.
Real-time polymerase chain reaction
Total RNA was isolated from kidney homogenate using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions. Two micrograms of total RNA were reverse-transcribed using the iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Target gene expression (the primers used are found in Supplementary data) was quantified using the iQ5 Multicolor Real-Time Polymerase Chain Reaction (RT-PCR) Detection System (BioRad). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as the housekeeping gene and was amplified in parallel with the genes of interest. The Δ threshold cycle (ΔCT) technique was applied for the quantitative cDNA analysis [28]. Statistical analyses were carried out for at least six to eight replicate experimental samples in each set. All the measurements were performed in duplicate. Results for double-distilled water controls were negative in all the runs.
Serum myeloperoxidase
Myeloperoxidase (MPO) activity was measured in serum using the MPO Colorimetric Activity Assay Kit (Sigma-Aldrich, St Louis, MO, USA) according to the manufacturer’s instructions.
Measurement of serum cytokines
Serum TNFα and interleukin-6 (IL-6) were measured using the Rat TNFα ELISA Kit (Thermo Fisher Scientific, Waltham, MA, USA) and Rat IL-6 ELISA Kit (Gene Probe, Diaclone, Besançon, France), respectively, according to the manufacturer’s instructions.
Serum monocyte chemoattractant protein (MCP-1) and transforming growth factor β (TGFβ) were measured using microbead-based assays developed and validated in J.V.B.'s laboratory (see Supplementary data for further details) [31].
Measurement of cytokines in urine
MCP-1, IL-6, IL-10 and TGFβ levels were measured in urine using microbead-based assays developed and validated in J.V.B.'s laboratory (see Supplementary data for further details) [31].
Statistical analysis
Quantitative variables were summarized as the mean ± standard error of the mean (SEM). Equality of variances was tested using Levene’s test. Normally distributed continuous variables were analyzed using analysis of variance (ANOVA), with the least significant difference test as a post hoc approach test to determine specific group differences. Differences were considered statistically significant if bilateral α-values were <0.05. Tests were performed using Statistical Package for Social Sciences (SPSS) version 11.5 (SPSS, Chicago, IL, USA).
RESULTS
Cilastatin ameliorates cisplatin-induced kidney injury in rats
The administration of cisplatin increased serum creatinine and BUN and decreased values of GFR in the test groups, compared with the control groups (Figure 1A–C). Treatment with cilastatin significantly reduced the effects of cisplatin for both parameters. Moreover, cilastatin was able to reduce the renal hypertrophy induced by cisplatin (Figure 1D). Cilastatin alone did not produce changes for any of the parameters in the test groups compared with the control groups.
FIGURE 1.

Effects of cilastatin on cisplatin-induced nephrotoxicity in rats. Parameters of renal function on day 5 after administration of cisplatin. (A) Serum creatinine, (B) blood urea nitrogen, (C) glomerular filtration rate, (D) renal hypertrophy. Cisplatin administration increases serum creatinine, blood urea nitrogen and renal hypertrophy, and reduces glomerular filtration rate, compared with control groups. Cilastatin significantly improved all the parameters. Results are expressed as mean ± SEM; n = 7–8 animals per group. cil, cilastatin; kidney w/BW, kidney weight:body weight. *P < 0.05 versus control groups; †P ≤ 0.0001 versus cisplatin group; ‡P < 0.05 versus cisplatin group; §P < 0.02 versus all groups.
The acute kidney injury (AKI) biomarker kidney injury molecule (KIM)-1 was measured to confirm the nephrotoxic effect of cisplatin (Figure 2). Cilastatin alone had no effects on renal KIM-1 expression. KIM-1 levels were significantly increased in relation to serum creatinine and BUN at both the mRNA level (Figure 2H) and at the protein level (Figure 2A–G) after administration of cisplatin. Cilastatin reduced protein expression compared with control levels, although while gene expression was reduced, it did not reach statistical significance (Figure 2).
FIGURE 2.

Effects of cisplatin and cilastatin in acute kidney injury biomarkers. (A–D) Localization of kidney injury molecule (KIM)-1 in kidney sections. (A) Control, (B) cisplatin, (C) control + cilastatin, (D) cisplatin + cilastatin (magnification 10×), bar = 100 µm. (E) Densitometric analysis of immunohistochemistry of KIM-1. (F) Representative western blot of KIM-1 in renal cortex. (G) Densitometric analysis of western blot of KIM-1. (H) Renal mRNA expression of KIM-1. Cilastatin significantly reduced the increase of protein but not mRNA expression of KIM-1 induced by cisplatin. Data are expressed as mean ± SEM; n = 7–8 animals per group. cil, cilastatin; a.u., arbitrary units. *P < 0.001 versus all groups; †P ≤ 0.05 versus control groups.
Cilastatin attenuates cisplatin-induced morphological tubular damage
Histological examination revealed blebbing of apical membranes, epithelial necrosis, protein casts formation, vacuolization and presence of mitotic nuclei in the proximal tubules of the cisplatin-administered group (Figure 3A–D). Treatment with cilastatin significantly improved cisplatin-induced tubular damage, as shown in the semiquantitative tubular damage score (Figure 3E). Cilastatin alone had no effects on tubular morphology in comparison with the control groups (Figure 3A, C and E).
FIGURE 3.
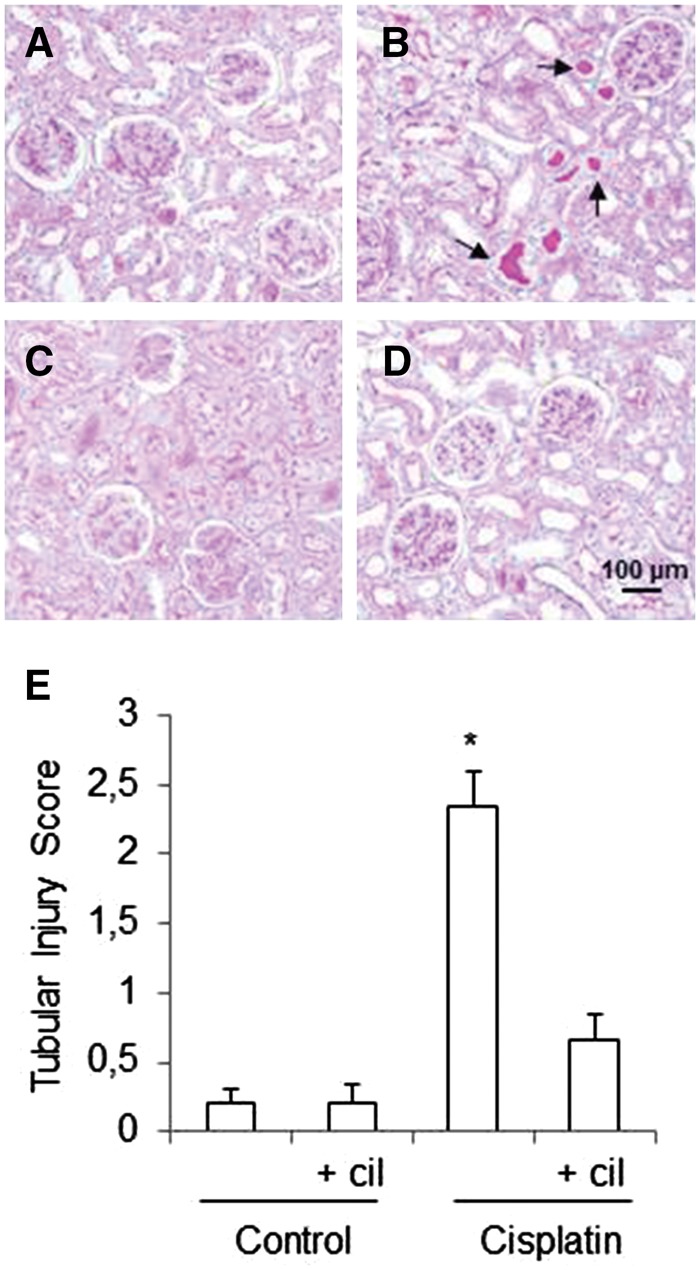
Effects of cilastatin on cisplatin-induced tubular injury. Representative images of the renal pathology (periodic acid–Schiff staining, magnification 20×) on day 5 after administration of cisplatin. (A) Control rats, (B) cisplatin, (B) control + cilastatin, (D) cisplatin + cilastatin. Control groups show normal renal structure; cisplatin-injected kidneys show marked injury with blebbing of apical membranes, epithelial necrosis, protein casts formation and vacuolization in the proximal tubules (arrows). These changes were significantly reduced by treatment with cilastatin. (E) Semiquantitative tubular injury score. Results are expressed as mean ± SEM; n = 7–8 animals per group. cil, cilastatin. *P < 0.01 versus all groups.
Effects of cisplatin and cilastatin on activation of NF-ĸB in the kidneys
Compared with control groups, the pro-inflammatory effect of cisplatin was associated with activation of NF-κB in the kidneys (Figure 4A). Cilastatin significantly decreased cisplatin-induced NF-κB activity, as shown by the semiquantitative NF-κB activity score (Figure 4B).
FIGURE 4.

Effects of cilastatin on cisplatin-induced renal nuclear factor (NF)-κB activation. (A) Representative electrophoretic mobility shift assay experiment from renal tissue. (B) Densitometric analysis of electrophoretic mobility shift assay on kidney tissue. Administration of cisplatin increases NF-κB activity on renal tissue. Cilastatin significantly reduced the increase of NF-κB activity induced by cisplatin. All results are expressed as mean ± SEM; n = 7–8 animals per group. cil, cilastatin. *P < 0.02 versus all groups.
Effects of cisplatin and cilastatin on urine and serum cytokines
NF-κB regulates the expression of many cytokines that participate in inflammatory processes. Figure 5 shows that administration of cisplatin increases the concentration of MCP-1 and IL-6 both in serum and in urine. Furthermore, cisplatin-administered rats have increased concentrations of TNFα in serum and decreased levels of IL-10 in urine. Treatment with cilastatin partially or totally restored the urinary and serum cytokine levels compared with the controls (Figure 5). Cilastatin alone had no effects on urine and serum cytokine concentration compared with the control groups.
FIGURE 5.

Effects of cisplatin and cilastatin on urine and serum cytokines. Serum and urine cytokine levels in studied groups. (A) Serum MCP-1, (B) serum IL-6, (C) serum TNFα, (D) urine MCP-1, (E) urine IL-6, (F) urine IL-10. Administration of cisplatin increased serum and urine levels of MCP-1 and IL-6, and serum levels of TNFα. Thus, cisplatin diminished urine levels of IL-10. These changes where significantly improved with cilastatin treatment. All results are expressed as mean ± SEM; n = 7–8 animals per group. cil, cilastatin. *P ≤ 0.02 versus all groups; †P ≤ 0.05 versus all groups; ‡P ≤ 0.05 versus control groups; §P< 0.01 versus cisplatin; ¶P ≤ 0.05 versus cisplatin.
Cilastatin ameliorates cisplatin-induced inflammatory cell infiltration
The immunohistochemical studies revealed that recruitment of monocytes/macrophages (as evidenced by labeling with CD68) in the renal cortices and outer medulla of cisplatin-administered rats was more intense than in controls. This finding was consistent with elevated MCP-1 levels (Figure 6A–E). Cilastatin-treated rats had fewer CD68-positive cells than the cisplatin-administered group (Figure 6B, D and E).
FIGURE 6.

Effects of cilastatin on cisplatin-induced inflammatory cell infiltration. Localization of CD68 (monocyte/macrophage) in kidney sections of (A) control rats, (B) cisplatin, (C) control + cilastatin and (D) cisplatin + cilastatin. Note increased staining in cisplatin-injected rats (arrows) compared with cisplatin + cilastatin and control rats (magnification 20×); bar = 100 µm. (E) Quantification of CD68 immunostaining in renal cells. (F) Serum myeloperoxidase (MPO) activity in the groups of rats studied. Administration of cisplatin increased systemic MPO activity. This change was significantly improved with cilastatin. All results are expressed as mean ± SEM; n = 7–8 animals per group. cil, cilastatin. *P < 0.05 versus all groups; †P ≤ 0.01 versus control groups; ‡P < 0.05 versus cisplatin group.
Moreover, cisplatin significantly increased systemic neutrophil activation (evidenced by increased MPO activity), which was partially attenuated by cilastatin (Figure 6F). Cilastatin alone had no effects on CD68-positive cells or systemic MPO activity when compared with vehicle-treated control groups (Figure 6F).
Effects of cisplatin and cilastatin on expression of adhesion molecules in the kidneys
To further understand the ability of cilastatin to inhibit inflammatory cell infiltration into the injured kidney, we measured the mRNA expression of intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 using quantitative RT-PCR. Figure 7F and G shows the relative expression of ICAM-1 and VCAM-1 normalized to GAPDH mRNA. The results revealed that administration of cisplatin increased the expression of both ICAM-1 and VCAM-1 compared with the control groups. Treatment with cilastatin significantly reduced cisplatin-induced changes. These results were confirmed by analysis of VCAM-1 protein levels using immunohistochemistry (Figure 7A–E). Cisplatin-administered rats presented higher levels of VCAM-1 in renal tissue than the control groups. Treatment with cilastatin significantly reduced the frequency of cisplatin-induced effects. Cilastatin alone had no effects on ICAM-1 and VCAM-1 mRNA expression and the presence of VCAM-1 in renal tissue (Figure 7).
FIGURE 7.

Effects of cisplatin and cilastatin on renal expression of intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1. Localization of VCAM-1 in kidney sections of (A) control rats, (B) cisplatin, (C) control + cilastatin and (D) cisplatin + cilastatin. Note increased renal staining in cisplatin-injected rats (arrows) compared with cisplatin + cilastatin and control groups (magnification 20×). (E) Semiquantification of VCAM-1 immunostaining in renal cells. (F) Renal mRNA expression of VCAM-1. (G) Renal mRNA expression of ICAM-1. Cilastatin significantly reduced the increase of both VCAM-1 and ICAM-1 mRNA expression induced by cisplatin. All results are expressed as mean ± SEM; n = 7–8 animals per group. cil, cilastatin. *P ≤ 0.001 versus all groups; †P < 0.05 versus all groups.
Cilastatin reduces cisplatin-induced increased expression of TGFβ1
TGFβ is a pro-inflammatory and profibrotic cytokine that exacerbates kidney damage. Immunohistochemistry revealed higher levels of TGFβ1 in the renal tissue of cisplatin-administered rats than in control groups (Figure 8A–E). This result was consistent with cisplatin-induced increases in levels of TGFβ1 in urine and serum (Figure 8F and G). In addition, TGFβ1 mRNA expression was also higher after administration of cisplatin than in the control groups (Figure 8H). Treatment with cilastatin significantly decreased TGFβ1 mRNA and protein levels in cisplatin-administered animals.
FIGURE 8.

Effects of cilastatin on the increase of cisplatin-induced transforming growth factor β1 (TGFβ1). Localization of TGFβ1 in kidney sections of (A) control rats, (B) cisplatin, (C) control + cilastatin and (D) cisplatin + cilastatin. Note increased tubular staining in cisplatin-injected rats (arrows) compared with cisplatin + cilastatin and control rats (magnification 20×); bar = 100 µm. (E) Semiquantification of TGFβ1 immunostaining in renal cells. (F) Serum TGFβ1 levels. (G) Urine TGFβ1 levels. (H) Renal mRNA expression of TGFβ1. Administration of cisplatin increased levels of TGFβ1 in kidney tissue, as well as serum and urine concentration. Also, cisplatin elevated TGFβ1 mRNA expression. These changes where significantly improved with cilastatin. All results are expressed as mean ± SEM; n = 7–8 animals per group. cil, cilastatin. *P < 0.001 versus all groups; †P ≤ 0.05 versus all groups.
DISCUSSION
An increased inflammatory response (together with oxidative stress and apoptosis) has been implicated in the development of cisplatin-induced AKI [1, 5, 6, 32]. Many studies have shown that induction of pro-inflammatory cytokines together with infiltration by macrophages and neutrophils, contributes to the pathogenesis and exacerbation of renal tissue damage in cisplatin-administered animals, resulting in a vicious cycle [1, 5, 12, 33–35]. Therefore, modulation of the renal inflammatory reaction may help to prevent cisplatin-induced AKI. Although several in vivo approaches have been proposed to reduce inflammation in cisplatin-induced nephrotoxicity, it is unclear whether such approaches limit the anticancer efficacy of cisplatin [1].
Several cytokines (e.g. TNFα, IL-1β, MCP-1 and IL-6) are upregulated in the kidney in the inflammatory cascade triggered by cisplatin [1, 11, 32, 36]. TNFα plays a key role in the nephrotoxicity of cisplatin by inducing a variety of signaling pathways with cellular responses ranging from activation of inflammation to cell death [1, 11, 32, 37]. In fact, TNFα is able to activate other cytokines and/or chemokines such as ICAM-1 and MCP-1 [11, 36, 37], in turn leading to activation of the NF-κB pathway [36, 38]. There is strong evidence for the pivotal role of NF-κB activation in the pathogenesis of cisplatin-induced renal inflammation [10, 34, 39]. NF-κB regulates neutrophil, macrophage and lymphocyte biology, as well as the synthesis of hundreds of pro-inflammatory genes (cytokines, growth factors and adhesion molecules), including TNFα, thus amplifying the kidney injury caused by inflammation.
Using pharmacological and genetic approaches, many authors have shown that inhibition of activation of TNFα and NF-κB after administration of cisplatin was associated with reduced damage and improved kidney function and morphology [1, 7, 12, 13, 40].
In the present study, we show for the first time that cilastatin reduces cisplatin-induced inflammation by attenuating of NF-κB and synthesis of major pro-inflammatory cytokines including TNFα and the recruitment of the inflammatory cells into renal tissue. Thus, consistent with previous reports, we found that levels of serum TNFα and NF-κB in renal tissue are markedly increased in cisplatin-administered rats. However, we found that treatment with cilastatin results in a significant decrease in the levels in both molecules, leading to a reduction in inflammatory damage.
We also evaluated the expression of chemokines, interleukins and adhesion molecules with a known major role in the inflammatory process. ICAM-1 and VCAM-1 play an important role in leukocyte recruitment, and MCP-1 recruits monocytes, memory T cells and dendritic cells to the inflamed tissues [32, 41]. Consistent with the results reported by other groups, we showed increased mRNA and/or protein levels of VCAM-1, ICAM-1 and MCP-1 in tissue, urine or serum in cisplatin-administered rats [36, 40–42]. Studies based on various strategies (D-Ribose, sulforaphane or anti-CD54) have shown that the decrease in levels of these molecules in cisplatin-administered animals improves kidney damage caused by inflammation [6, 36, 41]. Cilastatin inhibited the increase in these factors and consequently prevented infiltration of monocyte/macrophages into renal tissue and decreased neutrophil stimulation, as evidenced by inhibition of MPO activity at the systemic level. Macrophages and neutrophils intensify the cytotoxic effects of cisplatin, because they are reservoirs of pro-inflammatory cytokines, which they release together with reactive oxygen species (ROS) into damaged kidneys [6, 11, 40, 43]. Thus, inhibition of tissue infiltration by these cells can help to protect kidneys against cisplatin-induced renal damage.
Several studies have shown that cytokines such as IL-10 counterbalance or inhibit the increase in other pro-inflammatory cytokine levels in order to protect tissue against injury in various pathophysiological conditions. Tadagavadi and Reeves [44] showed increased inflammation in cisplatin-induced renal damage in the absence of IL-10 by using IL-10-deficient mice. IL-10 exerts anti-inflammatory and anticytotoxic effect in kidney injury associated with lupus nephritis, immune complex glomerulonephritis and ischemic or cisplatin-induced nephrotoxicity [44, 45]. In our study, animals receiving both cisplatin and cilastatin maintained IL-10 levels similar to those of the control groups, whereas levels decreased in rats receiving only cisplatin, thus confirming findings reported elsewhere [44, 46].
Fibrosis associated with inflammation has been also demonstrated in cisplatin-induced kidney injury [12, 47]. In particular, studies have demonstrated that cisplatin increases levels of the fibrogenic cytokine TGFβ, which stimulates extracellular matrix protein production and deposition, leading to glomerulosclerosis and tubulointerstitial fibrosis [48]. In fact, Bayomi et al. [47] reported that inhibition of the TGFβ receptor protects kidneys after administration of cisplatin by inhibition of apoptotic effects, pro-inflammatory cytokines and renal fibrosis markers. In the present study, we found that cisplatin-administered animals had elevated renal and systemic levels of TGFβ, which were effectively suppressed by cilastatin.
Cilastatin significantly decreased serum creatinine and BUN, and improved GFR and the morphologic and structural features that are characteristic of declining renal function. One such feature is the renal hypertrophy, an adaptive growth of kidney tissue to restore the lost renal function that finally leads to tubular atrophy and interstitial fibrosis [49]. Several studies have shown an increase in the kidney:body weight ratio after administration of cisplatin [24, 50]. Similarly, TGFβ is also a major mediator of hypertrophic cellular changes in kidney diseases (see above), and it is known that anti-TGFβ therapy attenuates renal hypertrophy, thus improving renal dysfunction [49, 51, 52]. Our results confirm these findings and the finding that treatment with cilastatin reduced levels of kidney interstitial immune cells and TGFβ, which may have helped to decrease the renal hypertrophy previously induced by cisplatin.
Cilastatin also decreased levels of KIM-1, which were increased by administration of cisplatin. KIM-1 is a well-recognized biomarker for kidney injury and is upregulated in the proximal tubule of the injured kidney [53]. Yang et al. [54] recently showed that KIM-1-mediated epithelial cell phagocytosis of apoptotic cells after administration of cisplatin or ischemia protects the kidney after acute injury by downregulating inflammation and, specifically, by decreasing NF-κB activity in a p85-dependent manner. In contrast, Nozaki et al. [55] showed that KIM-1 signaling helps activate T cells, which mediate cisplatin-induced AKI leading to increased leukocyte recruitment and inflammatory injury. The authors showed that the administration of anti-KIM-1 antibodies protected against renal damage by decreasing cisplatin-induced NF-κB activation [55]. In this study, we did not examine whether or not the role of cilastatin on KIM-1 function mediated injury or protection in cisplatin-administered kidneys, although the downregulation of renal KIM-1 expression observed in our model after treatment with cilastatin was associated with decreased NF-κB activation and improvement of kidney damage.
Our results show that cilastatin is able to decrease inflammation in cisplatin-induced nephrotoxicity and that is nephroprotective. However, Linkermann et al. [56] reported that cisplatin-induced kidney nephropathy is mediated through Fas ligand (FasL), concluding that cisplatin-induced kidney injury may be mediated through FasL expressed in tubular cells rather than inflammatory cells. In addition, several studies have shown that both ROS and caspase-8-dependent Fas receptor (FasR) can activate NF-κB in cisplatin-induced nephrotoxicity [42, 57–60]. We previously showed that cilastatin significantly decreased expression of FasR, FasL, cleaved caspase-8 and ROS, thereby decreasing cell death in the kidneys of the animals that received cisplatin [21]. Consequently, although our new results suggest a major cilastatin-induced nephroprotective effect against inflammation, we cannot assert that this effect is the cause or simply a consequence of decreasing apoptosis, oxidative stress and, ultimately, cell death. Additional experiments should be performed to establish the role of decreasing inflammation by cilastatin in the protection of cisplatin-induced kidney injury. Such knowledge is essential, as many other renal inflammatory conditions could benefit from the potential protective effects of cilastatin.
In conclusion, we provide evidence that cilastatin, a renal DHP-I inhibitor, decreases renal toxicity in cisplatin-induced kidney injury. The nephroprotective effect observed was associated with suppression of TNFα and NF-κB and the subsequent decreased inflammatory response. However, it remains to be determined whether this is the cause or a consequence of protection. Cilastatin could prove useful as an adjuvant in the treatment of cancer and protect against cisplatin-induced renal injury.
SUPPLEMENTARY DATA
Supplementary data are available online at http://ndt.oxfordjournals.org
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by Spanish grants from the Ministry of Economy and Competitiveness ISCIII-FIS (PI11/01132 and PI14/01195 cofinanced by FEDER Funds from the European Commission, ‘A way of making Europe’), ISCIII-RETIC REDinREN/RD16/0009/0026 and Comunidad de Madrid S2010/BMD2378 (CIFRA). We thank Merck Sharp and Dhome for providing cilastatin used in the study.
CONFLICT OF INTEREST STATEMENT
S.C., A.T. and A.L. are coinventors of cilastatin patents as a renal protector against toxic injuries, that are assigned to Fundación para la Investigación Biomédica del Hospital Gregorio Marañón (FIBHGM) and licensed by FIBHGM to Spherium Biomed S.L. The manuscript has not been published and is not being considered for publication elsewhere, in whole or in part, in any language, except in abstract form.
REFERENCES
- 1. Pabla N, Dong Z.. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 2008; 73: 994–1007 [DOI] [PubMed] [Google Scholar]
- 2. Wang D, Lippard SJ.. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 2005; 4: 307–320 [DOI] [PubMed] [Google Scholar]
- 3. Ries F, Klastersky J.. Nephrotoxicity induced by cancer chemotherapy with special emphasis on cisplatin toxicity. Am J Kidney Dis 1986; 8: 368–379 [DOI] [PubMed] [Google Scholar]
- 4. Schrier RW. Cancer therapy and renal injury. J Clin Invest 2002; 110: 743–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yao X, Panichpisal K, Kurtzman N. et al. Cisplatin nephrotoxicity: a review. Am J Med Sci 2007; 334: 115–124 [DOI] [PubMed] [Google Scholar]
- 6. Kelly KJ, Meehan SM, Colvin RB. et al. Protection from toxicant-mediated renal injury in the rat with anti-CD54 antibody. Kidney Int 1999; 56: 922–931 [DOI] [PubMed] [Google Scholar]
- 7. El-Garhy AM, Abd El-Raouf OM, El-Sayeh BM. et al. Ellagic acid anti-inflammatory and antiapoptotic potential mediate renoprotection in cisplatin nephrotoxic rats. J Biochem Mol Toxicol 2014; 28: 472–479 [DOI] [PubMed] [Google Scholar]
- 8. Ueki M, Ueno M, Morishita J. et al. Curcumin ameliorates cisplatin-induced nephrotoxicity by inhibiting renal inflammation in mice. J Biosci Bioeng 2013; 115: 547–551 [DOI] [PubMed] [Google Scholar]
- 9. Guijarro C, Ejido J.. Transcription factor-ĸB (NF-ĸB) and renal disease. Kidney Int 2001; 59: 415–424 [DOI] [PubMed] [Google Scholar]
- 10. Sanz AB, Sanchez-Niño MD, Ramos AM. et al. NF-ĸB in renal inflammation. J Am Soc Nephrol 2010; 21: 1254–1262 [DOI] [PubMed] [Google Scholar]
- 11. Ramesh G, Reeves WB.. TNF-α mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity. J Clin Invest 2002; 110: 835–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Francescato HD, Costa RS, Scavone C. et al. Parthenolide reduces cisplatin-induced renal damage. Toxicology 2007; 230: 64–75 [DOI] [PubMed] [Google Scholar]
- 13. Ramesh G, Reeves WB.. Salicylate reduces cisplatin nephrotoxicity by inhibition of tumor necrosis factor-alpha. Kidney Int 2004; 65: 490–499 [DOI] [PubMed] [Google Scholar]
- 14. Parkin ET, Turner AJ, Hooper NM.. Differential effects of glycosphingolipids on the detergent-insolubility of the glycosilphosphatidylinositol-anchored membrane dipeptidase. Biochem J 2001; 358: 209–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Norrby SR, Alestij K, Björnegård B. et al. Urinary recovery of N-formimidoyl thienamycin (MK0787) as affected by coadministration of N-formimidoyl thienamycin dehydropeptidase inhibitors. Antimicrob Agents Chemother 1983; 23: 300–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Birnbaum J, Kahan FM, Kropp H. et al. Carbapenems, a new class of beta-lactam antibiotics. Discovery and development of imipenem/cilastatin. Am J Med 1985; 78: 3–21 [DOI] [PubMed] [Google Scholar]
- 17. Carmellini M, Matteucci E, Boggi U. et al. Imipenem/cilastatin reduces cyclosporin-induced tubular damage in kidney transplant recipients. Transplant Proc 1998; 30: 2034–2035 [DOI] [PubMed] [Google Scholar]
- 18. Markewitz A, Hammer C, Pfeiffer M. et al. Reduction of cyclosporine-induced nephrotoxicity by cilastatin following clinical heart transplantation. Transplantation 1994; 57: 865–870 [DOI] [PubMed] [Google Scholar]
- 19. Tejedor A, Torres AM, Castilla M. et al. Cilastatin protection against cyclosporin A-induced nephrotoxicity: clinical evidence. Curr Med Res Opin 2007; 19: 505–513 [DOI] [PubMed] [Google Scholar]
- 20. Camano S, Lazaro A, Moreno-Gordaliza E. et al. Cilastatin attenuates cisplatin-induced proximal tubular cell damage. J Pharmacol Exp Ther 2010; 334: 419–429 [DOI] [PubMed] [Google Scholar]
- 21. Humanes B, Lazaro A, Camano S. et al. Cilastatin protects against cisplatin-induced nephrotoxicity without compromising its anticancer efficiency in rats. Kidney Int 2012; 82: 652–663 [DOI] [PubMed] [Google Scholar]
- 22. Lazaro A, Humanes B, Jado JC. et al. The authors reply. Kidney Int 2013; 83: 1201–1202 [DOI] [PubMed] [Google Scholar]
- 23. Moreno-Gordaliza E, Giesen C, Lázaro A. et al. Elemental bioimaging in kidney by LA-ICP-MS as a tool to study nephrotoxicity and renal protective strategies in cisplatin therapies. Anal Chem 2011; 83: 7933–7940 [DOI] [PubMed] [Google Scholar]
- 24. Do Amaral CL, Francescato HD, Coimbra TM. et al. Resveratrol attenuates cisplatin-induced nephrotoxicity in rats. Arch Toxicol 2008; 82: 363–370 [DOI] [PubMed] [Google Scholar]
- 25. Lee KW, Jeong JY, Lim BJ. et al. Sildenafil attenuates renal injury in an experimental model of rat cisplatin-induced nephrotoxicity. Toxicology 2009; 257: 137–143 [DOI] [PubMed] [Google Scholar]
- 26. Sido B, Hammer C, Mraz W. et al. Nephroprotective effect of imipenem/cilastatin in reducing cyclosporine toxicity. Transplant Proc 1987; 19: 1755–1758 [PubMed] [Google Scholar]
- 27. Hammer C, Thies JC, Mraz W. et al. Reduction of cyclosporin (CSA) nephrotoxicity by imipenem/cilastatin after kidney transplantation in rats. Transplant Proc 1989; 21: 931. [PubMed] [Google Scholar]
- 28. Lazaro A, Gallego-Delgado J, Justo P. et al. Long-term blood pressure control prevents oxidative renal injury. Antiox Redox Signal 2005; 7: 1285–1293 [DOI] [PubMed] [Google Scholar]
- 29. Suzuki Y, Gomez-Guerrero C, Shirato I. et al. Pre-existing glomerular immune complexes induce polymorphonuclear cell recruitment through an Fc receptor-dependent respiratory burst: Potential role in the perpetuation of immune nephritis. J Immunol 2003; 170: 3243–3253 [DOI] [PubMed] [Google Scholar]
- 30. Gallego-Delgado J, Lazaro A, Gomez-Garre D. et al. Long-term organ protection by doxazosin and/or quinapril as antihypertensive therapy. J Nephrol 2006; 19: 588–598 [PubMed] [Google Scholar]
- 31. Sabbisetti VS, Ito K, Wang C. et al. Novel assays for detection of urinary KIM-1 in mouse models of kidney injury. Toxicol Sci 2013; 131: 13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ozkok A, Edelstein CL.. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int 2014; 2014: 967826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sahu BD, Kuncha M, Sindhura GJ. et al. Hesperidin attenuates cisplatin-induced acute renal injury by decreasing oxidative stress, inflammation and DNA damage. Phytomedicine 2013; 20: 453–460 [DOI] [PubMed] [Google Scholar]
- 34. Arjumand W, Seth A, Sultana S.. Rutin attenuates cisplatin induced renal inflammation and apoptosis by reducing NKkB, TNF-α and caspase-3 expression in wistar rats. Food Chem Toxicol 2011; 49: 2013–2021 [DOI] [PubMed] [Google Scholar]
- 35. Sanchez-Gonzalez PD, Lopez-Hernandez FJ, Perez-Barriocanal F. et al. Quercitin reduces cisplatin nephrotoxicity in rats without compromising its anti-tumor activity. Nephrol Dial Transplant 2011; 26: 3484–3495 [DOI] [PubMed] [Google Scholar]
- 36. Ueki M, Ueno M, Morishita J. et al. D-ribose ameliorates cisplatin-induced nephrotoxicity by inhibiting renal inflammation in mice. Tohoku J Exp Med 2013; 229: 195–201 [DOI] [PubMed] [Google Scholar]
- 37. Dong Z, Atherton SS.. Tumor necrosis factor-alpha in cisplatin nephrotoxicity: a homebred foe? Kidney Int 2007; 72: 5–7 [DOI] [PubMed] [Google Scholar]
- 38. Li Q, Verma IM.. NF-kappaB regulation in the immune system. Nat Rev Immunol 2002; 2: 725–734 [DOI] [PubMed] [Google Scholar]
- 39. Akcay A, Nguyen Q, Edelstein CL.. Mediators of inflammation in acute kidney injury. Mediators Inflamm 2009; 2009: 137072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kang KP, Kim DH, Jung YJ. et al. Alpha-lipoic acid attenuates cisplatin-induced acute kidney injury in mice by suppressing renal inflammation. Nephrol Dial Transplant 2009; 24: 3012–3020 [DOI] [PubMed] [Google Scholar]
- 41. Guerrero-Beltrán CE, Mukhopadhyay P, Horváth B. et al. Sulforaphane, a natural constituent of broccoli, prevents cell death and inflammation in nephropathy. J Nutr Biochem 2012; 23: 494–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee S, Moon SO, Kim W. et al. Protective role of L-2-oxothiazolidine-4-carboxylic acid in cisplatin-induced renal injury. Nephrol Dial Transplant 2006; 21: 2085–2095 [DOI] [PubMed] [Google Scholar]
- 43. Lu LH, Oh DJ, Dursun B. et al. Increased macrophage infiltration and fractalkine expression in cisplatin-induced acute renal failure in mice. J Pharmacol Exp Ther 2008; 324: 111–117 [DOI] [PubMed] [Google Scholar]
- 44. Tadagavadi RK, Reeves WB.. Endogenous IL-10 attenuates cisplatin nephrotoxicity: role of dendritic cells. J Immunol 2010; 185: 4904–4911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Deng J, Kohda Y, Chiao H. et al. Interleukin-10 inhibits ischemic and cisplatin-induced acute renal injury. Kidney Int 2001; 60: 2118–2128 [DOI] [PubMed] [Google Scholar]
- 46. Kim MG, Yang HN, Kim HW. et al. IL-10 mediates rosiglitazone-induced kidney protection in cisplatin nephrotoxicity. J Korean Med Sci 2010; 25: 557–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bayomi HS, Elsherbiny NM, El-Gayar AM. et al. Evaluation of renal protective effects of inhibiting TGF-β type I receptor in a cisplatin-induced nephrotoxicity model. Eur Cytokine Netw 2013; 24: 139–147 [DOI] [PubMed] [Google Scholar]
- 48. Border WA, Noble NA.. Transforming growth factor beta in tissue fibrosis. N Engl J Med 1994; 331: 1286–1292 [DOI] [PubMed] [Google Scholar]
- 49. Loeffler I, Wolf G.. Transforming growth factor-β and the progression of renal disease. Nephrol Dial Transplant 2014; 29: i37–i45 [DOI] [PubMed] [Google Scholar]
- 50. Ali BH, Al Moundhri MS, Tag Eldin M. et al. The ameliorative effect of cysteine prodrug L-2-oxothiazolidine-4-carboxylic acid on cisplatin-induced nephrotoxicity in rats. Fundam Clin Pharmacol 2007; 21: 547–553 [DOI] [PubMed] [Google Scholar]
- 51. Spurgeon KR, Donohoe DL, Basile DP.. Transforming growth factor-beta in acute renal failure: receptor expression, effects on proliferation, cellularity, and vascularization after recovery from injury. Am J Physiol Renal Physiol 2005; 288: F568–F577 [DOI] [PubMed] [Google Scholar]
- 52. Ziyadeh FN. Mediators of diabetic renal disease: the case for tgf-Beta as the major mediator. J Am Soc Nephrol 2004; 15: S55–S57 [DOI] [PubMed] [Google Scholar]
- 53. Ichimura T, Hung CC, Yang SA. et al. Kidney injury molecule-1: a tissue and urinary biomarker for nephrotoxicant-induced renal injury. Am J Physiol Renal Physiol 2004; 286: F552–F563 [DOI] [PubMed] [Google Scholar]
- 54. Yang L, Brooks CR, Xiao S. et al. KIM-1-mediated phagocytosis reduces acute injury to the kidney. J Clin Invest 2015; 125: 1620–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nozaki Y, Nikolic-Paterson DJ, Yagita H. et al. Tim-1 promotes cisplatin nephrotoxicity. Am J Physiol Renal Physiol 2011; 301: F1098–F1104 [DOI] [PubMed] [Google Scholar]
- 56. Linkermann A, Himmerkus N, Rölver L. et al. Renal tubular Fas ligand mediates fratricide in cisplatin-induced acute kidney failure. Kidney Int 2011; 79: 169–178 [DOI] [PubMed] [Google Scholar]
- 57. Sung MJ, Kim DH, Jung YJ. et al. Genistein protects the kidney from cisplatin-induced injury. Kidney Int 2008; 74: 1538–1547 [DOI] [PubMed] [Google Scholar]
- 58. Kreuz S, Siegmund D, Rumpf JJ. et al. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J Cell Biol 2004; 166: 369–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bentele M, Lavrik I, Ulrich M. et al. Mathematical modeling reveals threshold mechanism in CD95-induced apoptosis. J Cell Biol 2004; 166: 839–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lavrik IN, Golks A, Riess D. et al. Analysis of CD95 threshold signaling: triggering of CD95 (FAS/APO-1) at low concentrations primarily results in survival signaling. J Biol Chem 2007; 282: 13664–13671 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.