

Abstract
The term "aging-related tau astrogliopathy" (ARTAG) describes pathological accumulation of abnormally phosphorylated tau protein in astrocytes. We evaluated the correlates of ARTAG types (i.e., subpial, subependymal, white and gray matter, and perivascular) in different neuroanatomical regions. Clinical, neuropathological, and genetic (eg, APOE ε4 allele, MAPT H1/H2 haplotype) data from 628 postmortem brains from subjects were investigated; most of the patients had been longitudinally followed at the University of Pennsylvania. We found that (i) the amygdala is a hotspot for all ARTAG types; (ii) age at death, male sex, and presence of primary frontotemporal lobar degeneration (FTLD) tauopathy are significantly associated with ARTAG; (iii) age at death, greater degree of brain atrophy, ventricular enlargement, and Alzheimer disease (AD)-related variables are associated with subpial, white matter, and perivascular ARTAG types; (iv) AD-related variables are associated particularly with lobar white matter ARTAG; and (v) gray matter ARTAG in primary FTLD-tauopathies appears in areas without neuronal tau pathology. We provide a reference map of ARTAG types and propose at least 5 constellations of ARTAG. Furthermore, we propose a conceptual link between primary FTLD-tauopathy and ARTAG-related astrocytic tau pathologies. Our observations serve as a basis for etiological stratification and definition of progression patterns of ARTAG.
Keywords: Aging-related tau astrogliopathy, Alzheimer disease, ARTAG, Chronic traumatic encephalopathy, Dementia, Tau, Tauopathy
INTRODUCTION
Aging-related tau astrogliopathy (ARTAG) is a recently described term that refers to a morphological spectrum of astroglial pathology detected by immunohistochemical staining for tau, especially with antiphosphorylation-dependent (p-tau) antibodies (1). Two tau-astrogliopathy morphologies that are also detectable by immunostaining for the 4 repeat isoform of tau, i.e., thorn-shaped astrocytes (TSA) and granular/fuzzy astrocytes (GFA), are distinguished morphologically and by their locations, e.g. subpial, subependymal, perivascular, white matter, and gray matter (1). Some studies have suggested that constellations of gray and white matter ARTAG might be associated with cognitive decline or focal cortical manifestations (2–4); however, the clinical significance of ARTAG is currently poorly understood. Earlier descriptions focused on the medial temporal lobe (particularly on subpial, perivascular, and white matter ARTAG) and failed to demonstrate a convincing association with dementia (5–8). A recent study on 4 supercentenarians applying the harmonized evaluation strategy of ARTAG confirmed that the basal forebrain is a common site for this pathology but showed that additional regions can also be affected (9).
A distinct aspect of ARTAG is its relation to the tau pathology described in chronic traumatic encephalopathy (CTE). Subpial and subependymal clusters of astrocytic tangles have been described in CTE (10, 11). A recent consensus meeting on the neuropathological criteria for the diagnosis of CTE indicated that the pathognomonic CTE lesions consist of p-tau aggregates in neurons, astrocytes, and cell processes around small vessels in an irregular pattern at the depths of the cortical sulci (12). Astroglial tau pathology is considered to be a supportive feature of CTE if TSA are seen at the glial limitans, most commonly found in the subpial and periventricular regions; on the other hand, TSA in the subcortical white matter or in the mediobasal region, including amygdala and hippocampus, are considered to be non-diagnostic and do not support the diagnosis of CTE (12). A recent study on the frequency of ARTAG in the basal forebrain supported the concept that CTE and ARTAG may share a common etiological pathway; this was also corroborated by the observation of male predominance in both pathologies (13).
Following the publication of consensus statements, ARTAG and CTE pathology have been reported in various neurodegenerative conditions in addition to Alzheimer disease (AD) (2, 3); these include α-synucleinopathies, including multiple system atrophy (MSA) (14), Parkinson disease (PD), and Lewy body dementia (LBD) (13), and in the prion disease Creutzfeldt-Jakob disease (15). The complexity of overlapping CTE/ARTAG pathologies and the effect of age on these was highlighted by studies reporting various proportions of pathologies interpreted as representing CTE (16, 17). These studies have shown that primary frontotemporal lobar degeneration (FTLD) tauopathies (i.e. disorders in which tau protein deposition is the predominant feature) also show ARTAG/CTE type pathologies. Importantly, non-ARTAG type astrocytic tau pathology is a hallmark of some primary FTLD-tauopathies (FTLD-tau), such as progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), and globular glial tauopathy (GGT). In other FTLD-tau subtypes such as Pick disease (PiD), argyrophilic grain disease (AGD), or neurofibrillary tangle (NFT) predominant senile dementia (NFT-dementia or "tangle-only" dementia; now included in the category of primary age-related tauopathy [PART]), astrocytic tau pathology is less prominent (18, 19). In particular, it is not clear whether the gray matter ARTAG seen in the elderly has any relation to the characteristic astrocytic tau morphologies like tufted astrocytes in PSP, astrocytic plaques in CBD, and ramified astrocytes in PiD.
In the present study we systematically mapped ARTAG types in different anatomical regions and correlated these with clinicopathological and genetic variables. We show that several ARTAG types may coexist in the same brain and specific patterns and constellations that are associated with certain clinicopathological variables can be recognized. These observations enhance our understanding of ARTAG types and suggest common or overlapping pathogenic aspects with CTE and primary FTLD-tauopathies.
MATERIALS AND METHODS
Selection of Cases
Six hundred and twenty-eight cases were systematically evaluated. Five hundred and seventy cases were examined in the collection of brains in the Center for Neurodegenerative Disease Research (CNDR) Brain Bank at the University of Pennsylvania, Philadelphia, PA (20); an additional 58 were from the longitudinal VITA study collection, Vienna, Austria (2).
Immunohistochemistry
Formalin fixed, paraffin-embedded tissue blocks of frontal, anterior cingulate, parietal, temporal, occipital, and entorhinal cortex, hippocampus, amygdala, basal ganglia, thalamus, mesencephalon, pons, medulla oblongata, and cerebellum were evaluated. Immunostaining for tau was performed with anti-tau PHF-1 (Ser396/Ser404, 1:2000; Gift of Peter Davies) and anti-tau AT8 (pS202, 1:200; Pierce Biotechnology, Rockford, IL). The Vectashield ABC detection kits, peroxidase/DAB, rabbit/mouse/rat (BA1000/BA2000/BA4001, 1:1000; Vector Laboratories, Burlingame, CA) were used for visualization of antibody reactions. Double immunolabeling was performed using anti-tau monoclonal antibody (MAb) AT8 (1:200) and MAb anti-GFAP 2.2B10 (21) (1:10 000). The fluorescence-labeled secondary antibodies were used as described earlier (18, 22). We evaluated double immunofluorescent labeling with a Leica TCS SPE-II scanning laser confocal microscope, as described earlier (18, 22).
Evaluation of Tau Pathologies
Following the consensus recommendations (1) we evaluated the presence (yes/no) and severity of ARTAG using a 3-tiered scoring scheme, as follows: 1, occasional and focal (i.e., mild); 2, numerous focal or occasional but scattered throughout an anatomical region; and 3, numerous widespread. Gray and white matter, subpial, and perivascular ARTAG types were determined in the (1) hippocampus pyramidal layers, dentate gyrus, inferior temporal gyrus, and amygdala: Together representing medial temporal lobe (MTL) structures; (2) the middle frontal gyrus, anterior cingulate, inferior parietal gyrus, superior temporal gyrus, and occipital cortex (area peristriata and striata): Together representing lobar structures; (3) the caudate-putamen, globus pallidus, thalamus, and basal forebrain: Together representing subcortical structures); (4) the midbrain tegmentum, substantia nigra, locus coeruleus, pontine base, tegmentum and base of the medulla oblongata: Together representing brainstem structures. Subependymal (inferior, anterior, and posterior horns of the lateral ventricle, 3rd ventricle and aqueduct at different brainstem levels were also evaluated. The percentage of examined regions showing different ARTAG types was calculated by dividing the number of regions showing ARTAG and the number of regions evaluated for ARTAG.
In all regions, a 3-grade score (mild, moderate, severe) was applied to describe neuronal tau pathology and the primary FTLD-tauopathy-related astroglial tau pathology. In addition, the presence of oligodendroglial tau pathology in these regions and tau grain pathology in the MTL were documented.
Genetic Analysis
Genomic DNA was extracted from brain tissue using commercial reagents (Qiagen, Hilden, Germany) and genotyped for APOE rs429358 and rs7412 (which define the ɛ2, ɛ3, and ɛ4 alleles) as well as MAPT rs1052553 (which distinguishes H1 and H2 haplotypes), as previously described (23).
Clinicopathological Variables
In addition to the documentation of tau pathologies, the following variables were included in the database: Gender, age at death, clinical diagnosis, neuropathological diagnosis, APOE genotype, and MAPT H1/H2 haplotype. We grouped the cases either based on clinical or neuropathological diagnoses. Five clinical groups were defined as follows: (1) cognitive decline/dementia, which included cases with clinical diagnosis of possible and probable AD, dementia of undetermined origin and frontotemporal dementia; (2) cases with motor neuron disease; (3) cases with movement disorder (e.g. parkinsonism, corticobasal syndrome, PSP syndrome), with or without dementia; (4) cases with psychiatric symptoms including chronic schizophrenia and major depression; and (5) cases without neuropsychiatric symptoms.
Neuropathological groupings were based on the primary histopathological diagnosis. AD and PART were included in 1 group but separate analyses were also performed (see below). Only 3 cases showed pure AGD; thus, a separate analysis was performed in which AGD was listed as a secondary diagnosis. Furthermore, a 3-grade score of ventricular enlargement, atherosclerosis in basal cerebral arteries, and brain atrophy (mild, moderate, severe) based on gross inspection; the presence or lack of α-synuclein, TDP-43 pathology, primary FTLD-tauopathy (further stratified as AGD, CBD, PSP, PiD), cerebral amyloid angiopathy (CAA), vascular disease and hippocampal sclerosis, was recorded. Furthermore, CERAD criteria (score A–C) (24), Braak stages of neurofibrillary degeneration (I–VI) (25, 26), and phases of Aβ parenchymal deposits grouped into 3 categories (phases 1–2, 3, and 4–5 separately) (27), were documented.
Statistical Methods
SPSS Statistics Version 23 was used for statistical analysis. Chi-square test and Spearman correlation coefficient were used to evaluate associations and correlations between variables of interest. Logistic regression models (binary or ordinal) were used to generate odds ratios (OR) and 95% confidence intervals, where the presence of ARTAG, their types and regional distributions and their severity scores were the dependent variables, and various clinical and pathological data were the independent variables. To reduce the chance of false positive discovery due to the relatively large number of statistical tests performed, we lowered the standard significance level down to 0.01. All statistical tests are 2-sided.
RESULTS
Overview
Clinicopathological characteristics of the cohort are summarized in Table 1. Clinical data were not available for 22 cases (3.5% of the cohort). Mean age at death of the clinical cohort was 77.85 years (range 71.69–83.27). Males were overrepresented in the group with movement disorders. Age-matched control cases did not show any neuropathology, including a lack of NFTs (i.e. cases with Braak stage I were included in the PART group). The mean age at death varied from 66.8 years (MSA) to 87 years (AGD).
TABLE 1.
Overview of Examined Cases and Frequencies of ARTAG
| Clinical Group | Number | % | Mean Age at Death | SE | Male | % | Female | % | ARTAG % |
|---|---|---|---|---|---|---|---|---|---|
| Cognitive decline/dementia | 319 | 50.7 | 79.08 | 0.47 | 170 | 53.3 | 149 | 46.7 | 63 |
| Motor neuron disease | 64 | 10.2 | 71.69 | 1.02 | 37 | 57.8 | 27 | 42.2 | 43.8 |
| Movement disorder | 135 | 21.5 | 75.81 | 0.82 | 87 | 64.4 | 48 | 35.6 | 68.9 |
| Psychiatric symptoms | 26 | 4.1 | 83.27 | 1.42 | 11 | 42.3 | 15 | 57.7 | 69.2 |
| No symptoms | 62 | 9.9 | 80.10 | 1.02 | 30 | 48.4 | 32 | 51.6 | 53.2 |
| Clinical data not available | 22 | 3.5 | – | – | – | – | – | – | – |
| Summary | 628 | 100.0 | 77.85 | 0.36 | 335 | 55.3 | 271 | 44.7 | 61.6 |
| Neuropathology Group | |||||||||
| AD/PART | 322 | 51.3 | 80.96 | 0.42 | 137 | 42.5 | 185 | 57.5 | 63 |
| ALS | 60 | 9.6 | 71.13 | 1.00 | 37 | 61.7 | 23 | 38.3 | 41.7 |
| AGD (pure) | 3 | 0.5 | 87.00 | 1.53 | 2 | 66.7 | 1 | 33.3 | 100 |
| AGD (secondary diagnosis) | 54 | 8.6 | 79.30 | 1.07 | 29 | 53.7 | 25 | 46.3 | 94.4 |
| CBD | 21 | 3.3 | 70.38 | 2.36 | 9 | 42.9 | 12 | 57.1 | 100 |
| LBD | 117 | 18.6 | 78.20 | 0.71 | 86 | 73.5 | 31 | 26.5 | 56.4 |
| MSA | 35 | 5.6 | 66.80 | 1.39 | 24 | 68.6 | 11 | 31.4 | 42.9 |
| PICK | 14 | 2.2 | 68.21 | 2.60 | 8 | 57.1 | 6 | 42.9 | 35.7 |
| PSP | 49 | 7.8 | 78.41 | 1.08 | 34 | 69.4 | 15 | 30.6 | 98 |
| Control | 7 | 1.1 | 75.00 | 2.49 | 6 | 1 | 17 | 48.6 | 28.6 |
| Summary | 628 | 100.0 | 77.84 | 0.36 | 343 | 54.6 | 285 | 45.4 | 61.8 |
Distribution Patterns of ARTAG
TSA with astrocytic processes reaching the surface of the brain, the ependyma, or vessel walls were observed in subpial, subependymal, and perivascular locations, respectively. Subpial and subependymal ARTAG showed focal accumulations or was widespread. Particularly in lobar regions, only limited foci of TSA with or without tau pathology in the underlying cortex were observed (Fig. 1A–C). Occasionally, lobar subpial ARTAG was associated with perivascular ARTAG in the cortex or white matter just below where subpial ARTAG was observed (Fig. 1D–F). In those cases, we did not observe accumulations of perivascular neuronal tau pathology. Importantly, in CBD cases only there were widespread subpial tau-positive astrocytic processes with less prominent cytoplasmic tau immunoreactivity (i.e., the typical TSA morphology) in lobar regions (Fig. 1G–I). Similarly, perivascular astrocyte processes were frequent in CBD but did not always show the TSA morphology seen in all other disease groups including PSP (Fig. 1J). This also differed from the accumulation of tau immunoreactive pathology around vessels showing CAA in AD cases since these were not related to astrocytic processes but represented mostly dystrophic neuritic components (Fig. 1K, L).
FIGURE 1.

Patterns of subpial and perivascular ARTAG. (A–C) Distinct foci of subpial thorn-shaped astrocytes (TSA) in the anterior cingulate cortex in a patient with PSP. (D–F) Perivascular ARTAG (D; indicated by an arrow and enlarged in E) was rarely associated with lobar subpial ARTAG (D; indicated by an arrowhead and enlarged in F). (G–I) In CBD, we observed subpial tau-positive astrocytic processes with less prominent cytoplasmic tau immunoreactivity in the parietal lobe (G; enlarged in H and I). Perivascular ARTAG in the basal forebrain as exemplified in a PSP case (J). Accumulations of tau immunoreactivity around vessels showing amyloid angiopathy in AD (K) were related to dystrophic neuritic components (L, green represents GFAP, red phospho-tau, immunoreactivity).
White matter ARTAG was characterized by TSA with or without fine granular immunoreactivity in the processes; this was widespread in the white matter or accentuated focally (Fig. 2A–C). White matter ARTAG was associated frequently with perivascular, subependymal ARTAG and also with gray matter ARTAG in adjacent areas (Fig. 2D–F). Gray matter ARTAG was represented mostly by GFA. In the amygdala, cortex and basal ganglia GFA showed the typical fine granular tau immunoreactivity in processes associated with variable accumulation of tau immunoreactivity in the cytoplasm (1). In the brainstem, the cytoplasm of GFA was plump and less tau immunoreactivity extended into the processes (Fig. 2G). TSA were also seen in the dentate gyrus and hippocampal CA4 (Fig. 2H); they also appeared in the amygdala and lobar regions as clusters (Fig. 2I). Importantly, in addition to the classical astrocytic tau-morphologies such as astrocytic plaques, tufted astrocytes and ramified astrocytes in primary FTLD-tauopathies (i.e. CBD, PSP, PiD), several astrocytic profiles that were indistinguishable from GFA also were seen in anatomical regions where characteristics of these primary FTLD-tauopathies were lacking (Fig. 2J–L).
FIGURE 2.

Patterns of white and gray matter ARTAG. (A–C) White matter ARTAG was characterized by thorn-shaped astrocytes in many brain regions (A, B) as well as in focal accentuations (C; indicated by asterisks). (D–F) White matter (WM) ARTAG was associated frequently with perivascular, subependymal (D; indicated by Ep and enlarged in E) ARTAG and also with gray matter (GM) ARTAG (D; enlarged in F). (G–L) Gray matter ARTAG was represented mostly by granular/fuzzy astrocytes (GFA) or TSA. In the brainstem the cytoplasm was plump (G; inset shows double immunolabeling for GFAP indicated by green and phospho-tau indicated by red). Accumulation of TSA in the dentate gyrus and CA4 in a case with clinically probable AD (H). Clusters of GFA in the amygdala in a case with schizophrenia (I; inset shows enlarged area). GFA-like astrocytic tau immunoreactivities were seen in CBD (J; parietal cortex), PSP (K; here frontal cortex), and in PiD (L; here occipital cortex: Left enlarged inset indicates a typical ramified astrocyte and the right, an enlarged image a GFA-type morphology).
ARTAG was seen in several anatomic locations (Fig. 3). Subpial ARTAG was noted in lobar areas, hippocampus, amygdala, basal forebrain, lateral parts of the midbrain, pons, medulla oblongata, and spinal cord. Subependymal ARTAG was present in the third ventricle, the inferior horn of the lateral ventricle, aqueduct of the midbrain and medulla oblongata and only in single cases and focally in the anterior and posterior horn of the lateral ventricle. White mater ARTAG appeared in the juxtacortical areas of the cortical lobes, subinsular area, hippocampal and peri-amygdaloid white matter, white matter areas surrounding the basal forebrain, cerebral peduncles, peri-olivary white matter, midline structures of the medulla oblongata, pyramids, and lateral and posterior tracts of the spinal cord. Gray matter ARTAG was observed in all examined lobar cortical regions, amygdala, hippocampus, dentate gyrus, caudate-putamen, thalamus, globus pallidus, claustrum, tectum and periaqueductal gray of the midbrain, locus coeruleus, substantia nigra, hypoglossal nucleus, dorsal vagus nucleus, inferior olives, and in the anterior horn of the spinal cord. In the medulla oblongata white and gray matter ARTAG was frequently asymmetric; in cases showing both white and gray matter ARTAG they did not always appear in the same sides. In the present cohort we did not observe white matter ARTAG in the corpus callosum, internal capsule, white matter of the pons base, or cerebellar peduncles. Perivascular ARTAG was seen where white and gray matter ARTAG was present.
FIGURE 3.
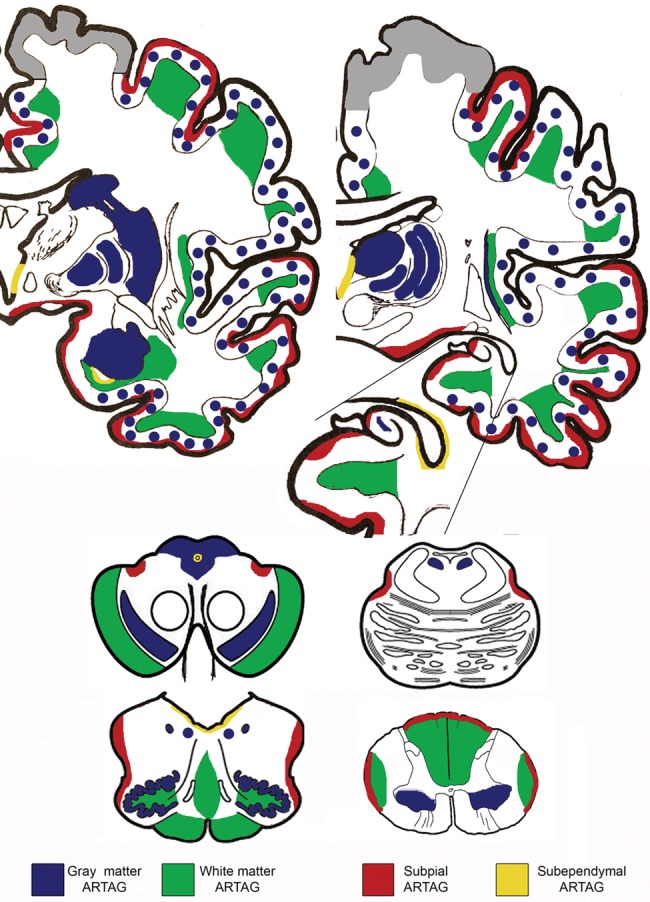
Distribution patterns of ARTAG in this study. Note that perivascular is not indicated here because it was seen together with white matter ARTAG. Gray color in the cortex indicates areas that were not evaluated.
Variables With Effects on the Presence of ARTAG
Logistic regression models were used to assess the univariate and multivariable effects of age at death, gender, variables related to the morphological diagnosis of AD (CERAD, BB stage, Aβ score), the presence of the APOE ε4 allele, and other pathological variables (the presence of TDP-43, α-synucleinopathy, vascular lesions, CAA, macroscopic ventricular enlargement, atherosclerosis score of basal arteries, and degree of brain atrophy) on the presence of ARTAG (Table 2). As single variables, age at death, Braak stage of NFT pathology, CERAD score, degree of brain atrophy, ventricular enlargement and basal atherosclerosis, and the presence of primary FTLD-tauopathy showed significant effects (p < 0.01), while male gender and the presence of TDP-43 proteinopathy showed somewhat weaker effects (p < 0.05). When age at death, gender and Braak stage of NFT pathology were added to the model, only age at death, male gender, and primary FTLD-tauopathy remained significant (p < 0.01); a smaller effect was noted for ventricular enlargement, basal atherosclerosis and brain atrophy (p < 0.05) (Table 2). When all examined pathological variables were added to the model, only age at death (OR: 1.11; 95 COI: 1.05–1.17), and the presence of primary FTLD-tauopathy (OR: 16.4; COI: 2.01–134.29) showed significant effects (p < 0.001). MAPT haplotype and APOE genotype did not have significant effects on the presence of ARTAG.
TABLE 2.
Logistic Regression Models for the Assessment of the Univariate and Multivariable Effect of Variables on the Presence of ARTAG in the Whole Study Cohort
| Presence of ARTAG | Univariate Logistic Regression Models |
Multiple Logistic Regression Model (With Age at Death) |
Multiple Logistic Regression Model (Age at Death, Gender, Braak Stage) |
|||
|---|---|---|---|---|---|---|
| Variables | Odds Ratio | p Value | Odds Ratio | p Value | Odds Ratio | p Value |
| (95% CI) | (95% CI) | (95% CI) | ||||
| Age at death | 1.06 | 0.0001 | – | – | 1.08 | 0.0001 |
| (1.04–1.088) | – | (1.05–1.10) | ||||
| Ventricular enlargement | 1.34 | 0.0001 | 1.25 | 0.009 | 1.24 | 0.033 |
| (1.14–1.5) | (1.05–1.49) | (1.01–1.53) | ||||
| Basal atherosclerosis | 1.55 | 0.0001 | 1.27 | 0.01 | 1.28 | 0.012 |
| (1.31–1.83) | (1.05–1.53) | (1.05–1.55) | ||||
| Brain atrophy | 1.3 | 0.003 | 1.27 | 0.009 | 1.33 | 0.015 |
| (1.09–1.55) | (1.06–1.52) | (1.05–1.69) | ||||
| TDP-43 pathology | 1.63 | 0.035 | 1.23 | 0.39 | 1.09 | 0.72 |
| (1.03–2.57) | (0.76–199) | (0.65–1.83) | ||||
| α-Syn pathology | 1.14 | 0.46 | 1.03 | 0.87 | 1.05 | 0.80 |
| (0.79–1.64) | (0.71–1.49) | (0.71–1.55) | ||||
| Primary FTLD-tauopathy | 6.16 | 0.0001 | 9.42 | 0.0001 | 55.19 | 0.0001 |
| (3.12–12.14) | (4.56–19.42) | (7.44–409.13) | ||||
| Aβ parenchymal deposit | 1.22 | 0.06 | 1.08 | 0.46 | 1.21 | 0.19 |
| (0.98–1.51) | (0.86–1.36) | (0.90–1.62) | ||||
| Braak NFT stage | 1.12 | 0.006 | 1.04 | 0.35 | 1.06 | 0.19 |
| (1.03–1.22) | (0.95–1.13) | (0.97–1.16) | ||||
| CERAD score | 1.17 | 0.01 | 1.03 | 0.63 | 1.02 | 0.81 |
| (1.03–1.33) | (0.9–1.18) | (0.84–1.24) | ||||
| Vascular lesions | 1.02 | 0.95 | 0.72 | 0.33 | 0.73 | 0.37 |
| 0.54–1.9 | (0.38–1.38) | (0.37–1.44) | ||||
| CAA | 2.23 | 0.088 | 1.7 | 0.26 | 2.00 | 0.17 |
| (.088–5.61) | (0.67–4.33) | (0.72–5.53) | ||||
| ApoE e4 allele | 0.89 | 0.54 | 0.95 | 0.82 | 0.94 | 0.76 |
| (0.61–1.29) | (0.64–1.41) | (0.62–1.41) | ||||
| Male gender | 1.42 | 0.031 | 1.73 | 0.002 | 1.87 | 0.001 |
| (1.03–1.97) | (1.23–2.44) | (1.30–2.69) | ||||
Examination of the effects of these variables on the presence of ARTAG types showed distinct results (Supplementary Data File S1). Age at death, together with the degree of brain atrophy, ventricular enlargement, basal atherosclerosis, and CERAD score showed weak but significant effects only for subpial, white matter, and perivascular ARTAG. In contrast, the effect of primary FTLD-tauopathy was significant for subependymal and gray matter ARTAG only.
Further evaluation of these variables on the appearance of ARTAG types in different anatomical regions revealed additional complex patterns (Supplementary Data File S2). Age at death had a weak but significant effect on the majority of ARTAG types in all examined anatomical regions except in lobar areas (i.e. subpial, subependymal, perivascular, and gray matter). Ventricular enlargement was associated mostly with lobar perivascular and white matter ARTAG, while the degree of atherosclerosis in basal arteries showed effect on the presence of subependymal ARTAG in subcortical areas (i.e., 3rd ventricle). The presence of primary FTLD-tauopathy showed strong associations with the presence of ARTAG types except for some (i.e. white matter, subpial and subependymal) appearing in the brainstem. AD-related variables were associated with the appearance of lobar white matter ARTAG. Interestingly, MAPT H2/H2 haplotype showed an association with lobar white matter ARTAG. CAA was associated with lobar subependymal (i.e. lateral ventricle) and subcortical subpial ARTAG; cerebrovascular disease (without specific anatomical location) was associated with brainstem perivascular ARTAG. Male gender showed an effect on the presence of MTL and lobar gray matter ARTAG. Finally, the presence of TDP-43 pathology was associated with the presence of subpial ARTAG in the MTL and subcortical (basal forebrain) regions. Lower Braak stage of NFT pathology and CERAD score were associated with MTL gray matter ARTAG. The presence of α-synucleinopathy showed weaker association (p < 0.05 and >0.01) with the lack of lobar subpial, and gray matter ARTAG as well as subcortical and brainstem gray matter ARTAG.
Frequency of ARTAG Types in Clinical and Pathological Groups
The frequency of ARTAG was significantly higher in individuals presenting with movement disorder than in those without neuropsychiatric symptoms (Table 1). This remained significant in logistic regression when age at death, gender, and Braak stage of NFT pathology were added to the model; however, significance was lost when the presence of primary FTLD-tauopathy was also included in the model. Furthermore, when evaluating ARTAG types in clinical groups, only the presence of white matter (OR: 2.81, 95 COI: 1.39–5.69, p = 0.004 for cognitive decline and OR: 2.57, 95 COI: 1.19–5.56, p = 0.016 for movement disorder) and perivascular (OR: 2.78, 95 COI: 1.28–6.07, p = 0.01 for cognitive decline and OR: 3.19, 95 COI: 1.38–7.40, p = 0.007 for movement disorder) ARTAG remained significant after correction for the presence of primary FTLD-tauopathies (Table 3; Fig. 4). When evaluating ARTAG types in subregions, only weaker effects were noted (Table 3; Supplementary Data Files S3 and S4).
TABLE 3.
Frequencies of ARTAG Types in Different Anatomical Regions in Clinical and Neuropathological Diagnostic Groups
| Clinical Group | MTL- SP | Lobar- SP | Subcort- SP | BST- SP | MTL- SE | Lobar- SE | Subcort- SE | BST- SE | MTL- PV | Lobar- PV | Subcort- PV | BST- PV | MTL- GM | Lobar- GM | Subcort- GM | BST- GM | MTL- WM | Lobar- WM | Subcort- WM | BST- WM |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cognitive decline/ dementia | 28.9 | 9.1 | 27.3 | 9.9 | 12.2 | 0.7 | 4.8 | 1.7 | 26.0 | 7.1 | 20.3 | 3.6 | 20.6 | 16.2 | 18.6 | 6.9 | 32.5 | 18.1 | 16.6 | 8.3 |
| Motor neuron disease | 17.5 | 7.8 | 12.7 | 3.2 | 11.1 | 0.0 | 0.0 | 9 | 15.9 | 1.6 | 6.3 | 0.0 | 23.8 | 9.4 | 6.3 | 3.2 | 15.9 | 1.6 | 6.3 | 1.6 |
| Movement disorder | 23.5 | 11.7 | 24.0 | 8.5 | 13.6 | 1.6 | 6.2 | 9 | 21.2 | 7.8 | 21.5 | 3.9 | 48.5 | 27.9 | 29.2 | 7 | 23.5 | 8.6 | 12.3 | 6.2 |
| Psychiatric symptoms | 30.8 | 15.4 | 30.8 | 12.0 | 24.0 | 0.0 | 7.7 | 9 | 30.8 | 3.8 | 30.8 | 0.0 | 30.6 | 15.4 | 19.2 | 8 | 42.3 | 7.7 | 23.1 | 4 |
| No symptoms | 24.6 | 1.7 | 11.5 | 5.4 | 9.8 | 3.7 | 1.9 | 1.8 | 11.5 | 1.7 | 15.4 | 3.6 | 24.6 | 3.3 | 7.7 | 10.7 | 19.7 | 0 | 9.4 | 7.5 |
| All cases | 26.1 | 9.0 | 23.8 | 8.5 | 13.2 | 1.0 | 4.5 | 1 | 22.6 | 6.0 | 19.1 | 3.1 | 28.0 | 16.7 | 18.7 | 6.9 | 27.8 | 11.9 | 14.2 | 6.8 |
| Neuropathology Group | ||||||||||||||||||||
| AD/PART | 29.5 | 6.2 | 25.5 | 10.9 | 12.7 | 1.3 | 5.2 | 1.7 | 24.5 | 7.4 | 20.3 | 5.3 | 17.7 | 9.6 | 16 | 8.6 | 30.9 | 19.2 | 15.5 | 8 |
| ALS | 11.9 | 6.7 | 11.7 | 3.3 | 10.2 | 0 | 0 | 1.7 | 13.6 | 1.7 | 6.7 | 0 | 22 | 5 | 6.7 | 3.3 | 13.6 | 1.7 | 6 | 3.3 |
| AGD (pure) | 0 | 33.3 | 0 | 0 | 0 | 0 | 0 | 0 | 33.3 | 0 | 0 | 0 | 33.3 | 66.7 | 0 | 0 | 33.3 | 0 | 0 | 0 |
| AGD (second diagnosis) | 33.3 | 19.6 | 34.6 | 11.3 | 20.4 | 2.2 | 7.5 | 0 | 31.5 | 7.8 | 32.7 | 3.8 | 90.4 | 54.9 | 44.2 | 9.6 | 37 | 7.9 | 18.9 | 3.9 |
| CBD | 55 | 100 | 50 | 16.7 | 35 | 7.1 | 18.8 | 0 | 45 | 46.7 | 58.8 | 11.8 | 90 | 93.3 | 94.1 | 23.5 | 50 | 14.3 | 41.2 | 5.9 |
| LBD | 25.6 | 3.4 | 24.8 | 6 | 9.4 | 1.7 | 3.4 | 3.4 | 21.9 | 1.7 | 13.7 | 3.4 | 34.2 | 10.3 | 4.3 | 1.7 | 26.5 | 3.4 | 14.5 | 8.5 |
| MSA | 11.4 | 0 | 11.8 | 0 | 5.7 | 0 | 2.9 | 0 | 5.7 | 2.9 | 11.8 | 0 | 17.1 | 5.7 | 0 | 0 | 5.7 | 2.9 | 2.9 | 0 |
| PiD | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 7.1 | 0 | 0 | 0 | 0 | 28.6 | 0 | 0 | 7.1 | 0 | 0 | 0 |
| PSP | 26.5 | 20.8 | 34.8 | 12.8 | 30.6 | 4.2 | 8.7 | 2.1 | 32.7 | 8.3 | 43.5 | 6.4 | 77.6 | 77.1 | 89.1 | 23.4 | 46.9 | 18.8 | 19.6 | 8.5 |
| Control | 14.3 | 14.3 | 14.3 | 0 | 14.3 | 0 | 0 | 0 | 14.3 | 0 | 0 | 0 | 0 | 0 | 14.3 | 0 | 14.3 | 0 | 14.3 | 0 |
| All cases | 26.2 | 9 | 23.8 | 8.5 | 13.5 | 1.2 | 4.7 | 1.2 | 22.8 | 5.9 | 19.3 | 3.5 | 28.1 | 17.1 | 19.1 | 7.5 | 28.3 | 12.6 | 14.1 | 6.9 |
MTL, medial temporal lobe; Subcort, subcortical; BST, brainstem; SP, subpial; SE, subependymal; PV, perivascular; GM, gray matter; WM, white matter; AD, Alzheimer disease; PART, primary age-related tauopathy, AGD: argyrophilic grain disease; CBD, corticobasal degeneration; LBD, Lewy body disease; MSA, multiple system atrophy; PiD, Pick disease; PSP, progressive supranuclear palsy.
FIGURE 4.

Percent of cases with (gray shading) or without (no shading) ARTAG types in clinical and neuropathological diagnostic groups. MND, motor neuron disease; MovDis, movement disorder (e.g. parkinsonism, corticobasal syndrome, PSP syndrome).
Next we performed logistic regression analysis to evaluate the effect of neuropathological diagnostic groups. After exclusion of primary FTLD tauopathies and other disorders (i.e., PD, LBD, MSA), we compared cases with definitive PART (28) and those with AD-related pathologies (Aβ and tau). The presence of Aβ pathology did not show a significant effect on the appearance of ARTAG (OR: 1.22, 95 COI: 0.98–1.51, p = 0.068; after correction for age at death, p = 0.46). Except for white matter ARTAG (after correction for age at death; OR: 1.26, 95 COI: 1.01–1.58, p = 0.038), there was a lack of effect of Aβ on the presence of ARTAG types. Regarding evaluation of regions, lobar white matter ARTAG was associated with the presence of Aβ plaques (after correction for age at death; OR: 2.11, 95 COI: 1.3–3.42, p = 0.002). Lobar white matter ARTAG was seen in advanced AD cases in the frontal, parietal, cingulate, temporal, and hippocampal white matter (6%–15% of cases depending on the region, the most in the parietal white matter).
We then asked whether cases with AD or PART pathology or no pathology showed differences when compared with other neuropathological diagnoses (Table 1). Logistic regression (with age at death and gender added to the model) revealed a strong association of ARTAG with PSP when compared with AD/PART (OR: 34.83, 95 COI: 4.66–260.09, p = 0.001) and the control group (OR: 140.53, 95 COI: 10.23–1929.67, p < 0.0001). Gray matter ARTAG was significantly associated with CBD (OR: 25.12, 95 COI: 2.37–266.25, p = 0.007) and less with PSP (OR: 13.95, 95 COI: 1.53–127.24, p = 0.019) when compared with controls. When the AD/PART group was compared with other neuropathological diagnostic groups, subpial and perivascular ARTAG was associated with CBD (OR: 4.84, 95 COI: 1.81–12.91, p = 0.002 for subpial and OR: 4.51, 95 COI: 1.74–12.68, p = 0.002 for perivascular ARTAG); subependymal ARTAG with CBD (OR: 5.65, 95 COI: 2.13–14.98, p < 0.001) and PSP (OR: 3.26, 95 COI: 1.63–6.53, p = 0.001); furthermore, gray matter ARTAG with CBD (OR: 8.23, 95 COI: 2.83–23.919, p < 0.001) and PSP (OR: 4.57, 95 COI: 2.36–8.87, p < 0.0001).
Comparisons of ARTAG types in major regions and subregions revealed significant differences for CBD and PSP (Table 3; Supplementary Data Files S4 and S5). Some comparisons were not statistically interpretable because of the lack of specific ARTAG types in some of the diagnostic groups (e.g., PiD, MSA, or controls). The presence of lobar gray matter ARTAG was particularly related to primary FTLD-tauopathies. Lobar white matter ARTAG appeared frequently in AD/PART cases (Table 3). LBD showed more MTL gray matter ARTAG than AD/PART but subcortical and brainstem gray matter ARTAG was seen more in AD/PART.
Next we focused only on the group of primary FTLD-tauopathies. We evaluated all clinicopathological and genetic variables (see above) and found that none of these showed a significant effect on ARTAG and its types in FTLD-tau disorders except for age at death, which showed a weak effect on the presence of perivascular ARTAG. Furthermore, when evaluating the presence of regional ARTAG types logistic regression models revealed a lack of effect of these variables.
Correlation of the Extent of ARTAG With Different Variables
Spearman correlation test revealed that the percentage of the investigated regions showing a subpial, perivascular or white matter ARTAG correlated weakly with age at death, the degree of ventricular enlargement, atherosclerosis of basal arteries, brain atrophy, and Braak NFT stage (the range of R was 0.1–0.16; p < 0.03 for all). Linear regression analysis (corrected for gender, age at death, presence of primary FTLD-tauopathy) showed that Braak NFT stage had an effect on the percent of examined regions showing white matter (OR: 1.46, 95 COI: 0.465–2.47, p = 0.004) ARTAG. In this model, age at death did not show any significant effect. In a similar model the degree of ventricular enlargement showed effect on the percentage of examined regions showing subpial (OR: 2.91, 95 COI: 0.76–5.06, p = 0.008) and white matter (OR: 3.03, 95 COI: 1.10–4.97, p = 0.002) ARTAG. The degree of atherosclerosis in basal arteries exhibited only a weak effect on the extent of subpial (OR: 2.64, 95 COI: 0.37–4.92, p = 0.023) and perivascular (OR: 2.00, 95 COI: 0.13–3.86, p = 0.036), while the degree of brain atrophy on perivascular (OR: 2.09, 95 COI: 0.19–3.99, p = 0.031) and white matter (OR: 3.44, 95 COI: 1.35–5.52, p = 0.001) ARTAG.
Correlation of the Severity of ARTAG With Different Variables
Spearman correlation test revealed that the mean severity scores for all regions showing subpial, perivascular or white matter ARTAG, but not subependymal and gray matter, correlated weakly with age at death, the degree of ventricular enlargement, atherosclerosis of basal arteries, brain atrophy, and Braak NFT stage (the range of R was 0.1–0.16; p < 0.03 for all). Applying a linear regression model (with gender, age at death, presence of primary FTLD-tauopathy) revealed that age at death showed only a weak effect for white matter (OR: 0.26, 95 COI 0.043–0.48, p = 0.019) and perivascular (OR: 0.20, 95 COI 0.007–0.39, p = 0.042) ARTAG. Ventricular enlargement and brain atrophy showed variable effects for subpial (ventricular enlargement, OR: 2.91, 95 COI 0.76–5.06, p = 0.008) and white matter (ventricular enlargement and brain atrophy, OR: 3.03 and 3.44, 95 COI 1.09–4.97 and 1.35–5.52, p = 0.002 and 0.001) ARTAG. Braak NFT stage showed significant results for white matter (OR: 1.46, 95 COI 0.46–2.47, p = 0.004) ARTAG. Importantly, in the AD/PART group the severity of lobar white matter ARTAG showed correlation with the CERAD score (R = 0.199, p < 0.0001; after correction for age at death, in linear regression model: OR: 4.85, 95% COI: 0.78–8.38; p = 0.018).
Relationship of Gray Matter Astroglial Tau Pathology in Primary FTLD Tauopathies and ARTAG
In addition to the presence of primary FTLD-tauopathy-related astroglial tau pathology (i.e. tufted astrocytes, astrocytic plaques, ramified astrocytes), in PSP, CBD, and PiD, we observed astrocytic tau immunoreactive lesions, which could not be classified as such. These showed fine granular immunoreactivity in the astrocytic processes more predominant in their distal (i.e. CBD) or proximal (i.e. PSP, PiD) segments, and were more compatible with GFA morphology. This is why the frequency of gray matter ARTAG was high in our PSP and CBD cohort. Accordingly, we compared the score of neuronal tau pathology with the score of GFA morphologies and the score of primary FTLD-tauopathy-related astroglial tau pathology. Heat maps revealed distinct patterns in PSP, CBD, and PiD (Fig. 5). In CBD cases, astrocytic plaques predominated in cortical and subcortical regions. In cortical regions, the astrocytic plaque score was higher than the neuronal tau load, whereas in the basal ganglia it was similar and in the brainstem neuronal tau pathology predominated. In addition to astrocytic plaques, GFA-like morphologies were also recognized. Indeed, the astrocytic morphology in the amygdala was more GFA than typical astrocytic plaque. In the occipital cortex, classical astrocytic plaques were only occasionally seen whereas GFA-type morphologies could be detected. In PSP cases, tufted astrocytes were more frequent in the striatum than neuronal tau pathology; however, neuronal tau predominated in the globus pallidus and brainstem; similar amounts were seen in cortical areas. Regarding astrocytic tau pathology, PSP cases showed similar patterns as CBD. Hence, in the amygdala and occipital cortex, as well as in the substantia nigra, astrocytic pathology was more GFA-type than typical tufted astrocytes. Both types were seen in cortical regions and in the striatum. In the amygdala, striatum, substantia nigra, and cortical areas except the occipital cortex, the GFA score correlated weakly with the neuronal tau pathology score (range of R values was 0.3–0.4, p < 0.03). In contrast to PSP and CBD, areas with moderate to severe neuronal pathology did not show unequivocal GFA in PiD cases. Furthermore, in the striatum in PiD cases, neuronal tau pathology correlated weakly with the amount of ramified astrocytes (R: 0.56, p = 0.037). Only 2 out of the 14 cases showed occasional neuronal inclusions in the occipital cortex. However, 4 cases showed GFA in the occipital cortex, including 3 in which there was a lack of neuronal tau inclusions.
FIGURE 5.

Heat maps and anatomical chart representation of tau pathologies in primary FTLD-tauopathies. Gray color indicates areas that were not systematically evaluated. In the radar chart, 5 anatomical regions are highlighted in all examined cases of PSP (n = 49), CBD (n = 21), and PiD (n = 14). Note that in the occipital cortex astroglial tau pathology predominates; furthermore, the proportions of astroglial (gray matter ARTAG or primary FTLD-tauopathy-related astrogliopathy, such as tufted astrocytes, astrocytic plaques and ramified astrocytes) and neuronal tau pathology differ considerably among PSP, CBD, and PiD cases.
In summary, a common feature of CBD, PSP and PiD was the presence of GFA-type astrocytes in areas with prominent involvement by neuronal and astroglial (i.e. typical tufted astrocytes, astrocytic plaques or ramified astrocytes) tau pathology but also in regions without prominent neuronal tau pathology or the presence of primary FTLD-tauopathy-related astrocytic morphologies, as exemplified by the occipital cortex. Furthermore, the amygdala always showed many GFA type morphologies and variably astrocytic plaques and tufted astrocytes, respectively, in CBD and PSP cases.
Peculiar Constellations of ARTAG
Evaluation of this cohort allowed us to recognize several peculiar constellations of ARTAG. First, a subset of cases in all disease groupings (clinical or neuropathological) showed prominent involvement of the amygdala in the form of gray matter ARTAG (consisting of clusters of TSA or GFA). Six out of 23 (26%) patients with psychiatric manifestations, 15/39 (38.4%) with PD, 12/59 (20%) of ALS, but only 17/180 (9%) clinically possible or probable AD, and 7/62 (11%) of individuals without documented clinical manifestations showed moderate or severe degrees of this. Second, a single case (a 75-year-old man with ALS) showed prominent gray matter ARTAG in the anterior horn of the spinal cord at several levels, but mostly where neurons did not show dramatic loss (Fig. 6A–C). Third, 2 elderly patients (81 and 79 years old) with PSP syndrome showed widespread white and gray matter ARTAG, particularly in the brainstem, associated with less prominent subcortical neuronal tau pathology and lacking tufted astrocytes in the cortex or striatum (Fig. 6D–F). Fourth, a 92-year-old woman with 6-years duration of progressive cognitive decline compatible with AD had the white matter predominant form (29) of GGT (type I) (29, 30), associated with severe white matter ARTAG pathology in the amygdala and basal forebrain (Fig. 6G–L).
FIGURE 6.

Peculiar patterns of ARTAG. (A–C) Gray and white matter ARTAG with thorny astrocytes in the anterior horn of the spinal cord (A; arrow indicates anterior horn enlarged in B; arrowhead indicates the anterior pyramidal tract enlarged in C). (D–F) Widespread white and gray matter ARTAG in the medulla oblongata in an 81-year-old man with atypical parkinsonism (D; arrow indicates medial lemniscus on the left and hilum of the inferior olive on the right, enlarged in E; arrowhead indicates hypoglossal nucleus enlarged in F). (G–L) Combined presence of globular oligodendroglial inclusions and thorn-shaped astrocytes in the peri-amygdaloid white matter (G; globular inclusions indicated by an arrowhead and enlarged in H and ARTAG indicated by an arrow and enlarged in I) and in the basal forebrain (J; perivascular ARTAG indicated by an arrowhead and enlarged in K and oligodendroglial globular inclusions indicated by an arrow and enlarged in L).
Finally, we were also interested in whether there are cases with gray matter ARTAG seen in several locations in the same brain without unequivocal features of primary FTLD-tauopathies. Indeed, out of the 487 cases where several regions were available for examination and primary FTLD tauopathies were excluded, 65 (13.3%) showed gray matter ARTAG only in the amygdala. Sixteen cases (3.2%) showed gray matter ARTAG only in subcortical regions, 12 (2.5%) only in the brainstem, and 7 (1.4%) only in lobar areas. Furthermore, combined forms were also observed: 13 (2.7%) with MTL and lobar, 6 (1.2%) with MTL and subcortical, 4 (0.8%) with MTL and brainstem, 13 (2.7%) with MTL and subcortical and lobar, 3 (0.6%) with MTL and subcortical and brainstem, 1 (0.2%) with MTL and lobar and brainstem, 5 (1.02%) with subcortical and lobar, 4 (0.82%) with subcortical and brainstem, and a single case (0.2%) with lobar and brainstem. All regions showed gray matter ARTAG in 2 cases (0.4%); both were 84-year-old men showing Braak NFT stage 2 pathology, one without documented cognitive decline (psychiatric symptoms not commented) and the other with schizophrenia. In summary, 35 cases (7.1%) showed gray matter ARTAG either in lobar, subcortical or brainstem regions and 52 (10.6%) various combinations of regional involvement by gray matter ARTAG.
DISCUSSION
TSA in the MTL and basal forebrain, especially in subpial, perivascular, and white matter location has long been considered to be an aging-related alteration (6, 7, 13, 31). Based on reports on elderly individuals, with or without clinical symptoms (2–4), the concept of ARTAG expanded this viewpoint and suggested that astroglial tau pathologies distinct from those seen in CBD, PSP, and PiD (i.e., primary FTLD-tauopathies) are present in many anatomical regions (1). Therefore, the umbrella term "ARTAG" was defined to include subpial, subependymal, perivascular, white and gray matter locations of 2 morphologies: TSA and GFA (1). Because of the paucity of data applying the conceptual approach of ARTAG, we performed a systematic study focusing on 3 levels: First, we sought to determine which variables associate with the general presence of any type of ARTAG; next, we evaluated associations of different types of ARTAG; and third, we determined whether these ARTAG types occurred in different anatomic locations. Similar to a recent report (8), our study confirms that ARTAG is not restricted to the MTL and basal forebrain and that the occurrence of ARTAG types and their regional distribution varies considerably in different neuropathological or clinical diagnostic groups.
The study on the neuropathology cohort of the MRC-CFAS reported that TSA showed a weak correlation between TSA and Braak stages of neurofibrillary degeneration (8). Another recent study in a younger cohort of patients with PD and dementia focused on the basal forebrain and showed increased frequency of ARTAG with age and with male gender but a lack of correlation with cognitive decline (13). Our cohort included young and aged individuals; this study expands these findings by providing a detailed map on the distribution and constellations of various ARTAG types. In addition to studies reporting ARTAG in community-based studies (2, 8), we focused on a large cohort of various disorders collected in CNDR Brain Bank (20).
We show that ARTAG is to be expected with higher probability if the individual is an elderly male, if brain atrophy and ventricular enlargement, and higher degree of atherosclerosis of basal vessels are noted in the postmortem macroscopic examination; but it is particularly to be expected when there are neuropathological features of primary FTLD-tauopathy (Table 2). These findings mostly remained significant in logistic regression tests in which, based on the results of previous studies (8, 13), we included gender, Braak NFT stage and age at death to the model. The strong effect of the presence of primary FTLD-tauopathy might be the reason why clinically documented movement disorders associated significantly with ARTAG (i.e. corticobasal syndrome and PSP syndrome with atypical parkinsonism were included in this clinical diagnostic group). Importantly, ARTAG types were associated with distinct clinicopathological variables. We do not interpret these findings to indicate that ARTAG causes the cognitive decline, psychiatric symptom, or movement disorder. However, some constellations, such as the prominent gray matter ARTAG in the amygdala in schizophrenia patients, lobar gray matter ARTAG in patients with cognitive decline or movement disorder even without concomitant presence of neuronal tau pathology might reflect an early response to neurodegeneration or dysfunction. Interestingly, subpial, white matter and perivascular ARTAG seemed to be independent of primary FTLD-tauopathy (Supplementary Data File S1); however, when region-specific effects were evaluated, the presence of primary FTLD-tauopathy became a significant factor (Supplementary Data File S2). These results suggest that disturbance of the tau homeostasis predisposes to the formation of ARTAG. Indeed, prominent ARTAG was documented in a young patient with familial FTLD-tauopathy (32), who was later confirmed to carry an MAPT duplication (33); however, there currently is a paucity of data on ARTAG in MAPT mutation carriers.
The basal forebrain and basal parts of the amygdala region are frequently involved by subpial and concomitant perivascular and white matter ARTAG. This supports the concept that exposure to cerebrospinal fluid or to extravasated plasma proteins, possibly due to defects in blood–brain barrier permeability, contributes to the generation of ARTAG (6, 7, 13), particularly because senescence is associated with significant, though often subtle, changes in blood–brain barrier (34). Notably, ventricular enlargement and brain atrophy, i.e., alterations also seen in CTE (10, 35), were associated with subpial, white matter, and perivascular ARTAG, including variable effects in lobar locations. Indeed, lobar subpial ARTAG is strongly reminiscent of that reported in CTE, particularly when perivascular ARTAG is seen in the adjacent cortex. Importantly, however, we did not see neuronal tau pathology in perivascular locations, which is thought to be pathognomonic of CTE (12). Subpial/perivascular ARTAG, especially in lobar areas or in the dorsolateral brainstem could also be considered as a preceding alteration to the full tau pathology seen in CTE, or it is possible that the inducing effect of ARTAG was not sufficient to generate additional neuronal tau accumulations. While TSA in the aged brain have been considered to be a nonspecific finding (5, 36, 37), our observations here raise the possibility that a subset of cases with various neurodegenerative conditions share a common pathogenesis with CTE. This concept was discussed in a recent study on CTE type pathology in neurodegenerative conditions, which documented frequent subpial tau pathology (17). Another study focusing on MSA attempted to distinguish CTE type from ARTAG (14). Our study also emphasizes that caution is needed in the interpretation of subpial and perivascular ARTAG. Interestingly, tufted astrocytes of PSP and astrocytic plaques of CBD seem to be spatially related to blood vessels (38), which, together with the frequent perivascular ARTAG-like tau pathology in primary FTLD tauopathies suggests that the presence of yet unidentified factors inducing the astrocytic tau pathology are related to blood vessels, including their permeability. Based on these observations and reports, for the etiology of subpial, perivascular and white matter ARTAG, physical (trauma) effects, blood–brain barrier dysfunction, blood- or vessel-related factors need to be considered. Indeed, the degree of basal atherosclerosis also showed variable effect on these ARTAG types. In addition, although subependymal ARTAG was mostly associated with the presence of primary FTLD tauopathies, basal atherosclerosis was noted in subcortical (i.e. 3rd ventricle) subependymal ARTAG. These observations support the concept that multiple pathologies, neurodegenerative and vascular, play complementary roles in brain pathologies of the elderly (39, 40). Hippocampal sclerosis of aging is frequently associated with arteriolosclerosis and TDP-43 pathologies (39). Interestingly, together with basal atherosclerosis, the presence of TDP-43 pathology frequently associated with subpial ARTAG in the basal forebrain and amygdala (MTL) in our study. Interestingly, ARTAG was not present in white matter areas prone to be involved in traumatic axonal injury (e.g. corpus callosum and cerebellar peduncles) (41), or in white matter areas involved by vascular disease or neurodegenerative pathologies (i.e. internal capsule or basis pontis). Because our cohort can be used as a reference cohort for future studies of ARTAG, the finding of ARTAG in these locations might be interpreted as an unusual phenomenon requiring the consideration of a distinct pathogenesis.
A notable observation was the strong association of AD-related variables with the presence of lobar white matter ARTAG, which was independent of primary FTLD-tauopathies. Interestingly, the H2/H2 MAPT haplotype was more frequent in these cases; this haplotype is rare in PSP and CBD and even in AD where the H1c haplotype is associated with an increased risk (42). ARTAG was seen in all lobes (particularly in the parietal lobe), but in only approximately 15% of advanced AD cases. The severity showed correlation with the scores of AD-related variables, particularly neuritic plaque scores. The distribution pattern of lobar white matter ARTAG in AD is reminiscent of lobar white matter arterial border zones, suggesting an astroglial response to hypoperfusion. White matter abnormalities are frequently detected in dementia by neuroimaging and they are thought to be partly secondary to neurodegeneration or primary damage of the white matter (43). Incomplete infarcts with rarefaction and gliosis confined to the deep white matter, tapering off toward the cortex, have been described in AD (44), and have been interpreted as an independent disorder of cerebrovascular, hypoperfusional/hypoxic origin (45). Indeed, white matter abnormalities detected by neuroimaging in the parietal and occipital white matter are thought to be caused by white matter hypoperfusion (46). We did not detect histopathological evidence of infarcts or extravasation of red blood cells in most of our cases with ARTAG; thus, a possible explanation could be chronic or repetitive hypoperfusion and/or impaired drainage of the interstitial fluid (47). The presence of lobar white matter ARTAG was associated with Aβ deposition, and less in PART (28). These observations support the notion of the interrelationships of AD and cerebrovascular disease (48). Thus, lobar white matter ARTAG might be the consequence of the process of impaired elimination of Aβ and other metabolites related to cerebrovascular alterations (49). Moreover, we found that CAA showed a strong association with subependymal ARTAG in the lateral ventricles and subpial ARTAG in the basal forebrain area.
Another important aspect of this study is related to the presence of gray matter ARTAG. Because we recognized overlap of GFA morphologies with some of the tau immunoreactive astrocytes in primary FTLD tauopathies together with tufted astrocytes in PSP (50), astrocytic plaques in CBD (51), and ramified astrocytes in PiD (52), we used a permissive strategy to evaluate gray matter ARTAG in primary FTLD-tauopathies. The classical astrocytic alterations in primary FTLD-tauopathies are considered to be related to degenerative rather than reactive processes (53). The concept that some astrocytic tau morphologies could be antecedent forms of tufted astrocytes, astrocytic plaques and ramified astrocytes could be viewed analogously to the maturation process of NFTs, from mostly nonargyrophilic pretangles to argyrophilic structures (54). This has previously discussed in PSP (55), and AGD (56), and for ARTAG in aged demented individuals (3). The fact that gray matter ARTAG in elderly individuals without unequivocal features of primary FTLD tauopathies show considerable overlap of the anatomical involvement observed for tufted astrocytes and astrocytic plaques (i.e. amygdala, striatum, frontal and temporal cortex, substantia nigra) suggests that at least a subset of gray matter ARTAG in the elderly might represent antecedent forms of primary FTLD tauopathies. It is reported that argyrophilic grains are observed frequently in CBD and PSP (57, 58). Therefore, the GFA-like morphologies in the amygdala in CBD and PSP may reflect concomitant AGD. On the other hand, the presence of ARTAG, including GFA in the amygdala without argyrophilic grains in other cases (i.e. non-CBD or PSP cases) argues for an independent process of GFA formation. Interestingly, GFA-type astrocytes in PSP and the aging brain show common immunostaining patterns with anti-tau antibodies (i.e. lack of truncated and conformationally modified forms, different phospho-epitopes), distinct from the tufted astrocytes (59). Importantly, we show that GFA-like tau positive astrocytes in primary FTLD-tauopathies appear also in regions without prominent neuronal tau pathology such as the occipital lobe; this has also been reported for phases of PiD (22). Similarly, evaluation of incidental CBD cases also suggests that tau astrogliopathy predominates the early stages of CBD (60). Accordingly, the frequency of early forms of primary FTLD tauopathies might be higher in the elderly than assumed. This has been addressed already in studies focusing on aging, reporting PSP type neuronal tau pathology in anatomically restricted areas, interpreted as early forms of disease (2, 61). In fact, PSP shows different phenotypes based on the distribution of neuronal and astrocytic tau pathology (62). Therefore, the presence of a few subcortical NFTs might be the predecessor of a different PSP type than presence of GFA-type astrocytic tau pathology in the striatum or cortical areas. Taking these aspects into consideration, at the light microscopic level, it is very difficult to decide what GFA represent. Scattered astrocytes in the gray matter with fine granular tau immunoreactivity in the processes might develop into astrocytic plaques, ramified astrocytes, globular astroglial inclusions, tufted astrocytes, or into GFA in a primary FTLD-tauopathy or in an aging brain (Fig. 7). Theoretically, GFA can also further develop into other tau-immunoreactive morphologies. Accordingly, the fine dot-like tau-immunoreactivity in astrocytic processes (as well as GFA) might represent the earliest steps of pathological tau accumulation in astrocytes. This also implies that certain epitopes may be accessible for antibodies and some may not be accessible. Thus, for example, a premature form of tufted astrocyte with GFA-like morphology in PSP might show different immunoreactivities using diverse tau-antibodies vs. the mature tufted astrocytes (59). Analogously to the concept of pretangles that most likely (but not yet proven in any way) evolve into mature NFTs, it is very difficult to predict in human postmortem studies how a GFA (or scattered fine tau-positive dots in astrocytic processes) would evolve. Evaluation of temporal alterations of astrocytic tau immunoreactive morphologies in experimental tauopathy models could contribute to a better understanding. Accordingly, it is meaningful to ask what is the significance of gray matter ARTAG? Based on the concept that tau pathology spreads in the nervous system and involves brain areas in a hierarchical manner (63), it is possible that astrocytes have an important role in this process. Indeed, cell-to-cell propagation of pathological tau, in addition to neurons, also involves astrocytes (64, 65). Interestingly, a similar phenomenon can be observed in Parkinson disease where α-synuclein accumulates in non-argyrophilic astrocytes correlating with the neuronal α-synuclein immunoreactivity (66, 67).
FIGURE 7.

Summary of the concept of astroglial tau pathology in the gray matter. Scattered and frequent dots in astrocytic processes can be seen in various primary FTLD-tauopathies and in the aging brain. Granular/fuzzy astrocytes (GFA) can also be seen in the aging brain and in primary FTLD-tauopathies, while in CBD there are astrocytic plaques, and in PSP there are tufted astrocytes. This concept suggests that the fine dot-like astrocytic tau-immunoreactivity can progress to any of these morphologies and in some circumstances together with the GFA might represent the earliest steps of pathological tau accumulation in astrocytes.
Conclusions
Our study provides a detailed map of different ARTAG types in the human brain in a large cohort of cases with various neurodegenerative conditions or without significant pathologies. Our data can be used as a reference when other disorders (e.g. traumatic brain injury) are evaluated and deviations from these patterns need to be highlighted. Our observations suggest that ARTAG types might have different etiologies, which might also differ depending on the anatomic location. For example the presence of the most frequent subpial, white matter, and perivascular ARTAG in basal areas is suggestive of a reactive change to cumulative age-related disturbances (i.e. dysfunction of the blood–brain barrier, interstitial fluid drainage, vascular components, local physical effects), whereas other ARTAG types, such as the lobar subpial or lobar white matter ARTAG, could reflect a different pathogenesis. In this respect our study further highlights the considerable overlap of ARTAG with CTE- and primary FTLD-tauopathy-related tau pathologies. The concept that some of the gray matter ARTAG may represent early forms of primary FTLD-tauopathies, at least in a subset of cases, suggests a higher incidence of the latter in the elderly than assumed. On the other hand, specific constellations of gray matter ARTAG in a subset of dementia cases (3) supports the notion that these might represent a distinct entity. The current concept of ARTAG, representing a wide spectrum of types in diverse anatomic locations, most likely evolves into the definition of better-demarcated constellations and, eventually, entities. This can lead to the understanding of the pathogenesis. Based on data from the published literature and our observations, we propose to distinguish at least 5 constellations of ARTAG because these may be present independently from each other as follows: (1) subpial ± white matter ± perivascular ARTAG in basal brain areas and medial temporal lobe; (2) subpial ±white matter ± perivascular ARTAG in the brainstem; (3) subpial ± perivascular ± gray matter ARTAG in lobar areas; (4) white matter ± perivascular ARTAG in lobar areas; and (5) gray matter ARTAG in limbic ± neocortical ± subcortical ± brainstem areas with or without features of primary FTLD-tauopathies.
Our study emphasizes the importance of astrocytes in neurodegenerative diseases (68). Whether accumulation of hyperphosphorylated tau in astrocytes reflects their contribution to disease spreading, to a phagocytic or scavenger effect, or only a mere reaction to local neurodegeneration (i.e., excitotoxicity) even preceding neuronal tau pathology (i.e., substantia nigra, hypoglossal nucleus, or amygdala) merits further studies. The constant involvement of the amygdala provides further evidence that this region is a hotspot for neuropathological alterations in the aging and neurodegenerative diseases and may also have relevance to psychiatric conditions and non-cognitive symptoms. Finally, it should be clarified whether ARTAG pathologies can be detected in vivo either by neuroimaging or by body-fluid examinations.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Center for Neurodegenerative Disease Research at the University of Pennsylvania (Philadelphia, PA) who contributed to this work, and the many patients studied and their families, for making the research reported here possible. We acknowledge the work of Dr Alice Chen-Plotkin for helping with the recruitment of human subjects.
REFERENCES
- 1. Kovacs GG, Ferrer I, Grinberg LT, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 2016;131:87–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kovacs GG, Milenkovic I, Wohrer A, et al. Non-Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community-based autopsy series. Acta Neuropathol 2013;126:365–84 [DOI] [PubMed] [Google Scholar]
- 3. Kovacs GG, Molnar K, Laszlo L, et al. A peculiar constellation of tau pathology defines a subset of dementia in the elderly. Acta Neuropathol 2011;122:205–22 [DOI] [PubMed] [Google Scholar]
- 4. Munoz DG, Woulfe J, Kertesz A.. Argyrophilic thorny astrocyte clusters in association with Alzheimer's disease pathology in possible primary progressive aphasia. Acta Neuropathol 2007;114:347–57 [DOI] [PubMed] [Google Scholar]
- 5. Ikeda K, Akiyama H, Kondo H, et al. Thorn-shaped astrocytes: possibly secondarily induced tau-positive glial fibrillary tangles. Acta Neuropathol 1995;90:620–5 [DOI] [PubMed] [Google Scholar]
- 6. Lace G, Ince PG, Brayne C, et al. Mesial temporal astrocyte tau pathology in the MRC-CFAS ageing brain cohort. Dement Geriatr Cogn Disord 2012;34:15–24 [DOI] [PubMed] [Google Scholar]
- 7. Schultz C, Ghebremedhin E, Del Tredici K, et al. High prevalence of thorn-shaped astrocytes in the aged human medial temporal lobe. Neurobiol Aging 2004;25:397–405 [DOI] [PubMed] [Google Scholar]
- 8. Wharton SB, Minett T, Drew D, et al. Epidemiological pathology of Tau in the ageing brain: application of staging for neuropil threads (BrainNet Europe protocol) to the MRC cognitive function and ageing brain study. Acta Neuropathol Commun 2016;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Takao M, Hirose N, Arai Y, et al. Neuropathology of supercentenarians - four autopsy case studies. Acta Neuropathol Commun 2016;4:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McKee AC, Stein TD, Kiernan PT, et al. The neuropathology of chronic traumatic encephalopathy. Brain Pathol 2015;25:350–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McKee AC, Stern RA, Nowinski CJ, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain 2013;136:43–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McKee AC, Cairns NJ, Dickson DW, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016;131:75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liu AK, Goldfinger MH, Questari HE, et al. ARTAG in the basal forebrain: widening the constellation of astrocytic tau pathology. Acta Neuropathol Commun 2016;4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koga S, Dickson DW, Bieniek KF.. Chronic traumatic encephalopathy pathology in multiple system atrophy. J Neuropathol Exp Neurol 2016;75:963–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kovacs GG, Rahimi J, Strobel T, et al. Tau pathology in Creutzfeldt-Jakob disease revisited. Brain Pathol 2016; doi: 10.1111/bpa.12411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bieniek KF, Ross OA, Cormier KA, et al. Chronic traumatic encephalopathy pathology in a neurodegenerative disorders brain bank. Acta Neuropathol 2015;130:877–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ling H, Holton JL, Shaw K, et al. Histological evidence of chronic traumatic encephalopathy in a large series of neurodegenerative diseases. Acta Neuropathol 2015;130:891–3 [DOI] [PubMed] [Google Scholar]
- 18. Irwin DJ, Cairns NJ, Grossman M, et al. Frontotemporal lobar degeneration: defining phenotypic diversity through personalized medicine. Acta Neuropathol 2015;129:469–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kovacs GG. Invited review: neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 2015;41:3–23 [DOI] [PubMed] [Google Scholar]
- 20. Toledo JB, Van Deerlin VM, Lee EB, et al. A platform for discovery: the University of Pennsylvania Integrated Neurodegenerative Disease Biobank. Alzheimers Dement 2014;10:477–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trojanowski JQ, Obrocka MA, Lee VM.. A comparison of eight different chromogen protocols for the demonstration of immunoreactive neurofilaments or glial filaments in rat cerebellum using the peroxidase-antiperoxidase method and monoclonal antibodies. J Histochem Cytochem 1983;31:1217–23 [DOI] [PubMed] [Google Scholar]
- 22. Irwin DJ, Brettschneider J, McMillan CT, et al. Deep clinical and neuropathological phenotyping of Pick disease. Ann Neurol 2016;79:272–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McMillan CT, Toledo JB, Avants BB, et al. Genetic and neuroanatomic associations in sporadic frontotemporal lobar degeneration. Neurobiol Aging 2014;35:1473–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991;41:479–86 [DOI] [PubMed] [Google Scholar]
- 25. Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Braak H, Braak E.. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991;82:239–59 [DOI] [PubMed] [Google Scholar]
- 27. Thal DR, Rub U, Orantes M, et al. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002;58:1791–800 [DOI] [PubMed] [Google Scholar]
- 28. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kovacs GG, Majtenyi K, Spina S, et al. White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol 2008;67:963–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ahmed Z, Bigio EH, Budka H, et al. Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 2013;126:537–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Uchikado H, Fujino Y, Lin W, et al. Frequency and relation of argyrophilic grain disease and thorn-shaped astrocytes in Alzheimer's Disease Advances in Alzheimer's and Parkinson's Disease: Insights, Progress, and Perspectives, New York: Springer, 2008:375–9 [Google Scholar]
- 32. Alexander J, Kalev O, Mehrabian S, et al. Familial early-onset dementia with complex neuropathologic phenotype and genomic background. Neurobiol Aging 2016;42:199–204 [DOI] [PubMed] [Google Scholar]
- 33. Le Guennec K, Quenez O, Nicolas G, et al. 17q21.31 duplication causes prominent tau-related dementia with increased MAPT expression. Mol Psychiatry 2016; doi: 10.1038/mp.2016.226. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 34. Mooradian AD. Effect of aging on the blood–brain barrier. Neurobiol Aging 1988;9:31–9 [DOI] [PubMed] [Google Scholar]
- 35. Hay J, Johnson VE, Smith DH, et al. Chronic traumatic encephalopathy: the neuropathological legacy of traumatic brain injury. Annu Rev Pathol 2016;11:21–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ikeda K. Glial fibrillary tangles and argyrophilic threads: classification and disease specificity. Neuropathology 1996;16:71–7 [Google Scholar]
- 37. Ikeda K, Akiyama H, Arai T, et al. Glial tau pathology in neurodegenerative diseases: their nature and comparison with neuronal tangles. Neurobiol Aging 1998;19:S85–91 [DOI] [PubMed] [Google Scholar]
- 38. Shibuya K, Yagishita S, Nakamura A, et al. Perivascular orientation of astrocytic plaques and tuft-shaped astrocytes. Brain Res 2011;1404:50. [DOI] [PubMed] [Google Scholar]
- 39. Nelson PT, Trojanowski JQ, Abner EL, et al. “New Old Pathologies”: AD, PART, and Cerebral Age-Related TDP-43 With Sclerosis (CARTS). J Neuropathol Exp Neurol 2016;75:482–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rahimi J, Kovacs GG.. Prevalence of mixed pathologies in the aging brain. Alzheimers Res Ther 2014;6:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Smith C, Margulies S, Duhaime A-C, Trauma In: Love S, Budka H, Ironside JW, Perry A eds. Greenfield's Neuropathology, Vol. 1 Boca Raton, FL: CRC Press, 2015:637–82 [Google Scholar]
- 42. Myers AJ, Pittman AM, Zhao AS, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis 2007;25:561–70 [DOI] [PubMed] [Google Scholar]
- 43. Love S, Miners JS.. White matter hypoperfusion and damage in dementia: post-mortem assessment. Brain Pathol 2015;25:99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brun A, Englund E.. A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol 1986;19:253–62 [DOI] [PubMed] [Google Scholar]
- 45. Englund E, Brun A, Alling C.. White matter changes in dementia of Alzheimer's type biochemical and neuropathological correlates. Brain 1988;111:1425–39 [DOI] [PubMed] [Google Scholar]
- 46. Hattori T, Orimo S, Aoki S, et al. Cognitive status correlates with white matter alteration in Parkinson's disease. Hum Brain Mapp 2012;33:727–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Weller RO, Hawkes CA, Kalaria RN, et al. White matter changes in dementia: role of impaired drainage of interstitial fluid. Brain Pathol 2015;25:63–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yarchoan M, Xie SX, Kling MA, et al. Cerebrovascular atherosclerosis correlates with Alzheimer pathology in neurodegenerative dementias. Brain 2012;135:3749–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weller RO, Hawkes CA, Carare RO, et al. Does the difference between PART and Alzheimer's disease lie in the age-related changes in cerebral arteries that trigger the accumulation of Abeta and propagation of tau? Acta Neuropathol 2015;129:763–6 [DOI] [PubMed] [Google Scholar]
- 50. Hauw JJ, Verny M, Delaere P, et al. Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer's disease and aging. Neurosci Lett 1990;119:182–6 [DOI] [PubMed] [Google Scholar]
- 51. Dickson DW, Bergeron C, Chin SS, et al. Office of rare diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 2002;61:935–46 [DOI] [PubMed] [Google Scholar]
- 52. Dickson DW. Pick's disease: a modern approach. Brain Pathol 1998;8:339–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Togo T, Dickson DW.. Tau accumulation in astrocytes in progressive supranuclear palsy is a degenerative rather than a reactive process. Acta Neuropathol 2002;104:398–402 [DOI] [PubMed] [Google Scholar]
- 54. Bancher C, Brunner C, Lassmann H, et al. Accumulation of abnormally phosphorylated tau precedes the formation of neurofibrillary tangles in Alzheimer's disease. Brain Res 1989;477:90–9 [DOI] [PubMed] [Google Scholar]
- 55. Santpere G, Ferrer I.. Delineation of early changes in cases with progressive supranuclear palsy-like pathology. Astrocytes in striatum are primary targets of tau phosphorylation and GFAP oxidation. Brain Pathol 2009;19:177–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Botez G, Probst A, Ipsen S, et al. Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta Neuropathol 1999;98:251–6 [DOI] [PubMed] [Google Scholar]
- 57. Dugger BN, Adler CH, Shill HA, et al. Concomitant pathologies among a spectrum of parkinsonian disorders. Parkinsonism Relat Disord 2014;20:525–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tatsumi S, Mimuro M, Iwasaki Y, et al. Argyrophilic grains are reliable disease-specific features of corticobasal degeneration. J Neuropathol Exp Neurol 2014;73:30–8 [DOI] [PubMed] [Google Scholar]
- 59. Ferrer I, Lopez-Gonzalez I, Carmona M, et al. Glial and neuronal tau pathology in tauopathies: characterization of disease-specific phenotypes and tau pathology progression. J Neuropathol Exp Neurol 2014;73:81–97 [DOI] [PubMed] [Google Scholar]
- 60. Ling H, Kovacs GG, Vonsattel JP, et al. Astrogliopathy predominates the earliest stage of corticobasal degeneration pathology. Brain 2016; 139:3237–52 [DOI] [PubMed] [Google Scholar]
- 61. Dugger BN, Hentz JG, Adler CH, et al. Clinicopathological outcomes of prospectively followed normal elderly brain bank volunteers. J Neuropathol Exp Neurol 2014;73:244–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yoshida M. Astrocytic inclusions in progressive supranuclear palsy and corticobasal degeneration. Neuropathology 2014;34:555–70 [DOI] [PubMed] [Google Scholar]
- 63. Brettschneider J, Del Tredici K, Lee VM, et al. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 2015;16:109–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Clavaguera F, Akatsu H, Fraser G, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 2013;110:9535–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Boluda S, Iba M, Zhang B, et al. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer's disease or corticobasal degeneration brains. Acta Neuropathol 2015;129:221–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kovacs GG, Breydo L, Green R, et al. Intracellular processing of disease-associated alpha-synuclein in the human brain suggests prion-like cell-to-cell spread. Neurobiol Dis 2014;69:76–92 [DOI] [PubMed] [Google Scholar]
- 67. Braak H, Sastre M, Del Tredici K.. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson's disease. Acta Neuropathol 2007;114:231–41 [DOI] [PubMed] [Google Scholar]
- 68. Garwood CJ, Ratcliffe LE, Simpson JE, et al. Review: astrocytes in Alzheimer's disease and other age-associated dementias; a supporting player with a central role. Neuropathol Appl Neurobiol 2016;doi:10.1111/nan.12338 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.