

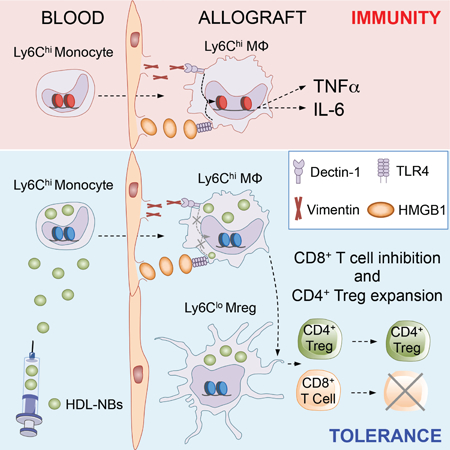
SUMMARY
Inducing graft acceptance without chronic immunosuppression remains an elusive goal in organ transplantation. Using an experimental transplantation mouse model, we demonstrate that local macrophage activation through dectin-1 and toll-like receptor 4 (TLR4) drives trained immunity-associated cytokine production during allograft rejection. We conducted nanoimmunotherapeutic studies and found that a short- term mTOR-specific high-density lipoprotein (HDL) nanobiologic treatment (mTORi-HDL) averted macrophage aerobic glycolysis and the epigenetic modifications underlying inflammatory cytokine production. The resulting regulatory macrophages prevented alloreactive CD8+ T cell-mediated immunity and promoted tolerogenic CD4+ regulatory T cell (Treg) expansion. To enhance therapeutic efficacy, we complemented the mTORi-HDL treatment with a CD40-TRAF6 specific nanobiologic (TRAF6i-HDL) that inhibits co-stimulation. This synergistic nanoimunnotherapy resulted in indefinite allograft survival. Together, we show that HDL- based nanoimmunotherapy can be employed to control macrophage function in vivo. Our strategy, focused on preventing inflammatory innate immune responses, provides a framework for developing targeted therapies that promote immunological tolerance.
Keywords: Nanoimmunotherapy, transplantation, trained immunity
Graphical Abstract
eTOC BLURB
An unresolved problem in organ transplantation is to establish graft acceptance in the absence long-term immunosuppressive therapy. Braza et al. unravel important molecular mechanisms underlying myeloid cell activation in an experimental organ transplantation model and develop a combined nanoimmunotherapy that targets myeloid cells in hematopoietic organs and the allograft. Short-term nanobiologic immunotherapy prevents inflammation and induces indefinite allograft survival.
INTRODUCTION
Successful organ transplantation is made possible by pharmacologic immunosuppression. Patients undergoing organ transplantation usually receive an immunosuppressive drug mixture that has dramatically improved the short-term results of organ transplantation (Gardiner et al., 2016; Lien, 2015). However, due to the detrimental effects of life-long continuous immunosuppression (Meier-Kriesche et al., 2004a), including infections, cancer and considerable metabolic toxicity (Naesens et al., 2009), a pressing need exists to reduce toxicity and improve long-term allograft survival. Despite efforts to use currently available immunosuppressive agents in less toxic ways, no alternative regimen has seriously challenged these drugs’ almost universal use.
Historically, transplant immunologists have attempted to develop novel tolerogenic protocols by targeting adaptive immune response mechanisms. Such work has been based on the observation that T cells are both necessary and sufficient to induce graft rejection (Miller, 1961; Pantelouris, 1971). Accordingly, several therapeutic agents have been developed against signal 1 (T cell receptor/CD3 complex), signal 2 (co- stimulatory receptors) and signal 3 (cytokine production), which are required to successfully activate the effector lymphocytes that mediate allograft rejection. While these methodologies have produced promising results (Page et al., 2012), long-term graft survival rates are suboptimal (Meier-Kriesche et al., 2004b), which underlines the need for alternative tolerance-inducing approaches.
Recent advances in our understanding of the graft-reactive immune response have demonstrated that innate immune cells initiate allograft rejection (Liu et al., 2012; Oberbarnscheidt et al., 2014; Zecher et al., 2009). This is consistent with clinical data from over three decades ago, which indicated that macrophages represent the majority of cells that infiltrate an allograft during severe rejection episodes (Hancock et al., 1983). Despite critical progress on the pathways by which macrophages distinguish between self and allogeneic non-self and promote organ rejection (Dai et al., 2017), the mechanisms by which these innate immune cells mediate graft loss are not fully understood.
Although regulating macrophage immunological function represents a potential target to control the immune response (Kranz et al., 2016), this strategy remains unexplored clinically (Mantovani et al., 2014; Martinez and Gordon, 2015). Here, we reveal a macrophage activation pathway that contributes to allograft rejection and introduce a myeloid-specific nanoimmunotherapy that synergistically targets mTOR and CD40-TRAF6, resulting in long-term organ transplant acceptance.
RESULTS
Donor allograft expresses vimentin and HMGB1 and promotes local training of macrophages
To decipher macrophage activation pathways that promote allograft immunity, we focused on the functional state of macrophages with increased inflammatory cytokine production caused by non-permanent epigenetic reprogramming associated with trained immunity (Saeed et al., 2014). We evaluated the possible role for dectin-1 and TLR4 agonists vimentin and the high mobility group box 1 (HMGB1) (Thiagarajan et al., 2013; Yang et al., 2010) that may be present under sterile inflammation. BALB/c (H2d) hearts were transplanted into fully allogeneic C57BL/6 (H2b) recipients as previously described (Corry et al., 1973) and our data (Figures 1A-1C) indicate that both proteins were upregulated in the donor allograft following organ transplantation. This suggests that vimentin and HMGB1 may be able to promote training of graft-infiltrating macrophages locally. To investigate this possibility, we first confirmed that graft-infiltrating macrophages expressed dectin-1 and TLR4 by flow cytometry (Figure 1D). Absence of dectin-1 and TLR4 expression using deficient recipient mice prevented the accumulation of graft-infiltrating inflammatory Ly6Chi macrophages (Figure 1E). Conversely, dectin-1 or TLR4-deficiency promoted the accumulation of Ly6Clo macrophages in the allograft, which we have reported to promote allograft tolerance (Braza et al., 2018; Conde et al., 2015).
Figure 1. Vimentin and HMGB1 are upregulated following organ transplantation and promote training of graft infiltrating macrophages.

(A-C) Immunostaining, real-time PCR and western blot analysis of vimentin and HMGB1 expression in donor and non-transplanted hearts (n=3/mice per group of three independent experiments, t-test; **P<0.01).
(D) Dectin-1 and TLR4 expression in graft infiltrating macrophages (n=3 mice/group of two independent experiments).
(E) Ly-6C expression in graft infiltrating macrophages from WT, dectin1 KO and TLR4 KO untreated recipient mice (n=3 mice/group of two independent experiments).
(F) Inflammatory cytokine production and chromatin immunoprecipitation of mouse monocytes trained with vimentin and HMGB1 (n=3 independent experiments, one-way ANOVA, **P<0.01; dashed line displays control non-trained conditions).
(G) Cytokine and lactate production of graft-infiltrating macrophages (n=4 mice/group of 2 independent experiments, one-way ANOVA, **P<0.01).
(H) Chromatin immunoprecipitation of graft-infiltrating macrophages (n=4 mice/group of 2 independent experiments, one-way ANOVA, *P<0.05; **P<0.01).
Having demonstrated that donor allografts upregulated vimentin and HMGB1, we next investigated whether vimentin and HMGB1 promoted macrophage training. Using an established in vitro trained immunity model, in which purified monocytes are exposed to β-glucan followed by re-stimulation with LPS, we observed a similar increase in the production of the pro-inflammatory cytokines TNFα and IL-6 upon vimentin and HMGB1 stimulation (Figure 1F), indicative of these proteins’ ability to induce macrophage training. To validate that vimentin and HMGB1 induced local training of graft infiltrating macrophages, we flow sorted these cells from heart allografts and evaluated their ability to produce pro-inflammatory cytokines and glycolytic products. We found that dectin-1 or TLR4 deficiency significantly lowered pro-inflammatory TNFα and IL-6 expression and lactate production by graft-infiltrating macrophages after ex vivo LPS stimulation (Figure 1G). In line with the protein expression, absence of dectin-1 or TLR4 prevented H3K4me3 epigenetic changes in the promoter of the pro-inflammatory cytokines TNFα and IL-6 and the glycolytic enzymes hexokinase (HK) and phosphofructokinase (PFKP) in graft-infiltrating macrophages (Figure 1H). Collectively, our data suggests that monocyte precursors in the bone marrow (Figure S1A) migrate to the allograft early after transplantation and become trained following vimentin/HMGB1 exposure locally.
mTORi-HDL nanoimmunotherapy prevents trained immunity in vitro
We developed a nanoimmunotherapy based on high-density lipoprotein (HDL) nanobiologics, which we have previously shown to target myeloid cells (Duivenvoorden et al., 2014; Perez-Medina et al., 2015; Tang et al., 2015). Since the mammalian target for rapamycin (mTOR) has been shown to regulate cytokine production (signal 3) through trained immunity (Netea et al., 2016; Saeed et al., 2014), we encapsulated the mTOR inhibitor rapamycin (Figure S1B) in a corona of natural phospholipids and apolipoprotein A-I (apoA-I) isolated from human plasma (Zamanian-Daryoush et al., 2013), to render mTORi-HDL nanobiologics as recently described (Mulder et al., 2018). The resulting nanobiologics had a drug encapsulation efficiency of 62 ± 11% and a mean hydrodynamic diameter of 12.7 ± 4.4 nm, as determined by high performance liquid chromatography and dynamic light scattering, respectively. Transmission electron microscopy revealed mTORi-HDL to have the discoidal structure (Figures 2A and S2C; STAR Methods) that is typical of HDL- based nanobiologics (Duivenvoorden et al., 2014).
Figure 2. mTORi-HDL nanoimmunotherapy prevents trained immunity in vitro and distributes systemically in vivo.
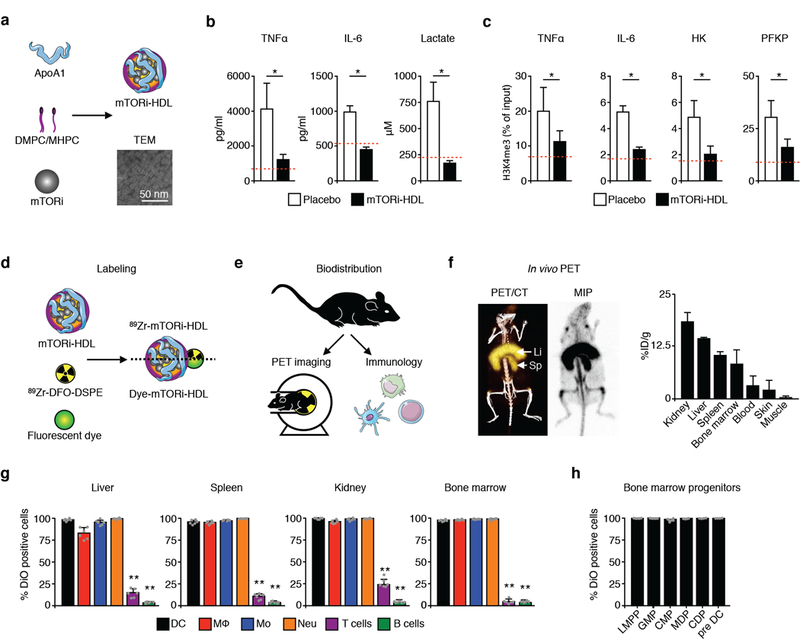
(A) Transmission electron microscopy (TEM) of mTORi-HDL nanobiologics.
(B) Cytokine and lactate production of human macrophages trained in vitro (n=3 independent experiments, t-test, *P<0.05; dashed line displays control non-β-glucan trained condition).
(C) Chromatin immunoprecipitation of human macrophages trained in vitro (n=3 independent experiments, t-test, *P<0.05; dashed line displays control non-β-glucan trained condition).
(D) Labeling of mTORi-HDL with either the radioisotope 89Zr or the fluorescent dyes DiO or DiR.
(E) micro-PET/CT and cellular specificity of mTORi-HDL nanobiologics.
(F) Representative micro-PET/CT 3D fusion image and PET maximum intensity projection (MIP) (mean ± SEM, n=3).
(G) Uptake of fluorescently labeled DiO mTORi-HDL by myeloid and lymphoid cells (n=5 mice/group, one-way ANOVA, **P<0.01).
(H) Uptake of fluorescently labeled DiO mTORi-HDL by bone marrow progenitors (mean ± SEM, n=5).
Using an established in vitro trained immunity model, in which purified human monocytes are exposed to β- glucan, we observed increased cytokine and lactate production upon re-stimulation with LPS. Conversely, β- glucan-trained human monocytes treated with mTORi-HDL during the training period displayed significantly less cytokine and lactate production upon LPS re-stimulation (Figure 2B). This is consistent with our previously reported work, which showed trained immunity to be mTOR-dependent (Cheng et al., 2014). As the higher cytokine and glycolytic responses may be the result of macrophages’ epigenetic reprogramming (Saeed et al., 2014), we assessed trimethylation of the histone H3K4, which designates open chromatin (Figure 2C; STAR Methods). mTORi-HDL treatment prevented epigenetic changes at the promoter level of four inflammatory genes associated with trained immunity in human monocytes.
We next evaluated the biodistribution and immune cell specificity of fluorescent-dyed (DiO or DiR) or zirconium-89 radiolabeled mTORi-HDL (89Zr-mTORi-HDL; Figure 2D; STAR Methods), using a combination of in vivo positron emission tomography with computed tomography (PET-CT) imaging, ex vivo near infrared fluorescence (NIRF) imaging and flow cytometry in C57BL/6 wild-type mice (Figure 2E). We detected 89Zr- mTORi-HDL accumulation in the kidney, liver and spleen (Figure 2F and Figures S1D-S1E), preferentially associated with myeloid cells, but not with T or B cells (Figure 2G). Importantly, we also observed strong mTORi-HDL accumulation in the bone marrow (Figures 2F-2G), associated with several myeloid cells and their progenitors (Figure 2H), potentially facilitates the induction of prolonged therapeutic effects.
mTORi-HDL nanoimmunotherapy prevents trained immunity in vivo
Next, we applied mTORi-HDL treatment to an experimental heart transplant mouse model (Figure 3A) and determined allograft targeting and immune cell specificity as described above. Six days after receiving heterotopic heart transplants, mice were treated with intravenous 89Zr-mTORi-HDL. The nanoimmunotherapy was allowed to circulate and distribute for 24 hours before mice were subjected to PET-CT. We saw marked 89Zr-mTORi-HDL presence in the heart allografts (Figures 3B and S1F; STAR Methods). After mice were sacrificed, the native heart and allograft were collected for ex vivo 89Zr quantification. We noted radioactivity (25.2 ± 2.4 × 103 counts/unit area) in the heart allograft (Tx) to be 2.3-fold higher than in native hearts (N) (11.1 ± 1.9 × 103 count/unit area) (Figure 3C).
Figure 3. mTORi-HDL nanoimmunotherapy targets myeloid cells in the allograft and prevents trained immunity.
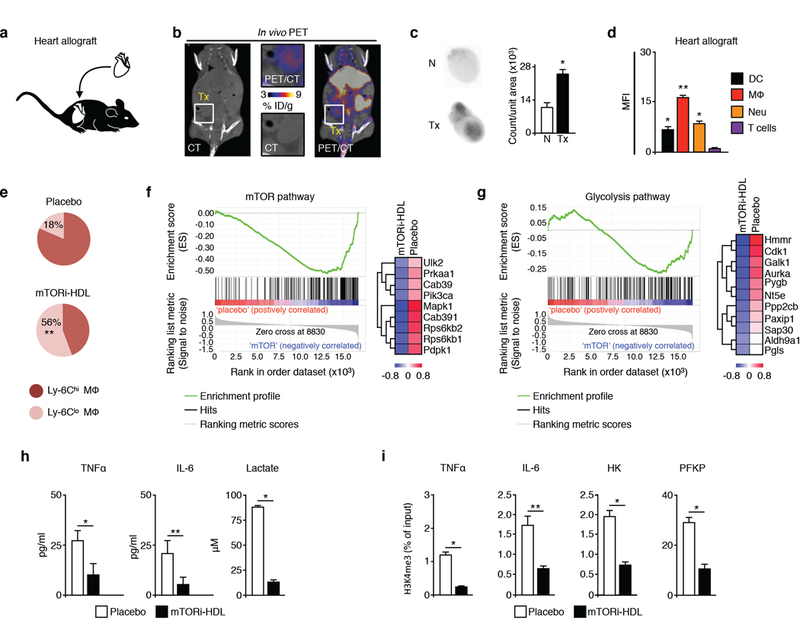
(A) BALB/c donor hearts (H2d) were transplanted into fully allogeneic C57BL/6 recipients (H2b).
(B) micro-PET/CT 3D fusion image 24 hours after intravenous administration of 89Zr-mTORi-HDL (n=3 mice/group of 2 independent experiments).
(C) Ex vivo autoradiography in native (N) and transplanted hearts (Tx) at 24 hours after intravenous 89Zr-mTORi-HDL (n=3 mice/group of 2 independent experiments, t-test, *P<0.05).
(D) Uptake of fluorescently labeled DiO mTORi-HDL by myeloid and lymphoid cells in the allograft (n=4 mice/group of 3 independent experiments; one-way ANOVA, *P<0.05; **P<0.01).
(E) Ly-6Chi / Ly-6Clo MΦ ratio in the allograft from either placebo or mTORi-HDL-treated recipients at day 6 post-transplantation (n=4 mice/group of 3 independent experiments; one-way ANOVA, *P 0.05; **P <0.01).
(F-G) GSEA gene array analysis for the mTOR and glycolysis pathways in intra-graft MΦ from placebo or mTORi-HDL-treated recipients (n=3 mice/group).
(H) Cytokine and lactate production of graft-infiltrating macrophages from either placebo or mTORi-HDL-treated recipients (n=4 mice/group of 3 independent experiments, t-test, *P<0.05; **P<0.01).
(I) Chromatin immunoprecipitation of graft-infiltrating macrophages from either placebo or mTORi-HDL-treated recipients (n=4 mice/group of 3 independent experiments, t-test, *P<0.05; **P<0.01).
Motivated by the favorable organ distribution pattern and heart allograft uptake, we evaluated the immune cell specificity of mTORi-HDL that had been labeled with the fluorescent dye DiO. 24 hours after intravenous administration, we collected the heart allograft, as well as blood and spleen, and measured mTORi-HDL distribution in DC, macrophages, neutrophils and T cells by flow cytometry. We observed mTORi-HDL cellular preference towards myeloid cells, with significantly higher uptake by macrophages than either DC or neutrophils in the allograft, blood and spleen (Figures 3D and S2A-S2B). T cells exhibited poor mTORi-HDL uptake (Figures S2C and S2D), which highlights the mTORi-HDL’s preferential targeting of myeloid cells.
We next assessed a treatment regimen involving three intravenous mTORi-HDL injections at 5mg/kg rapamycin per dose, at the day of transplantation as well as on postoperative days 2 and 5. We profiled the myeloid cell compartment in the allograft, blood and spleen of mice receiving either mTORi-HDL treatments or placebo. In line with our targeting data, we found that the overall numbers of macrophages, neutrophils and DC were significantly lower in the allograft, blood and spleen (Figure S3A) of mTORi-HDL-treated recipients, in comparison with either placebo or mice treated with oral rapamycin (5mg/kg on postoperative days 0, 2, and 5). We then determined mTORi-HDL nanoimmunotherapy’s effect on the distribution of two different macrophage subsets (Ly-6Chi and Ly-6Clo), which have distinct immune regulatory properties (Conde et al., 2015). Six days after transplantation, untreated recipient mice had increased numbers of inflammatory Ly-6Chi macrophages in the allograft, blood and spleen (Figures 3E and S3B). By contrast, mTORi-HDL- treated recipients had increased numbers of Ly-6Clo macrophages. The data indicate that while Ly-6Chi macrophages comprised the majority of macrophages during transplant rejection, our mTORi-HDL nanoimmunotherapy promotes the accumulation of Ly-6Clo macrophages. We did not observe this change in animals treated with oral rapamycin (Figure S3B).
To study the molecular pathways targeted by our mTORi-HDL nanoimmunotherapy, we used Gene Set Enrichment Analysis (GSEA) of mRNA isolated from flow-sorted macrophages from the allografts of animals treated with either placebo or mTORi-HDL. Gene array results indicated that the trained immunity-related mTOR and glycolysis pathways (Cheng et al., 2014) were negatively regulated by mTORi-HDL (Figures 3F and 3G). To substantiate these results, we flow sorted macrophages from heart allografts and evaluated their ability to produce inflammatory cytokines (signal 3) and glycolytic products. We observed that mTORi-HDL treatment significantly lowered TNFα and IL-6 protein expression and lactate production by graft-infiltrating macrophages after ex vivo LPS stimulation (Figure 3H). In line with the in vitro observations (Figures 2B and 2C), mTORi-HDL treatment also prevented H3K4me3 epigenetic changes in graft-infiltrating macrophages (Figure 3I; STAR Methods).
mTORi-HDL nanoimmunotherapy promotes organ transplant acceptance
We next assessed the immunological function of graft-infiltrating macrophages as recently described (Ochando and Conde, 2017). Ly-6Clo macrophages’ suppressive function was measured by their capacity to inhibit in vitro proliferation of carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled CD8+ T cells, as previously reported (Conde et al., 2015). We found that Ly-6Clo macrophages obtained from the allografts of mTORi-HDL-treated recipient mice inhibit T cell proliferation in vitro (Figure 4A). The same mTORi-HDL- treated allograft Ly-6Clo macrophages expand immunosuppressive Foxp3-expressing regulatory T cells (Treg). In accordance with these data, we observed significantly more CD4+CD25+ T cells in the allografts of mTORi-HDL-treated recipients (Figures 4B and S3C). These results suggest that mTORi-HDL treatment supports transplantation tolerance by promoting the development of Ly-6Clo regulatory macrophages (Mreg).
Figure 4. mTORi-HDL nanoimmunotherapy promotes organ transplant acceptance.
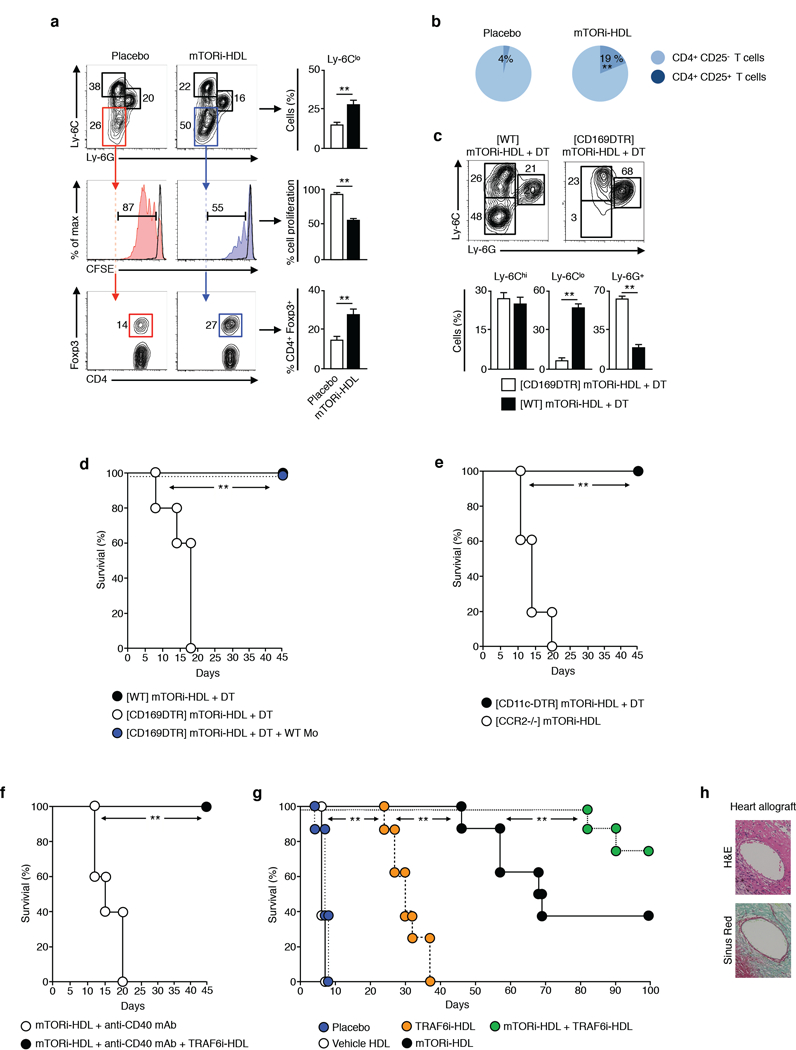
(A) Functional characterization of graft-infiltrating MΦ from placebo and mTORi-HDL-treated recipients using CD8 T cell suppressive and CD4 Treg expansion assays (n=4 mice/group of 3 independent experiments, t-test, **P≤0.01).
(B) Percentage of graft-infiltrating CD4+CD25+ Treg cells from placebo and mTORi-HDL-treated recipients (n=4 mice/group of 3 independent experiments, t-test, **P≤0.01).
(C) Depletion of CD169+ graft-infiltrating Mreg in placebo and mTORi-HDL-treated recipients (n= 5 mice/group of 3 independent experiments, t-test, **P<0.01).
(D) Graft survival following depletion CD169+ graft-infiltrating Mreg (n= 5 mice/group; Kaplan-Meier **P≤0.01).
(E) Graft survival following depletion of CD11c+ cells and in CCR2 deficient recipient mice (n=5 mice/group, Kaplan-Meier, **P<0.01).
(F) Graft survival of mTORi-HDL-treated recipients receiving agonistic stimulatory CD40 mAb in vivo with or without TRAF6i-HDL nanoimmunotherapy (n=5 mice/group, Kaplan-Meier, **P<0.01).
(G) Graft survival of placebo, vehicle HDL, mTORi-HDL, TRAF6i-HDL and mTORi-HDL/TRAF6i-HDL treated recipients (n=7–8 mice/group, Kaplan-Meier, **P<0.01).
(H) Immunohistochemistry of heart allografts from mTORi-HDL/TRAF6i-HDL-treated recipients on day 100 after transplantation (n = 5 mice/group; magnification X200).
To investigate the functional role of Ly-6Clo Mreg in transplant recipients, we depleted Ly-6Clo Mreg in vivo, as previously described (Conde et al., 2015; Miyake et al., 2007). Briefly, BALB/c (H2d) donor cardiac allografts were transplanted into C57BL/6 fully allogeneic CD169 diphtheria toxin (DT) receptor (DTR) (H2b) recipient mice treated with mTORi-HDL. We depleted regulatory Ly-6Clo Mreg by DT administration on the day of transplantation (Figure 4C), which resulted in early graft rejection (12.3 ± 1.8 days) despite mTORi-HDL treatment (Figure 4D). Adoptive transfer of wild-type monocytes restored allograft survival, thereby demonstrating that the nanoimmunotherapy exerts its effects through Mreg (Figure 4D). This was further confirmed using CD11c-DTR mice as transplant recipients, in which administration of DT in these mice depletes CD11c+ DC. We observed that graft survival prolongation was independent of CD11c+ DC. On the contrary, graft survival in CCR2-deficient recipient mice, with fewer Ly-6Chi circulating monocytes, was not prolonged (Figure 4E). Overall, these experiments demonstrate that macrophages are required for mTORi- HDL nanoimmunotherapy-facilitated organ transplant acceptance.
Previous work demonstrated that activated macrophages produce large amounts of IL-6 and TNFα that promote T cell graft-reactive alloimmunity (Shen and Goldstein, 2009). This study also demonstrated that absence of recipient IL-6 and TNFα synergizes with the administration of CD40-CD40L co-stimulatory blockade to induce permanent allograft acceptance. Based on this premise, we used concurrent co- stimulatory blockade (signal 2) to augment mTORi-HDL’s efficacy. To that aim, we employ a second nanoimmunotherapy treatment consisting of a CD40-TRAF6 inhibitory HDL (TRAF6i-HDL) (Figures S4A and S4B) (Lameijer et al., 2018). We confirmed its specificity for CD40 signaling inhibition using an agonistic CD40 mAb (clone FGK4.5), which induced rejection in mTORi-HDL treated recipients. We found that the TRAF6i-HDL nanobiologic treatment prevented the detrimental effects of stimulatory CD40 mAb and restored mTORi-HDL-mediated allograft survival (Figure 4F).
We then evaluated our nanoimmunotherapy’s ability to prolong graft survival of fully allogeneic donor hearts. Using the aforementioned three-dose regimen of 5mg/kg per dose on postoperative days 0, 2, and 5, the mTORi-HDL treatment significantly increased heart allograft survival as compared to placebo, HDL vehicle and oral/intravenous rapamycin treatments (Figures 4G and S4C). We subsequently tested a treatment regimen combining mTORi-HDL (signal 3) and TRAF6i-HDL (signal 2) nanobiologics. This mTORi- HDL/TRAF6i-HDL treatment synergistically promoted organ transplant acceptance and resulted in >70% allograft survival 100 days post-transplantation. The combined treatment dramatically outperformed the mTORi-HDL and TRAF6i-HDL monotherapies (Figure 4G) without histopathological evidence for toxicity or chronic allograft vasculopathy (Figures 4H and S4D).
Collectively, our data show that HDL-based nanoimmunotherapy prevents macrophage-derived inflammatory cytokine production associated with trained immunity. Further, HDL-based nanoimmunotherapy presented less toxicity than an oral rapamycin resulting in prolonged therapeutic benefits without off-target side effects (Figure S4E).
DISCUSSION
Inflammation is triggered by innate immune cells as a defense mechanism against tissue injury. An ancient mechanism of immunological memory, named trained immunity (Netea and van der Meer, 2017), is induced by self-derived damage-associated molecular patterns (DAMPs) following sterile inflammatory stimuli after tissue damage. In transplantation, the DAMPs vimentin and HMGB1 are upregulated in the donor organ following ischemia reperfusion injury, which activate myeloid cells through the pattern recognition receptors (PRRs) dectin-1 and TLR4. Consequently, innate immune cells upregulate co-stimulatory molecules, such as CD40 (signal 2) and secrete of pro-inflammatory cytokines, such as TNFα and IL-6 (signal 3) (Azimzadeh et al., 2005; dos Santos et al., 2015; Huang et al., 2007; Krawczyk et al., 2010).
Activation of myeloid cells though PRRs induces a metabolic reprograming towards aerobic glycolysis and the generation of ATP, which necessary for these cells to perform their function, including cytokine production (Pearce and Pearce, 2013). This metabolic switch in conjunction with an epigenetic rewiring represents the hallmark of trained immunity (Netea et al., 2016). The analysis of metabolic and epigenetic reprogramming of myeloid cells provides new insights into the regulation of the immune response and the mammalian target of rapamycin (mTOR) represents a central signaling pathway that controls myeloid cell epigenetic rewiring and anabolic metabolism associated with trained immunity (Cheng et al., 2014; Saeed et al., 2014).
Pharmacological inhibition of mTOR has been shown to selectively blunt myeloid cell activation and the expression of pro-inflammatory cytokines, such as TNFα and IL-6 in vitro (Hackstein et al., 2003; Turnquist et al., 2007; Weinstein et al., 2000). Building upon these observations, we designed a myeloid cell-specific nanoimmunotherapy, based on HDL nanobiologics functionalized with the mTOR inhibitor rapamycin (mTORi-HDL), which prevents epigenetic and metabolic modifications underlying trained immunity. In vivo, we found these nanobiologics associated with myeloid cells in the allograft as well as in the bone marrow and to prolong allograft survival. This therapeutic approach enables targeting graft infiltrating myeloid cells, whose role in initiating and sustaining graft-reactive immune response has recently become apparent (Zhuang et al., 2016). The focused in vivo delivery not only ‘redirects’ rapamycin to myeloid cells, but also has the potential to limit its associated side effects, such as impaired wound healing (Lakkis and Li, 2018).
Although mTORi-HDL, designed to prevent trained immunity (signal 3), significantly prolonged allograft survival, indefinite survival was achieved by simultaneous treatment with TRAF6-HDL, a nanobiologic therapy that inhibits CD40 costimulation (Signal 2). This is consistent with previous work, which demonstrated that the absence of macrophage derived TNF-α and IL-6 synergizes with CD40-CD40L costimulatory blockade to induce allograft acceptance (Shen and Goldstein, 2009).
We conclude that our short-term mTORi-HDL/TRAF6i-HDL combination therapy induces long-term allograft survival and therefore is an innovative translational treatment modality with the potential to facilitate successful organ transplantation without the need for continuous immunosuppression. Identifying trained immunity as a therapeutic target provides a compelling framework for developing new treatment paradigms with a focus on promoting immune tolerance by impeding macrophages’ epigenetic programming beyond organ transplantation. Targeting trained immunity’s specific mechanisms, such as metabolic and epigenetic pathways, is a novel therapeutic approach that can be developed to treat excessive immune activation in autoimmune disorders, chronic inflammatory conditions, cardiovascular diseases and allergies.
STAR METHODS
Contact For Reagent And Resource Sharing
Information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jordi Ochando (jordi.ochando@mssm.edu). Some restrictions apply to the use of mTORi-HDL and TRAF6i-HDL nanobiologics and a material transfer agreement (MTA) may be needed between the participant institutions.
Experimental Model and Subject Details
Mice
Female C57BL/6J (B6 WT, H-2b) and BALB/c (H-2d) mice were purchased from the Jackson Laboratory. Eight-week-old C57BL/6J (Foxp3tm1Flv/J), CCR2-deficient, and CD11c-DTR mice were purchased from the Jackson Laboratory. C57BL/6J CD169DTR mice were acquired from Masato Tanaka (Kawaguchi, Japan) (Miyake et al., 2007). Animals were enrolled at 8 to 10 weeks of age (body weight, 20–25 g). All experiments were performed with matched 8- to 12-week-old female mice in accordance with protocols approved by the Mount Sinai Animal Care and Utilization Committee.
Human samples
Buffy coats from pooled unspecified gender healthy donors were obtained after written informed consent (Sanquin blood bank, Nijmegen, The Netherlands). Gender and age of healthy donors was not collected and is therefore unavailable.
Method Details
Vascularized heart transplantation
BALB/c hearts were transplanted as fully vascularized heterotopic grafts into C57BL/6 mice as previously described (Corry et al., 1973). Hearts were transplanted into recipients’ peritoneal cavities by establishing end-to-side anastomosis between the donor and recipient aortae and end-to-side anastomosis between the donor pulmonary trunk and the recipient inferior vena cava. Cardiac allograft survival was subsequently assessed through daily palpation. Rejection was defined as the complete cessation of cardiac contraction and was confirmed by direct visualization at laparotomy. Graft survival was compared among groups using Kaplan-Meier survival analysis.
ApoA1 isolation
Human ApoA1was isolated from human HDL concentrates (Bioresource Technology) following a previously described procedure (Zamanian-Daryoush et al., 2013). Briefly, a potassium bromide solution (density: 1.20 g/mL) was layered on top of the concentrate and purified HDL was obtained by ultracentrifugation. The purified fraction was added to a chloroform/methanol solution for delipidation. The resulting milky solution was filtered and the ApoA1 precipitate was allowed to dry overnight. The protein was renatured in 6 M guanidine hydrochloride, and the resulting solution dialyzed against PBS. Finally, the ApoA1 PBS solution was filtered through a 0.22 µm filter and the protein’s identity and purity were established by gel electrophoresis and size exclusion chromatography.
HDL nanobiologics synthesis
mTORi-HDL nanoparticles were synthesized using a modified lipid film hydration method (Mulder et al., 2018). Briefly, 1,2-dimyristoyl-sn-glycero-3-phosphatidylcholine (DMPC), 1-myristoyl-2-hydroxy-sn-glycero- phosphocholine (MHPC) (both purchased from Avanti Polar Lipids) and rapamycin (Selleckchem) were dissolved in a chloroform/methanol (10:1 v/v) mixture at a 3:1:0.5 weight ratio. After evaporating the solvents, human APOA1 in PBS was added to hydrate the lipid film, in a phospholipid to APOA1 5:1 weight ratio, and left to incubate for 20 minutes in an ice bath. The resulting mixture was homogenized using a probe sonicator in an ice bath for 15 minutes to yield mTORi-HDL nanoparticles. mTORi-HDL was washed and concentrated by centrifugal filtration using 10 kDa molecular weight cut-off (MWCO) filter tubes. Aggregates were removed using centrifugation and filtration (0.22 µm). For the therapeutic studies, animals received oral doses or intravenous tail injections (for mTORi-HDL or intravenous Ra) at a rapamycin dose of 5 mg/kg on the day of transplantation, as well as days two and five post-transplantation.
TRAF6i-HDL nanoparticles were synthesized using a procedure similar to that described above. DMPC, MHPC and the TRAF6-inhibitor (2E)-1-phenyl-3-(2,5-dimethylanilino)-2-propen-1one (Zarzycka et al., 2015) were dissolved in a chloroform/methanol mixture (10:1 v/v) at a 8.7:1:0.6 weight ratio and then dried under vacuum to create a thin lipid film. PBS containing APOA1 was added to the lipid film, in a phospholipid to APOA1 9.5:1 weight ratio, and left to incubate at 37 °C for three hours until the film was hydrated an d a homogenous solution was formed. The solution was then sonicated for one hour to form TRAF6i-HDL nanoparticles. Subsequently, the solution was purified by multiple centrifugation and filtration steps as recently described (Lameijer et al., 2018). For the therapeutic studies, animals received TRAF6i-HDL at 5mg/kg on the day of transplantation, as well as days two and five post-transplantation.
HDL nanobiologics size and surface charge was determined by dynamic light scattering (DLS) and Z- potential measurements. The final composition after purification was determined by standard protein and phospholipid quantification methods (bicinchoninic acid assay and malachite green phosphate assay), whereas drug concentration was established by HPLC against a calibration curve of the reference compound. A variability of ± 15 % between batches was considered acceptable.
Radiolabeling mTORi-HDL nanoparticles
mTORi-HDL was radiolabeled with 89Zr according to previously described procedures (Perez-Medina et al., 2015). Briefly, ready-to-label mTORi-HDL was obtained by adding 1 mol % of the phospholipid chelator DSPE-DFO at the expense of DMPC in the initial formulation. Radiolabeling with 89Zr was achieved by reacting the DFO-bearing nanoparticles with 89Zr-oxalate in PBS (pH = 7.1) at 37 ºC for one hour. 89Zr- mTORi-HDL was isolated by centrifugal filtration using 10 kDa MWCO tubes. The radiochemical yield was 75 ± 2 % (n = 2).
Micro-PET/CT imaging and biodistribution studies
Mice (n = 6; 3 with heart transplants [weight: 18.8 ± 1.0 g]) were injected with a single 89Zr-mTORi-HDL (0.17 ± 0.01 mCi, ~0.25 mg APOA1) dose in 0.2 mL PBS solution via their lateral tail vein six days post graft transplantation. 24 hours later, animals were anesthetized with isoflurane (Baxter Healthcare, Deerfield, USA)/oxygen gas mixture (2% for induction, 1% for maintenance), and a scan was then performed using an Inveon PET/CT system (Siemens Healthcare Global, Erlangen, Germany). Whole body PET static scans, recording a minimum of 30 million coincident events, were performed for 15 minutes. The energy and coincidence timing windows were 350−700 keV and 6 ns, respectively. The image data were normalized to correct for PET response non-uniformity, dead-time count losses, positron branching ratio and physical decay to the time of injection, but no attenuation, scatter or partial-volume averaging correction was applied. The counting rates in the reconstructed images were converted to activity concentrations (percentage injected dose [%ID] per gram of tissue) using a system calibration factor derived from imaging a mouse-sized water- equivalent phantom containing 89Zr. Images were analyzed using ASIPro VMTM software (Concorde Microsystems, Knoxville, USA) and Inveon Research Workplace (Siemens Healthcare Global, Erlangen, Germany) software. Whole body standard low magnification CT scans were performed with the X-ray tube setup at a voltage of 80 kV and current of 500 µA. The CT scan was acquired using 120 rotational steps for a total of 220 degrees to yield an estimated scan time of 120 s with an exposure of 145 ms per frame. Immediately after the PET/CT scan, animals were sacrificed and tissues of interest – kidney, heart, liver, spleen, blood, bone, skin and muscle – were collected, weighed and counted on a Wizard2 2480 automatic gamma counter (Perkin Elmer, Waltham, USA) to determine radioactivity content. The values were decay- corrected and converted to percentage of injected dose per gram (%ID/g). To determine radioactivity distribution within the transplanted hearts, the native and grafted specimens were placed in a film cassette against a phosphorimaging plate (BASMS-2325, Fujifilm, Valhalla, USA) for 4 hours at −20 °C. The plate was read at a pixel resolution of 25 µm with a Typhoon 7000IP plate reader (GE Healthcare, Pittsburgh, USA). The images were analyzed using ImageJ software.
Immunofluorescence microscopy
Transplanted hearts were harvested, subdivided, frozen directly in Tissue-Tek OCT (Sakura), and stored at – 80°C in preparation for immunological studies. Sect ions of 8µm were cut using a Leica 1900CM cryomicrotome mounted on polylysine-coated slides, and fixed in acetone (at −20C degrees for 20 minutes) and then incubated with blocking buffer containing 1% BSA and 5% goat or rabbit serum. The slides were then incubated overnight at 4C with 1/100 rat anti-muse dectin1 (clone 2A11) or rabbit anti-mouse vimentin (clone EPR3776) from Abcam. After overnight incubation the slides were washed in PBS and then incubated with conjugated goat monoclonal anti-rabbit Cy-3 (1/800) or a goat monoclonal anti-rat Cy-2 (1/500) purchased from Jackson Immunoresearch. All slides were mounted with Vectashield with Dapi (Vector Laboratories) to preserve fluorescence. Images were acquired with a Leica DMRA2 fluorescence microscope (Wetzlar) and a digital Hamamatsu charge-coupled device camera. Separate green, red, and blue images were collected and analyzed with ImageJ software (NIH).
Isolation of graft-infiltrating leukocytes
Mouse hearts were rinsed in situ with HBSS with 1% heparin. Explanted hearts were cut into small pieces and digested for 40 minutes at 37 °C with 400 U/ml collagenase A (Sigma-Aldrich), 10 mM HEPES (Cellgro) and 0.01% DNase I (MP Biomedicals) in HBSS (Cellgro). Digested suspensions were passed through a nylon mesh and centrifuged, and the cell pellet was re-suspended in complete HBSS, stained and analyzed by flow cytometry (BD LSR-II; BD Biosciences).
Flow cytometry and cell sorting
For myeloid cell staining, fluorochrome-conjugated mAbs specific to mouse CD45 (clone 30-F11), CD11b (clone M1/70), CD11c (clone N418), F4/80 (clone CI:A3.1), Ly-6C (clone HK1.4) and corresponding isotype controls were purchased from eBioscience. Ly-6G (clone 1A8) mAb was purchased from Biolegend. For T- cell staining, antibodies against CD3 (clone 2C11), CD4 (clone GK1.5), CD8 (clone 53–6.7), and CD25 (clone PC61.5) were purchased from eBioscience. The absolute cell counting was performed using countbright beads (Invitrogen). For progenitor, myeloid and lymphoid cell staining in the bone marrow, spleen, kidney and liver, fluorochrome-conjugated mAbs specific to mouse B220/CD45R (clone RA3–6B2), CD34 (clone RAM34), CD16/32 (clone 93), CD90 (clone 53–2.1), CD19 (clone 1D3), CD115 (clone AFS98) and CD135 (clone A2F10) from eBioscience; CD49b (clone DX5), MHCII (clone M5/114.15.2) and Sca-1 (clone D7) were purchased from Biolegend; CD64 (clone X54–5/7.1), CD117 (clone 2B8), and CD172α (clone P84) were purchased from BD Biosciences. Flow cytometric analysis was performed on LSR II (BD Biosciences) and analyzed with FlowJo software (Tree Star, Inc.). Results are expressed as percentage of cells staining or cells counting (cells per milliliter) above background. To purify graft-infiltrating myeloid cells, donor heart single cell suspensions were sorted with an InFlux cell sorter (BD) to achieve >96% purity at the Flow Cytometry Shared Resource Facility at Icahn School of Medicine at Mount Sinai.
Human monocyte trained immunity experiments
Human monocytes were isolated and trained as previously described (Cheng et al., 2014). PBMC isolation was performed by dilution of blood in pyrogen-free PBS and differential density centrifugation over Ficoll- Paque (GE Healthcare, UK). Subsequently, monocyte isolation was performed by hyper-osmotic density gradient centrifugation over Percoll (Sigma). Monocytes (1×107) were plated to 10 cm Petri dishes (Greiner) in 10 ml medium volumes and incubated with either culture medium only as a negative control or 5 µg/ml of β- glucan with or without mTORi-HDL (1 µg/ml) for 24 hours (in 10% pooled human serum). At day six, cells were detached from the plate, and 1×105 macrophages were reseeded in 96-well flat bottom plates to be re- stimulated for 24 hours with 200 µl of either RPMI or Escherichia coli LPS (serotype 055:B5, Sigma-Aldrich, 10 ng/ml), after which supernatants were collected and stored at −20o C. Cytokine production was determined in supernatants using commercial ELISA kits for TNFα and IL-6 (R&D systems) following the instructions of the manufacturer. The remaining cells were fixed in 1% methanol-free formaldehyde and sonicated. Immunoprecipitation was performed using an antibody against H3K4me3 (Diagenode, Seraing, Belgium). DNA was isolated with a MinElute PCR purification kit (Quiagen) and was further processed for qPCR analysis using the SYBR green method. Samples were analyzed by a comparative Ct method according to the manufacturer’s instructions. The following primers were used: myoglobulin forward AGCATGGTGCCACTGTGCT; myoglobulin reverse GGCTTAATCTCTGCCTCATGAT; H2B forward TGTACTTGGTGACGGCCTTA; H2B reverse CATTACAACAAGCGCTCGAC; TNF forward GTGCTTGTTCCTCAGCCTCT; TNF reverse ATCACTCCAAAGTGCAGCAG; IL-6 forward AGGGAGAGCCAGAACACAGA; IL-6 reverse GAGTTTCCTCTGACTCCATCG; HK2 forward GAGCTCAATTCTGTGTGGAGT; HK2 reverse ACTTCTTGAGAACTATGTACCCTT; PFKP forward CGAAGGCGATGGGGTGAC; PFKP reverse CATCGCTTCGCCACCTTTC.
Mouse monocyte trained immunity experiments
Bone marrow monocytes were isolated using a monocyte isolation kit (Miltenyi). Monocytic precursors (1×106/well in a 48-well plate) were differentiated in vitro with 10ng/ml of recombinant murine GM-CSF (peprotech) for 6 days. On day 6, either 10 µg/ml of β-glucan (Sigma) or 100 µg/ml of vimentin (R&D systems) was added to the cultures for 24h. After 3 days of resting, macrophages were restimulated with either 10ng/ml of LPS (Sigma) or 20 µg/ml of HMGB1 (R&D systems) for 24h. Cytokine production was determined in supernatants using commercial ELISA kits for TNFα and IL-6 (R&D systems) while the remaining cells were used in chromatin immunoprecipitation (ChIP) assays.
Mouse Chromatin Immunoprecipitation (ChIP)
In vitro bone marrow derived trained macrophages or graft-infiltrating macrophages were used in this assay. The following antibodies were used: anti-H3K4me3 (39159; Active Motif), and anti-IgG (ab171870; Abcam). For experiments with ChIP followed by qPCR, crosslinking was performed for 10 min. For sonication, we used a refrigerated Bioruptor (Diagenode), which we optimized to generate DNA fragments of approximately 200–1,000 base pair (bp). Lysates were pre-cleared for two hours using the appropriate isotype-matched control antibody (rabbit IgG; Abcam). The specific antibodies were coupled with magnetic beads (Dynabeads® M-280 Sheep Anti-Rabbit IgG; ThermoFisher Scientific) overnight at 4°C. Antibody-bound beads and chromatin were then immunoprecipitated overnight at 4°C with rotation. After washing, revers e crosslinking was carried out overnight at 65°C. Aft er digestion with RNase and proteinase K (Roche), DNA was isolated with a MinElute kit (Qiagen) and used for downstream applications. qPCR was performed using the iQ SYBR Green Supermix (Bio-Rad) according to manufacturer’s instructions. Primers were designed using the Primer3 online tool; cross-compared to a visualized murine mm10 genome on the Integrated Genomics Viewer (IGV; Broad). Sequences of murine primers used for ChIP-qPCR were as follows: Actb promoter forward, 5′-GTTGGCTGTGCCAGTGTC-3, and Actb promoter reverse, 5′- CAGCTTCTTTGCAGCTCCTT-3; Tnf-alpha promoter forward, 5′-GCCACAAGCAGGAATGAGA-3, and Tnf- alpha promoter reverse, 5′-CCACATCTCCCTCCAGAA-3; Il1b promoter forward, 5′- GAGAGAGAGAGAGACTTACTTGCACA-3, and Il1b promoter reverse, 5′-TTTCACAGCTCTTCACTTCTGC- 3; Il-6 promoter forward, 5′-AATGTGGGATTTTCCCATGA-3, and Il-6 promoter reverse, 5’- GCAAGGAACTGCCTTCACTTA-3; Hk1 promoter forward, 5’-TTCCCCCGAAGACACTTTAC, and Hk1 promoter reverse, 5’-GAGGCAGAACAGGAACTCCA; Pfkp promoter forward, 5’- GCTGGTCAGGACACCGATAG, and Pfkp promoter reverse, 5’-GCCAGGGCTTCAGTGCTT.
Suppression assay
Spleens of C57BL/6 (H-2b) mice were gently dissociated into single-cell suspensions, and red blood cells were removed using hypotonic ACK lysis buffer. Splenocytes were labeled with CFSE at 5 µM concentration (using molecular probes from Invitrogen) followed by staining with anti-CD8 mAb for 30 minutes on ice. Responder CFSE+CD8+ T-cells were sorted using FACS Aria II (BD Biosciences) with >98% purity. CFSE+CD8+ T-cells were used together with anti-CD3/CD28 microbeads as stimulators. Stimulated CFSE+CD8+ T-cells were cultured with graft-infiltrating Ly-6Clo macrophages, mTORi-HDL or placebo for 72 hours at 37 °C in a 5% CO 2 incubator. T-cell proliferation was measured by flow cytometric analysis of CFSE dilution on CD8+ T-cells as recently described (Ochando and Conde, 2017).
Treg expansion assay
Spleens of C57BL/6-Foxp3tm1Flv/J (H-2b) mice were gently dissociated into single-cell suspensions, and red blood cells were removed using hypotonic ACK lysis buffer. Splenocytes were stained with anti-CD4 mAb for 30 minutes on ice. Responder CD4+ were sorted using FACS Aria II (BD Biosciences) with a purity of >98%. CD4+ T-cells were used together with anti-CD3/CD28 microbeads as stimulators. Stimulated CD4+ T-cells were cultured with graft-infiltrating Ly-6Clo macrophages, mTORi-HDL or placebo for 72 hours at 37 °C in a 5% CO2 incubator. Treg expansion was measured by flow cytometric analysis of Foxp3-RFP on CD4+ T-cells as recently described (Ochando and Conde, 2017).
Enzyme-linked immunosorbent assay (ELISA)
Bone marrow derived macrophages were trained as above. Graft-infiltrating macrophages were isolated as above. TNF-α and IL-6 cytokines produced by trained macrophages in vitro and by graft-infiltrating macrophages was assessed by ELISA (R&D Systems) according to the manufacturer protocol.
Microarray analysis
Graft-infiltrating recipient Ly-6Clo macrophages were sorted from mTORi-HDL-treated and placebo-rejecting recipients at day six after transplantation. Cells were sorted twice with a FACS Aria II sorter (BD Biosciences) to achieve >98% purity. Microarray analysis of sorted cells was performed with a total of six Affymetrix Mouse Exon GeneChip 2.0 arrays (Thermo Fisher Scientific) and samples of interest were run in triplicate. Raw CEL file data was normalized using Affymetrix Expression Console Software. Gene expression was filtered based on IQR (0.25) filter using gene filter package. The log2 normalized and filtered data (adjusted P <0.05) were used for further analysis. Gene signature comparisons were performed between intra-graft Ly6Clo macrophages from mTORi-HDL- and placebo-treated recipients. GSEA was performed using GSEA version 17 from Gene pattern version 3.9.6. Parameters used for the analysis were as follows. Gene sets c2.cp.biocarta.v5.1.symbols.gmt; c2.cp.kegg.v5.1.symbols.gmt; c2.cp.reactome.v5.1.symbols.gmt; c6.all.v5.1.symbols.gmt (Oncogenic Signatures); c7.all.v5.1.symbols.gmt (Immunologic signatures) and h.all.v5.1.symbols.gmt (Hallmarks) were used for running GSEA. To select the significant pathways from each gene set result, fdr q-value of 0.25 was set as cutoff. Only genes that contributed to core enrichment were considered.
In vivo macrophage depletion
To deplete CD169-expressing Ly-6Clo macrophages, heterozygous CD169-DTR recipients were injected intraperitoneally with 10 ng/g body weight of DT (Sigma-Aldrich) 24, 48 and 72 hours after transplantation as previously described (Conde et al., 2015; Miyake et al., 2007).
Quantification And Statistical Analysis
Statistical analyses
Results are expressed as mean ± SEM. Statistical comparisons between two groups were evaluated using the Mann-Whitney test or the Wilcoxon signed-rank test for paired measurements. Comparisons among three or more groups were analyzed using the Kruskal-Wallis test followed by Dunn’s multiple comparisons test. Kaplan-Meier curves were plotted for allograft survival analysis, and differences between the groups were evaluated using a log-rank test. A value of P ≤ 0.05 was considered statistically significant. GraphPad Prism 7 was used for statistical analysis.
Supplementary Material
HIGHLIGHTS.
-
1-
Vimentin and HMGB1 promote training of graft infiltrating macrophages
-
2-
HDL-nanobiologics target myeloid cells in hematopoietic organs and the allograft
-
3-
mTORi-HDL prevents trained immunity and TRAF6i-HDL inhibits CD40 costimulation
-
4-
Nanoimmunotherapy with both mTORi- and TRAF6i-HDL induces organ transplant acceptance
ACKNOWLEDGMENTS
We thank James Hutchinson (University Hospital Regensburg) and Heth Turnquist (University of Pittsburgh) for critical review of the manuscript. We thank the technical contributions of the flow cytometry and microsurgery shared resource facilities at Mount Sinai. We also acknowledge Marcy Kuenzel and Sridar Chittur at the University of Albany Center for Functional Genomics microarray core facility for their assistance in generating the microarray data. This work was supported by National Institutes of Health grants R01 HL118440, R01 HL125703 and P01 HL131478 (all to W.J.M.M.), R01 AI139623 (to J.O.), R01 EB009638 and EB009638 (to Z.A.F.) and R01 HL144072 (to W.J.M.M. and Z.A.F.); NIH Program of Excellence in Nanotechnology (PEN) Award (HHSN368201000045C, to Z.A.F.), K25 EB016673 (T.R.) and P30 CA008748, as well as the Harold S. Geneen Charitable Trust Award (Z.A.F.), the Netherlands Organization for Scientific Research (NWO) grant ZonMW Veni 016156059 (R.D.), ZonMW Vidi 91713324 (W.J.M.M.) ZonMW Vici 91818622 (W.J.M.M.), the Spanish Ministry of Science grant SAF2016–80031-R (J.O.) and the Comunidad de Madrid grant B2017/BMD-3731 (J.O.). C.C. is supported by a Scientist Development Grant from the American Heart Association (16SDG27250090). M.G.N. is supported by an ERC Consolidator Grant (#310372) and a Spinoza prize of the Netherlands Organization for Scientific Research. We thank Kaley Joyes for editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing interests.
Data And Software Availability
The microarray data discussed in this publication have been deposited at NCBI and are accessible through GEO Series accession number GSE119370:
REFERENCES
- Azimzadeh AM, Pfeiffer S, Wu GS, Schroder C, Zhou H, Zorn GL 3rd, Kehry M, Miller GG, Rose ML, and Pierson RN 3rd (2005). Humoral immunity to vimentin is associated with cardiac allograft injury in nonhuman primates. Am J Transplant 5, 2349–2359. [DOI] [PubMed] [Google Scholar]
- Braza MS, Conde P, Garcia M, Cortegano I, Brahmachary M, Pothula V, Fay F, Boros P, Werner SA, Ginhoux F, et al. (2018). Neutrophil derived CSF1 induces macrophage polarization and promotes transplantation tolerance. Am J Transplant 18, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao NA, Aghajanirefah A, et al. (2014). mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde P, Rodriguez M, van der Touw W, Jimenez A, Burns M, Miller J, Brahmachary M, Chen HM, Boros P, Rausell-Palamos F, et al. (2015). DC-SIGN(+) Macrophages Control the Induction of Transplantation Tolerance. Immunity 42, 1143–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corry RJ, Winn HJ, and Russell PS (1973). Primarily vascularized allografts of hearts in mice. The role of H-2D, H-2K, and non-H-2 antigens in rejection. Transplantation 16, 343–350. [DOI] [PubMed] [Google Scholar]
- Dai H, Friday AJ, Abou-Daya KI, Williams AL, Mortin-Toth S, Nicotra ML, Rothstein DM, Shlomchik WD, Matozaki T, Isenberg JS, et al. (2017). Donor SIRPalpha polymorphism modulates the innate immune response to allogeneic grafts. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos G, Rogel MR, Baker MA, Troken JR, Urich D, Morales-Nebreda L, Sennello JA, Kutuzov MA, Sitikov A, Davis JM, et al. (2015). Vimentin regulates activation of the NLRP3 inflammasome. Nat Commun 6, 6574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duivenvoorden R, Tang J, Cormode DP, Mieszawska AJ, Izquierdo-Garcia D, Ozcan C, Otten MJ, Zaidi N, Lobatto ME, van Rijs SM, et al. (2014). A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun 5, 3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner KM, Tett SE, and Staatz CE (2016). Multinational Evaluation of Mycophenolic Acid, Tacrolimus, Cyclosporin, Sirolimus, and Everolimus Utilization. Ann Transplant 21, 1–11. [DOI] [PubMed] [Google Scholar]
- Hackstein H, Taner T, Zahorchak AF, Morelli AE, Logar AJ, Gessner A, and Thomson AW (2003). Rapamycin inhibits IL-4--induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood 101, 4457–4463. [DOI] [PubMed] [Google Scholar]
- Hancock WW, Thomson NM, and Atkins RC (1983). Composition of interstitial cellular infiltrate identified by monoclonal antibodies in renal biopsies of rejecting human renal allografts. Transplantation 35, 458–463. [DOI] [PubMed] [Google Scholar]
- Huang Y, Yin H, Han J, Huang B, Xu J, Zheng F, Tan Z, Fang M, Rui L, Chen D, et al. (2007). Extracellular hmgb1 functions as an innate immune-mediator implicated in murine cardiac allograft acute rejection. Am J Transplant 7, 799–808. [DOI] [PubMed] [Google Scholar]
- Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC, Meng M, Fritz D, Vascotto F, Hefesha H, et al. (2016). Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401. [DOI] [PubMed] [Google Scholar]
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, and Pearce EJ (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkis FG, and Li XC (2018). Innate allorecognition by monocytic cells and its role in graft rejection. Am J Transplant 18, 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lameijer M, Binderup T, van Leent MMT, Senders ML, Fay F, Malkus J, Sanchez-Gaytan BL, Teunissen AJP, Karakatsanis N, Robson P, et al. (2018). Efficacy and safety assessment of a TRAF6-targeted nanoimmunotherapy in atherosclerotic mice and non-human primates. Nature Biomedical Engineering 2, 279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien YH (2015). Top 10 things primary care physicians should know about maintenance immunosuppression for transplant recipients. Am J Med [DOI] [PubMed]
- Liu W, Xiao X, Demirci G, Madsen J, and Li XC (2012). Innate NK cells and macrophages recognize and reject allogeneic nonself in vivo via different mechanisms. J Immunol 188, 2703–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Vecchi A, and Allavena P (2014). Pharmacological modulation of monocytes and macrophages. Curr Opin Pharmacol 17, 38–44. [DOI] [PubMed] [Google Scholar]
- Martinez FO, and Gordon S (2015). The evolution of our understanding of macrophages and translation of findings toward the clinic. Expert Rev Clin Immunol 11, 5–13. [DOI] [PubMed] [Google Scholar]
- Meier-Kriesche HU, Schold JD, and Kaplan B (2004a). Long-term renal allograft survival: have we made significant progress or is it time to rethink our analytic and therapeutic strategies? Am J Transplant 4, 1289–1295. [DOI] [PubMed] [Google Scholar]
- Meier-Kriesche HU, Schold JD, Srinivas TR, and Kaplan B (2004b). Lack of improvement in renal allograft survival despite a marked decrease in acute rejection rates over the most recent era. Am J Transplant 4, 378–383. [DOI] [PubMed] [Google Scholar]
- Miller JF (1961). Immunological function of the thymus. Lancet 2, 748–749. [DOI] [PubMed] [Google Scholar]
- Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, and Tanaka M (2007). Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell-associated antigens. J Clin Invest 117, 2268–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder WJM, van Leent MMT, Lameijer M, Fisher EA, Fayad ZA, and Perez-Medina C (2018). High-Density Lipoprotein Nanobiologics for Precision Medicine. Acc Chem Res 51, 127–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naesens M, Kuypers DR, and Sarwal M (2009). Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 4, 481–508. [DOI] [PubMed] [Google Scholar]
- Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O’Neill LA, and Xavier RJ (2016). Trained immunity: A program of innate immune memory in health and disease. Science 352, aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, and van der Meer JW (2017). Trained Immunity: An Ancient Way of Remembering. Cell Host Microbe 21, 297–300. [DOI] [PubMed] [Google Scholar]
- Oberbarnscheidt MH, Zeng Q, Li Q, Dai H, Williams AL, Shlomchik WD, Rothstein DM, and Lakkis FG (2014). Non-self recognition by monocytes initiates allograft rejection. J Clin Invest 124, 3579–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochando J, and Conde P (2017). Functional Characterization of Regulatory Macrophages That Inhibit Graft-reactive Immunity. J Vis Exp [DOI] [PMC free article] [PubMed]
- Page EK, Dar WA, and Knechtle SJ (2012). Tolerogenic therapies in transplantation. Front Immunol 3, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelouris EM (1971). Observations on the immunobiology of ‘nude’ mice. Immunology 20, 247–252. [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, and Pearce EJ (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Medina C, Tang J, Abdel-Atti D, Hogstad B, Merad M, Fisher EA, Fayad ZA, Lewis JS, Mulder WJ, and Reiner T (2015). PET Imaging of Tumor-Associated Macrophages with 89Zr-Labeled High-Density Lipoprotein Nanoparticles. J Nucl Med 56, 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, Cheng SC, Ratter J, Berentsen K, van der Ent MA, et al. (2014). Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345, 1251086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, and Goldstein DR (2009). IL-6 and TNF-alpha synergistically inhibit allograft acceptance. J Am Soc Nephrol 20, 1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Lobatto ME, Hassing L, van der Staay S, van Rijs SM, Calcagno C, Braza MS, Baxter S, Fay F, Sanchez-Gaytan BL, et al. (2015). Inhibiting macrophage proliferation suppresses atherosclerotic plaque inflammation. Sci Adv 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan PS, Yakubenko VP, Elsori DH, Yadav SP, Willard B, Tan CD, Rodriguez ER, Febbraio M, and Cathcart MK (2013). Vimentin is an endogenous ligand for the pattern recognition receptor Dectin-1. Cardiovasc Res 99, 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist HR, Raimondi G, Zahorchak AF, Fischer RT, Wang Z, and Thomson AW (2007). Rapamycin-conditioned dendritic cells are poor stimulators of allogeneic CD4+ T cells, but enrich for antigen-specific Foxp3+ T regulatory cells and promote organ transplant tolerance. J Immunol 178, 7018–7031. [DOI] [PubMed] [Google Scholar]
- Weinstein SL, Finn AJ, Dave SH, Meng F, Lowell CA, Sanghera JS, and DeFranco AL (2000). Phosphatidylinositol 3-kinase and mTOR mediate lipopolysaccharide-stimulated nitric oxide production in macrophages via interferon-beta. J Leukoc Biol 67, 405–414. [DOI] [PubMed] [Google Scholar]
- Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, et al. (2010). A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A 107, 11942–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian-Daryoush M, Lindner D, Tallant TC, Wang Z, Buffa J, Klipfell E, Parker Y, Hatala D, Parsons-Wingerter P, Rayman P, et al. (2013). The cardioprotective protein apolipoprotein A1 promotes potent anti-tumorigenic effects. J Biol Chem 288, 21237–21252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecher D, van Rooijen N, Rothstein DM, Shlomchik WD, and Lakkis FG (2009). An innate response to allogeneic nonself mediated by monocytes. J Immunol 183, 7810–7816. [DOI] [PubMed] [Google Scholar]
- Zhuang Q, Liu Q, Divito SJ, Zeng Q, Yatim KM, Hughes AD, Rojas-Canales DM, Nakao A, Shufesky WJ, Williams AL, et al. (2016). Graft-infiltrating host dendritic cells play a key role in organ transplant rejection. Nat Commun 7, 12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.