

Abstract
A critical challenge in the study of botanical natural products is the difficulty of identifying multiple compounds that may contribute additively, synergistically, or antagonistically to biological activity. Herein, it is demonstrated how combining untargeted metabolomics with synergy-directed fractionation can be effective toward accomplishing this goal. To demonstrate this approach, an extract of the botanical goldenseal (Hydrastis canadensis) was fractionated and tested for its ability to enhance the antimicrobial activity of the alkaloid berberine (4) against the pathogenic bacterium Staphylococcus aureus. Bioassay data were combined with untargeted mass spectrometry-based metabolomics data sets (biochemometrics) to produce selectivity ratio (SR) plots, which visually show which extract components are most strongly associated with the biological effect. Using this approach, the new flavonoid 3,3′-dihydroxy-5,7,4′-trimethoxy-6,8-C-dimethylflavone (29) was identified, as were several flavonoids known to be active. When tested in combination with 4, 29 lowered the IC50 of 4 from 132.2 ± 1.1 μM to 91.5 ± 1.1 μM. In isolation, 29 did not demonstrate antimicrobial activity. The current study highlights the importance of fractionation when utilizing metabolomics for identifying bioactive components from botanical extracts and demonstrates the power of SR plots to help merge and interpret complex biological and chemical data sets.
Graphical Abstract
In traditional and modern alternative health care practices, botanical extracts are employed frequently for medicinal purposes as complex mixtures. The claim is often made that such mixtures are more effective than their constituents in isolation due to additive or synergistic interactions among compounds.1–4 However, the identification of the chemical constituents responsible for the observed activity of complex extracts remains a challenging pursuit.
The traditional approach to identify active compounds from botanical mixtures, bioassay-guided fractionation, is highly effective for studying natural product mixtures when the activity can be traced to single, highly potent active compounds. Unfortunately, it is more difficult to effectively employ bioassay-guided fractionation when the activity of the mixture results from multiple compounds with low potency. Recently, a modification of the bioassay-guided fractionation approach designed to aid in the identification of multiple active compounds in a mixture, “synergy-directed fractionation”, was developed.5 With synergy-directed fractionation, a botanical extract is fractionated and the resulting fractions are tested for activity in combination with a known active constituent of the original extract.5 This approach was shown to be effective for identifying synergists in Hydrastis canadensis L. (Ranunculaceae).5 However, one of the limitations of synergy-directed fractionation is that this technique, like bioassay-guided fractionation, is inherently biased toward the compounds that are most easily isolated. Even though the methodology focuses on the most active fractions for isolation efforts, these fractions are often so complex that it is not possible to isolate every compound that they contain. Characteristics such as the abundance of a given compound and/or its ease of separation typically guide the isolation process in the latter steps, rather than true biological activity. As a result, the “active” compounds identified may represent only a subset of those responsible for the activity of the original mixture.
There has recently been a great deal of interest among the scientific community in the application of untargeted metabolomics to identify active mixture compounds.6–12 With this approach, analytical methods (typically NMR or MS) are used to detect as many of the small molecules in a mixture as possible (the “metabolome”), without bias toward which are chemically or biologically interesting. For metabolomics data to be employed for identification of active mixture components, it is necessary to collect metabolite profiles and corresponding biological assay data for a series of mixtures. Statistical methods are then applied to correlate chemical profile to bioactivity, a process that has been termed “biochemometrics”.13
In theory, biochemometrics has the potential to overcome some of the aforementioned limitations of bioassay-guided fractionation and to provide a more comprehensive representation of which compounds are responsible for the activity of a botanical extract. However, the inherent complexity of botanical extracts makes the data analysis aspects of the biochemometrics challenging when employed for this purpose. Several data analysis approaches have been employed to address this challenge. Inui et al. applied Pearson correlation coefficients to 2D orthogonal chromatography and incorporated biological data to identify mass spectral ions for which variance positively correlated with biological activity.9 Pearson correlation coefficients are a univariate statistical approach to measure the strength of linear correlation between single pairs of variables (i.e., concentration and biological activity) in isolation. A limitation of this approach is that the Pearson correlation does not consider potential interactions between variables and is easily skewed by such outliers.14 In contrast, Kulakowski et al. utilized unsupervised principal component analysis (PCA) and supervised orthogonal partial least-squares discriminatory analysis (OPLS-DA) to correlate bioactivity to LC-MS profiles.6 PCA can be used to group samples based upon covariance, so fractions with similar profiles will be clustered, while others will be in a different location in the PCA scores plot. PCA is useful for describing chemical differences among samples, but does not provide insight into which of the constituents of these samples are biologically active. OPLS-DA allows for the incorporation of biological data, which were in this case binary classifications, i.e., active/nonactive fractions.6 OPLS-DA plots appear similar to PCA plots, and an OPLS-DA model can be represented as an S-plot, where each axis represents the covariance and correlation loading variables.15 Biomarkers with intensities that correlate and covary with the bioactivity appear the furthest from the origin.6,7 An important limitation of this approach is that covariance increases with increased concentration of a constituent, such that the approach is biased toward selection of abundant constituents at the expense of low-abundant compounds that may have high biological activity.12
Selectivity ratio plots are derived from the ratio of explained and residual variance on a single multivariate component that incorporates bioactivity information in the multivariate PLS model and can thus be employed to identify mixture components that are most strongly associated with an observed biological effect independent of concentration.12 Selectivity ratios were first applied to identifying biomarkers of diseases from complex cerebrospinal fluid data sets.16 Recently, the applicability of this biochemometrics approach using selectivity ratios to identify biologically active compounds from fungal extracts was demonstrated.12 As of yet, this approach has not, however, been applied to botanical extracts. Compared to botanicals, fungi produce relatively simple mixtures of chemical compounds. Thus, it is expected that the application of this biochemometric approach to botanical extracts, which are more complex, would be more challenging. Furthermore, an added challenge with botanical extracts is that their greater complexity means that synergistic, additive, or antagonistic interactions are more likely to occur.17
With the study presented herein, an approach combining biochemometrics with synergy-directed fractionation to identify active compounds and/or synergists from a botanical extract was developed. As a case study, the botanical H. canadensis was selected, which is used widely in alternative healthcare practices for the treatment of infections.18,19 Activity against the common Gram-positive bacterium Staphylococcus aureus was evaluated, given that H. canadensis has previously been shown to possess activity against this pathogen.20
Earlier studies with H. canadensis have identified several compounds that interact synergistically to effect antimicrobial activity; thus, this botanical serves as a useful test case for evaluating a new approach to identify synergists. Specifically, H. canadensis is known to contain a number of flavonoids (sideroxylin, 6-desmethyl-sideroxylin, 8-desmethyl-sideroxylin, 3,5,3′-trihydroxy-7,4′-dimethoxy-6,8-di-C-dimethylflavone, 5,4′-dihydroxy-6-C-methyl-7-methoxyflavone, 5,4′-dihydroxy-6,8-di-C-methyl-7-methoxyflavone, 6,8-di-C-methylluteolin-7-methyl ether, and 6-C-methylluteolin-7-methyl ether [1–3, 5–9, respectively]), alkaloids, and other compounds (berberine, oxyberberine, berberastine, tetrahydroberberastine, canadine, canadaline, canadinic acid, 8-oxocanadine, β-hydrastine, hydrastidine, hydrastinine, oxyhydrastinine, noroxyhydrastinine, chilenine, 4′,5′-dimethoxy-4-methyl-3′-oxo(1,2,5,6-tetrahydro-4H-1,3-dioxolo-[4′,5′:4,5]-benzo[1,2-e]-1,2-oxazocin)-2-spiro-1′-phthalan, 8-oxotetrahydrothalifendine, corypalmine, isocorypalmine, tetrahydropalmatine, and 3,4-dimethoxycarbonylbenzoic acid [4, 10–28]) with diverse structures.21,22 Recently, it was shown that the flavonoids from H. canadensis sideroxylin (1), 6-desmethyl-sideroxylin (2), and 8-desmethyl-sideroxylin (3) enhance synergistically the antimicrobial activity of the alkaloid berberine (4) against S. aureus by acting as bacterial efflux pump inhibitors.5 In a separate report, the flavonoid 3,5,3′-trihydroxy-7,4′-dimethoxy-6,8-C-dimethylflavone (5) was also shown to act as an efflux pump inhibitor.21 H. canadensis also contains several alkaloids that do not act as efflux pump inhibitors (13, 17, 24) or possess biologically relevant antimicrobial activity (10, 13–15, 17, 24, 25).21–31 Finally, a number of compounds have also been identified from H. canadensis for which biological activity has not been evaluated (7, 11, 12, 16, 18, 19, 23, 27).21,30,32–37 Relying on existing knowledge about the chemistry and biological activity of H. canadensis, the goal with this investigation was to apply synergy-directed fractionation combined with biochemometric data analysis to an H. canadensis extract. The ultimate objective of these studies was to evaluate the effectiveness of this methodology to identify known active constituents and/or identify new bioactive compounds from a botanical extract.
RESULTS AND DISCUSSION
For this study, a leaf extract was chosen for evaluation because H. canadensis leaves have a higher flavonoid content than the roots. Leaf material also tends to be highly complex; therefore, it was anticipated that reducing the complexity by fractionating an H. canadensis extract would be necessary to provide useful data for biochemometrics analysis. To determine how much fractionation would be necessary, the results of biochemometric analysis were compared on a sequential series of H. canadensis extract fractions and subfractions with decreasing complexity. Specifically, the original extract was subjected to liquid–liquid partitioning initially and then separated chromatographically in a series of four stages (Figure S1, Supporting Information). The most active fraction from each stage was moved forward for further separation. To evaluate biological activity of the fractions produced by each stage of separation, S. aureus cultures were treated with varying concentrations of the alkaloid berberine (a known antimicrobial constituent of H. canadensis) in combination with constant concentrations of the H. canadensis fractions. The minimum concentration of berberine necessary to completely inhibit bacterial growth [minimum inhibitory concentration (MIC)] was compared in the presence of each H. canadensis fraction. With this approach, fractions containing compounds that enhance the antimicrobial activity of berberine (synergistically or additively) will lower the effective MIC of berberine against S. aureus (Figure 1). Metabolite profiles of the fractions from each stage of separation were collected using high resolving power mass spectrometry (Figure 2), as described in the Experimental Section, and biochemometric analyses (using selectivity ratio plots) were employed to integrate the metabolite profile data with the results of the bioassay data (Figure 3).
Figure 1.

Minimum inhibitory concentration (MIC) of each fraction + berberine against S. aureus. Berberine was tested alone, and berberine + piperine served as the positive control. Each fraction was combined at a constant concentration of fraction (75 μg/mL) with varying concentrations of berberine. (A) First-stage normal-phase fractions, where GS-4 lowered the MIC the greatest amount. (B) Secondary fractionation of GS-4 via normal-phase chromatography yielded fractions GS-4-1 through 16, and the fractions therefrom were also tested in combination with berberine. (C) This process was repeated for stage 3, which was prepared using reversed phase chromatographic separation of GS-4-4. Data from which the MIC values were derived, with its associated uncertainty and error bars, can be found in Figure S2 (Supporting Information). Note that error bars are not included in this figure because MIC is defined as the concentration of test compound necessary to completely inhibit bacteria growth, which is the same concentration for all three biological replicates.
Figure 2.

LC-MS chromatograms (collected with ultraperformance liquid chromatography coupled to a Q-Exactive Orbitrap mass spectrometer) of a series of H. canadensis extract fractions. (A) Positive-ion electrospray mass spectrometry chromatogram of the crude extract after liquid–liquid partitioning. (B) The most active fraction (GS-4) of the crude extract shown in A after separation with flash chromatography over silica gel using a hexane–chloroform–methanol gradient (stage 1 separation). GS-4 was further fractionated using a second stage of flash chromatography over silica gel with a hexane–ethyl acetate–methanol gradient with GS-4-4 being the most active (C). GS-4-4-2 generated with stage 3 fractionation (using reversed-phase preparative HPLC with a water–acetonitrile gradient) (D). (E–H) The same H. canadensis extract and fractions analyzed using LC-MS in the negative-ion mode. Arrows represent the region of the chromatogram in which a given ion corresponding to a known constituent of H. canadensis (indicated by compound numbers) could be detected. Compounds 1–7, 10, 13, 16, 17, 20–24, and 28 were identified by comparison of retention time and fragmentation with authenticated standards,20 while compounds 8, 9, 11, 12, 14, 15, 18, 19, and 25–27 were tentatively identified by comparison of accurate mass and molecular formula with literature reports.22,24,27,29,33,36,37 29 is a new flavonoid that was isolated and identified based on NMR and MS data as part of this report. Red dotted arrows represent alkaloids and other compounds, while green dashed arrows represent flavonoids. In cases where a specific peak is not apparent in the chromatogram, the ion was identified based on mass spectrometric data.
Figure 3.

Selectivity ratio plots for first, second, and third stages of fractionation [(A), (B), and (C), respectively] with green dashed arrows representing flavonoids and red dotted arrows representing alkaloids and other known constituents of H. canadensis. The signals of flavonoids and the alkaloid berberine, all known to be or predicted to be active in the assay being used, are not prominent in first-stage fractionation (A) and second-stage fractionation (B). (C) Third-stage fractionation yielded seven flavonoids (1, 2, 3, 5, 6, 8, 29) and three alkaloids (10, 22, 23) with negative selectivity ratios (suggesting these compounds to contribute additively or synergistically to antimicrobial activity). The bar labeled 29* represents the 13C isotope of 29. The identities and activities of compounds represented by other prominent peaks are not known. Selectivity ratio plots were generated by integrating the bioassay data and chemical profile for each stage of fractionation separately. (Data from multiple stages of fractionation were not combined.)
Biochemometrics to Guide Isolation
The H. canadensis extract and multiple fractions therefrom demonstrated the desired biological effect, an ability to enhance the antimicrobial activity of berberine (i.e., reduce its MIC value) against S. aureus (Figure 1; Figure S2, Supporting Information). Importantly, the positive control, a known efflux pump inhibitor38 (piperine), also reduced the MIC of berberine. From the first stage of fractionation, the strongest activity was observed for fraction 4, which reduced the MIC of berberine from 150 μg/mL to 18.8 μg/mL (Figure 1A). Thus, GS-4 was selected as starting material for subsequent fractionation, yielding a series of subfractions, from among which fraction GS-4-4 demonstrated the best activity (Figure 1B). This process was repeated to produce a subfraction set where GS-4-4-2 possessed the most potent activity in combination with berberine (Figure 1C) (see Figure S2, Supporting Information for fraction labeling scheme).
A necessary first step in the application of biochemometrics to identify active mixture components is that the compounds must be detectable by the analytical approach employed (in this case, LC-MS). Thus, the data resulting from LC-MS analysis of the H. canadensis extract (Figure 2A and F) were inspected to determine whether known H. canadensis constituents were detectable. Indeed, a number of flavonoids and alkaloids known to be present in H. canadensis were identifiable (Figure 2) based on LC-MS data (1–28) or by isolation and structure elucidation (29). Pursuit of the most biologically active fractions (Figure 1) throughout the isolation process resulted in selection of fractions with greater flavonoid abundance with each successive stage of isolation (Figure 2). This result is to be expected given that flavonoids are known to enhance the antimicrobial activity of alkaloids,4,39–41 and the biological assay used as a basis for this study was enhancement of antimicrobial activity of the alkaloid berberine.
A known limitation of the application of mass spectrometric analysis to evaluate chemical composition of mixtures is the selectivity of the technique. Relative abundances in LC-MS chromatograms reflect what is most easily ionizable rather than what is truly present in the mixture at highest concentration. Inspection of the data resulting from analysis of the H. canadensis extract evaluated herein illustrates this limitation. Alkaloids, which are easily protonatable, are highly amenable to analysis with electrospray ionization mass spectrometry in the positive-ion mode. Thus, positive-ion mode analysis of the H. canadensis resulted in chromatograms dominated by alkaloids (Figure 2A–D), at least in the earlier stages of fractionation before these alkaloids were separated into inactive fractions. Conversely, data collected in the negative-ion mode were dominated by flavonoids, which are more easily deprotonated (Figures 2E–H). It is important when conducting metabolomics profiling of a botanical extract to collect data in both ionization modes. Additionally, one must always be cognizant of the possibility that certain important mixture components may not be detected and that the relative abundances in LC-MS chromatograms may not reflect the actual relative abundances in the mixture.
An advantage of LC-MS is that it enables the simultaneous detection of multiple constituents that have the same chromatographic retention time. This phenomenon is most apparent from Figure 2A, where eight goldenseal compounds were found within one chromatographic peak that eluted around 4.1 min. The peak shown represents the relative abundance of the most abundant ion during this time span, not the collective signal of all ions. For metabolomics analysis, these chromatograms are deconvoluted (using MZmine) to identify multiple retention time–mass pairs, even those that coelute. Thus, all detectable ions (above a set noise threshold) are compared with bioactivity and correlations are determined, not just those that are apparent in the base peak chromatograms.
The selectivity ratio plots shown in Figure 3 are the output of biochemometric analysis combining the data obtained from biological evaluation (Figure 1) and chemical evaluation (Figure 2) of the H. canadensis extract and fractions. To generate these plots, a series of unique marker ions (m/z–retention time pairings) obtained from analysis of the LC-MS data were used from each stage of fractionation (595, 612, and 149 ions for stages one through three, respectively). The selectivity ratio plots represent the m/z of each ion detected on the x-axis and the selectivity ratio (the ratio of explained variance to residual variance, which is derived from the PLS model) on the y-axis for each independent stage of fractionation. Since the biological assay data used for this analysis was growth inhibition, the ions with the most negative (pointing down in Figure 3) selectivity ratios represent those associated with the desired biological effect (antimicrobial activity). Ions with very small selectivity ratios or positive selectivity ratios (either no bar or pointing up, respectively, in Figure 3) are not expected to have synergistic or additive activity when combined with berberine.
Selectivity ratio plots (Figure 3) have the advantage over LC-MS or LC-UV chromatograms in that the size of bars is representative of constituents possessing the desired effect (biological activity) rather than unrelated effects such as high concentration, ionizability, or molar absorptivity. Thus, in theory, selectivity ratio plots should be extremely helpful for guiding the isolation of bioactive compounds. It is worth mentioning here that scientists engaged in bioassay-guided fractionation typically do make decisions about which fractions to pursue for isolation based on which fractions possess the greatest biological activity. The challenge, of course, is in deciding which components of these fractions to isolate. Typically, a researcher would compare the chromatographic profile of active and inactive fractions to determine which peaks differ between them. However, when hundreds or thousands of components are present, and when more than one fraction is being compared, it becomes very difficult to do this with simple visual inspection. Also, visually one tends to be biased by the tallest peaks of those most distinctly separated from other peaks (even though these may be neither the most abundant nor the most active ions). The use of selectivity ratio plots improves the efficiency of the process by comparing all of the detected ions (both active and inactive) and corresponding peak areas of all fractions in a given stage of separation with their associated biological activity. The result is a quantitative value (the selectivity ratio) that describes how well each ion is associated with the observed biological activity independent of concentration bias (again, assuming that the ion is detected by the analytical method being employed).
How Much Fractionation Is Necessary?
It is important to recognize an inherent limitation of the biochemometric approach, in that associations between activity and chemical composition are correlative, rather than causal. It is always possible that ions identified as bioactive in a selectivity ratio plot may covary in fractions with active ions, but may not possess activity themselves. Such ions would represent false positives. The likelihood of observing such false positives increases as the chemical complexity of the fractions evaluated increases. Conversely, it is also possible that particular bioactive mixture components might go undetected by the analytical approach used or be at a concentration too low to register a biological effect (i.e., antimicrobial activity). Such a scenario would result in a false negative. Using H. canadensis as a test case, one of the goals of this study was to evaluate empirically the likelihood of occurrence of such false positives and false negatives by comparing the results of biochemometric analysis on three different stages of fractionation (Figure 3).
Stage 1 fractions were subjected to subsequent biochemometric analysis, and multivariate partial least-squares (PLS) modeling of the combined bioassay and spectral data matrix yielded the selectivity ratio plot shown in Figure 3A. The selectivity ratio plot based on the first stage of separation indicated several false negatives. Although four confirmed active flavonoids (compounds 1–3 and 5) and the alkaloid berberine (4) were detected among the ions from this first stage of fractionation (Figure 2A), none of these ions were highlighted as major active constituents in the selectivity plot (i.e., ions with large negative selectivity ratios). The flavonoids sideroxylin (1), 6-desmethyl-sideroxylin (2), and 8-desmethyl-sideroxylin (3) were detected in the most active fraction from the first stage of fractionation (GS-4, which lowered the MIC of berberine from 150 μg/mL to 18 μg/mL), but did not display the expected large, negative selectivity ratios. This failure to identify the active flavonoids among other mixture components can be explained by the high level of complexity of these fractions and inability to resolve active and inactive compounds. These findings demonstrate an inherent limitation of metabolomic studies in which investigators attempt to identify active compounds from mixtures without prior fractionation.
Notably, a number of ions were identified in this first-stage fractionation with highly negative selectivity ratios (Figure 1A). It is unclear whether these ions (for which activity and structure are unknown) constitute a series of false positives, or whether they are truly active but hitherto unidentified constituents of H. canadensis. The data do suggest that at least one of the ions identified in Figure 1A is a false negative. The ion with the most negative SR (m/z 527.3156 [M + H]+) was most abundant (relatively, based upon MS signal) in fraction GS-8, which did not prove to be an active fraction (Figure 1B). This highlights the potential for false positives when using this biochemometrics approach and further supports the assertion that some fractionation is required to obtain useful selectivity ratio plots. In addition, ions that appear to be “active” in the stage 1 fractions are not present in later-stage selectivity ratio plots, which indicates that they are false positives and not true antimicrobials or antimicrobial synergists.
GS-4 was chosen for further purification based upon its lowering of the MIC of berberine from 150 μg/mL to 18 μg/mL (Figure 1A). The resulting 16 fractions, excluding GS-4-1, lowered the MIC of berberine to values ranging between 2.34 and 75 μg/mL, and the resulting selectivity ratio plot is shown in Figure 3B. As with stage 1 fractions, the known flavonoid synergists (1–3) and the antimicrobial alkaloid berberine (4) were detected in these fractions. In this less complex series of stage 2 fractions, berberine (known to be active) displayed the expected negative selectivity ratio. However, the second stage of fractionation still failed to show activity for the known active flavonoids (1–3). Finally, as with the first-stage fractionation, several ions of unknown identity displayed large negative selectivity ratios. The possible biological activity of these ions was not further pursued, but could be the subject of future investigations.
Fraction GS-4-4 was the most bioactive from the second stage of fractionation, decreasing the MIC of berberine from 150 μg/mL to 2.3 μg/mL (Figure 1B). The nine subfractions of GS-4-4 were subjected to biochemometric analysis, resulting in the selectivity ratio plot shown in Figure 3C. Compounds 5 (m/z 359.1121 and 359.1122 [M + H]+) and 8 (m/z 329.1017 [M + H]+ and 327.0879 [M − H]−) had the third and fourth largest negative selectivity ratios, with the first and second largest negative selectivity ratios representing m/z 373.1279 [M + H]+ and its 13C isotope, respectively. This ion represented a compound not yet known to H. canadensis and was targeted for isolation. Additionally, chilenine (22) and 4′,5′-dimethoxy-4-methyl-3′-oxo(1,2,5,6-tetrahydro-4H-1,3-di-oxolo-[4′,5′:4,5]-benzo[1,2-e]-1,2-oxazocin)-2-spiro-1′-phthalan (23) were identified as potential active components based on biochemometric analysis of the third-stage fractions (Figure 3C). Chilenine has previously been reported as a constituent of H. canadensis, but its biological activity is unknown.21 The data presented here suggest that it might possess antimicrobial activity (alone or in combination with berberine), a possibility that could be investigated in a future study.
For the studies reported here, statistical analysis to compare chemical composition and biological activity was conducted separately for each stage of fractionation. The possibility of combining the bioassay and metabolomics data for all three stages of fractionation and analyzing as a single data set was also explored (data not shown), but the use of three stages in combination did not improve the ability to detect known active compounds. Previous reports suggest that the inclusion of mass spectrometric and biological data from multiple stages of fractionation can improve assignment of bioactive mixture compounds by increasing statistical power.11,12 For these previous studies, however, natural product mixtures were derived from bacteria and fungi, which are chemically less complex than the botanical extracts evaluated here. It appears that in situations such as this one, where the initial fractions are extremely complex, the inclusion of these data sets in the overall analysis may be detrimental rather than helpful.
Verification of Activity for a Compound Predicted to be Active Based on Biochemometrics
Guided by the results of the biochemometric analysis (Figure 2C), 29 was isolated and identified as the new flavonoid 3,3′-dihydroxy-5,7,4′-trimethoxy-6,8-C-dimethylflavone (Figures S3–S7, Supporting Information). The isolated mass (0.84 mg) was insufficient for completing a full synergy assessment, so 29 was tested at a constant concentration (75 μg/mL or 201.4 μM) in combination with concentrations of berberine ranging from 0 to 100 μg/mL (297.5 μM) (Figure 4). The known efflux pump inhibitor piperine38 was also included in this assay (at a constant concentration of 200 μM) as a positive control.
Figure 4.
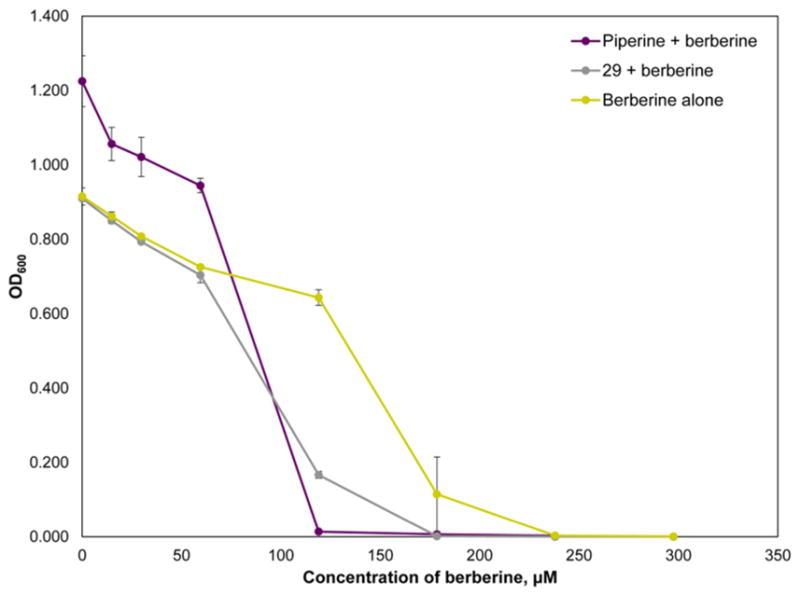
Dose–response curve of berberine ranging from 0 to 298 μM in combination with piperine (positive control, fixed concentration of 263 μM) and 29 (fixed concentration of 200 μM). Error bars represent standard error (error bars are not visible for some data points because they are smaller than the point size). The expected shift to the left (increased potency) in the berberine dose–response curves in the presence of both piperine and 29 was observed. Notably, 29 did not have any antimicrobial effect when tested individually at 200 and 400 μM, which suggests that the increased potency of berberine in combination with this compound is due to synergy and not additivity.
Both piperine and 29 influenced the activity of berberine (Figure 4). The MIC of berberine alone was 238 μM, but in combination with piperine and 29, the MIC was lowered to 119 and 179 μM, respectively. When tested in combination with 29, the IC50 of berberine was lowered from 132.2 ± 1.1 μM to 91.5 ± 1.1 μM. Piperine demonstrated a similar activity, resulting in a lowered berberine IC50 of 72.3 ± 1.0 μM. These data suggest that 29 does, indeed, enhance the antimicrobial activity of berberine. Importantly, 29 alone at 402.8 and 201.4 μM demonstrated no antimicrobial activity (data not shown), suggesting that the increased efficacy of berberine in combination with 29 is due to synergy, not additivity. Thus, 29, which has no antimicrobial activity alone, potentiates the antimicrobial efficacy of berberine. The potency of 29 in combination with berberine is not high as that of the precursor fraction (GS-4-4-2, which lowered the MIC of berberine from 150 μg/mL to 4.68 μg/mL), which further supports that the activity of fraction GS-4-4-2 is due to the presence of multiple constituents, including compounds 1–3, 5, and 6.
Did Biochemometrics Improve the Synergy-Directed Fractionation Process?
Previous work using synergy-directed fractionation on H. canadensis (without biochemometrics) resulted in the identification of three flavonoids (1–3) that synergistically enhanced the antimicrobial activity of berberine (4).5 The application of biochemometrics enabled the list of putative active compounds to be increased to nine compounds. Specifically, based on the biochemometrics data for the third stage of fractions (Figure 3C), the flavonoids 1, 2, 3, 5, 8, and 29 and the alkaloids 10, 22, and 23 were all predicted to act as additives or synergists to the antimicrobial activity of berberine. How accurate were these predictions? Compounds 1–3 and 5 are known efflux pump inhibitors, as demonstrated in several previous studies.5,21 Compound 8 has not previously been evaluated for biological activity against S. aureus, but has been shown to have antimicrobial activity against Streptococcus mutans and Fusobacterium nucleatum.22 Based upon its structural similarity to 1, which has one less hydroxy group at the C-3′ position, 8 is likely to be active as an efflux pump inhibitor. Compound 29 is a new compound, isolated as part of these studies, and demonstrated to increase synergistically the antimicrobial activity of berberine (Figure 4). It is likely that, like other flavonoids, the activity of 29 is due to bacterial efflux pump inhibition, although the efflux pump inhibitory activity of this compound was not evaluated here. Alkaloid 10 was shown previously in the literature to not have antimicrobial activity against S. aureus when tested at 25.6 μg using a disk diffusion assay.31 Further studies would be needed to show if 10 is antimicrobial under other conditions and if it exhibits synergy in combination with berberine. Similarly, alkaloids 22 and 23 have no reported biological activity, and their activity as synergists or antimicrobials could be confirmed with further studies. As a side note, alkaloid 23 was found to be a product of β-hydrastine-N-oxide under reflux conditions and is likely present here as an isolation artifact.21,42
A major challenge faced by any investigator conducting bioassay-guided fractionation is to decide which mixture components to target for isolation. The magnitude of this challenge is illustrated by the sheer number of ions detectable with mass spectrometric analysis of a botanical extract. In this study, a total of 595, 612, and 149 features were detected (above the selected signal threshold) in the first, second, and third stage of H. canadensis fractionation, respectively. When addressing this challenge, the inclination of the investigator is to focus on the most detectable ions (which may or may not be the most abundant) in the most biologically active fraction. The application of biochemometrics to guide isolation helps reduce that inherent bias. Relevant to the study here, 29 was identified for isolation in this project because it has the largest negative selectivity ratio in the third-stage fractionation. Notably, this compound was not the ion detected with highest abundance in the most active fraction (GS-4-4-2, Figure 2D and H). Visual inspection of the data from that fraction would have led the observer to overlook 29 and instead focus on 1, 2, and 3, which appear to be highly abundant and have already been isolated and identified as synergists as part of a previous study. Ultimately, isolation and subsequent NMR structure elucidation will always be needed to confirm absolute configuration and solve structures of unknown compounds. Furthermore, given the correlative rather than causal nature of activity predictions based on biochemometric data analysis, isolation and biological activity evaluation are very necessary as a means of validation. As demonstrated here, the use of biochemometrics to integrate bioassay data with mass spectrometric metabolite profiles can help guide the isolation process toward constituents that may possess relevant biological activity. Importantly, however, as was the case in this study, the complexity of botanical extracts may necessitate several stages of fractionation to simplify the mixture before biochemometric analysis yields useful results.
EXPERIMENTAL SECTION
General Experimental Procedures
Extracts were suspended in 1:1 MeOH–dioxane (LC/MS grade, Fisher Scientific, Hampton, NH, USA) at either 1 or 0.1 mg/mL and subjected to ultraperformance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) analysis via a Waters Acquity UPLC with an Acquity UPLC column (BEH C18, 1.7 μm, 2.1 × 50 mm, Waters Corporation, Milford, MA, USA) and a Thermo Q-Exactive Plus orbitrap mass spectrometer with heated electrospray ionization (Thermo Fisher Scientific, Milford, MA, USA). Analysis was performed at a flow rate of 0.3 mL/min using the following binary solvent system with A consisting of water with 0.1% formic acid additive and solvent B consisting of acetonitrile with 0.1% formic acid additive (LC/MS grade, Fisher Scientific). The gradient was as follows: 95:5 (A:B) from 0 to 1 min, increasing to 90:10 (A:B) from 1 to 2 min, 80:20 (A:B) from 2 to 3 min, 60:40 (A:B) from 3 to 4 min, 70:30 (A:B) from 4 to 5 min, 0:100 (A:B) from 5 to 6 min and held from 6 to 7 min, 95:5 (A:B) from 7 to 8 min and held from 8 to 9 min. The Thermo Q-Exactive Plus was operated in both positive and negative polarities using the following settings: spray voltage, 3.7 kV; capillary temperature, 350 °C; sheath gas, 25; auxiliary gas, 5; S-lens RF level, 50. Known constituents of goldenseal were listed in an inclusion list for fragmentation via high-energy collision-induced dissociation (HCD) and used for tentative identification based upon matching retention time, accurate mass, and accurate mass fragments with isolated standards (compounds 1–7, 10, 13, 16, 17, 20–24, and 28).20 Compounds without standards (8, 9, 11, 12, 14, 15, 18, 19, and 25–27) were identified tentatively by comparison of accurate mass and molecular formula with literature reports.21,23,26,28,32,35 Data were visualized using Xcalibur (v. 2.3.26, Thermo Fisher Scientific). Phenomenex Gemini-NX C18 analytical (5 μm; 250 × 4.6 mm) and preparative (5 μm; 250 × 21.2 mm) and Phenomenex Luna PFP(2) 100A analytical (5 μm; 150 × 4.6 mm) and preparative (5 μm; 250 × 21.2 mm, Phenomenex, Torrance, CA, USA) columns were used on a Varian Prostar HPLC system equipped with Prostar 210 pumps and a Prostar 335 photodiode array detector (Agilent Technologies, Santa Clara, CA, USA), with data collected and analyzed using Galaxie Chromatography Workstation software (version 1.9.3.2, Agilent Technologies). Flash chromatography was performed on a Teledyne ISCO CombiFlash Rf 200 (Teledyne-Isco, Lincoln, NE, USA) using Gold silica columns (330, 120, and 40 g columns) and monitored by UV and evaporative light-scattering detectors. 1D and 2D NMR spectra were recorded using a JEOL ECA-500 NMR spectrometer operating at 400 MHz for 1H and 100 MHz for 13C (JEOL, Peabody, MA, USA) or an Agilent 700 MHz NMR spectrometer (Agilent Technologies) equipped with a cryoprope, operating at 175 MHz for 13C. NMR data were visualized using MestReNova (v 11.0.4, Mestrelab Research, Santiago de Compostela, Spain). IC50 values were calculated using a four-parameter logistic standard curve analysis function in SigmaPlot (v.13, Systat Software, San Jose, CA, USA). All chemicals used unless otherwise stated were ACS grade and obtained from Fisher Scientific.
Plant Material
Hydrastis canadensis bulk leaf material was collected in June 2014 from William Burch in Hendersonville, North Carolina (NC, N 35°24.277′, W 082°20.993′, 702.4 m elevation). The plants were cultivated in their natural environment, a hardwood forest understory, and were a year old at the time of harvest. A voucher specimen for this H. canadensis plot has been deposited at the Herbarium of the University of North Carolina at Chapel Hill (NCU583414) and authenticated by Dr. Alan S. Weakly. Samples were dried at room temperature until crisp before extraction.
Extraction and Isolation
Dried aerial plant portions were ground mechanically using a Wiley mill standard model no. 3 (Arthur H. Thomas Co., Philadelphia, PA, USA) with a 2 mm mesh size and percolated in MeOH overnight three times. The MeOH extract was concentrated in vacuo before liquid–liquid extraction. The extract was defatted by partitioning between hexane and 10% aqueous MeOH (1:1). The dried aqueous MeOH was partitioned further between 4:5:1 EtOAc–MeOH–H2O, and the organic layer was washed with 1% saline solution to remove hydrosoluble tannins. The fractionation scheme is provided as Supporting Information. The first stage of normal-phase flash chromatography (330 g Gold silica gel column) was performed in two batches (58.49 g total) and conducted at a 200 mL/min flow rate with a 42.7 min hexane–CHCl3–MeOH gradient. The most active fraction from the first-stage separation (fraction 4, ~10 g) was subjected to a second stage of normal-phase flash chromatography (120 g Gold silica gel column and 40 g Gold silica gel column) and conducted at flow rates of 85 and 40 mL/min, respectively, with 57.6 and 33.4 min hexane–EtOAc–MeOH gradients, respectively. The most active fraction from the second-stage separation (subfraction 4, ~290 mg) was purified using reversed-phase HPLC with a Phenomenex Gemini-NX column at a 21.4 mL/min flow rate in three batches. A CH3CN–H2O with 0.1% formic acid gradient was employed, which was held at 30:70 for 5 min, increased to 60:40 from 5 to 10 min, increased to 0:100 from 10 to 25 min, and held at 100:0 from 25 to 40 min. It was determined that the ion of interest via biochemometric analysis of the third stage of fractionation, m/z 373.1279 [M + H]+, had the greatest relative abundance in fraction GS-4-5 and was therefore used for isolation. GS-4-5 was subjected to reversed-phase HPLC with a preparative Luna PFP column at a 21.4 mL/min flow rate. A CH3CN–H2O with 0.1% formic acid gradient was employed that increased from 35:65 to 50:50 over 8 min, held at 50:50 from 8 to 30 min, increased to 100:0 from 30 to 35 min, and then held at 100:0 from 35 to 55 min. Fractions 41 through 43 were recombined and subjected to reversed-phase HPLC with an analytical Luna PFP column at 1 mL/min. A MeOH–H2O with 0.1% formic acid gradient was employed where the solvent composition increased from 72:28 to 76:24 over 30 min and increased to 100:0 from 30 to 40 min. Fractions were collected manually into vials based upon UV absorption and LC-MS traces. One last stage of purification was performed to achieve 95.4% purity via UV (254 nm) using a Gemini analytical column with a CH3CN–H2O with 0.1% formic acid solvent system at 1 mL/min, where the gradient increased from 10:90 to 100:0 over 55 min and was held at 100:0 from 55 to 60 min. Compound 29 had a final yield of 0.84 mg (0.0014% of 58.49 g of EtOAc extract).
3,3′-Dihydroxy-5,7,4′-trimethoxy-6,8-C-dimethylflavone (29)
yellow solid; UV (MeOH) λmax (log ε) 254 (4.42) 363 (4.26) nm; 1H NMR (DMSO, 500 MHz, Figure S3, Supporting Information) and 13C NMR (DMSO, 175 MHz, Figure S4, Supporting Information), see Table 1; HRESIMS m/z 373.1279 [M + H]+ (calcd for C20H21O7, 373.1287). Because there were no HMBC correlations for the hydroxy protons attached to C-3 and C-3′, OH-3′ was assigned based upon literature precedent that hydroxy group protons attached to aromatic rings have sharper peaks than those that are not.43
Table 1.
NMR Spectroscopic Data (500 and 700 MHz, DMSO) for 3,3′-Dihydroxy-5,7,4′-trimethoxy-6,8-C-dimethylflavone (29)
| position | δC, type | δH (J in Hz) | HMBCa |
|---|---|---|---|
| 2 | 143.35, C | ||
| 3 | 138.49, C | ||
| 4 | 171.79, C | ||
| 5 | 155.28, C | ||
| 6 | 121.39, C | ||
| 7 | 160.96, C | ||
| 8 | 115.60, C | ||
| 9 | 153.47, C | ||
| 10 | 112.83, C | ||
| 1′ | 124.33, C | ||
| 2′ | 114.60, CH | 7.70, s | 2, 3′, 4′, 6′ |
| 3′ | 146.79, C | ||
| 4′ | 149.47, C | ||
| 5′ | 112.43, CH | 7.09, d (14) | 1′, 3′, 4′ |
| 6′ | 119.65, CH | 7.68, d (14) | 2, 2′, 4′ |
| OH-3 | 9.43, s | ||
| OCH3-5 | 61.47, | 3.75, s | 5 |
| CH3-6 | 9.17, | 2.18, s | 6, 5, 7 |
| OCH3-7 | 60.83, | 3.74, s | 7 |
| CH3-8 | 9.43, | 2.39, s | 7, 8, 9 |
| OH-3′ | 9.10, s | ||
| OCH3-4′ | 56.02, | 3.81, s | 4′ |
HMBC correlations are from the proton(s) stated to the indicated carbon.
Antimicrobial Assays
Minimum inhibitory concentrations were evaluated per the terms outlined by the Clinical Laboratory Standards Institute44 against wild-type Staphylococcus aureus (SA1199).45 A single colony inoculum was grown to log phase in Müeller Hinton broth and adjusted to a final assay concentration of 1.0 × 105 CFU/mL based on absorbance at 600 nm (OD600). The negative control consisted of 2% glycerol and 2% DMSO in broth (vehicle), and the known antimicrobial compound berberine46 served as the positive control. Triplicate wells were prepared with all treatments and controls. Duplicate plates were made without bacteria for background absorbance subtraction. OD600 was read after 18 h at 37 °C using a Synergy H1 microplate reader (Biotek, Winooski, VT, USA). The MIC was defined as the concentration at which no statistically significant difference was observed between the negative control and treated sample.
Combination Assays
Extracts were tested in combination with berberine over a concentration range of 2.3 to 150 μg/mL. Berberine alone served as a positive control and demonstrated an MIC value between 75 and 150 μg/mL, consistent with the literature.5 The vehicle consisted of 2% glycerol and 2% DMSO in broth. A simplified version of the synergy assay was performed after each stage of fractionation to quickly assess those fractions possessing additive and/or synergistic behavior. This involved testing berberine at a range of concentrations (2.3 to 150 μg/mL) in combination with a constant concentration of each of the extract fractions (75 μg/mL) or purified compound 29 (200 μM). The known efflux pump inhibitor piperine (75 μg/mL or 240 μM) was used as the positive control for these experiments.38,47 Fractions were classified as active if they enhanced the antimicrobial activity of berberine (lowered the MIC by 2-fold) and were advanced to the next stage of separation.
Biochemometric Analysis
LC-MS data sets were collected in positive and negative polarities and were analyzed, aligned, and filtered in independent batches using MZmine 2.17 software (http://mzmine.sourceforge.net/),48 as previously described by Kellogg et al.12 Briefly, raw data files were uploaded into MZmine for peak picking based upon mass spectral signals that were above the signal intensity of a mass spectrometric chromatogram of an injection of 1:1 MeOH–dioxane (solvent blank). Chromatograms were built based upon having a signal that lasted for longer than 0.01 min, minimum peak duration of 1.8 s (0.03 min), m/z variation tolerance of 0.05, and a m/z intensity variation tolerance of 20%. Peak list filtering and retention time alignment were also performed, and the join align algorithm compiled the final peak list. The mass spectral data matrix was exported into Excel (Microsoft, Redmond, WA, USA) where ions in common between the samples and solvent blank were manually removed from the sample mass spectral profiles, positive and negative polarity data sets were combined, and biological assay data were added in the form of MIC values of berberine in combination with certain fractions. Data matrices for each stage of fractionation were independently imported into Sirius version 10.0 (Pattern Recognition Systems AS, Bergen, Norway).49 An internally cross-validated PLS model of 100 iterations with a significance threshold set to 0.05 was constructed, and selectivity ratios were calculated using the Sirius software.50
Supplementary Material
Chart 1.

Acknowledgments
This work was supported by the National Center for Complementary and Integrative Health, National Institutes of Health [grants 1R01 AT006860 (NBC) and F31 AT009164 (ERB)]. Mass spectrometry data were collected in the Triad Mass Spectrometry Facility. We thank D. Todd for technical assistance and J. Rivera-Chávez for assisting with structure elucidation.
Footnotes
Notes
The authors declare no competing financial interest.
DEDICATION
Dedicated to Dr. Susan Band Horwitz, of Albert Einstein College of Medicine, Bronx, NY, for her pioneering work on bioactive natural products.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jnat-prod.7b00654.
Additional information (PDF)
References
- 1.Gilbert B, Alves LF. Curr Med Chem. 2003;10:13–20. doi: 10.2174/0929867033368583. [DOI] [PubMed] [Google Scholar]
- 2.Wagner H, Ulrich-Merzenich G. Phytomedicine. 2009;16:97–110. doi: 10.1016/j.phymed.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 3.Rasoanaivo P, Wright CW, Willcox ML, Gilbert B. Malar J. 2011;10(Suppl1):S4. doi: 10.1186/1475-2875-10-S1-S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stermitz FR, Lorenz P, Tawara JN, Zenewicz LA, Lewis K. Proc Natl Acad Sci U S A. 2000;97:1433–1437. doi: 10.1073/pnas.030540597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Junio HA, Sy-Cordero AA, Ettefagh KA, Burns JT, Micko KT, Graf TN, Richter SJ, Cannon RE, Oberlies NH, Cech NB. J Nat Prod. 2011;74:1621–1629. doi: 10.1021/np200336g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulakowski DM, Wu SB, Balick MJ, Kennelly EJ. J Chromatogr A. 2014;1364:74–82. doi: 10.1016/j.chroma.2014.08.049. [DOI] [PubMed] [Google Scholar]
- 7.Chan KM, Yue GGL, Li P, Wong ECW, Lee JKM, Kennelly EJ, Lau CBS. J Chromatogr A. 2017;1487:162–167. doi: 10.1016/j.chroma.2017.01.044. [DOI] [PubMed] [Google Scholar]
- 8.Li P, AnandhiSenthilkumar H, Wu SB, Liu B, Guo ZY, Fata JE, Kennelly EJ, Long CL. J Chromatogr B: Anal Technol Biomed Life Sci. 2016;1011:179–195. doi: 10.1016/j.jchromb.2015.12.061. [DOI] [PubMed] [Google Scholar]
- 9.Inui T, Wang Y, Pro SM, Franzblau SG, Pauli GF. Fitoterapia. 2012;83:1218–25. doi: 10.1016/j.fitote.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qiu F, Cai G, Jaki BU, Lankin DC, Franzblau SG, Pauli GF. J Nat Prod. 2013;76:413–9. doi: 10.1021/np3007809. [DOI] [PubMed] [Google Scholar]
- 11.Kurita KL, Glassey E, Linington RG. Proc Natl Acad Sci U S A. 2015;112:11999–12004. doi: 10.1073/pnas.1507743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kellogg JJ, Todd DA, Egan JM, Raja HA, Oberlies NH, Kvalheim OM, Cech NB. J Nat Prod. 2016;79:376–386. doi: 10.1021/acs.jnatprod.5b01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martens H, Bruun SW, Adt I, Sockalingum GD, Kohler A. J Chemom. 2006;20:402–417. [Google Scholar]
- 14.Wilcox RR. Statistical Modeling and Decision Science. 3. Academic Press; San Diego: 2012. Introduction to Robust Estimation and Hypothesis Testing; p. 608. [Google Scholar]
- 15.Wiklund S, Johansson E, Sjostrom L, Mellerowicz EJ, Edlund U, Shockcor JP, Gottfries J, Moritz T, Trygg J. Anal Chem. 2008;80:115–122. doi: 10.1021/ac0713510. [DOI] [PubMed] [Google Scholar]
- 16.Rajalahti T, Kroksveen AC, Arneberg R, Berven FS, Vedeler CA, Myhr KM, Kvalheim OM. J Proteome Res. 2010;9:3608–3620. doi: 10.1021/pr100142m. [DOI] [PubMed] [Google Scholar]
- 17.van Vuuren S, Viljoen A. Planta Med. 2011;77:1168–1182. doi: 10.1055/s-0030-1250736. [DOI] [PubMed] [Google Scholar]
- 18.McGuffin M, Hobbs C, Upton R, Goldberg A. American Herbal Products Association’s Botanical Safety Handbook. CRC Press; Boca Raton, FL: 1997. [Google Scholar]
- 19.Robbers JE, Tyler VE. Tyler’s Herbs of Choice: The Therapeutic Use of Phytomedicinals. Haworth Herbal Press; Binghampton, NY: 1999. [Google Scholar]
- 20.Cech NB, Junio HA, Ackermann LW, Kavanaugh JS, Horswill AR. Planta Med. 2012;78:1556–1561. doi: 10.1055/s-0032-1315042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leyte-Lugo M, Britton ER, Foil DH, Brown AR, Todd DA, Rivera-Chávez J, Oberlies NH, Cech NB. Phytochem Lett. 2017;20:54–60. doi: 10.1016/j.phytol.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang BY, Roberts SK, Chadwick LR, Wu CD, Kinghorn AD. Planta Med. 2003;69:623–6277. doi: 10.1055/s-2003-41115. [DOI] [PubMed] [Google Scholar]
- 23.Ettefagh KA, Burns JT, Junio HA, Kaatz GW, Cech NB. Planta Med. 2011;77:835–840. doi: 10.1055/s-0030-1250606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orhana I, Ozcelik B, Karaoglu T, Sener B. Z Naturforsch, C: J Biosci. 2007;62:19–26. doi: 10.1515/znc-2007-1-204. [DOI] [PubMed] [Google Scholar]
- 25.Wu WN, Beal JL, Mitscher LA, Salman KN, Patil P. Lloydia. 1976;39:204–212. [PubMed] [Google Scholar]
- 26.Gentry EJ, Jampani HB, Keshavarz-Shokri A, Morton MD, Vander Velde D, Telikepalli H, Mitscher LA, Shawar R, Humble D, Baker W. J Nat Prod. 1998;61:1187–1193. doi: 10.1021/np9701889. [DOI] [PubMed] [Google Scholar]
- 27.Costa EV, Marques FA, Pinheiro MLB, Vaz NP, Duarte MCT, Delarmelina C, Braga RM, Maia BHLNS. J Nat Prod. 2009;72:1516–1519. doi: 10.1021/np800788n. [DOI] [PubMed] [Google Scholar]
- 28.Scazzocchio F, Cometa MF, Tomassini L, Palmery M. Planta Med. 2001;67:561–564. doi: 10.1055/s-2001-16493. [DOI] [PubMed] [Google Scholar]
- 29.Galeffi C, Cometa MF, Tomassini L, Nicoletti M. Planta Med. 1997;63:194. doi: 10.1055/s-2006-957649. [DOI] [PubMed] [Google Scholar]
- 30.Pinho PMM, Pinto MMM, Kijjoa A, Pharadai K, Diaz JG, Herz W. Phytochemistry. 1992;31:1403–1407. [Google Scholar]
- 31.Wang L, Zhang SY, Chen L, Huang XJ, Zhang QW, Jiang RW, Yao F, Ye WC. Phytochem Lett. 2014;7:89–92. [Google Scholar]
- 32.Fajardo V, Elango V, Cassels BK, Shamma M. Tetrahedron Lett. 1982;23:39–42. [Google Scholar]
- 33.Messana I, La Bua R, Galeffi C. Gazz Chim Ital. 1981;110:539–543. [Google Scholar]
- 34.Rosa MC. Rev Bras Farm. 1939;20:191–196. [Google Scholar]
- 35.Wollenweber E, Wehde R, Dorr M, Lang G, Stevens JF. Phytochemistry. 2000;55:965–70. doi: 10.1016/s0031-9422(00)00348-4. [DOI] [PubMed] [Google Scholar]
- 36.Avula B, Wang YH, Khan IA. J AOAC Int. 2012;95:1398–405. doi: 10.5740/jaoacint.12-074. [DOI] [PubMed] [Google Scholar]
- 37.Weber HA, Zart MK, Hodges AE, White KD, Barnes SM, Moody LA, Clark AP, Harris RK, Overstreet JD, Smith CS. J AOAC Int. 2003;86:476–483. [PubMed] [Google Scholar]
- 38.Khan IA, Mirza ZM, Kumar A, Verma V, Qazi GN. Antimicrob Agents Chemother. 2006;50:810–2. doi: 10.1128/AAC.50.2.810-812.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morel C, Stermitz FR, Tegos G, Lewis K. J Agric Food Chem. 2003;51:5677–5699. doi: 10.1021/jf0302714. [DOI] [PubMed] [Google Scholar]
- 40.Guz NR, Stermitz FR, Johnson JB, Beeson TD, Willen S, Hsiang J, Lewis K. J Med Chem. 2001;44:261–268. doi: 10.1021/jm0004190. [DOI] [PubMed] [Google Scholar]
- 41.Stermitz FR, Tawara-Matsuda J, Lorenz P, Mueller P, Zenewicz L, Lewis K. J Nat Prod. 2000;63:1146–1149. doi: 10.1021/np990639k. [DOI] [PubMed] [Google Scholar]
- 42.Klötzer WEO. Helv Chim Acta. 1955;56:223–224. [Google Scholar]
- 43.Qiu Y, Yoshikawa M, Li Y, Dou D, Pei Y, Chen Y. J Shenyang Pharma Univ. 2000;17:267–268. [Google Scholar]
- 44.National Committee for Clinical Laboratory Standards. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. 10. Wayne, PA: 2015. [Google Scholar]
- 45.Kaatz GW, Seo SM. Antimicrob Agents Chemother. 1995;39:2650–2655. doi: 10.1128/aac.39.12.2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Amin AH, Subbaiah TV, Abbasi KM. Can J Microbiol. 1969;15:1067–1076. doi: 10.1139/m69-190. [DOI] [PubMed] [Google Scholar]
- 47.Nargotra A, Koul S, Sharma S, Khan IA, Kumar A, Thota N, Koul JL, Taneja SC, Qazi GN. Eur J Med Chem. 2009;44:229–238. doi: 10.1016/j.ejmech.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 48.Pluskal T, Castillo S, Villar-Briones A, Oresic M. BMC Bioinf. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kvalheim OM, Chan HY, Benzie IFF, Szeto YT, Tzang AHC, Mok DKW, Chau FT. Chemom Intell Lab Syst. 2011;107:98–105. [Google Scholar]
- 50.Rajalahti T, Arneberg R, Berven FS, Myhr KM, Ulvik RJ, Kvalheim OM. Chemom Intell Lab Syst. 2009;95:35–48. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.