

Abstract
Hydrogen-bonds (H-bonds) within the secondary coordination sphere are often invoked as essential noncovalent interactions that lead to productive chemistry in metalloproteins. Incorporating these types of effects within synthetic systems has proven a challenge in molecular design that often requires the use of rigid organic scaffolds to support H-bond donors or acceptors. We describe the preparation and characterization of a new hybrid tripodal ligand ([H2pout]3–) that contains two mono-deprotonated urea groups and one phosphinic amide: the urea groups serve as H-bond donors while the phosphinic amide group serves as a single H-bond acceptor. The [H2pout]3– ligand was utilized to stabilizes a series of Mn–hydroxido complexes in which the oxidation state of the metal center ranges from 2+ to 4+. The molecular structure of MnIII–OH complex demonstrates that three intramolecular H-bonds involving the hydroxido ligand are formed. Additional evidence for the formation of intramolecular H-bonds was provided by vibrational spectroscopy in which the energy of the O–H vibration supports its assignment as an H-bond donor. The step-wise oxidation of [MnIIH2pout(OH)]2- to its higher oxidized analogs was further substantiated by electrochemical measurements and results from electronic absorbance and electron paramagnetic resonance spectroscopies. Our findings illustrate the utility of controlling both the primary and secondary coordination spheres to achieve structurally similar Mn–OH complexes with varying oxidation states.
Graphical Abstract
Introduction
To determine correlations between the structure and function of a metal complex, it is necessary to understand the relationships between the primary and secondary coordination spheres of the metal ion(s).1–4 Control of the primary coordination sphere is often governed via relatively strong M–L covalent bonds which chemists have utilized in a variety of ways. Rather than using covalent bonds, the secondary coordination spheres of metal ions are regulated through networks of weaker non-covalent interactions that are often difficult to manipulate within synthetic systems.5,6 Hydrogen bonds (H-bonds) are the most versatile of these interactions and have been actively designed into a variety of ligand frameworks. The most common of these ligands incorporate either H-bond donors or acceptors to promote intramolecular H-bonds.3,7–17 However, there are fewer examples that utilize a mixture of H-bond donors/acceptors to target a specific type of M–L unit.18–21
Our group has specialized in the development of ligand platforms that promote intramolecular H–bonding networks to investigate their influence on the secondary coordination sphere. One design uses the symmetrical urea ligand [H3buea]3– (Figure 1A) that provides a cavity with three H-bond donors positioned to stabilize monomeric Mn, Fe, Co, and Ni complexes with terminal oxido/hydroxido ligands.2,22–28 We found that the strongly anionic ligand field provided by the mono-deprotonated urea groups also assists in stabilizing high valent species that includes MnV-oxido and MnIV–hydroxido complexes.28–30 We have also examined the chemistry of tripodal ligands such as [RST]3– (Figure 1B) which contains deprotonated sulfonamide units that also function as H-bond acceptors.31–34 Recently, hybrid tripods were introduced that contain both urea and sulfonamido groups to vary the intramolecular H-bonding network that surrounds Co–OH units (Figure 1C).18 These studies showed that deprotonated sulfonamido ligands are useful H-bond acceptors; however, they do not provide a sufficient primary coordination sphere to stabilize monomeric M–OH complexes with oxidation states greater than 3+.
Figure 1.

Metal complexes surround by ligand frameworks with various H-bond networks: [H3buea]3–(A), [RST]3– (B), and [H22tol]3– (C). H-bonds donating groups are shown in blue, while accepting moieties are highlighted in red. The H-bond interactions are illustrated with black dotted lines.
To circumvent this problem, we have turned to ligands that contain deprotonated phosphinic amides in place of sulfonamido groups. We reasoned that phosphinic amides would produce a stronger anionic ligand field while still providing H-bond acceptors through the P=O units. In this report, we describe the development and Mn chemistry of the hybrid ligand [H2pout]3– (Figure 2) that installs a combination of two H-bond donors and one H-bond acceptor within the secondary coordination sphere. The [H2pout]3– ligand was used to prepare the MnII–OH complex [MnIIH2pout(OH)]2– (Figure 2) which had sufficiently low redox potentials to synthetically prepare its corresponding Mnn–OH (n = III, IV) analogs.
Figure 2.

Metal complex within the [H2pout]3– framework where n = 2+, m = 2-; n = 3+, m = 1-; n = 4+, m = 0
Experimental Section
General Procedures.
All manipulations, unless otherwise stated, were completed under an argon atmosphere in a VAC drybox. Solvents were sparged with argon and dried over columns containing Q-5 and molecular sieves. All reagents were purchased from commercial suppliers and used as received unless otherwise noted. Potassium hydride as a 30% suspension in mineral oil was filtered and washed five times each with Et2O and pentane and dried under vacuum. The ligand precursors 1,1′-(((2-aminoethyl)azanediyl)bis(ethane-2,1-diyl))bis(3-(tert-butyl)urea) and ferrocenium tetrafluoroborate were synthesized using literature procedures.10,35
Physical Methods.
Electronic absorbance spectra were recorded in a 1 cm cuvette on an 8453 Agilent UV-vis spectrometer equipped with an Unisoku Unispeks cryostat. X-band (9.64 GHz) and S-band (3.50 GHz) EPR spectra were recorded on a Bruker spectrometer equipped with Oxford liquid helium cryostats. The quantification of all signals is relative to a CuEDTA spin standard. The concentration of the standard was derived from an atomic absorption standard (Aldrich). For all instruments, the microwave frequency was calibrated with a frequency counter and the magnetic field with an NMR gaussmeter. A modulation frequency of 100 kHz was used for all EPR spectra. The EPR simulation software (SpinCount) was written by one of the authors.36 EPR samples were prepared under an inert atmosphere, sealed with a septum, and cooled to 77K unless otherwise mentioned. The sign of D for Mn(II)–OH and Mn(IV)–OH were determined from simulations of perpendicular-mode EPR spectra collected at variable temperatures. The sign of D for the Mn(III)–OH complex has not be determined experimental and is based on the value obtained for the similar complexes, [Mn(III)H3buea(OH)]2-.1H and13C nuclear magnetic resonance (NMR) spectroscopies were conducted using a Bruker DRX500 spectrometer. Cyclic voltammetric experiments were conducted using a CHI600C electrochemical analyzer. A 2.0 mm glassy carbon electrode was used as the working electrode at scan velocities of 100 mV s−1. A cobaltocenium/cobaltocene couple (CoCp2+/CoCp2) (ΔEp = – 0.136 V vs Fc+/0) was used to monitor the Ag wire reference electrode, and all potentials are reference to the Fc+/0 couple. The DFT calculations were performed with Gaussian ‘0937 using the hybrid functional B3LYP and the basis set 6–311G. Geometry optimizations were performed using the structures determined from X-ray diffraction for the MnII and MnIII complexes. The calculations for the manganese(IV) complex were based on the crystal structure of the MnIII complex.
Preparative Methods.
H5pout.
The preparative route for this compound followed the method previous report for H22Tol in which only the last step was different.18 A solution of 1,1′-(((2-aminoethyl)azanediyl)bis(ethane-2,1-diyl))bis(3-(tert-butyl)urea) (2.00 g, 4.47 mmol) and triethylamine (2.55 g, 25.2 mmol) in 100 mL anhydrous THF was treated dropwise with diphenyl phosphinic chloride dissolved in 100 mL THF (~1 drops/second) while stirring and a white precipitate formed immediately. Once the addition was complete, 100 mL THF was added to rinse the addition funnel. The addition funnel was removed, and the round bottom flask was covered with a glass stopper. After stirring overnight, the white precipitate, Et3NHCl, was removed via filtration. The filtrate was dried under reduced pressure and the solid residue was further dried under vacuum. Diethyl ether (150 mL) was added to the residue to afford an off-white powder, which was collected on a medium porosity glass-fritted funnel, washed once with MeCN (50 mL), twice with Et2O (50 mL), and dried for several hours under reduced pressure to give the desired white product (2.7 g, 85 %).1H NMR (500 MHz, CDCl3, ppm): 2.50 (t, 6H), 3.11 (q, 6H), 6.13 (q, 3H), 7.45 (t, 12H), 7.50 (d, 6H), 7.61 (t, 12H);13C NMR (125 MHz, CDCl3, ppm): 29.7, 36.3, 45.7, 49.7, 128.9 (d), 130.8, 131.9, 132.0 (d), 132.4 (d), 158.5.31P NMR (162 MHz, CDCl3, ppm): 28.6; HRMS (ES+, m/z): Exact mass calculated for NaC42H45N4O9P3 [M + Na]: 567.3188, Found: 567.3186. FTIR (Nugol, cm–1): 3400, 3341, 3231, 2954, 2922, 2853, 1678, 1657, 1546, 1451, 1378, 1287, 1264, 1221, 1120, 1047, 814, 723, 690.
K 2[MnIIH2pout(OH)].
A solution of H5pout (0.100 g, 0.183 mmol) in anhydrous DMA (4 mL) was treated with potassium hydride (KH) (0.030 g, 0.75 mmol) and the reaction allowed to proceed until gas evolution ceased and all solids were dissolved. To the colorless solution was added MnII(OAc)2 (0.031 g, 0.18 mmol). After stirring for 90 min, water (3 μL, 0.2 mmol) was added via syringe and the reaction mixture was filtered after 15 min through a medium porosity glass-fritted funnel to afford a light-yellow filtrate and white solid on the glass frit (KOAc, 90%). Et2O was allowed to diffuse into DMA resulting in pale yellow crystals (80%) suitable for XRD studies. Anal. Calcd for K2[MnH2pout(OH)], C28H43K2MnN6O4P: C, 48.61; H, 6.27; N, 12.15. Found C, 48.79; H, 6.91; N, 12.48. FTIR (ATR, cm–1) 3656, 3261, 3124, 3071, 2955, 2797, 1645, 1585, 1510, 1435, 1390, 1350, 1310, 1245, 1205, 1185, 1115, 1045, 1015, 975, 885, 815, 755, 710, 630. UV-vis (DMF:THF, λmax nm, (εmax, M−1 cm−1)) 525 (sh). EPR (X-band Perpendicular, DMF:THF, 16 K, g = 6.0, 3.9, 2.4, 1.6, 1.4, 1.3, Aiso = 250). E1/2 (DMF, V vs [FeCp2]+/–): MnIII/II = –1.47.
K[MnIIIH2pout(OH)] was prepared by a similar method to K2[MnIIH2pout(OH)] with the following modifications: after the addition of water, half an equivalent of elemental I2 (0.028 g, 0.11 mmol) was added to the yellow filtrate that caused an immediately color change to dark green. The solution was left to stir for 30 min and then concentrated to dryness under reduced pressure. The residue was titrated with Et2O (20 mL) and again dried under vacuum. The free-flowing green solid was redissolved in MeCN and filtered through a fine porous-glass frit to remove a white solid (KI, 27 mg, 90%). Single crystals suitable for XRD studies were obtained by vapor diffusion of Et2O into the solution of DMA of the salt (60%). Anal. Calcd for K2[MnH2pout(OH)], C 28H43KMnN6O4P • 0.5KI • DMA: C, 46.71; H, 6.37; N, 11.92. Found C, 46.74; H, 6.38; N, 11.28. FTIR (ATR, cm–1) 3300, 3274, 3211, 2956, 2875, 2810, 1610, 1567, 1500, 1490, 1462, 1450, 1433, 1420, 1399, 1378, 1369, 1238, 1112, 1006, 1020, 978, 790, 743, 724, 696, 661, 616, 594. UV-vis (DMF:THF, λmax nm, (εmax, M−1 cm−1)) 440 (210), 740 (236). EPR (X-band Perpendicular, DMF:THF, 19 K, g = 8, Az = 270) E0 (DMF, V vs [FeCp2]+/–): MnIV/III = – 0.25.
Oxidation of [MnIIIH2pout(OH)]–.
A 20 mM stock solution of K[MnH2pout(OH)] was prepared in a 1:1 DMF:THF mixture at room temperature and kept in a −35 °C freezer for the duration of the experiment. Addition, via air tight syringe, of a 40 μL aliquot of stock metal complex to the solvent mixture (2 mL) in a 1 cm quartz cuvette, which was sealed with a rubber septum and precooled to the desired temperature in the 8453 Agilent UV−vis spectrophotometer equipped with an Unisoku Unispeks cryostat, to give the desired concentration for oxidation experiments (400 μM). The solution of metal complex was allowed to equilibrate to the desired temperature for at least 15 min. Additionally, a stock solution of [FeCp2]BF4 (40 mM) was prepared in 1:1 DMF:THF mixture and kept in a −35 °C freezer for the duration of the experiment. One equiv (20 μL) of [FeCp2]BF4 was added to the MnIII−OH solution via gastight syringe. The disappearance of the two low-energy d-d transitions corresponding to the MnIII–OH species were monitored spectrophotometrically, with the appearance of new transitions attributed to the MnIV–OH species. EPR samples were prepared in a similar manner and frozen via submersion into liquid N2. The value of D for [MnIIH2pout(OH)]2- and [MnIVH2pout(OH)] were determined from simulations of EPR spectra collected at variable temperatures. For [MnIIIH2pout(OH)]–, the value of D was determined from the temperature dependence of the signal as displayed in Figure 7.
Figure 7.

Parallel-mode EPR spectrum of a frozen solution of [MnIIIH2pout(OH)]– in solvent 1:1 DMF:THF. Sample temperature 19 K, microwaves 9.318 GHz, 2 mW. The black trace is a simulation for a S = 2 species. Inset: experimental signal intensity times temperature versus temperature (points) and the calculated % population of the EPR active doublet of S = 2 and D = +2.7 cm−1.
X-ray Crystallographic Methods.
A Bruker SMART APEX II diffractometer was used to collect all data. The APEX238 program package was used to determine the unit-cell parameters and for data collections. The raw frame data was processed using SAINT39 and SADABS40 to yield the reflection data files. Subsequent calculations were carried out using the SHELXTL40 program. The structures were solved by direct methods and refined on F2 by full-matrix least-squares techniques.
Results and Discussion
Synthesis.
The preparation of H5pout was accomplished in a five-step convergent synthesis adapted from the methods developed for previously reported H52tol ligand (Figure 1C).18 Central to the syntheses is the diurea compound, 1,1′-((2aminoethyl)azanediyl)bis(ethane-2,1-diyl))bis(3-(tert-butyl)urea which is combined in the final step with diphenylphosphinic chloride in THF and excess Et3N. Precipitation of the ammonium chloride salt was observed immediately, and H5pout was isolated as a white solid in 85% yield after purification.
The MnII–OH complex, [MnIIH2pout(OH)]2-, was synthesized following the procedure outline in Scheme 1, in which the ligand was initially formed via deprotonation with KH. Metal ion coordination was achieved using MnII(OAc)2 and the hydroxido ligand was derived from water; note that an extra equivalent of base was added to the reaction mixture which is needed to produce the hydroxido ligand in [MnIIH2pout(OH)]2-. One-electron oxidation of the MnII–OH complex with elemental iodine gave the corresponding MnIII–OH species, [MnIIIH2pout(OH)]– (Scheme 1).26
Scheme 1.

Preparation of Mn–OH complexes.
Structural Properties.
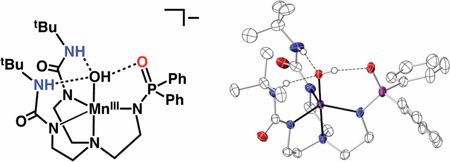
The molecular structures of K2[MnIIH2pout(OH)] and K[MnIIIH2pout(OH)] salts were characterized with X-ray diffraction methods (Figure 3, Tables 1 and 2). DFT calculations gave optimized structures for both that agreed with the molecular structures determined by XRD measurements (Table 1). The structures for both salts revealed that each Mn–OH anion is mononuclear with a distorted trigonal bipyramidal (tbp) coordination geometries. The primary coordination spheres around the Mn center in each complex is composed of a N4O donor set with three anionic N-atom donors forming the trigonal plane. The axial positions contain the apical amine N1-atom from the [H2pout]3- ligand and O1-atom from the hydroxido ligand. Statistically significant differences are found between the Mn–N and Mn–O1 bond lengths between the two structures. For instance, the Mn–O1 bond distance is contracted from 2.051(1) Å to 1.834(1) Å upon conversion to [MnIIIH2pout(OH)]–, a change which is consistent with oxidation at the metal center. A similar shortening in bond lengths after oxidation is observed for the bonds within the equatorial plane, in which the average Mn–N bond distance changed from 2.188(1) Å to 2.052(2) Å.
Figure 3.

Thermal ellipsoid plots of [MnIIH2pout(OH)]2– (A), K2[MnIIH2pout(OH)] (B), [MnIIIH2pout(OH)]– (C), K[MnIIIH2pout(OH)] (D). Thermal ellipsods are drawn at the 50% probability level, only urea and hyrdoxido hydrogen atoms are shown for clarity.
Table 1.
Selected Bond Distances and Angles for K2[MnIIH2poutOH)] and K[MnIIIH2pout(OH)] Determined by XRD and DFT Methods.
| Bond Distances (Å) or Angles (°) | XRD | DFT | XRD | DFT |
|---|---|---|---|---|
| K2[MnIIH2pout(OH)] | K[MnIIIH2pout(OH)] | |||
| Mn(1)-O(1) | 2.051(1) | 2.07 | 1.834(1) | 1.85 |
| Mn(1)-N(1) | 2.315(1) | 2.46 | 2.054(2) | 2.11 |
| Mn(1)-N(2) | 2.214(1) | 2.17 | 2.056(2) | 2.03 |
| Mn(1)-N(3) | 2.220(1) | 2.15 | 2.035(2) | 2.06 |
| Mn(1)-N(4) | 2.121(1) | 2.20 | 2.068(2) | 2.10 |
| O(1)⋯O(2) | 3.958(2) | 2.685(2) | 2.71 | |
| O(1)⋯N(5) | 2.768(2) | 2.79 | 2.776(2) | 2.78 |
| O(1)⋯N(6) | 3.053(2) | 2.83 | 2.773(2) | 2.77 |
| O(1)-Mn(1)-N(1) | 170.13(5) | 172 | 179.18(7) | 178 |
| O(1)-Mn(1)-N(2) | 103.60(5) | 100 | 97.36(6) | 99 |
| O(1)-Mn(1)-N(3) | 93.48(5) | 98 | 97.49(6) | 98 |
| O(1)-Mn(1)-N(4) | 108.76(5) | 111 | 98.82(6) | 97 |
| N(1)-Mn(1)-N(4) | 78.68(5) | 77 | 81.91(7) | 82 |
| N(1)-Mn(1)-N(2) | 78.62(5) | 77 | 81.96(6) | 83 |
| N(2)-Mn(1)-N(4) | 112.73(5) | 107 | 116.27(7) | 118 |
| N(3)-Mn(1)-N(1) | 77.22(5) | 77 | 82.53(6) | 81 |
| N(3)-Mn(1)-N(2) | 119.46(5) | 116 | 124.58(7) | 125 |
| N(3)-Mn(1)-N(4) | 115.48(5) | 122 | 113.59(7) | 111 |
Table 2.
EPR parameters of the [MnnH2pout(OH)]m complexes
| [MnnH2pout(OH)]m | S | Da | E/D | g | |A|b |
|---|---|---|---|---|---|
| MnII | 5/2 | −0.24 | 0.065 | 2.00, 2.00, 2.00 | Aiso = 250 |
| MnIII | 2 | +2.7 | 0.04 | -, -, 2.01 | Az = 270 |
| MnIV(1) | 3/2 | +0.8 | 0.33 | 2.00, 2.01, 1.98 | 152, 198, 183 |
| MnIV(2) | 3/2 | +0.7 | 0.17 | 2.06, 1.96, 2.02 | 224, 188, 201 |
| acm−1;bMHz |
The molecular structures also show that [H2pout]3- interacts with the Mn–OH unit through H-bonds to influence the secondary coordination sphere. In each structure, the urea NH groups serve as H-bond donors to the hydroxido ligand as judged by the average N…O distances of 2.910(2) Å in [MnIIH2pout(OH)]2- and 2.774(2) Å in [MnIIIH2pout(OH)]–. There is a third intramolecular H-bond in the MnIII–OH structure that is formed between the hydroxido ligand and the O2-atom of the phosphinic group (2.685(2) Å). The presence of this additional H-bond could account for the shorter Mn–O bond distance in [MnIIIH2pout(OH)]– when compared to that found in [MnIIIH3buea(OH)]– (Figure 1A, Mn–O bond length of 1.877(2) Å).41 Because the hydroxido acts as an H-bond donor, it should have more negative character, which would result in a shorter Mn1–O1 bond distance in [MnIIIH2pout(OH)]–. Similar structural results were found in Co–OH complexes and emphasized the structural effects of the H-bonds within the secondary coordination sphere.18 Notice also potassium ion does not interact with the Mn–OH unit in the lattice of K[MnIIIH2pout(OH)]; it only interact with the carbonyl groups of the ureas which are pointed away from the hydroxido ligand (Figure 3D).
Similar comparison for [MnIIH2pout(OH)]2- are hindered because the positioning of the potassium ions complicates interpretation of the secondary coordination sphere. One potassium ion (K2) interacts with the O2-atom of [H2pout]3- that distorts the cavity and prevents formation of an intramolecular H-bond (Figure 3B). However, the O–H bond is positioned toward the phosphinic unit in a similar manner as in [MnIIIH2pout(OH)]–, but it is now closer to one of the phenyl substitutients. This lack of a third intramolecular H-bonds results in a Mn1–O1 bond length that is the statistically the same as that found in the related MnII–OH complex, [MnIIH3buea(OH)]2-(Mn–O bond distance of 2.059(2) Å).26
Electronic Absorbance and Vibrational Properties.
The conversion of [MnIIH2pout(OH)]2- to [MnIIIH2pout(OH)]– was monitored by optical spectroscopy in 1:1 DMF:THF at −80˚C. The MnII–OH species had no observable features in the visible region but upon oxidation new bands appeared at λmax(εM) = 440 (210) and 740 (236) nm (Figure 4A). The energies of these features are similar to those found in the related MnIII–O(H) complexes [MnIIIH3buea(O)]2- and [MnIIIH3buea(OH)]–; however, there are noticeable differences in the values of the extension coefficients. For [MnIIIH3buea(OH)]–, bands were observed at λmax(ε#M) = 427 (ε1 = 390) and 720 (ε2 = 500) nm (ε1/ε2 = 0.78) that change in [MnIIIH3buea(O)]2- to λmax(eM) = 498 (ε1 = 490) and 725 (ε2 = 240) nm (ε1/ε2 = 2.0).42 In addition, an analogous MnIII–OH in tbp coordination geometry with no intramolecular H-bonds has a similar visible absorbance spectrum in which the lower energy band has the greater intensity (ε1/ε2 = 0.62).43 The comparable extinction coefficients of the absorbance bands in the spectrum of [MnIIIH2pout(OH)]– (ε1/ε2= 0.89) may be attributed to the Mn–OH unit being an H-bond donor to form a relatively strong H-bond with the phosphinic amide O-atom; this additional interaction would make the hydroxido ligand slightly more oxido-like, which would affect a change in the absorbance spectrum as observed.
Figure 4.

UV-visible spectra monitoring the conversion of [MnIIH2pout(OH)]2- (---) to [MnIIIH2pout(OH)]– (—) (A) and [MnIIIH2pout(OH)]– to [MnIVH2pout(OH)] (—) (B). Spectra were recorded in a 1:1 DMF:THF mixture at – 80 °C.
Analysis of vibrational properties using FTIR spectroscopy showed bands attributed to the n(MnO–H) at 3656 and 3300 cm−1 for the MnII–OH and MnIII–OH complexes. Because of the interactions of the potassium ions with [MnIIH2pout(OH)]2- (Figure 3A) it is not possible to directly compare this complex with other Mn–OH species. A more straightforward comparison can be made for [MnIIIH2pout(OH)]– and its n(MnO–H) band is boarder (fwhm = 90 cm–1) and at lower energy that the comparable peak for [MnIIIH3buea(OH)]– which was found at 3613 cm−1 (fwhm = 15 cm–1).41 These vibrational data are also consistent with presence of an intramolecular H-bond between the phosphinic arm and the hydroxido ligand that causes a weaken the O–H bond.
Electrochemical Properties.
Cyclic voltammetry (CV) was employed to analyze the redox properties of the Mn–OH complexes. Electrochemical data showed a reversible one-electron redox couple at −1.47 V vs [FeCp2]+/0 that is assigned to the MnIII/II–OH process (Figure 5B). This potential is similar to one found in [MnIIIH3buea(OH)]– that occurs at −1.50 V vs [FeCp2]+/0 that is the MnIII/II OH couple.44 A second redox event was observed at a more positive potential that is center around – 0.25 V vs [FeCp2]+/0 and assigned to the MnIV/III–OH couple (Figure 5B).
Figure 5.

Cyclic voltammograms of [MnIIH2pout(OH)] recorded in DMF: MnIII/II couple (A), MnIV/IIIcouple (B). Measurement were done at room temperature under Ar with a scan rate of 100 mV s−1.
The process is not reversible under our experimental conditions which we suggest is caused by the oxidized MnIV–OH species not being stable at room temperature. Again, a similar result was observed for the MnIV/III–OH couple (– 0.18 V vs [FeCp2]+/0) for [MnIVH3buea(OH)] at room temperature and subsequent studies found it was only stable at temperatures lower than −50˚C.30
Accessing a MnIV–OH Species: Spectroscopic Studies.
The electrochemical data of the MnII–OH complex suggested that accesses a higher valent MnIV–OH species should be possible with mild oxidants. We tested this premise by monitored the formation of this complex at –80˚C in 1:1 DMF:THF mixture using ferrocenium cation as the oxidant. Clean conversion was observed that initiated from [MnIIIH3buea(OH)]– to produce a new spectrum with a prominent band at λmax(εM) = 460 nm (1390) and a shoulder at 560 nm; a single isosbestic point was found at 675 nm (Figure 4B). The final spectrum closely resembled that of [MnIVH3buea(OH)] which has an absorbance band λmax (εM) = 466 nm (5600). This new oxidized species was stable for hours at −80˚C.
EPR Studies.
We have previously shown that EPR spectroscopy is an effective method for probing changes in the electronic structure of monomeric Mn–O(H) complexes.30 Our approach utilized both perpendicular- and parallel-modes at X-band to follow spin state changes upon oxidation (Table 2). The EPR spectrum of [MnIIH2pout(OH)]2- (Figure 6) in perpendicular-mode showed signals over a wide magnetic field range. The spectra are complicated by resonances from multiple overlapping transitions as has been described previously for [MnIIH3buea(OH)]2-.45 The simulation overlaid on the experimental spectrum in Figure 6 is for a S = 5/2 spin center using the parameters of described in Table 2. The simulation intensity is in quantitative agreement with the sample concentration determined from the weight of the complex dissolved in the solvent. The zero-field parameters are similar to the previously characterized [MnIIH3buea(OH)]2- and, as expected, [MnIIH2pout(OH)]2- showed an increased in rhombic parameter E/D owing to the unsymmetrical nature of the tripodal ligand.45
Figure 6.

Perpendicular-mode EPR spectrum of a frozen solution of [MnIIH2pout(OH)]2- in 1:1 DMF:THF. Sample temperature 16 K, microwaves, 9.645 GHz, 0.2 mW. The black trace is a simulation for a S = 5/2 species.
The addition of 1 equiv [FeIIICp2]+ resulted in the disappearance of the signals from the MnII–OH complex and the appearance of a 6-line hyperfine signal centered at g = 8 with Az = 270 MHz (a = 9.6 mT) in parallel-mode (Figure 7). The simulation overlaid on the experimental spectrum are for an S = 2 spin center (Figure 7, Table 2) and its intensity is in quantitative agreement with the sample concentration. The zero-field parameters determined from the spectra are statistically the same as those of [MnIIIH3buea(OH)]–.45 Taken together, these data are consistent with the formation of the high-spin MnIII–OH complex, [MnIIIH2pout(OH)]–.
The fully oxidized [MnIVH2pout(OH)] complex showed no features in parallel-mode, but perpendicular-mode signals were observed when measured at both S- and X-bands. Figure 8 displaces the spectra collected at both bands with the magnetic field ranges chosen to equate the g-value ranges for both frequencies. The positions of the resonances and the simulations are indicative of an S = 3/2 spin-state that is consistent with a high-spin MnIV–OH complex in local C3 symmetry (Table 2). Our analysis of the EPR data showed that two distinct MnIV–OH species are formed, a result that is similar to what we reported for [MnIVH3buea(OH)].30,45 The largest difference between the two species for [MnIVH2pout(OH)] is in their E/D values: species 1 (E/D = 0.33) is significantly more rhombic than species 2 (E/D = 0.17). The effective g-tensors for the |±1/2> and excited |±3/2> doublets for the two species are reported in Table 3 and indicated on Figure 8. For species 1, the effective g-tensors for each doublet are the same because of the large value of E/D. We have been successful in determining the hyperfine tensor for both species, even though the hyperfine splitting is not resolved in all directions. This determination was accomplished because we used two different frequencies in our analysis as previously described.28
Figure 8.

Perpendicular-mode EPR spectra of a 1:1 DMF:THF frozen solution [MnIVH2pout(OH)] at S-band (A) and X-band (B). Sample temperature was 5 K, microwave power was 0.03 mW for S-band and 0.2 mW for X-band. The black traces overlaid on the experimental spectra are the sum of the S = 3/2 simulations for the E/D = 0.17 and 0.33 species in a ratio of 70:30, respectively.
Table 3.
DFT Computed Metrical Parameters for Proposed Structures of [MnIVH2pout(OH)].
| Bond Distances (Å) or Angles (°) | Aa | Ba |
|---|---|---|
| Mn(1)-O(1) | 1.80 | 1.78 |
| Mn(1)-N(1) | 2.15 | 2.11 |
| Mn(1)-N(2) | 1.91 | 1.92 |
| Mn(1)-N(3) | 1.93 | 2.01 |
| Mn(1)-N(4) | 1.96 | 1.91 |
| O(1)⋯O(2) | 2.58 | 2.59 |
| O(1)⋯N(5) | 2.73 | 4.36 |
| O(1)⋯N(6) | 2.76 | 2.70 |
| O(1)-Mn(1)-N(1) | 177 | 177 |
| O(1)-Mn(1)-N(2) | 97 | 97 |
| O(1)-Mn(1)-N(3) | 98 | 95 |
| O(1)-Mn(1)-N(4) | 96 | 98 |
| N(1)-Mn(1)-N(4) | 82 | 84 |
| N(1)-Mn(1)-N(2) | 83 | 84 |
| N(2)-Mn(1)-N(4) | 132 | 114 |
| N(3)-Mn(1)-N(1) | 84 | 82 |
| N(3)-Mn(1)-N(2) | 110 | 130 |
| N(3)-Mn(1)-N(4) | 114 | 112 |
see Figure 9
The rhombic character of the two species is consistent with a MnIV ion in a C3 symmetry in which an appreciable Jahn-Teller distortion is expected. However, the reason(s) for the presence of two species and their exact structural differences are not known. The simulations showed that species 2 (the less rhombic species) was more abundant by a ratio of 2:1. We used density functional theory (DFT) to probe the possible structures of [MnIVH2pout(OH]]. The DFT calculations converged to two optimized structures shown in Figures 9 and S1 with selected bond lengths given in Table 3. Structure A had a lower energy and is similar to the structure of [MnIIIH2pout(OH)]– with three intramolecular H-bonds involving the hydroxido ligand. Structure B has one urea arm rotated such that the carbonyl O-atom was proximal to the Mn–O(H) unit. A similar reorientation of a urea arm has been proposed for the protonated form of [FeIVH3buea(O)]– .46 The computed equatorial Mn-N bond lengths were more similar for species A relative to those found for species B which suggests A corresponds to the species with E/D values of 0.17 and 0.33, respectively. DFT calculations of the spin-dipolar A-tensors for the two species gave (29, −2, −27) MHz (A) and (30, 6, −36) MHz (B), which were in approximate agreement with the experimental values of (17, 3, −20) MHz for A and (26, −5, −21) MHz for B. Finally, we note that the EPR spectrum of the related complex [MnIVH3buea(OH)]– also consisted of two species with similar values for E/D to that of [MnIVH2pout(OH)]–. However, one important difference between the two MnIV–OH complexes is that the more abundant species in [MnIVH3buea(OH)]– is the one that is most rhombic (E/D = 0.33). The reason(s) for this difference is under investigation.
Figure 9.

Possible structures for the two MnIV–OH complexes of [MnIVH2pout(OH)] determined from DFT.
Preparation and Properties of [MnIIIH2pout(O)]2- and [MnIVH2pout(O)]–.
We have also prepared the related MnIII/IV–oxido complexes of [H2pout]2- to compare their spectroscopic properties to the related Mn–OH complexes. The synthesis of [MnIIIH2pout(O)]2- was achieved via deprotonation of the MnIII–OH complex with KOBut (Scheme 2A). Conversion to its MnIV–oxido analog was accomplished using ferrocenium (Scheme 2B); [MnIVH2pout(O)]– was also be prepared via deprotonation of [MnIVH2pout(OH)] with KOBut (Scheme 2C). The electronic absorbance spectrum of [MnIIIH2pout(O)]2- contains two major features in the visible region at λmax(εM) = 500 (400) and 760 (190) nm with an ε1/ε2= 2.1 (Figure S2A). These values are reminiscent of those observed for [MnIIIH3buea(O)]2- (see above). In addition, the parallel-mode EPR spectrum for [MnIIIH2pout(O)]2- showed a six-lined signal at g = 7.91 with a hyperfine constant Az = 290 MHz that consistent with a monomeric MnIII complex having an S = 2 spin ground state (Figure S3A). Moreover, these values are distinct from those observed for [MnIIIH2pout(OH)]–. Similar differences were observed for [MnIVH2pout(O)]– when compared to [MnIVH2pout(OH)]: the MnIV– oxido complex had absorbance bands at λmax(εM) = 440 (1060) and 745 (200) nm (Figure S2B) that are similar to those found for [MnIVH3buea(O)]–. The perpendicular-mode EPR spectrum of [MnIVH2pout(O)]– was broad and only the resonance at g = 5.09 could be identified (Figure S3B). The hyperfine pattern at g = 5.09 and the other broad features were different from that of [MnIVH2pout(OH)] (Figure S3B). Taken together, these findings demonstrate that the spectroscopic differences between Mn–hydroxido and Mn–oxido complexes with tripodal ligands can be used to differentiate these types of complexes in solution.
Scheme 2.

Preparation of Mn–oxido complexes of [H2pout]3-.
Summary and Conclusions
Manganese complexes with terminal hydroxido ligands are relatively rare because of the strong propensity of the Mn–OH units to aggregate to form multinuclear species. However, they have been implicated as key intermediates in a variety of chemical processes,26,30,41,43,44,47–54 most notably as intermediates formed during biological water oxidation.17,55–59 Results from structural biology on the oxygen evolving complex (OEC) suggest that H-bonding networks to coordinate water molecules assist in forming Mn–OH complexes that can be in either the MnIII or MnIV oxidation states.60–64 One approach toward studying the structural outcomes of the secondary coordination sphere is to develop chelating ligands that can provide H-bond donors/accepts around the Mn–OH unit. Toward this goal, tripodal ligand [H2pout]3- was designed to accommodate three intramolecular H-bond to the hydroxido ligand. Spectroscopic and structural results on [MnIIH2pout(OH)]2- and [MnIIIH2pout(OH)]– indicate that this type of H-bonding network was achieved and the O-atom of the phosphinic amide arm can serve as a H-bond acceptor. In addition, our electrochemical studies demonstrate that [MnIIIH2pout(OH)]– can be converted to its MnIV– OH analog which was achieved using the mild oxidant ferrocenium. The formation of [MnIVH2pout(OH)] was monitored spectrophotometrically and further characterized using variable frequency EPR spectroscopy that showed a high-spin MnIV species is readily formed. These results illustrate the utility of this ligand design in forming mononuclear metal-hydroxido complexes that span a variety of different oxidation states.
Supplementary Material
Synopsis.
A new hybrid tripodal ligand led to a series of mononuclear Mn–OH complexes. The ligand framework was tailor to promote three intramolecular hydrogen bonds to the hydroxido ligand: two utilize urea groups as hydrogen bond donors and the third is a phosphinic amide that is a hydrogen bond acceptor. The step-wise oxidation from MnII–OH to MnIV–OH was achieved through control of the primary and secondary coordination spheres.
ACKNOWLEDGMENT
We thank the National Institutes of Health USA (GM050781 to ASB and GM077387 to MPH) for financial support
Footnotes
Supporting Information.
The details for the crystal data (Table S1).
Accession Codes CCDC 1849228–1849229 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interests.
REFERENCES
- (1).Shook RL; Borovik AS The Effects of Hydrogen Bonds on Metal-Mediated O 2 activation and Related Processes. Chem. Commun 2008, 46, 6095–6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).MacBeth CE; Hammes BS; Young VG; Borovik AS; Young VG; Borovik AS Hydrogen-Bonding Cavities about Metal Ions: Synthesis, Structure, and Physical Properties for a Series of Monomeric M–OH Complexes Derived from Water. Inorg. Chem 2001, 40, 4733–4741. [PubMed] [Google Scholar]
- (3).Borovik AS Bioinspired Hydrogen Bond Motifs in Ligand Design: The Role of Noncovalent Interactions in Metal Ion Mediated Activation of Dioxygen. Acc. Chem. Res 2005, 38, 54–61. [DOI] [PubMed] [Google Scholar]
- (4).Shook RL; Borovik AS Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg. Chem 2010, 49, 3646–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Cook SA; Hill EA; Borovik AS Lessons from Nature: A Bio-Inspired Approach to Molecular Design. Biochemistry 2015, 54, 4167–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cook SA; Borovik AS Molecular Designs for Controlling the Local Environments around Metal Ions. Acc. Chem. Res 2015, 48, 2407–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Szymczak NK; Oelkers AB; Tyler DR Detection of Hydrogen Bonding in Solution: A 2H Nuclear Magnetic Resonance Method Based on Rotational Motion of a Donor/Acceptor Complex. Phys. Chem. Chem. Phys 2006, 8, 4002–4008. [DOI] [PubMed] [Google Scholar]
- (8).Hartle MD; Delgado M; Gilbertson JD; Pluth MD Stabilization of a Zn(II) Hydrosulfido Complex Utilizing a Hydrogen-Bond Accepting Ligand. Chem. Commun 2016, 52, 7680–7682. [DOI] [PubMed] [Google Scholar]
- (9).Dahl EW; Dong HT; Szymczak NK Phenylamino Derivatives of Tris(2-Pyridylmethyl)Amine: Hydrogen-Bonded Peroxodicopper Complexes. Chem. Commun 2018, 54, 892–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lucas RL; Zart MK; Murkerjee J; Sorrell TN; Powell DR; Borovik AS A Modular Approach toward Regulating the Secondary Coordination Sphere of Metal Ions: Differential Dioxygen Activation Assisted by Intramolecular Hydrogen Bonds. J. Am. Chem. Soc 2006, 128, 15476–15489. [DOI] [PubMed] [Google Scholar]
- (11).Kendall AJ; Zakharov LN; Gilbertson JD Synthesis and Stabilization of a Monomeric Iron(II) Hydroxo Complex via Intra Molecular Hydrogen Bonding in the Secondary Coordination Sphere. Inorg. Chem 2010, 49, 8656–8658. [DOI] [PubMed] [Google Scholar]
- (12).Dauth A; Love JA Synthesis and Reactivity of 2-Azametallacyclobutanes. Dalton Trans 2012, 41, 7782–7791. [DOI] [PubMed] [Google Scholar]
- (13).Lacy DC; Mukherjee J; Lucas RL; Day VW; Borovik AS Metal Complexes with Varying Intramolecular Hydrogen Bonding Networks. Polyhedron 2013, 52, 261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Moore CM; Szymczak NK 6,6′-Dihydroxy Terpyridine: A Proton-Responsive Bifunctional Ligand and Its Application in Catalytic Transfer Hydrogenation of Ketones. Chem. Commun 2013, 49, 400–402. [DOI] [PubMed] [Google Scholar]
- (15).Matson EM; Bertke JA; Fout AR Isolation of Iron(II) Aqua and Hydroxyl Complexes Featuring a Tripodal H-Bond Donor and Acceptor Ligand. Inorg. Chem 2014, 53, 4450–4458. [DOI] [PubMed] [Google Scholar]
- (16).Matson EM; Park YJ; Fout AR Facile Nitrite Reduction in a Non-Heme Iron System: Formation of an Iron(III)-Oxo. J. Am. Chem. Soc 2014, 136, 17398–17401. [DOI] [PubMed] [Google Scholar]
- (17).Li X; Siegbahn PEM Alternative Mechanisms for O2 Release and O–O Bond Formation in the Oxygen Evolving Complex of Photosystem II. Phys. Chem. Chem. Phys 2015, 17, 12168–12174. [DOI] [PubMed] [Google Scholar]
- (18).Jones JR; Ziller JW; Borovik AS Modulating the Primary and Secondary Coordination Spheres within a Series of CoII-OH Complexes. Inorg. Chem 2017, 56, 1112–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Matson EM; Bertke JA; Fout AR Isolation of Iron(II) Aqua and Hydroxyl Complexes Featuring a Tripodal H-Bond Donor and Acceptor Ligand. Inorg. Chem 2014, 53, 4450–4458. [DOI] [PubMed] [Google Scholar]
- (20).Park YJ; Matson EM; Nilges MJ; Fout AR Exploring Mn-O Bonding in the Context of an Electronically Flexible Secondary Coordination Sphere: Synthesis of a Mn(III)-Oxo. Chem. Commun 2015, 51, 5310–5313. [DOI] [PubMed] [Google Scholar]
- (21).Matson EM; Park YJ; Fout AR Facile Nitrite Reduction in a Non-Heme Iron System: Formation of an Iron(III)-Oxo. J. Am. Chem. Soc 2014, 136, 17398–17401. [DOI] [PubMed] [Google Scholar]
- (22).Ray M; Golombek AP; Hendrich MP; Young VG; Borovik AS Synthesis and Structure of a Trigonal Monopyramidal Fe(II) Complex and Its Paramagnetic Carbon Monoxide Derivative. J. Am. Chem. Soc 1996, 118, 6084–6085. [Google Scholar]
- (23).Hammes BS; Ramos-Maldonado D; Yap GPA; Liable-Sands L; Rheingold AL; Young VG; Borovik AS C3-Symmetric Chiral Amidate Complexes: Effects of Ligand Binding on Cavity Structure. Inorg. Chem 1997, 36, 3210–3211. [DOI] [PubMed] [Google Scholar]
- (24).Hammes BS; Young VG Jr.; Borovik AS Hydrogen-Bonding Cavities about Metal Ions: A Redox Pair of Coordinatively Unsaturated Paramagnetic Co-OH Complexes. Angew. Chem. Int. Ed 1999, 38, 666–669. [DOI] [PubMed] [Google Scholar]
- (25).Shirin Z; Hammes BS; Young VG; Borovik AS Hydrogen Bonding in Metal Oxo Complexes: Synthesis and Structure of a Monomeric Manganese(III)−Oxo Complex and Its Hydroxo Analogue. J. Am. Chem. Soc 2000, 122, 1836–1837. [Google Scholar]
- (26).Gupta R; MacBeth CE; Young VGJ; Borovik AS Isolation of Monomeric MnIII/II-OH and MnIII-O Complexes from Water: Evaluation of O-H Bond Dissociation Energies. J. Am. Chem. Soc 2002, 124, 1136–1137. [DOI] [PubMed] [Google Scholar]
- (27).Gupta R; Borovik AS Monomeric MnIII/II and FeIII/II Complexes with Terminal Hydroxo and Oxo Ligands: Probing Reactivity via O–H Bond Dissociation Energies. J. Am. Chem. Soc 2003, 125, 13234–13242. [DOI] [PubMed] [Google Scholar]
- (28).Gupta R; Taguchi T; Lassalle-Kaiser B; Bominaar EL; Yano J; Hendrich MP; Borovik AS High-Spin Mn–Oxo Complexes and Their Relevance to the Oxygen-Evolving Complex within Photosystem II. Proc. Natl. Acad. Sci 2015, 112, 5319–5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Taguchi T; Gupta R; Lassalle-Kaiser B; Boyce DW; Yachandra VK; Tolman WB; Yano J; Hendrich MP; Borovik AS Preparation and Properties of a Monomeric High-Spin MnV–Oxo Complex. J. Am. Chem. Soc 2012, 134, 1996–1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Taguchi T; Stone KL; Gupta R; Kaiser-Lassalle B; Yano J; Hendrich MP; Borovik AS Preparation and Properties of an MnIV–Hydroxide Complex: Proton and Electron Transfer at a Mononuclear Manganese Site and Its Relationship to the Oxygen Evolving Complex within Photosystem II. Chem. Sci 2014, 5, 3064–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Park YJ; Ziller JW; Borovik AS The Effects of Redox-Inactive Metal Ions on the Activation of Dioxygen: Isolation and Characterization of a Heterobimetallic Complex Containing a MnIII–(μ-OH)–CaII Core. J. Am. Chem. Soc 2011, 133, 9258–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lacy DC; Park YJ; Ziller JW; Yano J; Borovik AS Assembly and Properties of Heterobimetallic CoII/III/CaII complexes with Aquo and Hydroxo Ligands. J. Am. Chem. Soc 2012, 134, 17526–17535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Park YJ; Cook SA; Sickerman NS; Sano Y; Ziller JW; Borovik AS Heterobimetallic Complexes with MIII -(μ-OH)-MII Cores (MIII = Fe, Mn, Ga; MII = Ca, Sr, and Ba): Structural, Kinetic, and Redox Properties. Chem. Sci 2013, 4, 717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sickerman NS; Peterson SM; Ziller JW; Borovik AS Synthesis, Structure and Reactivity of FeII/III–NH3 Complexes Bearing a Tripodal Sulfonamido Ligand. Chem. Commun 2014, 50, 2515–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Connelly NG; Geiger WE Chemical Redox Agents for Organometallic Chemistry. Chem. Rev 1996, 96, 877–910. [DOI] [PubMed] [Google Scholar]
- (36).Petasis DT; Hendrich MP Quantitative Interpretation of Multifrequency Multimode EPR Spectra of Metal Containing Proteins, Enzymes, and Biomimetic Complexes. Methods Enzymol 2015, 563, 171–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).See SI for full reference
- (38).Bruker AXS Inc, APEX2 Version 2014.11–0. Madison, WI: 2014. [Google Scholar]
- (39).Bruker AXS Inc, SA INT Version 8.34a. Madison, WI: 2013. [Google Scholar]
- (40).Sheldrick GM SADABS. Bruker AXS, Inc: Madison: 2014. [Google Scholar]
- (41).MacBeth CE; Gupta R; Mitchell-Koch KR; Young VG; Lushington GH; Thompson WH; Hendrich MP; Borovik AS Utilization of Hydrogen Bonds To Stabilize M−O(H) Units: Synthesis and Properties of Monomeric Iron and Manganese Complexes with Terminal Oxo and Hydroxo Ligands. J. Am. Chem. Soc 2004, 126, 2556–2567. [DOI] [PubMed] [Google Scholar]
- (42).Parsell TH; Behan RK; Green MT; Hendrich MP; Borovik AS Preparation and Properties of a Monomeric MnIV-Oxo Complex. J. Am. Chem. Soc 2006, 128, 8728–8729. [DOI] [PubMed] [Google Scholar]
- (43).Shirin Z; Young VG; Borovik AS Synthesis and Structure of a MnIII (OH) Complex Generated from Dioxygen Dioxygen Activation at Room Temperature by a New MnII Complex Having a Cavity Motif Yields a Monomeric MnIII Complex with a Terminally Bonded Hydroxo Ligand Whose Oxygen Atom Is. 1997, 36, 1967–1968. [Google Scholar]
- (44).Gupta R; Borovik AS Monomeric MnIII/II and FeIII/II Complexes with Terminal Hydroxo and Oxo Ligands: Probing Reactivity via O−H Bond Dissociation Energies. J. Am. Chem. Soc 2003, 125, 13234–13242. [DOI] [PubMed] [Google Scholar]
- (45).Gupta R; Taguchi T; Borovik AS; Hendrich MP Characterization of Monomeric Mn(II/III/IV)-Hydroxo Complexes from X- and Q-Band Dual Mode Electron Paramagnetic Resonance (EPR) Spectroscopy. Inorg. Chem 2013, 52, 12568–12575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hill EA; Weitz AC; Onderko E; Romero-Rivera A; Guo Y; Swart M; Bominaar EL; Green MT; Hendrich MP; Lacy DC; et al. Reactivity of an FeIV Oxo Complex with Protons and Oxidants. J. Am. Chem. Soc 2016, 138, 13143–13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Halbach RL; Gygi D; Bloch ED; Anderson BL; Nocera DG Structurally Characterized Terminal Manganese(IV)Oxo Tris(Alkoxide) Complex. Chem. Sci 2018, 9, 4524–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Leto DF; Jackson TA Mn K-Edge X-Ray Absorption Studies of Oxo- and Hydroxo-Manganese(IV) Complexes: Experimental and Theoretical Insights into Pre-Edge Properties. Inorg. Chem 2014, 53, 6179–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Leto DF; Massie AA; Colmer HE; Jackson TA X-Band Electron Paramagnetic Resonance Comparison of Mononuclear MnIV-Oxo and MnIV-Hydroxo Complexes and Quantum Chemical Investigation of MnIV Zero-Field Splitting. Inorg. Chem 2016, 55, 3272–3282. [DOI] [PubMed] [Google Scholar]
- (50).Rice DB; Massie AA; Jackson TA Manganese-Oxygen Intermediates in OO Bond Activation and Hydrogen-Atom Transfer Reactions. Acc. Chem. Res 2017, 50, 2706–2717. [DOI] [PubMed] [Google Scholar]
- (51).Wijeratne GB; Corzine B; Day VW; Jackson TA Saturation Kinetics in Phenolic O-H Bond Oxidation by a Mononuclear Mn(III)-OH Complex Derived from Dioxygen. Inorg. Chem 2014, 53, 7622–7634. [DOI] [PubMed] [Google Scholar]
- (52).Coggins MK; Brines LM; Kovacs JA Synthesis and Structural Characterization of a Series of MnIIIOR Complexes, Including a Water-Soluble MnIIIOH That Promotes Aerobic Hydrogen-Atom Transfer. Inorg. Chem 2013, 52, 12383–12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Pal S; Armstrong WH Products from Reactions of Manganese Oxo Complex [Mn2O2(O2CCH3)(Tpen)]2+ in Acidic and Neutral Aqueous Media: [Mn2(μ-O)2(μ-O2CCH3)(Tpen)]3+ and [{Mn3(μ-O)4(OH)(Tpen)}2(μ-Tpen)]6+. Inorg. Chem 1992, 31, 5417–5423. [Google Scholar]
- (54).Goldsmith CR; Cole AP; Stack TDP C-H Activation by a Mononuclear Manganese(III) Hydroxide Complex: Synthesis and Characterization of a Manganese-Lipoxygenase Mimic J. Am. Chem. Soc 2005, 127, 9904–9912. [DOI] [PubMed] [Google Scholar]
- (55).Sproviero EM; Gascon JA; McEvoy JP; Brudvig GW; Batista VS Computational Studies of the O2-Evolving Complex of Photosystem II and Biomimetic Oxomanganese Complexes. Coord. Chem. Rev 2008, 252, 395–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Siegbahn PEM Mechanisms for Proton Release during Water Oxidation in the S2 to S3 and S3 to S4 Transitions in Photosystem II. Phys. Chem. Chem. Phys 2012, 14, 4849. [DOI] [PubMed] [Google Scholar]
- (57).Siegbahn PEM A Structure-Consistent Mechanism for Dioxygen Formation in Photosystem II. Chem. Eur. J 2008, 14, 8290–8302. [DOI] [PubMed] [Google Scholar]
- (58).Rapatskiy L; Cox N; Savitsky A; Ames WM; Sander J; Nowaczyk MM; Rögner M; Boussac A; Neese F; Messinger J; et al. Detection of the Water-Binding Sites of the Oxygen-Evolving Complex of Photosystem II Using W-Band17O Electron-Electron Double Resonance-Detected NMR Spectroscopy. J. Am. Chem. Soc 2012, 134, 16619–16634. [DOI] [PubMed] [Google Scholar]
- (59).Meyer TJ; Huynh MHV; Thorp HH The Possible Role of Proton-Coupled Electron Transfer (PCET) in Water Oxidation by Photosystem II. Angew. Chem. Int. Ed 2007, 46, 5284–5304. [DOI] [PubMed] [Google Scholar]
- (60).Suga M; Akita F; Hirata K; Ueno G; Murakami H; Nakajima Y; Shimizu T; Yamashita K; Yamamoto M; Ago H; et al. Native Structure of Photosystem II at 1.95 Å Resolution Viewed by Femtosecond X-Ray Pulses. Nature 2014, 517, 1–17. [DOI] [PubMed] [Google Scholar]
- (61).Pantazis DA; Ames W; Cox N; Lubitz W; Neese F Two Interconvertible Structures That Explain the Spectroscopic Properties of the Oxygen Evolving Complex of Photosystem II in the S2 State. Angew. Chem., Int. Ed. Engl 2012, 51, 9935–9940. [DOI] [PubMed] [Google Scholar]
- (62).Umena Y; Kawakami K; Shen J-R; Kamiya N Crystal Structure of Oxygen-Evolving Photosystem II at a Resolution of 1.9 Å. Nature 2011, 473, 55–60. [DOI] [PubMed] [Google Scholar]
- (63).Siegbahn PEM Structures and Energetics for O2 Formation in Photosystem II. Acc. Chem. Res 2009, 42, 1871–1880. [DOI] [PubMed] [Google Scholar]
- (64).Shoji M; Isobe H; Nakajima T; Shigeta Y; Suga M; Akita F; Shen J-R; Yamaguchi K Large-Scale QM/MM Calculations of the CaMn4O5 Cluster in the S3 State of the Oxygen Evolving Complex of Photosystem II. Comparison between Water-Inserted and No Water-Inserted Structures. Faraday Discuss 2017, 198, 83–106. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.