

Abstract
Collapsing focal segmental glomerulosclerosis (cFSGS) in the native kidney is associated with heavy proteinuria and accelerated renal failure. However, cFSGS in the renal allograft is less well characterized. Here we report clinico-pathologic features and APOL1 donor risk genotypes in 38 patients with de novo post-kidney transplant cFSGS. Recipients were 34% female and 26% African-American. Concurrent viral infections and acute vaso-occlusion (including thrombotic microangiopathy, cortical necrosis, atheroembolization, and cardiac arrest with contralateral graft thrombosis) were present in 13% and 29% of recipients, respectively. Notably, 61% of patients had concurrent acute rejection and 47% received grafts from African-American donors, of which 53% carried APOL1 high-risk genotypes. These frequencies of acute rejection and grafts from African-American donors were significantly higher than in our general transplant population (35% and 16%, respectively). Patients had a median serum creatinine of 5.4mg/dl, urine protein/creatinine 3.5 g/g, and 18% had nephrotic syndrome. Graft failure occurred in 63% of patients at an average of eighteen months post-index biopsy. By univariate analysis, donor APOL1 high-risk genotypes, post-transplant time, nephrotic syndrome, and chronic histologic changes were associated with inferior graft survival while acute vaso-occlusion was associated with superior graft survival. Donor APOL1 high-risk genotypes independently predicted poor outcome. Compared to native kidney cFSGS, post-transplant cFSGS had more acute vaso-occlusion but less proteinuria. Thus, de novo cFSGS is associated with variable proteinuria and poor prognosis with potential predisposing factors of African-American donor, acute rejection, viral infection and acute vaso-occlusion. Additionally, donor APOL1 high risk genotypes are associated with higher incidence and worse graft survival.
Keywords: Collapsing glomerulopathy, Acute rejection, Acute vaso-occlusion, African-Americans, APOL1 genotypes, Kidney transplantation
Introduction
Collapsing focal segmental glomerulosclerosis (cFSGS), also called collapsing glomerulopathy, is defined by the presence of at least one glomerulus with segmental or global wrinkling and retraction of the glomerular basement membranes accompanied by hypertrophy and hyperplasia of overlying glomerular epithelial cells1. cFSGS is typically characterized by massive proteinuria, elevated serum creatinine, and rapid progression to end-stage renal disease (ESRD)2–5. While described as idiopathic in 39 to 77% of reported cases6, 7, cFSGS has recognized associations with viral infections [mainly HIV, and to a lesser extent parvovirus, cytomegalovirus (CMV), and Epstein-Barr virus (EBV)], medications [e.g., interferon (IFN) and pamidronate], vaso-occlusive disease [e.g. thrombotic microangiopathy, cortical necrosis and athero-embolization], hemophagocytic syndrome, and collagen vascular diseases8–13. Genetically, cFSGS has a strong association with APOL1 high-risk genotypes in African-Americans (AA)14and a probable association with HLA-B44 or HLA-DR4 in Caucasians2. IFN appears to be an important mediator, as supported by association of cFSGS with IFN treatment12 and conditions that promote IFN production such as intercurrent viral infections, collagen vascular diseases, and hemophagocytic syndrome15–17. Moreover, IFN has been shown to upregulate the expression and enhance the effects of APOL1 in podocytes and endothelial cells18. Oxidative stress also appears to play a pathogenetic role3, as evidenced by the association of acute vaso-occlusion with cFSGS9, 19. However, the latter pathway may not be completely independent from IFN given the causative association between IFN and several types of vaso-occlusive disease, including thrombotic microangiopathy20, 21.
In the kidney transplantation (KTx) setting, cFSGS can manifest as de novo or recurrent disease. Several studies have attempted to investigate post-KTx cFSGS (Supplemental Table 1). Most were small22–27 and the largest combined 20 cFSGS cases with other forms of post-KTx FSGS, where patients with cFSGS were not separately evaluated28. Although some studies suggested that post-KTx cFSGS may be different from cFSGS in the native kidney24–26, none directly compared demographic, clinical and pathologic parameters between these entities. This manuscript reports the natural history, presenting features, donor APOL1 genotypes and prognosis of de novo post-KTx cFSGS in 38 cases diagnosed at Columbia University Medical Center (CUMC). These cases are contrasted with presenting clinical and biopsy findings in 255 cases of cFSGS in the native kidney.
Results
Demographic, clinical, and pathologic features:
Between 2005 and 2017, 38 adult kidney allograft recipients (18 years or older) had one or more renal biopsies with de novo cFSGS (Figure 1A). Recipients had a median age of 57 years and included 34% women and 63% recipients of deceased donor allografts (Table 1). This cohort included AA (n=10, 26%), Caucasians (n=13, 34%), Hispanics (n=10, 27%), and others (n=5, 13%). Notably, 18 (47%) of the recipients received an allograft from a total of 17 AA donors. Remaining grafts were received from Caucasian (n=16, 42%) or Hispanic (n=4, 11%) donors. The presence of either HLAB44 or HLADR4 serotypes were noted in 24% of Caucasian recipients and 31% of Caucasian donors. Thirty-one donors were successfully genotyped for APOL1.Of the donors that could not be typed (n=8), three Caucasians were assumed to have APOL1 low-risk genotypes given the rare incidence of APOL1 high-risk genotypes in this population29. Altogether, APOL1 data were analyzed in grafts of 34/38 kidney allograft recipients, of which 9 had high-risk APOL1 genotypes (8 AA and 1 Hispanic). Therefore, of the 18 grafts from AA donors, 15 (83%) were successfully genotyped and 53% (8 of 15) had APOL1 high-risk genotypes (Table 1).
Figure 1: Representative photo-micrographs of post-KTx cFSGS and accompanying histologic lesions.
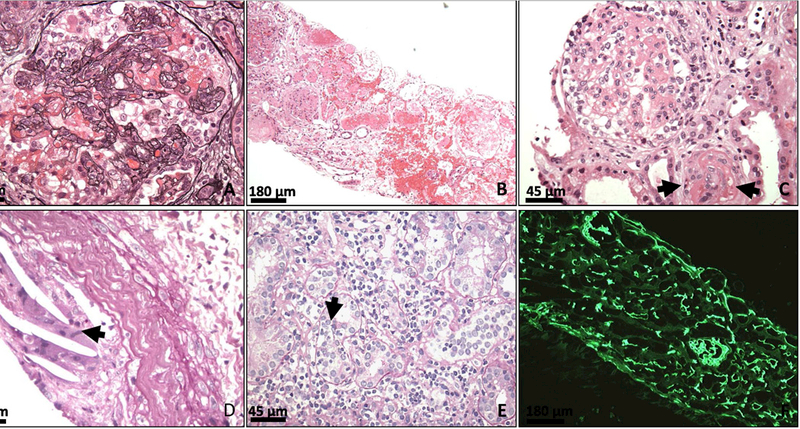
(A) A glomerulus showing features of cFSGS in an allograft biopsy. Note the glomerular tuft collapse and the prominence of epithelial cells (Jones’ Methenamine Silver stain; original magnification x600) (B) An allograft with cortical necrosis involving most of the low power view image and ~25% of total sampled cortex (hematoxylin and eosin, original magnification x100) (C) An allograft with thrombotic microangiopathy. The arteriole displays near occlusion of its lumen and shows segmental fibrinoid necrosis (arrows). cFSGS is seen in the adjacent glomerulus (hematoxylin and eosin stain, original magnification x400) (D) An allograft with arterial athero-emboli manifested as empty needle-shape spaces (because of the dissolving of cholesterol during preparation) that are surrounded by giant cells (arrow) (periodic acid–Schiff stain, original magnification x400) (E) An allograft with grade 1B acute T cell ediated rejection. Note the interstitial inflammation and severe tubulitis (arrow) (periodic acid–Schiff stain, original magnification x400). (F) An allograft with antibody-mediated rejection showing diffuse C4d staining in peritubular and glomerular capillaries (immunofluorescence C4d staining, original magnification x100).
Table 1:
Demographic and clinical characteristics of post-KTx cFSGS
| Characteristics | n=38 |
|---|---|
| Recipients age (years) | 57 (47, 65) |
| Recipients gender (female) | 13/38 (34%) |
| Recipients race (AA) | 10/38 (26%) |
| Recipient HLA (B44 or DR4) | 11/38 (29%) [3/13; 24% of Caucasians] |
| Allograft source (deceased donors) | 24/38 (63%) |
| Allograft from AA donor | 18/38 (47%) |
| Allograft from donor with APOL1 high-risk genotypes1 | 9/34 (26%) [8/15; 53% of AA] |
| HLA-B44 or HLA-DR4 in donors | 11/37 (30%) [5/16; 31% of Caucasians] |
| Etiology of ESRD - Hypertension - Diabetes - Glomerulonephritis2 - Polycystic kidney disease - Calcineurin inhibitor toxicity - Others - Unknown |
11/38 (29%) 7/38 (18%) 6/38 (16%) 4/38 (11%) 3/38 (8%) 4/38 (10%) 3/38 (8%) |
| Post-transplant interval to biopsy (days) | 231 (14, 1000) [21/38 (55%) within the 1st year post-transplantation] |
| Maintenance immunosuppression 3 - Calcineurin inhibitor - Mycophenolate mofetil - Corticosteroids - Sirolimus - Belatacept - Azathioprine |
36/38 (95%) 31/38 (82%) 16/38 (42%) 3/38 (8%) 2/38 (5%) 1/38 (3%) |
| Serum creatinine at biopsy (mg/dL) | 5.4 (3.4, 6.8) |
| Urine protein/creatinine at biopsy (g/g) | 3.5 (1.3, 6.5) |
| Serum albumin at biopsy (g/dL) | 3.2 (2.8, 3.7) |
| Nephrotic range proteinuria | 19/37 (51%) (NA in 1) |
| Nephrotic syndrome | 7/38 (18%) |
| Circulating donor-specific antibody4 | 8/26 (31%) |
| Traditional causes of collapsing FSGS5 - Viral infection - Acute vaso-occlusive disease |
15/38 (39%) 5/38 (13%) 11/38 (29%) |
- Abbreviations: AA; African American; ESRD, end stage renal disease
Data on APOL1 genotypes was not available for 7 donors; Given the extremely low positivity in Caucasians without kidney disease, APOL1 risk genotypes were considered absent in 3 Caucasian donors not genotyped
Glomerulonephritis included lupus nephritis (n=2), IgA nephropathy (n=2), ANCA-associated (n=1), and anti-glomerular basement membrane glomerulonephritis (n=1)
Calcineurin inhibitors include tacrolimus (n=33) and cyclosporine (n=3). Although our protocol is steroids-free, the high proportion of patients on corticosteroids may be attributed to early biopsies (patients still on taper corticosteroids) or to biopsies from patients with previous immunologic complication(s) (e.g. prior rejection) that necessitate re-initiation of corticosteroids. A few patients were maintained on sirolimus or Belatacept with concurrent low dose of calcineurin inhibitors
Data on circulating donor-specific antibody within 2 months of biopsy was available for 26 patients
Viral infections were assessed by PCR in the plasma. One patient had both EBV infection and cortical necrosis
The most common reported cause of ESRD was hypertension (29%) followed by diabetes (18%) and glomerulonephritis (16%) (Table 1). Biopsy samples were obtained a median of 231 [Interquartile (IQR): 14, 1000] days after transplantation. At the time of index biopsy, 95% of patients were maintained on calcineurin inhibitors and 82% on mycophenolate mofetil. Furthermore, 42% of patients were receiving corticosteroids and 8% sirolimus. At biopsy, the median serum creatinine was 5.4 mg/dl (IQR: 3.4, 6.8). Notably, 5 patients were still experiencing delayed graft function at the time of biopsy. Median urine protein/creatinine was 3.5 g/g (IQR: 1.3, 6.5) while full nephrotic syndrome was present in only 18% of patients. Traditional causes of cFSGS were noted in <40% of patients and included active viral infections [CMV (n=3), Parvovirus (n=1) and EBV (n=1)] and/or acute vaso-occlusive disease (n=11, of whom 1 had EBV infection) (Table 1).
Acute vaso-occlusion occurred at a median of 12 (IQR: 9–325) days post-transplantation and none of these 11 patients had received a graft from a donor with APOL1 high-risk genotypes. All but one (10/11; 91%) of these cases were diagnosed solely by allograft biopsy, which showed cortical necrosis (n=6), active thrombotic microangiopathy (n=2), and athero-embolization (n=2) (Figure 1B-D). In the remaining case, the diagnosis was rendered based on a combination of clinical, radiology and biopsy findings. Directly after transplantation, this patient who received a dual kidney transplant, experienced a respiratory and cardiac arrest lasting 5 minutes. Despite successful resuscitation, renal ultrasound with Doppler revealed no perfusion to the left allograft that failed and was removed, while a subsequent biopsy of the surviving allograft performed 9 days post-transplantation revealed cFSGS and acute tubular necrosis that were not evident on the post-reperfusion biopsy.
Detailed pathologic characteristics are presented in Table 2. Concurrent acute rejection was present in 23 (61%) patients and was characterized as borderline changes (n=7), overt acute T cell mediated rejection (n=9, Figure 1E), antibody-mediated rejection (n=2, Figure 1F), or mixed rejection (n=5). A median of 14% of sampled glomeruli had collapsing lesions and tubular microcysts were identified in 13% of biopsies. Median arteriolar hyalinosis score was 1 and only 4 biopsies demonstrated well developed “beaded” hyalinosis. The 8 allograft biopsies assessed by electron microscopy showed variable foot process effacement (median: 70%, range: 15– 100%)
Table 2:
Pathologic characteristics of post-KTx cFSGS
| Concurrent acute rejection - Mixed rejection - AMR - TCMR ࿀ Borderline ࿀ 1A ࿀ 1B ࿀ 2A |
23/38 (61%) 5/38 (13%) [TCMR: BL: 4, 2A: 1] 2/38 (5%) 16/38 (42%) 7/16 5/16 1/16 3/16 |
| Glomeruli (#) | 15 (12, 20) |
| Arteries (#) | 2 (2, 3) |
| Global glomerulosclerosis (%) | 11 (0, 25) |
| Collapsed glomeruli (%) | 14 (8, 19) |
| Non-collapsed segmental sclerosis (%) | 0 (0, 10) |
| Interstitial inflammation (i: 0–1) | 1 (0, 1) [0.9 ±0.9] |
| Tubulitis (t: 0–3) | 1 (0, 1) [0.8 ±0.9] |
| Intimal arteritis (v: 0–3) | 0 (0, 0) [0.1 ±0.3] |
| Peritubular capillaritis (ptc: 0–3) | 0 (0, 1) [0.6 ±1.0] |
| Glomerulitis (g: 0–3) | 0 (0, 0) [0.2 ±0.6] |
| Transplant glomerulopathy (cg: 0–3) | 0 (0, 0) [0.1 ±0.4] |
| Interstitial fibrosis (ci: 0–3) | 1 (0.3, 2) [1.4 ±1.1] |
| Tubular atrophy (ct: 0–3) | 1 (1, 2) [1.4 ±1.1] |
| Arteriosclerosis (0–3) | 1 (1, 2) [1.4 ±0.9] |
| Arteriolosclerosis (0–3) | 1 (1, 2) [1.3 ±0.9] |
| C4d staining in peritubular capillaries (ptc0–3) | 0 (0, 0) [0.3 ±0.9] |
| Protein droplets in glomerular epithelial cells | 31/38 (82%) |
| Prominent protein droplets in tubules | 16/38 (42%) |
| Tubular microcysts | 5/38 (13%) |
| Foot process effacement (%) 1 - Focal - Extensive - Diffuse |
70 (48, 93) 2/8 (25%) 3/8 (37.5%) 3/8 (37.5%) |
- Abbreviations: AMR, antibody-mediated rejection; TCMR, acute T cell mediated rejection Semi-quantitative values were presented as median (IQR1, IQR3) and mean ±SD
Evaluation of foot process effacement was available only for 8 biopsies.
Comorbidities/Risk factors
The cohort with de novo post-KTx cFSGS had a high percentage of acute rejection episodes and allografts from AA donors. To explore the potential that these were significant contributors to the development of post-KTx cFSGS, demographic information and acute rejection episodes were compared with a historic cohort of all patients who underwent renal transplantation at CUMC between 2006 and 2009 (n=773). As shown in Figure 2 and Supplemental Table 2, patients with de novo post-KTx cFSGS were more likely to have received allografts from an AA donor and had a much higher frequency of acute rejection episodes (the frequency of concurrent rejection episodes in de novo cFSGS was higher than that observed in general transplant controls during 59±32 months of post-transplant follow-up). In contrast, only 3 (8%) of de novo post-KTx cFSGS were maintained on sirolimus. This was comparable to the ~5% of our kidney transplant population.
Figure 2: Comparison of demographic data in cases with post-KTx cFSGS and the general transplant population at Columbia University Medical Center.
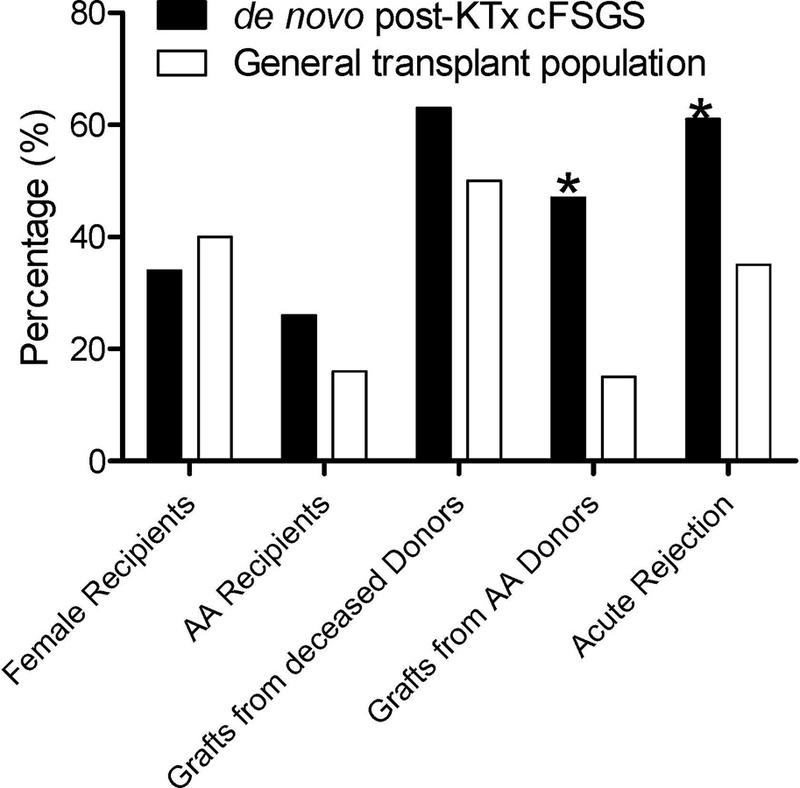
Demographic information were compared between our cohort and all patients who underwent kidney transplantation at CUMC between 2006 and 2009 including crossmatch positive and ABO-incompatible patients (n=773). Acute rejection in general transplant cohort was calculated during a follow-up of 59 ±32 months from transplantation while it was only considered at that time of index biopsy in the de novo post-KTx cFSGS cohort. (* P<0.05; for more detailed analysis, please refer to Supplemental Table 1).
Treatment and outcome
Treatment was variable and aimed often at concurrent conditions such as rejection or viral infection. Seventeen patients (45%) were treated conservatively while the remaining 21 received one or more of the following: corticosteroids, IVIG, plasmapheresis, lymphocyte depletion therapy, valganciclovir, or conversion from one maintenance immunosuppression regimen to another (Table 3). Data on short-term outcome (within 100 days post-biopsy) was available in 32 patients; none had complete remission and 34% had partial remission. Median follow-up after index biopsy was 16 (IQR: 5, 54) months. Allograft failure developed in 24/38 (63%) patients at a median of 8 months after index biopsies (IQR: 2, 22 months) (Figure 3A). Patients with partial remission had longer allograft survival compared to those with persistent renal dysfunction (Figure 3B). Follow-up biopsies were available in 25 patients and did not demonstrate lesions of cFSGS in 15. Notably, 13/15 (87%) of the latter patients had one or more of potentially reversible risk factors, including acute vaso-occlusion (n=8), acute rejection (n=8), and viral infection (n=2). Remarkably, 11/15 (73%) of the patients without cFSGS on follow-up biopsies maintained graft function until the end of follow-up [57 (IQR: 19, 66) months post-index biopsy] with a median last serum creatinine of 1.8 (IQR: 1.6, 3) mg/dL. Allograft survival in the latter subgroup was superior to that in patients with persistent cFSGS on follow-up biopsies (Figure 3C).
Table 3:
Treatment and clinico-pathologic follow-up of post-KTx cFSGS
| Concurrent conditions | Treatments |
|---|---|
| Mixed rejection (n=5) | 3/5: Plasmapheresis with IVIG (n=2) or thymoglobulin T cell depletion (n=1) 2/5: Conservative treatment1 |
| AMR (n=2) | 1/2: Plasmapheresis and conversion from cyclosporine to tacrolimus 1/2: IVIG and conversion from tacrolimus to Belatacept |
| TCMR (n=16) | 1/16: OKT3 T cell depletion and steroids 8/16: Steroids with IVIG (n=2), plasmapheresis (n=1 for diffuse FPE) or conversion from maintenance IS to another (n=3)2 1/16: Valganciclovir (for concurrent viral infection) 1/16: Plasmapheresis only (for diffuse FPE; patient was recently treated for previous acute rejection) 1/16: IVIG only 4/16: Conservative treatment (1 with concurrent viral infection)3 |
| Viral infection without acute rejection (n=3) |
2/3: IVIG with Valganciclovir (n=1) or steroids (n=1) 1/3: Conservative treatment |
| Others (n=12) | 10/12 Conservative treatment 1/12 Steroids 1/12 Conversion from tacrolimus to Belatacept |
Abbreviations: AMR, antibody-mediated rejection; FPE, foot process effacement; IS, immunosuppression; IVIG, intravenous immunoglobulin; TCMR, T cell mediated rejection
Conservative treatment in mixed rejection was given to 2 patients: Both had their TCMR characterized as “borderline changes”. With regard to AMR, the 1st had an ABO-incompatible graft and just completed peri-transplant desensitization treatment before the biopsy. The 2nd had C4d-negative AMR. At the time of biopsy, this category was not correctly as an AMR by Banff criteria
Conversion IS in TCMR included rapamycin to tacrolimus (n=1), rapamycin to lefunomide (n=1) and tacrolimus to Belatacept (n=1)
Conservative treatment in TCMR (n=4): all had borderline changes of whom 1 had a concurrent viral infection, 2 had been recently treated for previous acute rejection, and 1 had severe chronic changes
Figure 3: Graft survival in post-KTx cFSGS.
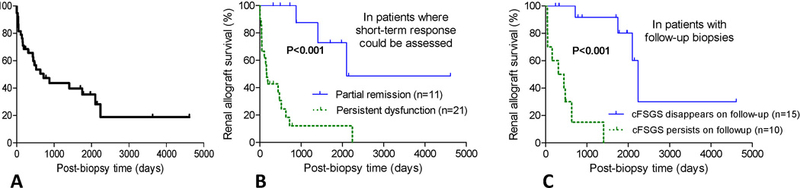
(A) Kaplan–Meier curve for post-biopsy cumulative kidney allograft survival in patients with de novo post-KTx cFSGS (n=38) (B) Curves for patients who had short term partial response vs. these who had persistent dysfunction (P<0.001) (C) Curves for patients who had absence or persistence of cFSGS on follow-up biopsies are also shown (P<0.001).
Prognostic indicators
Given the poor prognosis of de novo post-KTx cFSGS, variables associated with allograft outcome were analyzed. By univariate Cox regression, acute vaso-occlusive disease was significantly associated with superior allograft survival. In contrast, donor high-risk APOL1 genotypes, post-transplant interval to biopsy, nephrotic syndrome, percentage of global glomerulosclerosis, and histologic scores of interstitial fibrosis/tubular atrophy, arteriosclerosis, and arteriolar hyalinosis were all significantly associated with shorter allograft survival (Table 4). Using Cox multivariable analysis that included the 8 significant variables in univariate analyses, both acute vaso-occlusive disease and donor APOL1 high-risk genotypes were independently associated with allograft outcome (Table 4). To avoid potential overfitting in multivariable analysis, we also used a step-up method that would only include the variables that were significant. The latter analysis kept APOL1 high-risk genotypes [HR: 6.6 (1.9 – 23), P=0.003) and interstitial fibrosis scores [HR: 3.1 (1.8 – 5.3), P<C0.001) as the only significant predictors while acute vaso-occlusion did not appear in the stepwise model. Together, findings confirmed the prognostic importance of APOL1 high-risk genotypes in post-KTx cFSGS.
Table 4:
Univariate and multivariable analyses of the association of different variables with graft survival
| Variables | Univariate | Multivariate (n=34) * | ||
|---|---|---|---|---|
| HR (95% CI) | P value | HR (95% CI) | P value | |
| Recipient age | 1.0 (0.98 – 1.05) | 0.35 | ||
| Recipient female gender | 1.6 (0.72 – 3.66) | 0.25 | ||
| Recipient AA race | 1.0 (0.41 – 2.64) | 0.94 | ||
| Recipient HLA (B44 or DR4) | 0.7 (0.26 – 1.65) | 0.37 | ||
| Allograft deceased donor | 0.8 (0.34 – 1.70) | 0.51 | ||
| Allograft from AA donor | 1.6 (0.68 – 3.56) | 0.29 | ||
| Donors HLA (B44 or DR4) | 1.4 (0.59 – 3.33) | 0.44 | ||
| Post-transplant interval to biopsy (days) | 1.0 (1.0 – 1.001) | 0.03 | 1.0 (1.0 – 1.001) | 0.19 |
| Serum creatinine at biopsy | 0.9 (0.77 – 1.10) | 0.38 | ||
| Urine protein/creatinine | 1.1 (0.97 – 1.16) | 0.21 | ||
| Serum albumin | 0.6 (0.27 – 1.28) | 0.18 | ||
| Nephrotic range proteinuria | 2.2 (0.93 – 5.26) | 0.07 | ||
| Nephrotic syndrome | 5.3 (1.92 – 14.8) | 0.01 | 3.5 (0.68 – 17.5) | 0.13 |
| Viral infection | 0.8 (0.18 – 3.32) | 0.72 | ||
| Acute vaso-occlusive disease | 0.2 (0.05 – 0.62) | 0.007 | 0.2 (0.04– 0.98) | 0.047 |
| Concurrent rejection1 | 1.8 (0.73 – 4.31) | 0.20 | ||
| Global glomerulosclerosis (%) | 1.0 (1.01 – 1.06) | 0.002 | 1.0 (0.93 – 1.01) | 0.15 |
| Collapsed glomeruli (%) | 1.0 (0.96 – 1.06) | 0.69 | ||
| Interstitial inflammation (i0–3) | 1.2 (0.78 – 1.89) | 0.39 | ||
| Tubulitis (t0–3) | 1.3 (0.83 – 1.97) | 0.27 | ||
| Intimal arteritis (v0–3) | 0.7 (0.22 – 2.52) | 0.64 | ||
| Peritubular capillaritis (ptc0–3) | 1.2 (0.77 – 1.72) | 0.49 | ||
| Transplant glomerulitis (g0–3)2 | 1.6 (0.83 – 3.14) | 0.16 | ||
| Transplant glomerulopathy (cg0–3) | 1.3 (0.43 – 3.89) | 0.65 | ||
| Interstitial fibrosis (ci0–3) | 2.5 (1.67 – 3.83) | <0.001 | 1.5 (0.65 – 3.6) | 0.34 |
| Tubular atrophy (ct0–3) | 2.5 (1.66 – 3.90) | <0.001 | ||
| Arteriosclerosis (cv0–3) | 2.0 (1.17 – 3.33) | 0.01 | 2.2 (0.63 – 7.5) | 0.22 |
| Arteriolar hyalinosis (ah0–3) | 1.5 (1.03 – 2.29) | 0.04 | 0.8 (0.27 – 2.68) | 0.77 |
| C4d score (C4d0–3) | 0.4 (0.15 – 1.19) | 0.11 | ||
| Glomerular PDs | 1.6 (0.73 – 3.65) | 0.23 | ||
| Prominent PDs in tubules | 1.6 (0.73 – 3.65) | 0.23 | ||
| Tubular microcysts | 2.6 (0.82 – 8.00) | 0.11 | ||
| Allograft from donor with APOL1 high- risk genotypes3 |
5.6 (1.92 – 16.4) | 0.002 | 4.8 (1.04 – 22.3) | 0.04 |
- Abbreviations: AA; African American; PDs; protein droplets
n=34 because 4 patients were not assessed for APOL1 genotypes
- For multivariate analysis, only variables with P< 0.05 were considered. Given the co-linearity, only interstitial fibrosis and not tubular atrophy was considered
The results were not significant when rejection was assessed separately for TCMR [HR: 1.6 (0.68 – 3.60), P=0.29] or AMR [HR: 0.8 (0.26 – 2.24), P=0.62]
The results were not significant when sum of g and ptc (g+ptc) was used [HR: 1.2 (0.88 – 1.56), P=0.29]
Data on APOL1 typing was not available for 7 donors; given the extremely low positivity in Caucasians, APOL1 risk genotypes were considered absent in 3 Caucasian donors who could not be genotyped
APOL1 high-risk genotypes in the donors
Finally, the 9 patients with cFSGS who received allografts from donors with APOL1 high-risk genotypes were analyzed separately (Table 5). With regard to comorbidities, 7/9 (78%) of these patients had concurrent acute rejection (n=6) or viral infection (n=1) while the remaining two had a prior history of viral infection with/without acute rejection. In contrast, none of them had concurrent acute vaso-occlusive disease. These 9 patients were then compared to patients who received allografts harboring APOL1 low-risk genotypes (Supplemental Table 3). Despite the small sample size, recipients of grafts with APOL1 high-risk genotypes had less vaso-occlusion but more proteinuria and tubular protein reabpsortion droplets, and a trend toward higher percentage of collapsed glomeruli and nephrotic syndrome. When donors were stratified based on donor race and APOL1 genotypes, patients receiving grafts from AA donors with APOL1 high-risk genotypes had worse allograft survival than these receiving their allografts from AA donors with low-risk genotypes or from Caucasian donors (Figure 4).
Table 5:
Clinico-pathologic characteristics of patients who received renal allografts from donors with APOL1 high-risk genotypes
| Subjects | Patient #8 |
Patient #11 |
Patient #12 |
Patient #14 |
Patient #25 |
Patient #27 |
Patient #35 |
Patient #37 |
Patient #38 |
|---|---|---|---|---|---|---|---|---|---|
| Donor race | Hispanic | AA | AA | AA | AA | AA | AA | AA | AA |
| Allograft source | L | D | D | D | D | D | D | L | D |
|
APOL1 testing specimen |
Post-rep | Post-rep | Non- infl Bx |
Post-rep | Blood | Blood | Post-rep | Blood | Blood |
| Post-transplant interval to biopsy (days) |
602 | 173 | 653 | 86 | 423 | 229 | 505 | 1489 | 425 |
| Concurrent comorbidities, namely AR, viral infection, or acute VOD |
AR | AR | AR | AR | Prior AR Prior CMV |
CMV | AR | AR | Prior CMV |
| Serum Cr at biopsy (mg/dL) |
9.7 | 2.5 | 6.5 | 7.2 | 5.5 | 6.4 | 1.7 | 7.9 | 5.2 |
| Urine protein/Cr at biopsy (g/g) |
3.8 | 2.2 | 6.9 | 2 | 3.5 | 15 | 6 | 11.6 | 7.6 |
| Collapsed glomeruli (%) |
31 | 13 | 17 | 15 | 13 | 33 | 28 | 12 | 25 |
- Abbreviations: AA, African American; AR, acute rejection; Cr, creatinine; D, deceased donor; L, living donor; Non-infl Bx, non-inflamed biopsy performed at 1 month post-transplantation; Post-rep: post-reperfusion biopsy, VOD, vaso-occlusive disease
- Recipient #25 and #38 received their allografts from the same donor
Recipients #25 had CMV infection 230 days before index biopsies which persisted till 217 days before index biopsy and a biopsy with borderline changes 209 days prior to index biopsy
Recipients #38 had CMV infection 71 days before index biopsies which persisted till 57 days before index biopsy
Figure 4: Allograft survival in patients stratified by donor race and APOL1 genotype.
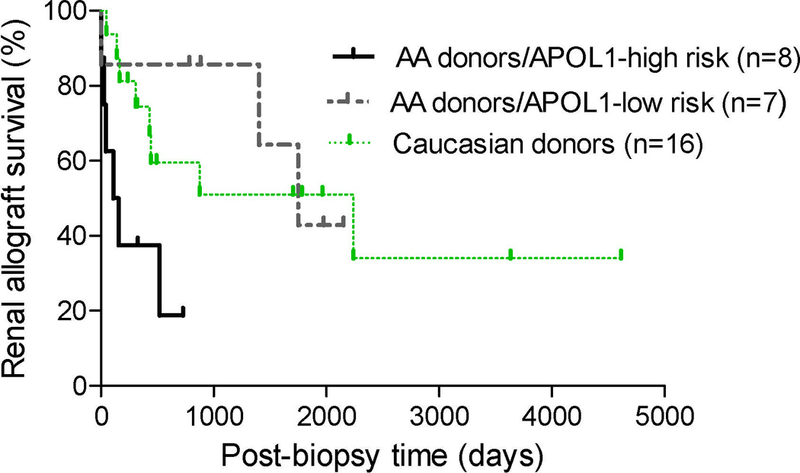
Kaplan–Meier survival curves for post-biopsy cumulative kidney allograft survival in patients stratified as receiving grafts from AA donors with APOL1 high-risk genotypes, AA donors with APOL1 low-risk genotypes, and Caucasian donors (P=0.03)
AA donors/APOL1 high-risk genotypes vs. AA donors/APOL1 low-risk genotypes (P=0.029)
AA donors/APOL1 high-risk genotypes vs. Caucasian donors (P=0.026)
AA donors/APOL1 low-risk genotypes vs. Caucasian donors (P=0.76)
Comparison with cFSGS in native biopsies
To assess whether cFSGS that occurs in the allograft differs from cFSGS in the native kidney, de novo post-KTx cFSGS (n=38) was compared with native cFSGS (n=255). De novo post-KTx cFSGS occurred in older patients who were less likely to be AA and was associated with less proteinuria, higher serum albumin, and lower incidence of full nephrotic syndrome (Table 6). De novo post-KTx cFSGS was more likely associated with acute vaso-occlusive disease and less likely associated with HIV infection (the latter largely the result of transplant restriction of HIV population). Post-KTx cFSGS showed less glomerulosclerosis and tubulointerstitial scarring than in native kidney disease, probably reflecting earlier disease in the allograft where renal function is frequently monitored (Table 6).
Table 6:
Comparison of demographic and clinico-pathologic parameters between cFSGS in allografts and native kidneys
|
De novo Post-KT cFSGS (n=38) |
native cFSGS controls (n=255) |
P values | |
|---|---|---|---|
| Demographic and clinical parameters | |||
| Age (years) | 57 (47, 65) | 46 (34, 60) | P=0.005 |
| Female gender | 13/38 (34%) | 119/255 (47%) | P=0.17 |
| AA recipients/patients1 | 10/38 (26%) | 157/225 (70%) | P<0.001 |
| Serum Cr at biopsy (mg/dL) | 5.4 (3.4, 6.8) | 4.0 (2.2, 6.8) | P=0.09 |
| Urine protein/Cr at biopsy (g/g)2 | 3.5 (1.3, 6.5) | 7.0 (4.1, 12.0) | P<0.001 |
| Serum albumin at biopsy (g/dL)3 | 3.2 (2.8, 3.7) | 2.4 (1.8, 3.0) | P<0.001 |
| Nephrotic proteinuria2 | 19/37 (51%) | 209/249 (84%) | P<0.001 |
| Nephrotic syndrome4 | 7/38 (18%) | 91/220 (41%) | P=0.007 |
| Traditional causes 5 Viral Acute VOD IFN treatment Pamidronate treatment |
15/38 (39%) 5/38 (13%) - HIV: 0/38 (0%) 11/38 (29%) 0/38 (0%) 0/38 (0%) |
87/255 (34%) 2 67/255 (26%) - HIV: 58/255 (23%) 13/255 (5%) 3/255 (1%) 4/255 (2%) |
P=0.592 P=0.11 P<0.001 P<0.001 P=1.00 P=1.00 |
| Pathology parameters | |||
| Global GS (%) | 11 (0, 25) | 21 (6, 41) | P=0.01 |
| Collapsed Glom (%) | 14 (8, 19) | 17 (8, 31) | P=0.15 |
| IFTA (%) | 25 (6, 44) | 40 (20, 65) | P=0.002 |
| Glom PDs | 31/38 (82%) | 193/255 (76%) | P=0.19 |
| FPE (%)6 - Focal - Extensive - Diffuse |
70 (48, 93) 2/8 (25%) 3/8 (37.5%) 3/8 (37.5%) |
90 (60, 100) 31/161 (19%) 40/161 (25%) 90/161 (56%) |
P=0.45 |
| Prominent tubular PDs | 16/38 (42%) | 78/255 (31%) | P=0.19 |
| Tubular microcysts | 5/38 (13%) | 90/255 (35%) | P=0.005 |
| Arteriosclerosis (0–3) | 1 (1, 2) [1.4 ±0.9] | 2 (1, 2) [1.5 ±0.9] | P=0.54 |
| Arteriolosclerosis (0–3) | 1 (1, 2) [1.3 ±0.9] | 2 (1, 2)][1.4 ±0.9] | P=0.25 |
Abbreviations: AA; African American; Cr, creatinine; Glom, glomeruli; GS, glomerulosclerosis; FPE, foot process effacement; IFN, Interferon; IFTA, interstitial fibrosis/tubular atrophy; NA, not available; PDs, protein droplets; VOD, vaso-occlusive disease
Semi-quantitative values were presented as median (IQR1, IQR3) and mean ±SD
Data on race is not available for 30 native controls
Data on proteinuria is unavailable for 1 post-KTx cFSGS and 6 native controls
Data on serum albumin is unavailable for 29 native controls
Data on nephrotic syndrome is not available for 35 native controls
Viral infections in post-KTx cFSGS (n=5) included 3 CMV 1 Parvovirus & 1 EBV while these in native controls (n=67) included 58 HIV, 7 Parvovirus, 1 CMV & 1 EBV
Data on FPE was available for 8 post-KTx cFSGS and 161 native controls
Discussion
Reports examining de novo cFSGS in the allograft setting are limited to small case series (Supplemental Table 1), which suggested that post-KTx cFSGS might differ from cFSGS in native kidneys given the relatively low incidence of nephrotic syndrome and high frequency of acute vaso-occlusive disease. The current study represents the largest clinico-pathologic study of de novo post-KTx cFSGS to date and confirms that this pattern of glomerular injury is associated with poor long-term allograft survival and variable degrees of proteinuria, typically lacking overt nephrotic syndrome. Compared to cFSGS in native kidneys, post-KTx cFSGS had a higher incidence of acute vaso-occlusive disease but less proteinuria and a lower incidence of nephrotic syndrome. While disparities in proteinuria may be partly attributable to post-transplant immunosuppression and lead time effect, the small sample of post-KTX cFSGS that underwent electron microscopic assessment precluded meaningful correlations with degree of foot process effacement.
We confirmed traditional risk factors for de novo post-KTx cFSGS in 39% of patients, including acute vaso-occlusion and/or concurrent viral infection. In addition, when compared to the general transplant population at CUMC, patients with post-KTx cFSGS were more likely to have received a kidney from an AA donor and to have acute rejection episodes, often concurrent with the onset of cFSGS (Figure 2). The high frequency of allografts received from AA donors in our cohort suggested a potential role for genetic factors in development of de novo post-KTx cFSGS. Shah et al. reported that two patients who developed de novo cFSGS received a kidney from the same deceased donor with an APOL1 high-risk genotype30. In our cohort, 15 allografts were donated from AA donors, 8 (53%) of which had APOL1 high-risk genotypes. This is considerably higher than the 13% frequency seen in the general AA population31 and suggests that donor APOL1 genotypes may contribute to post-KTx cFSGS. Despite the small sample size, patients with cFSGS who received allografts from donors with APOL1 high-risk genotypes had more proteinuria and a trend toward higher percentage of collapsed glomeruli compared to those who received allografts with APOL1 low-risk genotypes.
The mechanisms of APOL1-associated podocyte injury remain to be defined, but may involve enhanced expression of APOL1 risk variant proteins by IFN and other innate immune system signaling pathways. This could result in disrupted intracellular vesicle trafficking and autophagy with subsequent podocyte loss18, 32 which, if true, could trigger rapid parietal epithelial cell proliferation, producing a pattern of cFSGS33. Indeed, this mechanism has been proposed to explain the association between APOL1 high-risk genotypes and cFSGS secondary to HIV, lupus, and IFN treatment in native kidneys18, 34, 35. In the allograft setting, acute rejection could increase IFN36, 37 or other cytokine levels promoting cFSGS by a similar mechanism. Indeed all patients with post-KTx cFSGS who received allografts from APOL1 high-risk donors had a concurrent or recent history of acute rejection or active viral infection (Table 4).
A prior study in native cFSGS showed that 100% of Caucasian patients harbored either HLA-B44 or HLA-DR4 alleles2. In our post-KTx cFSGS cohort, only 24% of Caucasian recipients and 30% of Caucasian donors had HLA-B44 or HLA-DR4, similar to healthy Caucasians (B44: 32% and DR4: 25%). Although case reports proposed that sirolimus is associated with post-KTx cFSGS38, 39, this study was unable to detect an association. Prior studies suggested that chronic calcineurin inhibitor toxicity could lead to post-KTx cFSGS22, 23, 25. Although this may be confounded by the high percentage of early biopsies in this study (55% within the 1st year after transplantation), the low arteriolar hyalinosis scores and the comparable degree of vascular sclerosis in native kidneys with cFSGS do not support a primary role for chronic calcineurin inhibitor toxicity in our cohort.
To our knowledge, this is the first report to identify prognostic factors in patients with post-KTx cFSGS. Patients who had either partial clinical response or absence of cFSGS on follow-up biopsies had longer graft survival than those with persistent allograft dysfunction or persistence of cFSGS in subsequent biopsies. On univariate analysis, donor APOL1 high-risk genotypes, post-transplant interval to biopsy, nephrotic syndrome, extent of global glomerulosclerosis and tubulointerstitial scarring, and severity of vascular sclerosis were associated with inferior graft survival. In contrast, acute vaso-occlusion was associated with favorable graft survival. In these patients, the presumably localized pattern of injury (collapsed glomeruli were typically adjacent to zones of cortical infarction or in profoundly ischemic glomeruli) and the lack of donor APOL1 high-risk genotypes may explain their superior outcome. In two subsequent multivariable analyses, donor APOL1 high-risk genotypes remained significantly associated with graft outcome. Donor APOL1 high-risk genotypes account for much of the increased risk of graft failure from AA deceased donor kidneys relative to those from donors of European ancestry40, 41.
The current report is the largest study of de novo post-KTx cFSGS and the first to systematically assess APOL1 genotyping in kidney donors and to compare presenting clinical and pathologic features with a large cohort of cFSGS in native kidneys. Although this study is also the first to investigate prognostic variables in de novo cFSGS, the results of the multivariable analyses need to be confirmed by a larger study. Limitations of this study include the inadequate electron microscopic assessment in allograft biopsies and the lack of assessment of APOL1 genotyping and outcome in native cFSGS controls.
In conclusion, de novo post-KTx cFSGS is a pattern of glomerular injury associated with variable proteinuria and shortened graft survival. AA donors, especially those harboring APOL1 high-risk genotypes, may predispose recipients to cFSGS when exposed to second hits, such as acute rejection or viral infection that could activate podocyte innate immune pathways. Hence, cFSGS is associated with high proteinuria and inferior transplant outcomes in grafts from APOL1 high-risk donors. Prospective studies will be required to assess the true risk of cFSGS in kidney transplants from APOL1 high-risk donors prior to altering the allocation of donor kidneys. Additional studies are also needed to address potential roles of IFN and other molecular mediators of podocyte injury and cFSGS. That said, our data suggests that mitigation of these “second hits” has the potential to improve graft outcomes.
Material and Methods:
This study was performed under the approved guidelines of the institutional review board of CUMC. Allograft biopsies with cFSGS were identified retrospectively from the archives of the Renal Pathology Laboratory of CUMC from 1/2005 to 1/2017 by searching the “final diagnosis” for the words of “collapsing” or “collapse” and excluding biopsies with “immune complex glomerulonephritis”. Only patients >18 years of age at the time of biopsy who had de novo cFSGS and who were treated at CUMC were included. The first post-transplant biopsies with cFSGS were considered the “index biopsies”. These biopsies were re-reviewed jointly by two renal pathologists (DS and IB) and cases were included when consensus was achieved about meeting criteria for cFSGS as defined by the Columbia classification of FSGS1. Thirty-eight cases were identified. Demographic, clinical, and laboratory parameters were extracted from medical records. Nephrotic range proteinuria and hypoalbuminemia were defined as urine protein to creatinine ratio >3.5 g/day and serum albumin <3.0 g/dL, respectively. Nephrotic syndrome was defined as presence of nephrotic range proteinuria, hypoalbuminemia, and greater than trace peripheral edema.
Pathologic assessment
(A) All 38 biopsies were processed by standard techniques of light microscopy (stained with Hematoxylin and eosin, periodic acid–Schiff, Masson trichrome and Jones methenamine silver) and stained for C4d (37 by immunofluorescence and 1 by immunoperoxidase). Histologic parameters were evaluated according to the 2015 Banff criteria for renal allograft pathology42. Acute T cell mediated rejection was defined by the presence of borderline or greater changes (grades IA-III) (B) Immunofluorescence panel staining was performed in 27 (71%) index biopsies and in 7 (18%) allograft biopsies performed within 2 months of the index biopsy (C) Electron microscopy was performed in 9 biopsies. Foot process effacement was assessed in glomeruli that did not show collapsing or other segmental sclerotic lesions and as such could be evaluated in 8 of these 9 biopsies. Foot process effacement was reported as focal, extensive, and diffuse when involving <50%, 50–80%, and >80% of the glomerular capillary surface areas, respectively2.
APOL1 genotyping from kidney donors
Detailed methodology for DNA extraction and APOL1 genotyping are provided in the Supplemental material. Six kidney donors (one of whom donated kidneys to 2 recipients) had DNA available from whole blood for APOL1 genotyping. Twenty-four donors had APOL1 genotyping performed on DNA extracted from formalin-fixed paraffin-embedded allograft biopsies. High-risk genotypes were defined by the presence of 2 renal-risk alleles (G1/G1; G2/G2, or G1/G2). Due to the lack of donor cells, residual tissue, or the suboptimal quality of DNA extracted from biopsies, APOL1 genotyping could not be performed in 7 donors (3 Caucasians, 3 AA, and 1 Hispanic). Given the low probability (<0.4%) of Caucasians (including these with chronic kidney disease) to harbor APOL1 renal high-risk genotypes29, the three un-typed Caucasian donors were presumed to have APOL1 low-risk genotypes. In total, APOL1 data were analyzed in kidney allografts of 34/38 (89%) recipients who developed post-KTx cFSGS.
Follow-up and outcome
The primary end-point was graft failure, defined as re-initiation of renal replacement therapy. Renal replacement therapy was initiated a few days before the index biopsy in two patients, both of whom remain on chronic dialysis. For analysis, graft failure in these two patients was attributed to cFSGS. To assess short-term outcome (100 days post-biopsy), the following definitions were applied: (1) Complete remission: decreased proteinuria to <0.5 g/g with serum creatinine < 1.5 mg/dL (2) Partial remission: reduction in proteinuria by at least 50% and to <2 g/g with stable kidney function (<20% increase in serum creatinine) (3) Persistent renal dysfunction: failure to meet criteria for either complete or partial remission. Follow-up biopsies were assessed for the presence of cFSGS.
Controls
(1) To explore relevant comorbidities, the cohort of 38 patients with post-KTx cFSGS was compared to all 773 adult patients (>18 years) who underwent renal transplantation at CUMC between 1/2006 and 12/2009 (combining patients who received living and deceased allografts and including sensitized and non-sensitized recipients). This period of time was selected because of the availability of the data, which was partially published elswhere43.
(2) To assess whether cFSGS developing in the transplant versus in the native kidney differs with respect to demographic and clinico-pathologic characteristics, post-KTx cFSGS patients were compared to 255 controls with cFSGS in the native kidney retrieved from CUMC files during the same time period (2005–2017) using similar inclusion criteria (patients >18 years, immune complex-mediated glomerulonephritis excluded, and cases reviewed jointly by 2 renal pathologists).
Statistical analysis
Statistical analysis was performed using Prism 5 2007 (GraphPad Inc., San Diego CA) SPSS Statistics 24 (IBM, Armonk, NY), and Stata-11 (StataCorp, LP, College Station, Texas). Continuous data were presented as median and IQR (25th and 75th percentile). Semi-quantitative values were additionally presented as mean ±standard deviation (SD). Continuous variables were compared using Mann-Whitney test while categorical variables were compared using Fisher’s exact test. Non-significant values by Fisher exact test were confirmed by mid-p and Chi-square tests and discrepancies, if any, were noted in the tables. Graft survival was assessed by the Kaplan-Meier method and univariate comparisons were performed using log rank test. Cox proportional hazards (PH) univariate analyses for several demographic and clinico-pathologic variables were used to guide the selection of variables for multivariable analysis whenever these results were significant. To avoid the issue of overfitting, a step-up method was employed (forward stepwise likelihood ratio) that would only include the variables that were significant at the 0.05 level in the final model. P values <0.05 with two-sided hypothesis testing were considered statistically significant.
Supplementary Material
Acknowledgments
Acknowledgement: We thank Maria Lourdes Diaz Belvis at Columbia University Medical Center and Lou Craddock at Wake Forest School of Medicine for the excellent technical assistance.
SM is supported by funding from the NIH/NIDDK (R01-DK114893 and U01-DK116066)
BF is supported by NIH R01 DK084149 and R01 DK070941
Abbreviations
- AA
African-American
- APOL1
apolipoprotein L1 gene
- cFSGS
collapsing focal segmental glomerulosclerosis
- CMV
Cytomegalovirus
- CUMC
Columbia University Medical Center
- EBV
Epstein-Barr virus
- ESRD
end-stage kidney disease
- IFN
interferon
- KTx
kidney transplantation
- PCR
polymerase chain reaction
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosure: Wake Forest University Health Sciences and Barry Freedman have rights to a U.S. patent related to APOL1 genetic testing. Dr. Freedman is a consultant for Ionis and AstraZeneca.
References:
- 1.D’Agati VD, Fogo AB, Bruijn JA, et al. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis 2004; 43: 368–382. [DOI] [PubMed] [Google Scholar]
- 2.Laurinavicius A, Hurwitz S, Rennke HG. Collapsing glomerulopathy in HIV and non-HIV patients: a clinicopathological and follow-up study. Kidney Int 1999; 56: 2203–2213. [DOI] [PubMed] [Google Scholar]
- 3.Albaqumi M, Soos TJ, Barisoni L, et al. Collapsing glomerulopathy. Journal of the American Society of Nephrology : JASN 2006; 17: 2854–2863. [DOI] [PubMed] [Google Scholar]
- 4.Detwiler RK, Falk RJ, Hogan SL, et al. Collapsing glomerulopathy: a clinically and pathologically distinct variant of focal segmental glomerulosclerosis. Kidney Int 1994; 45: 1416–1424. [DOI] [PubMed] [Google Scholar]
- 5.Valeri A, Barisoni L, Appel GB, et al. Idiopathic collapsing focal segmental glomerulosclerosis: a clinicopathologic study. Kidney Int 1996; 50: 1734–1746. [DOI] [PubMed] [Google Scholar]
- 6.Nicholas Cossey L, Larsen CP, Liapis H. Collapsing glomerulopathy: a 30-year perspective and single, large center experience. Clinical Kidney Journal 2017; 10: 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferreira AC, Carvalho D, Carvalho F, et al. Collapsing glomerulopathy in Portugal: a review of the histological and clinical findings in HIV and non-HIV patients. Nephrol Dial Transplant 2011; 26: 2209–2215. [DOI] [PubMed] [Google Scholar]
- 8.Joshi A, Arora A, Cimbaluk D, et al. Acute Epstein-Barr virus infection-associated collapsing glomerulopathy. Clin Kidney J 2012; 5: 320–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buob D, Decambron M, Gnemmi V, et al. Collapsing glomerulopathy is common in the setting of thrombotic microangiopathy of the native kidney. Kidney Int 2016; 90: 1321–1331. [DOI] [PubMed] [Google Scholar]
- 10.Thaunat O, Delahousse M, Fakhouri F, et al. Nephrotic syndrome associated with hemophagocytic syndrome. Kidney Int 2006; 69: 1892–1898. [DOI] [PubMed] [Google Scholar]
- 11.Markowitz GS, Appel GB, Fine PL, et al. Collapsing focal segmental glomerulosclerosis following treatment with high-dose pamidronate. Journal of the American Society of Nephrology : JASN 2001; 12: 1164–1172. [DOI] [PubMed] [Google Scholar]
- 12.Markowitz GS, Nasr SH, Stokes MB, et al. Treatment with IFN-{alpha}, -{beta}, or -{gamma} is associated with collapsing focal segmental glomerulosclerosis. Clin J Am Soc Nephrol 2010; 5: 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nasr R, Johns C, Gertner E. Collapsing glomerulopathy in collagen vascular-like disease. Lupus 2014; 23: 75–80. [DOI] [PubMed] [Google Scholar]
- 14.Kasembeli AN, Duarte R, Ramsay M, et al. APOL1 Risk Variants Are Strongly Associated with HIV-Associated Nephropathy in Black South Africans. Journal of the American Society of Nephrology : JASN 2015; 26: 2882–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fensterl V, Sen GC. Interferons and viral infections. Biofactors 2009; 35: 14–20. [DOI] [PubMed] [Google Scholar]
- 16.Kim T, Kanayama Y, Negoro N, et al. Serum levels of interferons in patients with systemic lupus erythematosus. Clin Exp Immunol 1987; 70: 562–569. [PMC free article] [PubMed] [Google Scholar]
- 17.Chandrakasan S, Filipovich AH. Hemophagocytic lymphohistiocytosis: advances in pathophysiology, diagnosis, and treatment. J Pediatr 2013; 163: 1253–1259. [DOI] [PubMed] [Google Scholar]
- 18.Nichols B, Jog P, Lee JH, et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int 2015; 87: 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenberg A, Bastacky SI, Iqbal A, et al. Focal segmental glomerulosclerosis associated with nephrotic syndrome in cholesterol atheroembolism: clinicopathological correlations. Am J Kidney Dis 1997; 29: 334–344. [DOI] [PubMed] [Google Scholar]
- 20.Kavanagh D, McGlasson S, Jury A, et al. Type I interferon causes thrombotic microangiopathy by a dose-dependent toxic effect on the microvasculature. Blood 2016; 128: 2824–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kundra A, Wang JC. Interferon induced thrombotic microangiopathy (TMA): Analysis and concise review. Crit Rev Oncol Hematol 2017; 112: 103–112. [DOI] [PubMed] [Google Scholar]
- 22.Gupta R, Sharma A, Agarwal SK, et al. Collapsing glomerulopathy in renal allograft biopsies: A study of nine cases. Indian J Nephrol 2011; 21: 10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meehan SM, Pascual M, Williams WW, et al. De novo collapsing glomerulopathy in renal allografts. Transplantation 1998; 65: 1192–1197. [DOI] [PubMed] [Google Scholar]
- 24.Nadasdy T, Allen C, Zand MS. Zonal distribution of glomerular collapse in renal allografts: possible role of vascular changes. Hum Pathol 2002; 33: 437–441. [DOI] [PubMed] [Google Scholar]
- 25.Stokes MB, Davis CL, Alpers CE. Collapsing glomerulopathy in renal allografts: a morphological pattern with diverse clinicopathologic associations. Am J Kidney Dis 1999; 33: 658–666. [DOI] [PubMed] [Google Scholar]
- 26.Swaminathan S, Lager DJ, Qian X, et al. Collapsing and non-collapsing focal segmental glomerulosclerosis in kidney transplants. Nephrol Dial Transplant 2006; 21: 2607–2614. [DOI] [PubMed] [Google Scholar]
- 27.Canaud G, Bruneval P, Noel LH, et al. Glomerular collapse associated with subtotal renal infarction in kidney transplant recipients with multiple renal arteries. Am J Kidney Dis 2010; 55: 558–565. [DOI] [PubMed] [Google Scholar]
- 28.Patel RD, Vanikar AV, Nigam LA, et al. De Novo Focal Segmental Glomerulosclerosis in Renal Allograft-Histological Presentation and Clinical Correlation: Single Centre Experience. J Clin Diagn Res 2017; 11: EC39–EC42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooke JN, Bostrom MA, Hicks PJ, et al. Polymorphisms in MYH9 are associated with diabetic nephropathy in European Americans. Nephrol Dial Transplant 2012; 27: 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shah PB, Cooper JE, Lucia MS, et al. APOL1 Polymorphisms in a Deceased Donor and Early Presentation of Collapsing Glomerulopathy and Focal Segmental Glomerulosclerosis in Two Recipients. Am J Transplant 2016; 16: 1923–1927. [DOI] [PubMed] [Google Scholar]
- 31.Friedman DJ, Pollak MR. Apolipoprotein L1 and Kidney Disease in African Americans. Trends Endocrinol Metab 2016; 27: 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartleben B, Wanner N, Huber TB. Autophagy in glomerular health and disease. Semin Nephrol 2014; 34: 42–52. [DOI] [PubMed] [Google Scholar]
- 33.Hakroush S, Cebulla A, Schaldecker T, et al. Extensive podocyte loss triggers a rapid parietal epithelial cell response. J Am Soc Nephrol 2014; 25: 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. Journal of the American Society of Nephrology : JASN 2011; 22: 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsen CP, Beggs ML, Saeed M, et al. Apolipoprotein L1 risk variants associate with systemic lupus erythematosus-associated collapsing glomerulopathy. Journal of the American Society of Nephrology : JASN 2013; 24: 722–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bestard O, Crespo E, Stein M, et al. Cross-validation of IFN-gamma Elispot assay for measuring alloreactive memory/effector T cell responses in renal transplant recipients. Am J Transplant 2013; 13: 1880–1890. [DOI] [PubMed] [Google Scholar]
- 37.Najafian N, Salama AD, Fedoseyeva EV, et al. Enzyme-linked immunosorbent spot assay analysis of peripheral blood lymphocyte reactivity to donor HLA-DR peptides: potential novel assay for prediction of outcomes for renal transplant recipients. Journal of the American Society of Nephrology : JASN 2002; 13: 252–259. [DOI] [PubMed] [Google Scholar]
- 38.Dogan E, Ghanta M, Tanriover B. Collapsing glomerulopathy in a renal transplant recipient: potential molecular mechanisms. Ann Transplant 2011; 16: 113–116. [DOI] [PubMed] [Google Scholar]
- 39.Izzedine H, Brocheriou I, Frances C. Post-transplantation proteinuria and sirolimus. N Engl J Med 2005; 353: 2088–2089. [DOI] [PubMed] [Google Scholar]
- 40.Freedman BI, Pastan SO, Israni AK, et al. APOL1 Genotype and Kidney Transplantation Outcomes From Deceased African American Donors. Transplantation 2016; 100: 194–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Julian BA, Gaston RS, Brown WM, et al. Effect of Replacing Race With Apolipoprotein L1 Genotype in Calculation of Kidney Donor Risk Index. Am J Transplant 2017; 17: 1540–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loupy A, Haas M, Solez K, et al. The Banff 2015 Kidney Meeting Report: Current Challenges in Rejection Classification and Prospects for Adopting Molecular Pathology. Am J Transplant 2017; 17: 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Batal I, Mohan S, De Serres SA, et al. Analysis of dendritic cells and ischemia-reperfusion changes in postimplantation renal allograft biopsies may serve as predictors of subsequent rejection episodes. Kidney Int 2018; 93: 1227–1239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.