

Abstract
Homozygous recessive mutations in the PRICKLE1 gene were first described in three consanguineous families with myoclonic epilepsy. Subsequent studies have identified neurological abnormalities in humans and animal models with both heterozygous and homozygous mutations in PRICKLE1 orthologues. We describe a 7-year-old with a novel de novo missense mutation in PRICKLE1 associated with epilepsy, autism spectrum disorder, and global developmental delay.
Keywords: PRICKLE1, de novo mutation, prickle, epilepsy, autism spectrum disorder
Introduction
Homozygous recessive mutations in the PRICKLE1 gene were first described in three consanguineous families with myoclonic epilepsy (Bassuk et al., 2008). Subsequent studies have identified neurological abnormalities in humans and animal models with both heterozygous and homozygous mutations in PRICKLE1 orthologues (Bosoi et al., 2011; Ehaideb et al., 2014; Paemka et al., 2013; Tao et al., 2011). Inherited heterozygous PRICKLE1 mutations have been reported in several human conditions including epilepsy (Tao et al., 2011) autism (Paemka et al., 2013), and spina bifida (Bosoi et al., 2011). Additionally, de novo PRICKLE1 mutations have been reported in association with fetal agenesis of the corpus callosum (Bassuk & Sherr, 2015), Charcot Marie Tooth Disease and epilepsy(Pehlivan et al., 2015). We describe a 7-year-old with a novel de novo mutation in PRICKLE1 associated with epilepsy, autism spectrum disorder, and global developmental delay.
Materials and Methods
The family was consented by an IRB approved protocol at the University of Iowa, after informed consent was obtained, clinical whole exome sequencing was performed on DNA isolated from the child and from the parents. Clinical exome sequencing was performed by Gene Dx (Gaithersburg, MD) using standard techniques. Briefly, the clinical exome sequencing begins with massive parallel sequencing using the Illumia HiSeq sequencing system with 100bp paired-end reads. Bi-directional sequence was assembled, aligned to reference gene sequences based on human genome build GRCh37/UCSC hg19, and analyzed for sequence variants using a custom-developed analysis tool (Xome Analyzer). 100% of the coding region was covered at a minimum of 10× coverage.
Results
Case Report
Patient x is a 7-year-old boy. He presented at 11 months with abnormal body movements consistent with myoclonic seizures. EEG demonstrated generalized epileptiform discharges. MRI was normal (Figure 1). The patient was significantly delayed in meeting his motor milestones; he sat up at 18 months, and walked at 2 ½ years. At 6 years of age he was clinically evaluated for behavioral concerns and was diagnosed with autism spectrum disorder and a mild intellectual disability. After informed consent was obtained from an IRB approved protocol from the University of Iowa, clinical whole exome sequencing was performed on DNA isolated from Patient x and from the parents, revealing a de novo mutation in the PRICKLE1 gene c.1444 G>A, D482N (Figure 1). No other de novo mutations were identified, nor were any other mutations in known disease-causing genes uncovered, including known global developmental delay, autism spectrum disorder, and epilepsy genes (Supplementary table 1). Genes evaluated include those from the GeneDx epilepsy panel (https://www.genedx.com/test-catalog/available-tests/comprehensive-epilepsy-panel/). The de novo mutation was validated by Capillary sequencing. This variant was absent from over 6500 alleles in the Exome Variant Server (http://evs.gs.washington.edu/EVS/)) and absent from the 60,706 individuals from the ExAc Browser (http://exac.broadinstitute.org/). Sequence alignment shows that the D482N encoding PRICKLE1 mutation alters an amino acid conserved throughout vertebrate evolution.
Figure 1:
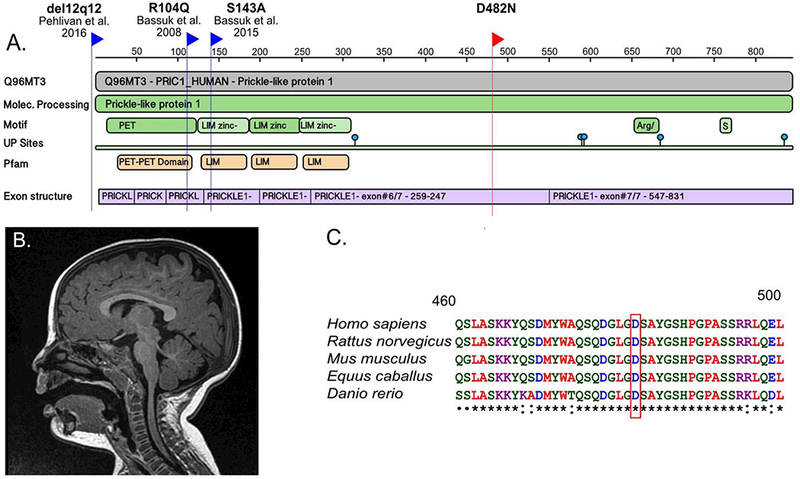
(a) Protein Feature View for PRICKLE1 from the RCSB Protein Data Bank (PDB) website. The blue flags indicate the location of human PRICKLE1 mutations that have been previously reported, including a de novo mutation associated with fetal agenesis of the corpus callosum and polymicrogyria (Bassuk & Sherr, 2015), a homozygous recessive mutation associated with myoclonic epilepsy (Bassuk et al., 2008), and a ~30-kb deletion involving the 5’ untranslated region and start of the PRICKLE1 coding sequence associated with Charcot Marie Tooth Disease and epilepsy (Pehlivan et al., 2015). The red flag indicates the novel de novo PRICKLE1 missense mutation. (b) Midline sagittal image from an MRI conducted at 1-year-old showing normal findings. (c) Multiple species protein alignment of the region surrounding the amino acid change caused by the novel de novo mutation in PRICKLE1. The red box indicates the reference amino acid, D, in humans and other vertebrates. The amino acid is completely conserved in vertebrates used in the alignment, and is within a highly conserved domain across all species. Peptide sequences were downloaded from UniProt BLAST and the alignment was performed using the Clustal Omega website (http://www.ebi.ac.uk/Tools/msa/clustalo/).
Discussion
PRICKLE1 mutations in humans were originally described as recessive mutations in families with syndromic myoclonic epilepsy (Bassuk et al., 2008). Subsequently variation in PRICKLE1 has been described in patients with non-syndromic epilepsy (Tao et al., 2011), autism (Paemka et al., 2013), and spina bifida (Bosoi et al., 2011). Additionally, de novo PRICKLE1 mutations have been reported in association with agenesis of the corpus callosum and polymicrogyria (Bassuk & Sherr, 2015), Charcot Marie Tooth Disease and epilepsy (Pehlivan et al., 2015). Our description of a novel de novo PRICKLE1 mutation associated with global developmental delay, autism spectrum disorder, and epilepsy in the absence of any other de novo mutation or known disease causing mutation strongly implicates the PRICKLE1 gene as a causative factor.
Like the original human PRICKLE1 mutant families (Bassuk et al., 2008) the current patient had normal MRI findings. This is in contrast to our previously reported PRICKLE1 de novo mutation with agenesis of the corpus callosum and polymicrogyria (Bassuk & Sherr, 2015). Thus there seems to be a spectrum of radiological findings among PRICKLE1 mutant patients. These findings strengthen and expand the clinical spectrum of abnormalities associated with PRICKLE1 mutations in humans.
Supplementary Material
Supplementary Table 1: Selected genes found to have no pathogenic variants through whole exome sequencing. Genes listed are associated with epilepsy, autism spectrum disorder, and global developmental delay.
Acknowledgments
Funding from NIH 5R01NS098590 (to AGB).
References
- Bassuk AG, & Sherr EH (2015). A de novo mutation in PRICKLE1 in fetal agenesis of the corpus callosum and polymicrogyria. J Neurogenet, 29(4), 174–177. doi: 10.3109/01677063.2015.1088847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassuk AG, Wallace RH, Buhr A, Buller AR, Afawi Z, Shimojo M, … El-Shanti HI (2008). A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome. Am J Hum Genet, 83(5), 572–581. doi: 10.1016/j.ajhg.2008.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosoi CM, Capra V, Allache R, Trinh VQ, De Marco P, Merello E, Kibar Z (2011). Identification and characterization of novel rare mutations in the planar cell polarity gene PRICKLE1 in human neural tube defects. Hum Mutat, 32(12), 1371–1375. doi: 10.1002/humu.21589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehaideb SN, Iyengar A, Ueda A, Iacobucci GJ, Cranston C, Bassuk AG, Manak JR (2014). prickle modulates microtubule polarity and axonal transport to ameliorate seizures in flies. Proc Natl Acad Sci U S A, 111(30), 11187–11192. doi: 10.1073/pnas.1403357111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paemka L, Mahajan VB, Skeie JM, Sowers LP, Ehaideb SN, Gonzalez-Alegre P, Bassuk AG (2013). PRICKLE1 interaction with SYNAPSIN I reveals a role in autism spectrum disorders. PLoS One, 8(12), e80737. doi: 10.1371/journal.pone.0080737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehlivan D, Beck CR, Okamoto Y, Harel T, Akdemir ZHC, Jhangiani SN, Lupski JR (2015). The role of combined SNV and CNV burden in patients with distal symmetric polyneuropathy. Genetics In Medicine, 18, 443. doi: 10.1038/gim.2015.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao H, Manak JR, Sowers L, Mei X, Kiyonari H, Abe T, Bassuk AG (2011). Mutations in prickle orthologs cause seizures in flies, mice, and humans. Am J Hum Genet, 88(2), 138–149. doi: 10.1016/j.ajhg.2010.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: Selected genes found to have no pathogenic variants through whole exome sequencing. Genes listed are associated with epilepsy, autism spectrum disorder, and global developmental delay.