

Abstract
Ferroptosis is a form of lipid peroxidation-induced cell death that can be regulated in many ways, from altering the activity of antioxidant enzymes to the level of transcription factors. The p53 tumor suppressor is ‘the guardian of the genome’ that participates in the control of cell survival and division under various stresses. Beyond its effects on apoptosis, autophagy, and cell cycle, p53 also regulates ferroptosis either through a transcriptional or posttranslational mechanism. On one hand, p53 can enhance ferroptosis by inhibiting the expression of SLC7A11 (solute carrier family 7 member 11) or by enhancing that of SAT1 (spermidine/spermine N1-acetyltransferase 1) and GLS2 (glutaminase 2). On the other hand, p53 suppresses ferroptosis through the direct inhibition of DPP4 (dipeptidyl peptidase 4) activity or by the induction of CDKN1A/p21 (cyclin dependent kinase inhibitor 1A) expression. Here, we review recent discoveries and emerging trends in the study of the ferroptosis network and highlight the context-dependent impact of p53 on ferroptosis and oxidative stress.
Introduction
Discovered in the 1970s, the tumor suppressor protein p53 (TP53) plays a critical role in the cellular response to various stresses, including DNA damage, hypoxia, nutrition starvation, and oncogene activation [1]. Activation of p53 can lead to survival or death, depending on the levels of stress [2]. Low levels of stress or damage trigger p53 activation to induce cell cycle arrest, DNA repair, and survival. p53 can protect against oxidative stress-induced DNA damage and death via downregulation of the production of reactive oxygen species (ROS) in cells. In contrast, high levels of stress or injury result in the activation of p53 to induce apoptosis and death. Unfortunately, p53 is usually mutated or depleted in many cancers, which limits the antitumor function of p53. Many studies have been focusing on the identification of p53 target genes that mediate tumor suppressor function. In addition to acting as a transactional factor in the nucleus, transcription-independent functions of cytosolic p53 are documented in the processes controlling cell death and metabolism, including apoptosis and autophagy [3]. For example, cytosolic p53 can directly bind to pro-apoptotic members of the BCL-2 family (BAX [BCL2 associated X, apoptosis regulator] and BBC3/PUMA [BCL2 binding component 3]) to increase mitochondrial membrane permeabilization and the release of pro-apoptotic factors from the mitochondria [4, 5]. Unlike nuclear p53, which acts as an autophagy-promoting transcription factor [6, 7], cytosolic p53 can block autophagy in response to nutrient starvation or mTOR inhibition [8]. These context-dependent roles of p53 in survival and death are regulated in a fine-tuned manner by its ubiquitination, phosphorylation, acetylation, and other modifications [9, 10]. Over the last three years, studies in both cell cultures and animal models have established that p53 represents a novel regulator of ferroptosis [11-14] (Fig. 1), a form of regulated cell death characterized by the accumulation of lethal iron or lipid hydroperoxides (e.g., PUFA-OOH) [15]. In this review, we will summarize the molecular mechanism of ferroptosis and focus on the current understanding of connections between p53 and ferroptosis and its potential as a target in cancer therapy.
Figure 1.
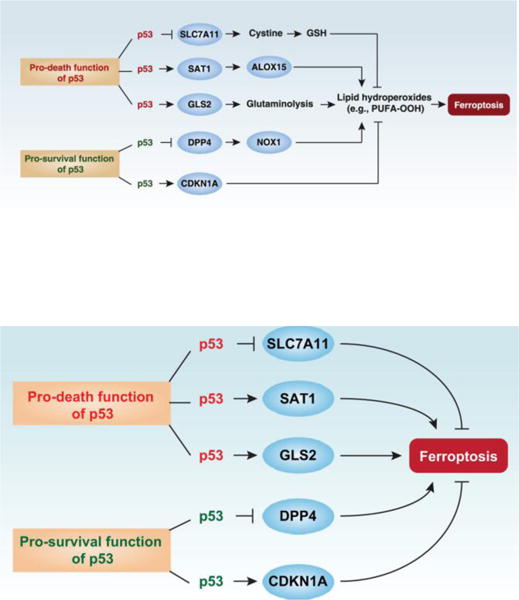
The dual role of p53 in the control of ferroptosis. Ferroptosis is characterized by lipid peroxidation. p53 plays a context-dependent role in the regulation of lipid peroxidation in ferroptosis. On one hand, p53 can enhance ferroptosis through the inhibition of SLC7A11 expression or promotion of SAT1 and GLS2 expression. On the other hand, p53 could suppress ferroptosis through the inhibition of DPP4 activity or induction of CDKN1A/p21 expression.
Ferroptosis basics
Since its discovery in 2012 [15], the study of ferroptosis has been a fast-growing field in cell death research [16]. The process and function of ferroptosis, as well as its impact in disease susceptibility, has been recently well-reviewed [17, 18]. We first briefly introduce the major inducers and regulators of ferroptosis.
Inducers
Erastin
Oncogenic RAS mutations, including K-RAS, H-RAS, and N-RAS, have been shown to drive the development of various cancers across different cells of origin and etiologies. These oncogenes therefore are highly attractive targets for anticancer drug discovery, colliding with the fact that thus far no direct RAS inhibitors have been introduced into clinical practice [19]. Erastin, the first inducer of ferroptosis, was identified through a high-throughput small molecule screening searching for agents that selectively killed RAS mutated cancer cells, including H-Ras-mutant engineered human foreskin fibroblasts (BJeLR), N-Ras-mutant HT-1080 cells (a fibrosarcoma cell line), and K-Ras-mutant Calu-1 cells (a lung cancer cell line) [20, 21]. The prosurvival RAS-RAF-MEK-ERK pathway in cancer cells is required for erastin-induced ferroptosis [20]. However, the previous conclusion that erastin fails to induce cell death in RAS wild type cells has been challenged by recent studies. This evidence includes: 1) Erastin can trigger leukemia cell death in a RAS-independent manner [22]; 2) Normal cells without RAS mutation such as kidney tubule cells and fibroblasts are sensitive to erastin [12, 23-26]; 3) Forced expression of mutant RAS limits the anticancer activity of erastin in RMS13 cells (a rhabdomyosarcoma cell line) [27]. In addition, an early report described that the direct target of erastin is the mitochondrial VDAC (voltage-dependent anion channel)-2 and -3, which cause mitochondrial injury with increased mitochondrial membrane permeabilization [20]. Indeed, knockdown of VDAC2 or VADC3 by RNAi limits erastin-induced ferroptosis [20]. However, increasing evidence indicates that the major target of erastin is the antiporter system Xc−, which localizes in the cell membrane [15]. Although the half-life of erastin is relatively short in vivo, it exhibits anticancer activity in multiple mouse models, indicating that yet-to-be-discovered metabolites produced from erastin may mediate antineoplastic effects [28, 29].
RSL3
RSL3 was also identified through a high-throughput small molecule screening campaign designed to identify agents that selectively killed H-Ras-mutant engineered BJeLR cells in a non-apoptotic manner [30]. Similar to erastin, the original study indicated that iron accumulation, ROS production, and RAS-RAF-MEK-ERK activation are required for RSL3-induced ferroptosis [30]. Unlike erastin, signaling through mitochondrial VDAC2 or VADC3 and membrane system Xc− do not seem to be required for RSL3-induced ferroptosis [30]. In contrast, GPX4 (glutathione peroxidase 4) is inhibited by RSL3 [31]. The early study suggests that RSL3 binds GPX4 to inhibit its enzymatic activity and hence to interfere with its capacity to prevent lipid peroxidation [31]. However, recent studies indicate that RSL3 as well as erastin can induce GPX4 protein degradation [32-34]. HSPA5 (heat shock protein family A [Hsp70] member 5), a molecular chaperone that contributes to endoplasmic reticulum (ER) homeostasis, can inhibit GPX4 degradation through a protein-protein interaction [34]. Moreover, the transcription factor ATF4 (activating transcription factor 4), which is involved in the ER stress response, is required for HSPA5 and SLC7A11 (solute carrier family 7 member 11) upregulation to inhibit ferroptosis [34, 35]. In contrast, ATF4-mediated expression of DDIT3/CHOP (DNA damage inducible transcript 3) contributes to the sensitization of cells to TNFSF10/TRAIL (TNF superfamily member 10)-induced apoptosis that is mediated by ferroptotic agents such as erastin and artesunate [36]. These findings build evidence supporting a complex interplay between ER stress, ferroptosis, and apoptosis.
Regulators
System Xc−
The cystine-glutamate antiporter system Xc− is composed of a substrate-specific subunit SLC7A11 and a regulatory subunit SLC3A2 (solute carrier family 3 member 2). System Xc−-mediated cystine uptake is required for the production of cysteine and subsequent synthesis of glutathione (GSH) [37]. In addition to erastin and glutamate, other drugs, including sulfasalazine and sorafenib, can block system Xc− activity and subsequently lead to GSH depletion and ferroptosis [38]. These system Xc− inhibitors are termed type I ferroptosis inducers. In contrast, β-mercaptoethanol-induced cystine uptake inhibits erastin-induced ferroptosis in HT-1080 cells [15]. Indeed, erastin can elicit negative feedback loop that limits ferroptosis. This feedback regulation is based on the capacity of erastin to induce SLC7A11 expression in an iron- and ROS-independent manner [15]. Mechanistically, ATF4 and p53 have been reported to promote or inhibit SLC7A11 gene transcription in ferroptosis, respectively [12, 35]. Of note, knockdown of SLC7A11 increases erastin-induced death, which indicates that another non-system Xc− pathway may contribute to its cytotoxicity. The molecular targets of erastin in the regulation of oxidative injury during ferroptosis remain to be further explored.
GPX4
Glutathione peroxidases (GPXs) including GPX1-8 play key roles in the regulation of oxidative stress. Among them, GPX4 seems to be special in protecting cells against membrane lipid peroxidation in ferroptosis [31]. GSH-dependent GPX4 activity catalyzes the reduction of lipid hydroperoxides to alcohols or free hydrogen peroxide to water [39]. Several GPX4 inhibitors such as RSL3 and FIN56 are categorized as type II ferroptosis inducers [31]. GPX4 degradation seems to be a universal event in the induction of ferroptosis [33, 34, 40]. GPX4 inhibitors induce ferroptosis even in conditions in which the GSH pool is intact, contrasting with the effects of type I ferroptosis inducers (system Xc− inhibitors) that cause GSH depletion [31]. Genetic depletion of GPX4 can trigger ferroptosis in an iron-, MEK-, and ROS-dependent manner in vitro or in vivo [23, 24, 31]. However, induction of ferroptosis is not the only reason for the increased tissue injury in mice in which GPX4 expression is abolished in a conditional or inducible fashion. In some cases, increased apoptosis and necroptosis also contribute to GPX4 deficiency-induced tissue injury in vivo [41-44]. Further investigation is needed to understand the function of GPX4 in various types of regulated cell death. Other GPXs such as GPX7 and GPX8 may participate in the control of ferroptosis [31]. Finally, functional relationship between GPXs and other antioxidant systems remains to be further explored.
NFE2L2
NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2) is a master transcription factor that controls the cellular response to oxidative or electrophilic stresses [45]. Embarking on the search for novel transcription factor in ferroptosis, we discovered that activation of NFE2L2 by type I ferroptosis inducers such as erastin and sorafenib can inhibit ferroptosis in liver cancer cells [46]. This process is generally divided into two phases: 1) Stabilization of NFE2L2 by SQSTM1/p62 (sequestosome 1) and 2) NFE2L2-mediated expression of anti-oxidant proteins and detoxifying enzymes. In normal, non-stressed conditions, NFE2L2 is constantly degraded by the ubiquitination-proteasome system. In response to erastin and sorafenib, SQSTM1 (a stress-inducible and multifunctional protein in autophagy) binds KEAP1 ([kelch-like ECH-associated protein 1], an adaptor protein of Cullin-3 ubiquitin ligase) and then arrests ubiquitination of NFE2L2 and increases its protein stability [46]. NFE2L2 then translocates to the nucleus and promotes the transcription of cytoprotective genes. We identified metallothionein-1G (a cysteine residue-rich protein) as a direct NFE2L2 target gene contributing to ferroptosis resistance [47]. NFE2L2 also promotes SLC7A11 and GPX4 expression in some cases [48, 49]. These findings suggest that NFE2L2 may be a central anti-ferroptosis transcription factor.
ACSL4
ACSL (Acyl-coenzyme A synthetase long-chain) family members, including ACSL1, ACSL3, ACSL4, ACSL5, and ACSL6, are expressed at the ER and mitochondrial outer membrane, where they can synthesize acyl-coenzyme A from fatty acids. Impaired ACSL pathways had previously been implicated in the regulation of apoptosis, depending on cell type [50-52]. We found that ACSL4 plays a key role in promoting erastin-induced ferroptosis through 5-HETE-mediated lipotoxicity by accumulation of lipid intermediates. ACSL4 (but not ACSL1, ACSL3, ACSL5, and ACSL6) expression correlates with cellular sensitivity to erastin-induced ferroptosis [53]. Suppression of ACSL4 expression by RNA interference increases ferroptosis resistance in HepG2 (human hepatoblastoma) and HL-60 (acute myeloid leukemia) cells, whereas overexpression of ACSL4 by gene transfection restores ferroptosis sensitization in LNCaP (a human prostate carcinoma cell line) and K562 cells (a chronic myeloid leukemia cell line) [53]. Two independent groups confirmed that ACSL4 is a critical driver of ferroptosis, and knockout of ASCL4 using CRISPR/Cas9 technology reverses ferroptosis induced by GPX4 deficiency [54, 55]. Collectively, these studies indicate that ACSL4 is not only a biomarker of, but also a contributor to, ferroptosis.
NCOA4
Autophagy is an intracellular degradation pathway that is regulated by the autophagy-related (ATG) proteins and their posttranslational modification [56]. It has been thought that ferroptosis differs from autophagy [15], but recent experimental evidence has revealed that ferroptosis may constitute a form of autophagic cell death that can be specifically regulated by ferritinophagy [57, 58]. The role of nonselective and selective autophagy in cellular homeostasis is complex, with opposite effects on survival and death, depending on the type of stressors and context of cargos [59-61]. Ferritinophagy is the process of autophagic degradation of ferritin that requires classic ATG proteins such as ATG5 and ATG7 and the specific cargo receptor NCOA4 (nuclear receptor coactivator 4) to control cellular iron homeostasis [62]. We found that erastin stimulates the formation of autophagosomes, the double-membrane vesicles that are responsible for delivering cytoplasmic material to lysosomes [58]. More strikingly, the interaction between NCOA4 and ferritin occurs within the autophagosomes and may depend on specific autophagy [58]. Finally, selective autophagy of ferritin driven by NCOA4 increases toxic iron-induced ROS production to induce ferroptosis [58]. It remains unknown whether other forms of selective autophagy such as mitophagy (removal of mitochondria via autophagy) and lipophagy (degradation of lipids via autophagy) may similarly favor ferroptosis.
HSPs
HSPs (heat shock proteins) are highly conserved in eukaryotes and function as molecular chaperones to participate in the regulation of protein assembly, folding, export, and turn-over [63]. HSPs can be rapidly induced under various stressful conditions such as heat shock, oxidative stress and oncogenic stress. Based on their molecular size, HSPs are divided into six distinct subfamilies: HSP100, HSP90, HSP70, HSP60, small HSP, and HSP10. Among them, HSPB1 (a member of the small HSPs) and HSPA5 (a member of HSP70 and the primary ER chaperone) are negative regulators of ferroptosis [28, 34]. Protein kinase C-mediated HSPB1 phosphorylation diminishes toxic iron-induced ROS production in ferroptosis [28], whereas HSPA5 confers protection against ferroptosis by increasing GPX4 protein stability [34]. HSF1 (heat shock transcription factor 1) plays a central role in the transcriptional activation of HSPs that also protect cells against ferroptosis [28]. Moreover, evidence is emerging that HSF1 and NFE2L2 engage in crosstalk by sharing overlapping transcriptional targets for cytoprotection in response to various stressors [64].
Pro-death function of p53 in ferroptosis
Inhibition of SLC7A11 expression
The first report of p53 induction in response to ferroptosis was published in 2015 [12]. In this study, the authors found that p53 promotes ferroptosis in fibroblasts and certain cancer cells (human breast cancer MCF7 and human osteosarcoma U2OS) due to the transrepression of SLC7A11 expression (Fig. 1). In particular, p533KR, an acetylation-defective mutant in which 3 lysine residues (in positions 117, 161 and 162) have been replaced by arginine residues, is highly effective in repressing SLC711A, yet does not affect the expression of other known p53 target genes involved in the regulation of cell cycle (e.g., CDKN1A/p21) or apoptosis (e.g., BAX) [12]. In contrast, p534KR98 (an acetylation-defective mutant in which an addition lysine in position 98 has been replaced) is unable to reduce SLC711A expression [65]. Moreover, induction of ferroptosis, but not cell-cycle arrest, apoptosis, or senescence, was found to be required for the tumor suppression function of p533KR in vitro and in vivo [12]. In human cancers, wild-type p53 is degraded by high levels of the oncogenic E3 ubiquitin protein ligase MDM2. Thus, the inhibition of MDM2-dependent proteasomal degradation of p53 presents an appealing therapeutic strategy for the treatment of cancer [66]. As expected, the level of p53 is increased in MDM2−/− cells. Ferroptosis also contributes to the embryonic lethality observed in MDM2−/− mouse embryos, which can be reversed by administration of ferroptosis inhibitors such as ferrostatin-1 [12]. However, the other study showed that ferrostatin-1 alone cannot prevent cell death induced by MDM2 deficiency [67], indicating different mechanisms in the regulation of the MDM2-p53 network in the determination of cell death. Of note, the anti-ferroptosis activation of ferrostatin-1 and liproxstatin-1 (another widely-used ferroptosis inhibitor) are mediated through their reactivity as radical-trapping antioxidants rather than their potency as inhibitors of lipoxygenases [68]. The site of levels of p53 acetylation is determined by six different histone acetyltransferases: CREBBP/CBP (CREB binding protein), EP300/p300 (E1A binding protein P300), KAT2B/PCAF (lysine acetyltransferase 2B), KAT5/Tip60 (lysine acetyltransferase 5), KAT8/MOF (lysine acetyltransferase 8), and KAT6A/MOZ (lysine acetyltransferase 6A) [69]. The ability of these acetyltransferases to regulate ferroptosis remains unclear.
Promotion of SAT1 expression
The low-molecular-weight polyamines, including putrescine, spermidine and spermine, are implicated in the regulation of cellular growth, proliferation, and differentiation. At the molecular level, SAT1 (spermidine/Spermine N1-acetyltransferase 1) is an important regulator in polyamine metabolism through acetylating spermidine and spermine using acetyl-coenzyme A [70]. Impaired polyamine metabolism and abnormal SAT1 expression is associated with various pathological conditions, including cancer [70]. The activity of SAT1 is increased in response to various stresses, including oxidative stress, heat shock, and inflammatory stimuli. Previous studies have observed that overexpression of SAT1 results in rapid depletion of cellular spermidine and spermine, which cause significant growth inhibition and mitochondrial apoptosis [71]. Recent research studies have found that SAT1 is a transcriptional target of p53 in MCF7, U2OS, A375 (a human melanoma cell line), and H1299 cells (a human lung cancer cell line) (Fig. 1) [13]. However, only ferrostatin-1, but not other cell death inhibitors (Z-VAD-FMK, necrostatin-1, and 3-methyladenine), can inhibit ROS-induced cell death in SAT1 Tet-on cells [13]. SAT1 depletion also inhibits p53- and p533KR-induced ferroptosis [13]. Mechanistically, SAT1 has no effects on the expression and activity of SLC7A11 and GPX4 [13]. In contrast, SAT1 induction correlates with the expression levels of ALOX15 (arachidonate 15-lipoxygenase), but not ALOX5 and ALOX12 [13]. Pharmacologic inhibition of ALOX15 by PD146176 attenuates SAT1-mediated ferroptosis, indicating that ALOX15 is a downstream effector of p53-induced SAT1 expression in ferroptosis [13]. However, how cancer cells activate this p53-SAT1-ALOX15 metabolic pathway and the molecular cues behind the ferroptosis have largely remained obscure.
Promotion of GLS2 expression
Glutamine metabolism is another target for alteration in ferroptosis. Glutamine is required for the induction of ferroptosis during serum-induced injury after amino acid starvation [72]. The first step of glutamine catabolism is its conversion to glutamate, which is catalyzed by cytosolic glutamine amidotransferases or by mitochondrial glutaminases [73]. Glutamate can be further converted into α-ketoglutarate, which is an important substrate for the citric acid cycle to produce ATP in the mitochondria [73]. As a core member of the mitochondrial glutaminases, GSL2 (glutaminase 2) has been recently identified as a transcriptional target of p53 and its expression is responsible for p53-mediated oxygen consumption, mitochondrial respiration, and ATP generation in cancer cells [74]. Moreover, GLS2 expression increases cellular antioxidant function through increased GSH production in HepG2, HCT116 (a human colorectal cancer cell line), and LN-2024 (a human glioblastoma cell line) cells [74]. Based on these findings [74], GLS2 should be a negative regulator of ferroptosis. However, a recent study observed that knockdown of GLS2 inhibits (but not promotes) serum-dependent ferroptosis in fibroblasts (Fig. 1) through control of glutaminolysis [72]. Whether the specific requirement of GLS2 in erastin- or RSL3-induced ferroptosis and whether GLS2 is responsible for p53-induced ferroptosis requires further investigation.
Pro-survival function of p53 in ferroptosis
Inhibition of DPP4 activity
Colorectal cancer (CRC) develops through a series of genetic modifications, including K-RAS mutation, p53 mutation, or p53 depletion, that transform the normal colonic epithelium to an adenoma and then ultimately adenocarcinoma. The status of K-RAS mutation did not affect ferroptosis sensitivity in CRC cells [14]. In contrast, we uncovered a pro-survival function of p53 in the inhibition of ferroptosis through the regulation of DPP4 (dipeptidyl peptidase-4) localization and activity, but not DPP4 expression (Fig. 1) [14]. Knockout, knockdown, or pharmacologic inhibition of p53 increases the anticancer activity of type I ferroptosis inducer (erastin and SAS), but not type II ferroptosis inducer (RSL3 and FIN56) [14]. Remarkably, DPP4 inhibitors (vildagliptin, alogliptin, and linagliptin), but not other protease inhibitors (doxycycline, ritonavir, atazanavir, VX-222, semagacestat, Z-FA-FMK, odanacatib, ZVAD-FMK, and DAPT), completely block erastin-induced cell death in p53-deficient CRC cells [14]. DPP4 has peptidase activity and its inhibitors are a relatively new class of oral diabetes drugs. Mechanistically, p53 depletion prevents nuclear accumulation of DPP4 and then triggers membrane-associated DPP4-mediated lipid peroxidation through binding to NOX1 (NADPH oxidase 1), which finally results in ferroptosis in CRC cells [14]. These results provide evidence of a unique metabolic role for p53, linking DPP4 activity and ROS homeostasis, which may contribute to an emerging anticancer strategy for improved antitumor efficacy in precision medicine.
Promotion of CDKN1A/p21 expression
The tumor suppressor CDKN1A/p21 (cyclin dependent kinase inhibitor 1A, also known as p21WAF1/Cip1) is a key mediator of p53-dependent cell cycle arrest after DNA damage [75]. CDKN1A also has pro-survival functions in response to oxidative stress by inhibiting apoptosis. A recent study reports that p53-mediated CDKN1A expression delays the onset of ferroptosis in response to subsequent cystine deprivation in cancer cells (Fig. 1) [11]. Increased p53 expression by using the MDM2 inhibitor nutlin-3 blocks system xc− inhibitor-induced ferroptosis in HT-1080 cells [11]. In contrast, CRISPR/Cas9 technology-mediated p53 depletion cells are sensitive to ferroptosis [11], supporting a pro-survival function of p53 in ferroptosis. This reduced sensitivity to ferroptosis in wild type p53 cells requires p53-dependent expression of CDKN1A and subsequently, the production of intracellular GSH [11]. CDKN1A mediates its activities in cell cycle arrest, primarily by binding to and inhibiting the kinase activity of the cyclin-dependent kinases (CDKs) [75]. Interestingly, CDKN1A-mediated cell cycle arrest is not enough to inhibit ferroptosis since CDK4/6 inhibitors cannot block ferroptosis [11]. Understanding the mechanism of action of CDKN1A in ferroptosis may shed new light on the role of CDKN1A in the development and treatment of cancer.
Conclusions and perspectives
It is clear that systemic or local iron overload can cause various pathological conditions and disease such as hemochromatosis [76-78]. Excess iron can be a risk for carcinogenesis and neurodegenerative diseases [79, 80]. However, the mechanism responsible for cell death in response to iron overload is not fully understood. Ferroptosis seems to be a unique form of the cell death pathway linked to iron overload, in accordance with its name. The original study indicated that ferroptosis is different from other types of regulated cell death including apoptosis, necroptosis, and autophagy [15]. However, this notion has been challenged by recent studies. For example, erastin can induce CASP9/Caspase 9-dependent mitochondrial apoptosis in cancer cells [29]. Necroptosis-deficient cells seem more sensitive to ferroptosis [81]. In addition, the activation of autophagy appears to be a universal event among the induction of ferroptosis [57, 58]. The molecular mechanism in ferroptosis is more complex than previously thought [17, 18]. Indeed, these so-called core regulators of ferroptosis such as SLC7A11, GPX4, ACSL4, NFE2L2, and p53 have been engaged in the control of other types of regulated cell death. Unfortunately, the unique effector in the network of ferroptosis remains unknown and needs to be identified. The bidirectional control of ferroptosis by p53 through transcription-dependent and -independent mechanisms is context-dependent [11-14]. Furthermore, p53 is a multifunctional protein with multiple potential modifications and biochemical properties from the regulation by single-nucleotide polymorphism, long non-coding RNAs, and SOCS1 (suppressor of cytokine signaling 1) in ferroptosis [82-86]. Although the molecular switch between apoptosis and ferroptosis in p53-mediated cell death are poorly understood, certain Bcl-2 family members such as BID (BH3 interacting domain death agonist) and BBC3/PUMA may play a role in the regulation of the crosstalk between these two types of cell death through induction of mitochondrial metabolism or ER stress [36, 87]. A better understanding of the mechanisms by which p53 controls ferroptosis in cancer and non-cancer cells could allow us to develop new treatments for human diseases.
Highlights.
Ferroptosis is a form of regulated cell death
p53 plays a dual role in ferroptosis
p53 enhances ferroptosis through targeting SLC7A11, SAT1 or GLS2
p53 suppresses ferroptosis through targeting DPP4 or CDKN1A
Activation of autophagy promotes ferroptosis
Acknowledgments
We apologize to the researchers who were not referenced due to space limitations. We thank Christine Heiner (Department of Surgery, University of Pittsburgh) for her critical reading of the manuscript. This work was supported by grants from the US National Institutes of Health (R01GM115366, R01CA160417, and R01CA211070), the American Cancer Society (Research Scholar Grant RSG-16-014-01-CDD), the Natural Science Foundation of Guangdong Province (2016A030308011), the National Natural Science Foundation of China (31671435 and 81772508), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2017), Lin He’s Academician Workstation of New Medicine and Clinical Translation (2017), and the International Scientific and Technology Cooperation Program of China (2015DFA31490). GK is supported by the Ligue contre le Cancer Comité de Charente-Maritime (équipe labelisée); Agence National de la Recherche (ANR) – Projets blancs; ANR under the frame of E-Rare-2, the ERA-Net for Research on Rare Diseases; Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Chancelerie des universités de Paris (Legs Poix), Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); Fondation Carrefour; Institut National du Cancer (INCa); Inserm (HTE); Institut Universitaire de France; LeDucq Foundation; the LabEx Immuno-Oncology; the RHU Torino Lumière, the Searave Foundation; the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); and the Paris Alliance of Cancer Research Institutes (PACRI).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kastenhuber ER, Lowe SW. Putting p53 in Context. Cell. 2017;170(6):1062–1078. doi: 10.1016/j.cell.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kruiswijk F, Labuschagne CF, Vousden KH. p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat Rev Mol Cell Biol. 2015;16(7):393–405. doi: 10.1038/nrm4007. [DOI] [PubMed] [Google Scholar]
- 3.Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458(7242):1127–30. doi: 10.1038/nature07986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303(5660):1010–4. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 5.Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309(5741):1732–5. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 6.Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126(1):121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 7.Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell death and differentiation. 2011 doi: 10.1038/cdd.2011.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio J, Carnuccio R, Moll U, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G. Regulation of autophagy by cytoplasmic p53. Nat Cell Biol. 2008;10(6):676–87. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16(11):528–36. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bode AM, Dong Z. Post-translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4(10):793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 11.Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, Dixon SJ. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018;22(3):569–575. doi: 10.1016/j.celrep.2017.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57–62. doi: 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113(44):E6806–E6812. doi: 10.1073/pnas.1607152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR, Lotze MT, Zeh HJ, 3rd, Kang R, Kroemer G, Tang D. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017;20(7):1692–1704. doi: 10.1016/j.celrep.2017.07.055. [DOI] [PubMed] [Google Scholar]
- 15.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, Laurenzi V De, Maria R De, Debatin KM, DeBerardinis RJ, Deshmukh M, Daniele N Di, Virgilio F Di, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, Garcia-Saez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jaattela M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Munoz-Pinedo C, Nagata S, Nunez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25(3):486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23(3):369–79. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascon S, Hatzios SK, Kagan VE, Noel K, Jiang X, Linkermann A, Murphy ME, Overholtzer M, Oyagi A, Pagnussat GC, Park J, Ran Q, Rosenfeld CS, Salnikow K, Tang D, Torti FM, Torti SV, Toyokuni S, Woerpel KA, Zhang DD. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170(1):17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, Smith R, Lessnick SL, Sahasrabudhe S, Stockwell BR. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–8. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003;3(3):285–96. doi: 10.1016/s1535-6108(03)00050-3. [DOI] [PubMed] [Google Scholar]
- 22.Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D. The Ferroptosis Inducer Erastin Enhances Sensitivity of Acute Myeloid Leukemia Cells to Chemotherapeutic Agents. Molecular & Cellular Oncolog. 2015 doi: 10.1080/23723556.2015.1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. 2015;212(4):555–68. doi: 10.1084/jem.20140857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Radmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Forster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O’Donnell VB, Kagan VE, Schick JA, Conrad M. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–91. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, Weinlich R, Vanden Berghe T, Vandenabeele P, Pasparakis M, Bleich M, Weinberg JM, Reichel CA, Brasen JH, Kunzendorf U, Anders HJ, Stockwell BR, Green DR, Krautwald S. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111(47):16836–41. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc. 2014;136(12):4551–6. doi: 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schott C, Graab U, Cuvelier N, Hahn H, Fulda S. Oncogenic RAS Mutants Confer Resistance of RMS13 Rhabdomyosarcoma Cells to Oxidative Stress-Induced Ferroptotic Cell Death. Frontiers in oncology. 2015;5:131. doi: 10.3389/fonc.2015.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, Wang H, Cao L, Tang D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene. 2015;34(45):5617–25. doi: 10.1038/onc.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huo H, Zhou Z, Qin J, Liu W, Wang B, Gu Y. Erastin Disrupts Mitochondrial Permeability Transition Pore (mPTP) and Induces Apoptotic Death of Colorectal Cancer Cells. PLoS One. 2016;11(5):e0154605. doi: 10.1371/journal.pone.0154605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234–45. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu Y, Xie Y, Cao L, Yang L, Yang M, Lotze MT, Zeh HJ, Kang R, Tang D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol Cell Oncol. 2015;2(4):e1054549. doi: 10.1080/23723556.2015.1054549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, Stockwell BR. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497–503. doi: 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu S, Zhang Q, Sun X, Zeh HJ, 3rd, Lotze MT, Kang R, Tang D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017;77(8):2064–2077. doi: 10.1158/0008-5472.CAN-16-1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen D, Fan Z, Rauh M, Buchfelder M, Eyupoglu IY, Savaskan N. ATF4 promotes angiogenesis and neuronal cell death and confers ferroptosis in a xCT-dependent manner. Oncogene. 2017;36(40):5593–5608. doi: 10.1038/onc.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong SH, Lee DH, Lee YS, Jo MJ, Jeong YA, Kwon WT, Choudry HA, Bartlett DL, Lee YJ. Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget. 2017;8(70):115164–115178. doi: 10.18632/oncotarget.23046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M, Smith SB, Ganapathy V, Maher P. The cystine/glutamate antiporter system x(c)(−) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18(5):522–55. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, Stockwell BR. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife. 2014;3:e02523. doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Savaskan NE, Ufer C, Kuhn H, Borchert A. Molecular biology of glutathione peroxidase 4: from genomic structure to developmental expression and neural function. Biol Chem. 2007;388(10):1007–17. doi: 10.1515/BC.2007.126. [DOI] [PubMed] [Google Scholar]
- 40.Wang D, Peng Y, Xie Y, Zhou B, Sun X, Kang R, Tang D. Antiferroptotic activity of non-oxidative dopamine. Biochem Biophys Res Commun. 2016;480(4):602–607. doi: 10.1016/j.bbrc.2016.10.099. [DOI] [PubMed] [Google Scholar]
- 41.Ran Q, Liang H, Ikeno Y, Qi W, Prolla TA, Roberts LJ, 2nd, Wolf N, Van Remmen H, Richardson A. Reduction in glutathione peroxidase 4 increases life span through increased sensitivity to apoptosis. J Gerontol A Biol Sci Med Sci. 2007;62(9):932–42. doi: 10.1093/gerona/62.9.932. [DOI] [PubMed] [Google Scholar]
- 42.Canli O, Alankus YB, Grootjans S, Vegi N, Hultner L, Hoppe PS, Schroeder T, Vandenabeele P, Bornkamm GW, Greten FR. Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood. 2016;127(1):139–48. doi: 10.1182/blood-2015-06-654194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoo SE, Chen L, Na R, Liu Y, Rios C, Van Remmen H, Richardson A, Ran Q. Gpx4 ablation in adult mice results in a lethal phenotype accompanied by neuronal loss in brain. Free Radic Biol Med. 2012;52(9):1820–7. doi: 10.1016/j.freeradbiomed.2012.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8(3):237–48. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sun X, Ou Z, Chen R, Niu X, Chen, Kang R, Tang D. Activation of the p62-Keap1-NRF2 Pathway Protects against Ferroptosis in Hepatocellular Carcinoma Cells. Hepatology. 2015 doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sun X, Niu X, Chen R, He W, Chen D, Kang R, Tang D. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology. 2016;64(2):488–500. doi: 10.1002/hep.28574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen D, Tavana O, Chu B, Erber L, Chen Y, Baer R, Gu W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol Cell. 2017;68(1):224–232 e4. doi: 10.1016/j.molcel.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Telorack M, Meyer M, Ingold I, Conrad M, Bloch W, Werner S. A Glutathione-Nrf2-Thioredoxin Cross-Talk Ensures Keratinocyte Survival and Efficient Wound Repair. PLoS Genet. 2016;12(1):e1005800. doi: 10.1371/journal.pgen.1005800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reinartz A, Ehling J, Leue A, Liedtke C, Schneider U, Kopitz J, Weiss T, Hellerbrand C, Weiskirchen R, Knuchel R, Gassler N. Lipid-induced up-regulation of human acyl-CoA synthetase 5 promotes hepatocellular apoptosis. Biochimica et biophysica acta. 2010;1801(9):1025–35. doi: 10.1016/j.bbalip.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 51.Saraswathi V, Hasty AH. Inhibition of long-chain acyl coenzyme A synthetases during fatty acid loading induces lipotoxicity in macrophages. Arteriosclerosis, thrombosis, and vascular biology. 2009;29(11):1937–43. doi: 10.1161/ATVBAHA.109.195362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gassler N, Roth W, Funke B, Schneider A, Herzog F, Tischendorf JJ, Grund K, Penzel R, Bravo IG, Mariadason J, Ehemann V, Sykora J, Haas TL, Walczak H, Ganten T, Zentgraf H, Erb P, Alonso A, Autschbach F, Schirmacher P, Knuchel R, Kopitz J. Regulation of enterocyte apoptosis by acyl-CoA synthetase 5 splicing. Gastroenterology. 2007;133(2):587–98. doi: 10.1053/j.gastro.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 53.Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochemical and biophysical research communications. 2016;478(3):1338–43. doi: 10.1016/j.bbrc.2016.08.124. [DOI] [PubMed] [Google Scholar]
- 54.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, Prokisch H, Trumbach D, Mao G, Qu F, Bayir H, Fullekrug J, Scheel CH, Wurst W, Schick JA, Kagan VE, Angeli JP, Conrad M. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, Kapralov AA, Amoscato AA, Jiang J, Anthonymuthu T, Mohammadyani D, Yang Q, Proneth B, Klein-Seetharaman J, Watkins S, Bahar I, Greenberger J, Mallampalli RK, Stockwell BR, Tyurina YY, Conrad M, Bayir H. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xie Y, Kang R, Sun X, Zhong M, Huang J, Klionsky DJ, Tang D. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11(1):28–45. doi: 10.4161/15548627.2014.984267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26(9):1021–32. doi: 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, 3rd, Kang R, Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–8. doi: 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15(2):81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501. doi: 10.1038/ncb2979. [DOI] [PubMed] [Google Scholar]
- 61.Hou W, Zhang Q, Yan Z, Chen R, Zeh HJ, III, Kang R, Lotze MT, Tang D. Strange attractors: DAMPs and autophagy link tumor cell death and immunity. Cell Death Dis. 2013;4:e966. doi: 10.1038/cddis.2013.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–9. doi: 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40(2):253–66. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 64.Dayalan Naidu S, Kostov RV, Dinkova-Kostova AT. Transcription factors Hsf1 and Nrf2 engage in crosstalk for cytoprotection. Trends Pharmacol Sci. 2015;36(1):6–14. doi: 10.1016/j.tips.2014.10.011. [DOI] [PubMed] [Google Scholar]
- 65.Wang SJ, Li D, Ou Y, Jiang L, Chen Y, Zhao Y, Gu W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016;17(2):366–373. doi: 10.1016/j.celrep.2016.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moll UM, Petrenko O. The MDM2-p53 interaction. Mol Cancer Res. 2003;1(14):1001–8. [PubMed] [Google Scholar]
- 67.Thomasova D, Bruns HA, Kretschmer V, Ebrahim M, Romoli S, Liapis H, Kotb AM, Endlich N, Anders HJ. Murine Double Minute-2 Prevents p53-Overactivation-Related Cell Death (Podoptosis) of Podocytes. J Am Soc Nephrol. 2014 doi: 10.1681/ASN.2014040345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, Pratt DA. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent Sci. 2017;3(3):232–243. doi: 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reed SM, Quelle DE. p53 Acetylation: Regulation and Consequences. Cancers (Basel) 2014;7(1):30–69. doi: 10.3390/cancers7010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thomas T, Thomas TJ. Polyamine metabolism and cancer. J Cell Mol Med. 2003;7(2):113–26. doi: 10.1111/j.1582-4934.2003.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mandal S, Mandal A, Park MH. Depletion of the polyamines spermidine and spermine by overexpression of spermidine/spermine N(1)-acetyltransferase 1 (SAT1) leads to mitochondria-mediated apoptosis in mammalian cells. Biochem J. 2015;468(3):435–47. doi: 10.1042/BJ20150168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell. 2015;59(2):298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16(10):619–34. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107(16):7455–60. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–14. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Siddique A, Kowdley KV. Review article: the iron overload syndromes. Aliment Pharmacol Ther. 2012;35(8):876–93. doi: 10.1111/j.1365-2036.2012.05051.x. [DOI] [PubMed] [Google Scholar]
- 77.Silva B, Faustino P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim Biophys Acta. 2015;1852(7):1347–59. doi: 10.1016/j.bbadis.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 78.Crielaard BJ, Lammers T, Rivella S. Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discov. 2017;16(6):400–423. doi: 10.1038/nrd.2016.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13(5):342–55. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014;13(10):1045–60. doi: 10.1016/S1474-4422(14)70117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Muller T, Dewitz C, Schmitz J, Schroder AS, Brasen JH, Stockwell BR, Murphy JM, Kunzendorf U, Krautwald S. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci. 2017;74(19):3631–3645. doi: 10.1007/s00018-017-2547-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li T, Liu X, Jiang L, Manfredi J, Zha S, Gu W. Loss of p53-mediated cell-cycle arrest, senescence and apoptosis promotes genomic instability and premature aging. Oncotarget. 2016;7(11):11838–49. doi: 10.18632/oncotarget.7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, Scott JP, Cai KQ, Campbell MR, Porter DK, Wang X, Bell DA, Li X, Garlick DS, Liu Q, Hollstein M, George DL, Murphy ME. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev. 2016;30(8):918–30. doi: 10.1101/gad.275891.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang GX, Tu HC, Dong Y, Skanderup AJ, Wang Y, Takeda S, Ganesan YT, Han S, Liu H, Hsieh JJ, Cheng EH. DeltaNp63 Inhibits Oxidative Stress-Induced Cell Death, Including Ferroptosis, and Cooperates with the BCL-2 Family to Promote Clonogenic Survival. Cell Rep. 2017;21(10):2926–2939. doi: 10.1016/j.celrep.2017.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, Shi Y, Shen Y, Liu X, Liai W, Yang R, Xiao D, Cheng Y, Liu S, Zhou H, Cao Y, Yu W, Muegge K, Yu H, Tao Y. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018 doi: 10.1158/0008-5472.CAN-17-3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Saint-Germain E, Mignacca L, Vernier M, Bobbala D, Ilangumaran S, Ferbeyre G. SOCS1 regulates senescence and ferroptosis by modulating the expression of p53 target genes. Aging (Albany NY) 2017;9(10):2137–2162. doi: 10.18632/aging.101306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neitemeier S, Jelinek A, Laino V, Hoffmann L, Eisenbach I, Eying R, Ganjam GK, Dolga AM, Oppermann S, Culmsee C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017;12:558–570. doi: 10.1016/j.redox.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]