

Abstract
Pharmacologic inhibition of acetyl-CoA carboxylase (ACC) enzymes, ACC1 and ACC2, offers an attractive therapeutic strategy for non-alcoholic fatty liver disease (NAFLD) via simultaneous inhibition of fatty acid synthesis and stimulation of fatty acid oxidation. However, the effects of ACC inhibition on hepatic mitochondrial oxidation, anaplerosis, and ketogenesis in vivo are unknown. Here, we evaluated the impact of a novel liver-directed allosteric inhibitor of ACC1 and ACC2 (Compound 1) on these parameters, as well as glucose and lipid metabolism, in control and diet-induced rodent models of NAFLD. Oral administration of Compound 1 preferentially inhibited ACC enzymatic activity in the liver, reduced hepatic malonyl-CoA levels and enhanced hepatic ketogenesis by 50%. Furthermore, administration for 6 days to high-fructose fed rats resulted in a 20% reduction in hepatic de novo lipogenesis. Importantly, long-term treatment (21 days) significantly reduced high-fat sucrose diet (HFSD)-induced hepatic steatosis, PKCε activation and hepatic insulin resistance. ACCi treatment was associated with a significant increase in plasma triglycerides (∼30 to 130%, depending on length of fasting). ACCi-mediated hypertriglyceridemia could be attributed to a ∼15% increase in hepatic VLDL production and ∼20% reduction in triglyceride clearance by lipoprotein lipase (LPL) (P ≤ 0.05). At the molecular level, these changes were associated with increases in LXR/SREBP1 and decreases in PPARα target activation and could be reversed with fenofibrate co-treatment in a high-fat diet mouse model. Collectively, these studies warrant further investigation into the therapeutic utility of liver-directed ACC inhibition for the treatment of NAFLD and hepatic insulin resistance.
Keywords: Hepatic steatosis, triglycerides, lipogenesis, fatty acid oxidation, diacylglycerol, malonyl-CoA, lipase/lipoprotein, protein kinase Cε
Introduction
Non-alcoholic fatty liver disease (NAFLD) affects 1 in 3 Americans and is strongly associated with obesity, insulin resistance, type 2 diabetes (T2D), and cardiovascular disease (1). NAFLD encompasses a spectrum of pathologies ranging from simple steatosis to non-alcoholic steatohepatitis (NASH) and is a key predisposing factor for the development of cirrhosis and hepatocellular carcinoma (2). NAFLD develops when the rate of fatty acid input (fatty acid uptake and de novo synthesis with subsequent esterification to triglycerides) exceeds the rate of fatty acid output (fatty acid oxidation and secretion of VLDL) (3). Genetic and physiological mechanisms regulating these processes have developed during evolution to cope with starvation but become dysregulated in the face of nutritional oversupply (4). In this setting, they converge to promote the accumulation of ectopic lipids leading to the development of NAFLD and hepatic insulin resistance, which increases the risk of fasting hyperglycemia and T2D (2). While the direction of causality for this relationship remains uncertain (5), numerous studies have demonstrated that hepatic accumulation of the lipid moiety, diacylglycerols (DAGs), leads to the activation of protein kinase C epsilon (PKCε) and impairment of hepatic insulin action (6). In parallel, dysregulated adipose tissue lipolysis as well as intrahepatic lipolysis can drive hepatic gluconeogenesis by increasing fatty acid flux to the liver, hepatic acetyl-CoA levels and pyruvate carboxylase (PC) activity (7). Collectively, these processes promote hepatic insulin resistance and increased rates of hepatic glucose production (HGP) that promote fasting and postprandial hyperglycemia in T2D. Moreover, they suggest that therapies targeted to prevent hepatic lipid accumulation may be efficacious for treating NAFLD, insulin resistance and T2D.
Acetyl-CoA carboxylase (ACC) catalyzes the ATP-dependent condensation of acetyl-CoA and carbonate to form malonyl-CoA and plays a crucial role in fatty acid metabolism (8). In mammals, there are two isoforms of the ACC enzyme: ACC1, which is primarily expressed in lipogenic tissues (liver, adipose, lactating mammary gland) and ACC2, which is primarily expressed in oxidative tissues (heart, skeletal muscle) (8). ACC1 is located in the cytosol and is responsible for the rate-controlling and first committed reaction in de novo lipogenesis (DNL). In contrast, the mitochondrial membrane-embedded ACC2 negatively regulates fatty acid oxidation by producing localized malonyl-CoA, which allosterically inhibits carnitine palmitoyltransferase I (CPT1) and the transfer of long-chain CoAs into the mitochondria (9).
The importance of ACC as a drug discovery target to lower liver triglycerides by simultaneously reducing fatty acid synthesis and stimulating fatty acid oxidation has been strongly supported by genetic and pharmacological studies (10). Specifically, we previously demonstrated that reducing hepatic expression of ACC1 and ACC2 using antisense oligonucleotides (ASOs) results in marked reductions in hypertriglyceridemia, hepatic triglyceride content and reversal of hepatic insulin resistance in a high-fat fed rodent model of NAFLD and hepatic insulin resistance (11). In addition, a novel liver-directed allosteric inhibitor of ACC1 and ACC2 (GS-0976) has been shown to improve hepatic steatosis and markers of liver injury in patients with NASH and to reduce hepatic steatosis and dyslipidemia in rodent models of obesity (12-14). Despite this, the effect of liver-directed ACC inhibition on hepatic mitochondrial oxidation, anaplerosis, and ketogenesis in vivo are unknown. Here, we performed a comprehensive set of metabolic flux analyses to assess these parameters in both chow-fed rats and in rat models of diet-induced obesity (DIO). We hypothesized that chronic inhibition of hepatic ACC by Compound 1 would lead to reduced hepatic fat content, due to increases in hepatic mitochondrial oxidation and reduced de novo lipogenesis (DNL), which in turn would lead to increased hepatic insulin sensitivity. The data presented demonstrate that liver-directed inhibition of ACC significantly reduces hepatic steatosis and hepatic insulin resistance; however long-term treatment was paradoxically associated with increased plasma triglycerides, consistent with a recent report on a prior clinical candidate (15). As such, we also evaluated the effect of ACC inhibition on triglyceride metabolism and the effect of co-administration of a fibrate to understand the mechanism for this observation.
Materials and Methods
Animal studies
Male Sprague-Dawley (SD) rats were purchased from Charles River Laboratories. For HFSD studies, male SD rats were placed on a safflower oil-based high-fat diet (Dyets #112245) and supplemented with 1% (wt/vol) sucrose (Domino Foods, Inc) drinking water. Following 3 days of diet, rats were given 10 mg/(kg BW) Compound 1 in ∼200 mg of peanut butter (Skippy® Brand) or vehicle control once daily for 21 days. Unless otherwise stated all rats were sacrificed by intravenous pentobarbital 2-6 h following the last dose of Compound 1. Tissue samples were collected as previously described (7) and stored at −80°C until subsequent analyses. All procedures were approved by the Institutional Animal Care and Use Committee of Yale University School of Medicine. Mouse studies were conducted at Covance Central Laboratories. C57BL/6 mice were administered either a standard chow diet (Harlan Teklad Global Diets 2014, TD2014) or a high fat, high cholesterol diet (Research Diets Inc, DB12079B), herein referred to as a fast-food diet (FFD) (16) for 5 months. Mice were administered fructose/glucose in drinking water. Compound 1 was dosed at 1-10 mg/kg/day PO. Fenofibrate (0.1%) was administered in chow ad libitum in the FFD.
Metabolic Flux and Hyperinsulinemic-Euglycemic Clamp Studies
Metabolic flux studies and clamp studies were performed as previously described (17) and in the Online Materials and Methods.
DNL Studies
Rats were fed a high fructose (60%; Research Diets D02022704) diet and treated with 10 mg/kg BW/day Compound 1 for 6 days. De novo lipogenesis (DNL) was assessed according to previously established methods (18).
Plasma Analysis and Lipoprotein Measurements
The lipid distribution in plasma lipoprotein fractions and plasma metabolites were assessed as previously described (17, 19) and in the Online Materials and Methods Section. βOHB enrichment and EGP were measured by GC/MS as previously described (17). Hepatic mitochondrial fluxes were assessed by a combined 13/C NMR/GC-MS method using PINTA as previously described (20).
Hepatic VLDL Secretion and Plasma Lipid Clearance
Hepatic VLDL-triglyceride secretion and plasma lipid clearance were measured as previously described (21). LPL activity was assessed in pre- and post-heparin plasma using the LPL Activity Assay Kit from Cell Biolabs, Inc. according to the manufacturer's instructions.
Hepatic Lipid Analysis
Rat liver triglycerides, DAGs, ceramides, long-chain acyl-CoAs, acylcarnitines, malonyl-CoA, and acetyl-CoA were extracted and measured by LC-MS/MS as described (17, 22). Quantification of mouse hepatic triglycerides and cholesterol was performed by Metabolon Inc.
RNA Isolation, RNA-seq, qRT-PCR, and Western blot analysis
epatic RNA isolation and qRT-PCR analysis were performed as previously described (22). qRT-PCR analysis of lipid metabolism related genes was performed using the Rat Lipid Metabolism Tier 1 and 2 Arrays (Bio-Rad) as per the manufacturer's instructions. RNA-seq, Western blot analysis, and PKCε translocation were performed as previously described (22) and in the Online Materials and Methods Section.
Statistics
Statistics were performed as described in the Online Materials and Methods section.
Results
Compound 1 Target Engagement
To determine the potential of Compound 1 to exhibit in vivo efficacy, we measured ACC engagement and malonyl-CoA levels in the tissues of chow-fed male SD rats after acute treatment (1.5 h) with 10 mg/kg Compound 1 (ACCi) or vehicle control. Compound 1 interacts within the dimerization site of the biotin carboxylase (BC) domain of ACC1 and ACC2 and specifically binds to the negative regulatory AMPK phosphorylation site (hACC1R172 and hACC2R277) (Figure 1A–B). By using a phospho-specific antibody that recognizes this site (pACCS79), we found that acute treatment with ACCi significantly inhibited our ability to detect hepatic pACCS79 (Figure 1C), thereby confirming Compound 1 engagement of ACC. Consistent with this, ACCi treatment significantly reduced hepatic malonyl-CoA levels (Figure 1D), while acetyl-CoA levels were unaffected (Figure S1A–D). Importantly, no appreciable differences were detected in the phosphorylation status of ACC or malonyl-CoA levels in other metabolic tissues, including gastrocnemius (muscle), heart, epididymal white adipose tissue (eWAT), or brain (Figure 1C–D).
Figure 1. Liver-directed inhibition of ACC by Compound 1.
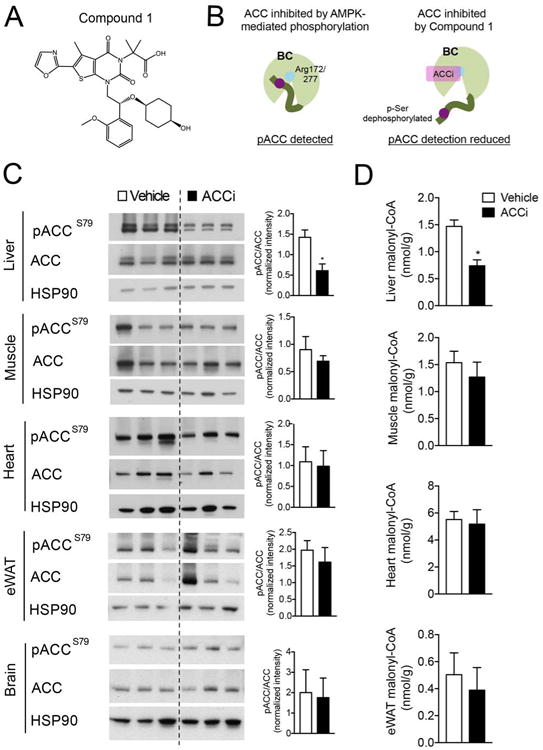
(A) Chemical structure of Compound 1. (B) Diagram depicting the use of AMPK phosphorylation sites as a biomarker of ACC engagement by Compound 1. (C-D) Representative Western blots depicting pACC (S79) and total ACC (C) and malonyl-CoA (D) in the liver, gastrocnemius muscle (muscle), heart, epididymal white adipose (eWAT) and brain of chow-fed male Sprague-Dawley (SD) rats treated with an intragastric bolus of 10 mg/kg Compound 1 (ACCi) or vehicle control for 1.5 h. HSP90 was used as a loading control. Quantification of blots shown to the right. Data are presented as mean ± SEM. n = 6-9 per treatment group. *P ≤ 0.05 by unpaired student's t-test compared to vehicle control.
Liver-Directed Inhibition of ACC Enhances Hepatic Ketogenesis and Reduces Hepatic Lipogenesis
To explore the functional consequence of inhibiting ACC1 and ACC2, we measured in vivo rates of hepatic mitochondrial oxidation, anaplerosis, ketogenesis, and lipogenesis in male SD rats. In accordance with ACCi-mediated reductions in hepatic malonyl-CoA content (Figure 1D), acute treatment with 10 mg/kg ACCi significantly increased plasma βOHB and whole-body βOHB turnover compared to vehicle controls (Figure 2A–B). While no significant differences were observed in whole-body glucose turnover or fasting (∼6 h) plasma glucose (Figure S2A–C), acute ACCi treatment significantly reduced plasma non-esterified fatty acids (NEFAs), most likely due to increased hepatic fat oxidation (Figure S2G). Plasma insulin, total cholesterol and triglycerides were similar between treatment groups (Figure S2D–F).
Figure 2. Inhibition of ACC enhances hepatic ketogenesis and reduces hepatic DNL in chow and high-fructose fed rats, respectively.
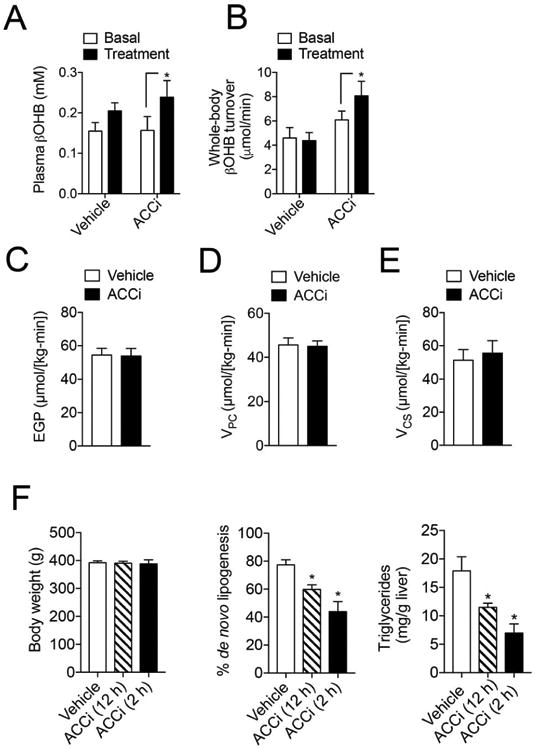
(A-B). Plasma βOHB (A) and whole-body βOHB turnover (B) in chow-fed male Sprague-Dawley (SD) rats fasted for 6 h, infused with 0.1 mg/(kg-min) [13C4]βOHB (basal) and treated with an intragastric bolus of 10 mg/kg Compound 1 (ACCi) or vehicle control for 1.5 h (treatment). n = 6-9 per treatment group. (C-E) Endogenous glucose production (C), hepatic pyruvate carboxylase flux (VPC) (D) and hepatic mitochondrial citrate flux (VCS) (E) in male SD rats fasted overnight, infused with 0.1 mg/(kg-min) [1,2,3,4,5,6,6-2H7]glucose and 40 uM/(kg-min) [3-13C]lactate and treated as in (A). n = 7 per treatment group. (F) Body weight, % de novo lipogenesis (DNL) and hepatic triglyceride content in male SD rats fed a high fructose diet (60%) for 6 days and treated with ACCi [10 mg/(kg BW per day)] or vehicle control. Rates of DNL were assessed in the hepatic fatty acid pool after 3 d treatment with deuterated water. Rats were sacrificed 12 h (n = 12-14 per treatment group) or 2 h (n = 6 per treatment group) after dosing. Data are presented as mean ± SEM. *P ≤ 0.05 by paired student's t-test compared to basal in each treatment group (A-B). *P ≤ 0.05 by unpaired student's t-test compared to vehicle control (C-F).
We recently developed a positional isotopomer NMR tracer analysis (PINTA) method to measure changes in hepatic mitochondrial flux rates in awake rodents using [3-13C]lactate and [1,2,3,4,5,6,6-2H7]glucose tracers (20). Using this approach, we examined rates of hepatic pyruvate carboxylase flux (VPC) and citrate synthase flux (VCS) in awake overnight fasted chow-fed rats after an intragastric bolus of 10 mg/kg ACCi or vehicle control. In agreement with results obtained from ∼6 h fasted rats, no significant differences were detected in endogenous glucose production (EGP) or rates of hepatic mitochondrial oxidative (VCS) and gluconeogenic fluxes from pyruvate carboxylase (VPC) (Figure 2C–E; Figure SH–J).
We next ascertained the physiological significance of inhibiting ACC1. To this end, hepatic DNL was assessed using deuterated water in high-fructose fed rats treated with ACCi or vehicle control [10 mg/(kg BW/day) × 6 days]. Consistent with previous reports (15, 23), ACCi treatment markedly reduced hepatic DNL and triglyceride content in a time-dependent manner by 23–36% and 43–61%, respectively (Figure 2F). Collectively, these studies demonstrate the in vivo efficacy of ACCi to significantly reduce hepatic ACC activity, increase fatty acid oxidation, and reduce hepatic DNL.
Long-term Inhibition of ACC Reduces Hepatic Steatosis but Increases Fasting Plasma Triglycerides and NEFAs
In order to determine the long-term effect of ACC inhibition on glucose and lipid metabolism, male SD rats were fed a high-fat diet supplemented with 1% sucrose drinking water (HFSD), a model that explores the ability of ACCi to reverse diet-induced NAFLD and insulin resistance by increasing fat oxidation and/or inhibiting DNL. To induce hepatic steatosis, rats were fed a HFSD for 3 days and then treated orally with 10 mg/kg/day ACCi or vehicle control for 21 days (Figure 3A). This dose led to Compound 1 liver exposure of ∼10 μM and inhibition of malonyl-CoA that approximated levels achieved clinically with GS-0976 (Figure 3B–C). Importantly, Compound 1 muscle levels were close to the limit of quantitation (0.03 μM), thus further supporting the liver specificity of Compound 1 (Figure 3C). Body weight, fasting plasma glucose, insulin, C-peptide, total cholesterol, and βOHB were similar between treatment groups, while plasma high-density lipoprotein cholesterol (HDL-C) was significantly increased in overnight fasted ACCi-treated rats compared to controls (Table 1). Additionally, long-term inhibition of hepatic ACC did not alter plasma aminotransferases (Table 1). Importantly, ACCi prevented HFSD-induced hepatic steatosis, as observed both macroscopically and microscopically by H&E and Oil Red O staining (Figure 3D). Paradoxically, and consistent with human clinical data, long-term inhibition of ACC significantly increased plasma triglycerides by 30–130% (Table 1), despite a ∼70% reduction in hepatic triglyceride content (Figure 3E). Liver-directed inhibition of ACC also caused a 25% increase in fasting plasma NEFA concentration (Table 1).
Figure 3. Compound 1 reduces hepatic steatosis in high-fat diet fed rats.
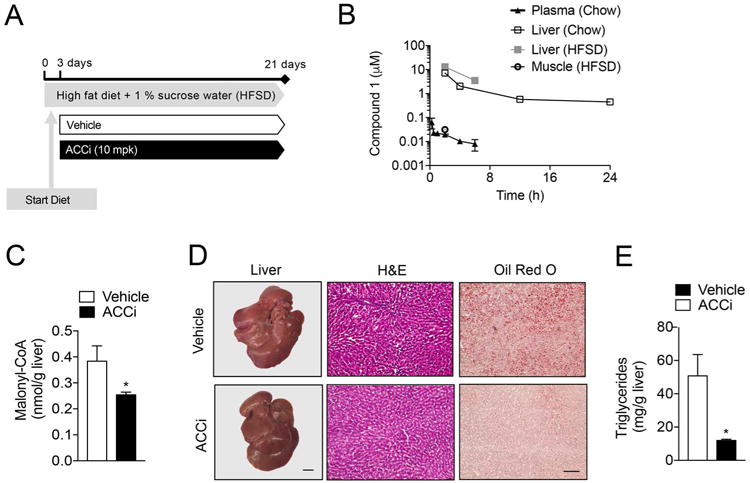
(A) Male Sprague-Dawley (SD) rats were fed a high-fat diet (60% Safflower oil) supplemented with 1% sucrose drinking water (HFSD) for 3 days and treated with 10 mg/kg/day Compound 1 (ACCi) or vehicle control for 21 days. (B) Plasma, liver and muscle pharmacokinetics following single oral administration of 10 mg/kg Compound 1 to male SD-rats fed a chow diet (Chow) or rats treated as in (A) and treated with ACCi 2 or 6 h before sacrifice (HFSD). n = 3-6 per treatment group. (C) Hepatic malonyl-CoA levels in overnight fasted rats treated as in (A). (D) Representative livers (left) and liver sections stained with H&E (middle) and Oil Red O (right) of overnight rats treated as in (A). Macroscopic scale bar = 6.4 mm; microscopic scale bar = 50 μm. (E) Hepatic triglyceride (TAG) content in overnight fasted rats treated as in (A). Data are presented as mean ± SEM. *P ≤ 0.05 by unpaired student's t-test compared to vehicle control. n = 5 per treatment group.
Table 1. Metabolic parameters.
| 6 h fast | Overnight fast | |||
|---|---|---|---|---|
| Vehicle | ACCi | Vehicle | ACCi | |
| Physiologic Parameters | ||||
| Body weight (g) | 454.6 ± 10.9 | 421 ± 21.8 | 416 ± 16.5 | 409 ± 14.8 |
| Plasma metabolites | ||||
| Glucose (mg/dL) | – | – | 104.4 ± 4.7 | 105.3 ± 5.2 |
| Insulin (μU/ml) | – | – | 15.9 ± 3.0 | 19.3 ± 3.1 |
| C-peptide (pmol/L) | – | – | 366.4 ± 58.5 | 361.1 ± 67.5 |
| Triglycerides (mg/dL) | 122.5 ± 15.2 | 280.7 ± 35.7* | 46.2 ± 4.1 | 61.7 ± 4.8* |
| Total cholesterol (mg/dL) | 79.8 ± 5.1 | 69.3 ± 4.6 | 60.1 ± 1.9 | 64.4 ± 2.7 |
| HDL-C (mg/dL) | 31.75 ± 2.3 | 32.5 ± 2.0 | 16.0 ± 1.1 | 19.0 ± 1.0* |
| NEFAs (mM) | 0.35 ± 0.1 | 0.45 ± 0.1 | 0.90 ± 0.05 | 1.13 ± 0.07* |
| βOHB (mM) | 0.22 ± 0.6 | 0.20 ± 0.02 | 1.54 ± 1.4 | 1.40 ± 0.09 |
| ALT (Units/L) | 30.6 ± 3.5 | 26.3 ± 0.93 | 30.2 ± 3.39 | 23.9 ± 0.28 |
| AST (Units/L) | 81.3 ± 10.4 | 80.2 ± 9.0 | 105.2 ± 38.7 | 86.3 ± 11.1 |
Male Sprague-Dawley (SD) rats were fed a high fat diet supplemented with 1% sucrose drinking water (HFSD) for 3 days and treated with 10 mg/kg/day GS-834356 (ACCi) or vehicle control for 21 days.HDL-C: high-density lipoprotein cholesterol; NEFAs: non-esterified fatty acids; βOHB: β-hydroxybutyrate; ALT: alanine aminotransferase; AST: aspartate aminotransferase
Data are presented as mean ± SEM; n = 8-9 per group (6 h) or 11-12 per group (12 h fast)
P≤ 0.05 compared to vehicle-treated rats by unpaired student's t-test.
Liver-Directed ACC Inhibition Improves Hepatic Insulin Sensitivity
Hepatic steatosis is strongly associated with hepatic insulin resistance, as least in part by diacylglycerol (DAG)-mediated activation of PKCε and subsequent impairment of insulin receptor kinase (IRK) activity through increased phosphorylation of IRK threonine 1160 (murine threonine 1150) (22). Given the ability of long-term ACC inhibition to significantly reduce hepatic triglycerides, we next assessed whole-body and tissue-specific insulin action in weight-matched HFSD-fed rats using hyperinsulinemia-euglycemic clamp studies (Figure S3A). Oral administration of ACCi (10 mg/kg/day × 21 days) significantly reduced hepatic malonyl-CoA levels while fasting plasma glucose concentrations were unchanged (Figure S3B–C). Under hyperinsulinemic conditions, the glucose infusion rate (GIR) required to maintain euglycemia was similar between treatment groups (Figure 4A, S3D) and insulin-mediated whole-body glucose disposal (Rd) was unchanged (Figure 4B). However, ACCi treatment increased basal rates of hepatic glucose production (23%, P ≤ 0.05), which could be attributed to a 16% increase in hepatic acetyl-CoA levels (Figure 4D). In contrast, insulin-mediated suppression of EGP, indicative of hepatic insulin sensitivity, was significantly improved (Figure 4E). Because adipose tissue lipolysis can indirectly alter hepatic gluconeogenesis, we next assessed plasma NEFA concentrations during the hyperinsulinemic-clamp. As shown in Figure 4F and Figure S3E, ACC inhibition did not significantly change plasma insulin levels or insulin-mediated suppression of circulating fatty acids (Figure 4F), suggesting a direct hepatocellular effect for ACCi-mediated alterations in EGP.
Figure 4. Liver-directed inhibition of ACC improves hepatic insulin sensitivity.
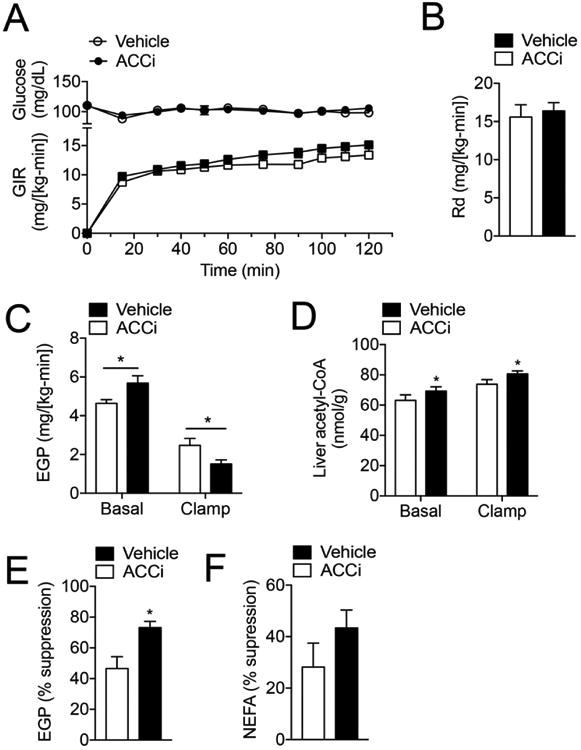
(A) Plasma glucose and glucose infusion rate (GIR) during a hyperinsulinemia-euglycemic clamp (4 mU/[kg-min]) insulin) in rats fed a high fat diet supplemented with 1% sucrose drinking water (HFSD) for 3 days and treated with 10 mg/kg/day Compound 1 (ACCi) or vehicle control for 21 days (n = 10 per group). (B) Insulin-stimulated glucose disposal (Rd). (C) Endogenous glucose production (EGP) during the basal and steady-state period of the clamp. (D) Hepatic acetyl-CoA levels in basal (overnight fasted) and clamped rats treated as in (A). (E) Insulin-mediated suppression of EGP during the clamp. (F) Insulin-mediated suppression of plasma non-esterified fatty acids (NEFAs) during the clamp. In all panels, n = 10 per treatment group. Data are presented as mean ± SEM. *P ≤ 0.05 by unpaired student's t-test compared to vehicle control.
To understand the mechanism by which ACCi improved hepatic insulin sensitivity, we measured hepatic DAGs, ceramides, long-chain acyl-CoAs (LCCoAs), acylcarnitines, and PKCε translocation in 6 h fasted HFSD-fed rats. Long-term inhibition of ACC resulted in a 27% reduction (P≤ 0.05) in hepatic DAG membrane content (Figure 5A and S5A), whereas hepatic lipid and cytosolic DAG content, LCCoAs, and acylcarnitines were unchanged (Figures S4 and S5B-C). The ACCi-mediated reduction in membrane DAGs was associated with a significant decrease in PKCε translocation to the plasma membrane and increased hepatic insulin action, as reflected by a 2–3–fold increase in insulin-stimulated AKT and IRK phosphorylation (Figure 5C–D). While total hepatic ceramide content was significantly decreased by ACCi treatment (Figure 5B), two ceramide species implicated in resistance (C16 and C18 ceramides (24, 25)) were not statistically different between treatment groups (Figure S5D). Taken together, these data demonstrate that long-term inhibition of hepatic ACC reverses diet-induced NAFLD and hepatic insulin resistance by reducing DAG-mediated PKCε activation and improving hepatic insulin responsiveness.
Figure 5. Inhibition of hepatic ACC reduces membrane DAG content and PKCε translocation and increases hepatic insulin action in rats fed a HFSD.
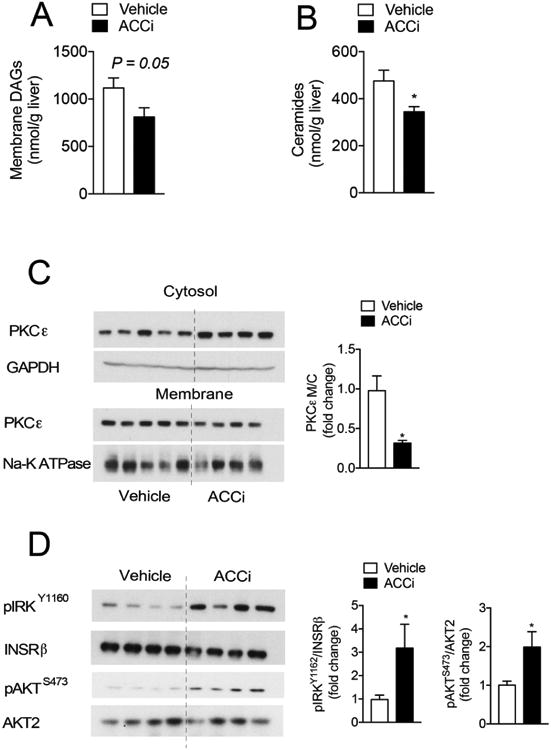
(A-C) Membrane-associated diacylglycerol (DAG) (A), total ceramides (B), and representative Western blot of protein kinase C epsilon (PKCε) membrane to cytosol translocation (C) in the livers of 6 h fasted rats that were fed a high-fat diet (60% Safflower oil) supplemented with 1% sucrose drinking water (HFSD) for 3 days and treated with 10 mg/kg/day Compound 1 (ACCi) or vehicle control for 21 days. GAPDH and NA-K ATPase were used as cytosolic and membrane loading controls, respectively. Quantification of blot shown in the right panel. (D) Representative Western blot of pAKTS473, AKT2, pIRKY1162, and INSRβ in clamped livers of rats treated as in (A). Quantification of blot shown to the right. n = 8-9 (A and C) or 12-13 (B) per treatment group. Data are presented as mean ± SEM. *P ≤ 0.05 by unpaired student's t-test compared to vehicle control.
Inhibition of Hepatic ACC Increases VLDL Secretion and Reduces LPL Activity
We next sought to define the mechanism responsible for the development of ACCi-mediated hypertriglyceridemia. To this end, we first measured plasma triglyceride concentrations in FPLC-separated lipoprotein fractions from overnight fasted HFSD-fed rats. Consistent with previous reports, long-term inhibition of ACC mainly increased plasma triglycerides in the VLDL fraction (Figure 6A–B). Hypertriglyceridemia may represent an increase in VLDL-triglyceride production that overcomes peripheral clearance and/or from a decrease in clearance without a compensatory decrease in production (26). As such, we measured triglyceride secretion in overnight fasted ACCi-treated rats after injection of Poloxamer 405 to inhibit lipolysis of triglyceride-rich lipoproteins (27). Surprisingly, long-term inhibition of ACC significantly increased hepatic VLDL-triglyceride release by 15% (Figure 6C–D), despite a marked reduction in hepatic DNL (Figure 2F). As lipoprotein lipase (LPL)-mediated triglyceride catabolism is another major determinant of circulating plasma triglycerides (28), we also assessed plasma lipid clearance in ACCi-treated HFSD-fed rats after an intravenous bolus of Intralipid. Long-term inhibition of ACC was also associated with a significant reduction in lipid clearance by LPL (Figure 6E–F).
Figure 6. Long-term inhibition of ACC increases hepatic TG secretion and reduces plasma lipid clearance in HFSD-fed rats.
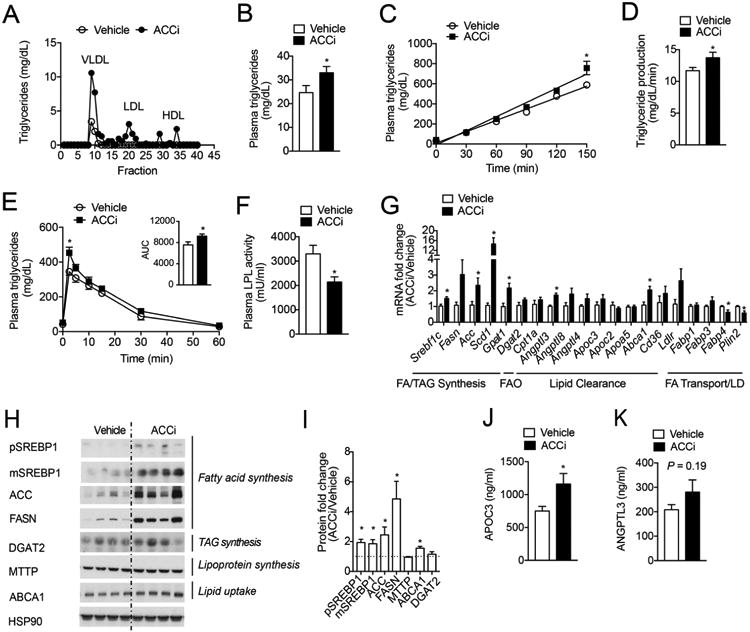
(A) Plasma triglyceride content of FPLC-fractionated lipoproteins from pooled plasma (n = 4 per group) of overnight fasted rats that were fed a high-fat diet (60% Safflower oil) supplemented with 1% sucrose drinking water (HFSD) for 3 days and treated with 10 mg/kg/day Compound 1 (ACCi) or vehicle control for 21 days. (B) Fasting plasma triglycerides in rats treated as in (A). n = 12 per group. (C-D) Hepatic triglyceride production in overnight fasted rats treated as in (A) and injected with Poloxamer 407 to inhibit lipolysis of triglyceride rich lipoproteins (TRL). n = 10-14 per treatment group. (E) Lipid clearance test in overnight fasted rats treated as in (A) and given an intravenous bolus of 20% Intralipid conjugated with 3H-labeled triolein. n = 6 per treatment group. (F) Post-heparin plasma LPL activity in rats treated as in (A). n = 7-9 per treatment group. (G–I) mRNA (G) and protein (H) expression of indicated genes in the livers of overnight fasted rats treated as in (A). n = 7–10 per treatment group. In panel (H), HSP90 was used as a loading control. Quantification of blot shown in panel (I). (J) Plasma APOC3 concentration in rats treated as in (A). n = 7 per treatment group. (K) Plasma ANGPTL3 concentration in rats treated as in (A). n = 12 per treatment group. Data are presented as mean ± SEM. In panels (B-G and J-K), *P ≤ 0.05 by unpaired student's t-test compared to vehicle control.
Disequilibrium of nuclear hormone receptor regulation in ACCi-treated rats
To ascertain the molecular mechanism by which long-term ACC inhibition leads to alterations in triglyceride production and clearance, we performed unbiased gene expression arrays in HFSD-fed rats treated with 10 mg/(kg BW per day) ACCi or vehicle control. These studies revealed that the mRNA levels of many genes regulated by the nuclear transcription factors, LXRα and SREBP1c, (Acc, Scd1, Pnpla3, Abca1) were significantly increased in the livers of ACCi-treated rats compared to controls (Figure S6). In contrast, the mRNA levels of genes regulated by PPARα, including Plin2, Fabp2, and Fabp4 tended to be reduced (Figure S6). Importantly, these findings were recapitulated at the mRNA level with independent primers and at the protein level by Western blotting. Of note, long-term ACCi treatment significantly increased SREBP1 protein expression and the mRNA and protein levels of SREBP1c target genes (29), including ACC, FASN, SCD1 and GPAT1 (Figure 6G–I). Consistent with this, liver-directed inhibition of ACC also significantly increased the hepatic 18:1/18:0 ratio, an indirect marker of SCD1 activity (Figure S4A). In contrast, ACCi-treatment did not alter the protein expression of other genes (DGAT2 and MTTP) involved in triglyceride and lipoprotein synthesis, respectively (Figure 6G–I). PPARα target genes also downregulate the expression of the LPL inhibitor APOC3 (30), which showed a marked increase (P ≤ 0.05) in the plasma of ACCi-treated rats compared to controls (Figure 6J). While ANGPTL3, a negative regulator of LPL activity and LXR target gene (31), was significantly increased at the mRNA level (Figure 6G), no significant differences were detected at the protein level (Figure 6K). Additionally, ACC inhibition significantly increased hepatic ABCA1 expression (Figure 7G–I), a key regulator of reverse cholesterol transport and plasma HDL-cholesterol levels (32). Collectively, this gene signature is consistent with previous reports documenting a derepression of LXR/SREBP1 target genes caused by reduced levels of polyunsaturated fatty acids (PUFAs) (15, 33). Given that ACCi markedly reduces hepatic malonyl-CoA levels (Figure 3C), it is possible that this leads to a decrease in PUFAs and subsequent upregulation of nuclear hormone signaling that ultimately drives increased VLDL-triglyceride secretion and reduced lipid clearance. Indeed, when we assessed PUFA levels in the triglyceride compartment of ACCi-treated livers, they were significantly reduced compared to controls (Figure S7).
Figure 7. Fenofibrate reverses ACCi-mediated hypertriglyceridemia in FFD-fed mice.
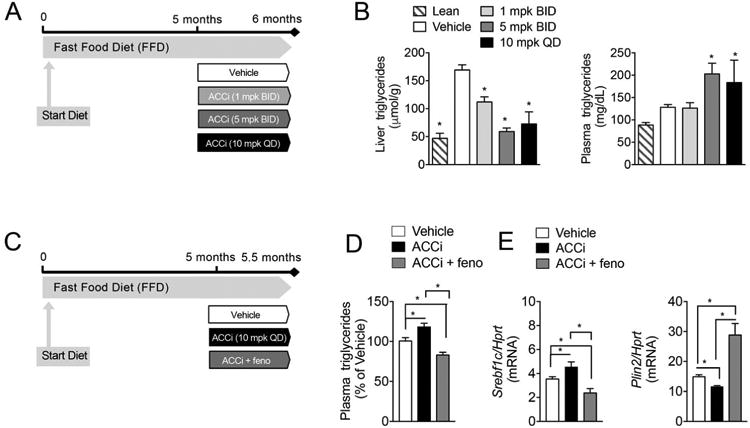
(A) Male B6 mice were fed a fast food diet (FFD) for 5 months and treated with 1 mg/kg (BID), 5 mg/kg (BID), 10 mg/kg (BID) Compound 1 (ACCi) or vehicle control for 28 days. (B–C) Liver (B) and plasma (C) triglycerides in mice treated as in (A). n = 8–13 per treatment group. (C) Male B6 mice were fed a fast food diet (FFD) for 5 months and treated with vehicle, 10 mg/kg (BID) Compound 1 (ACCi), or 10 mg/kg ACCi (BID) and fenofibrate (0.1% in chow, feno) for 14 days. (D) Plasma triglycerides (pooled samples collected at 2, 6, and 24 h post dose) in mice treated as in (C). Values are expressed as percent of vehicle-treated animals collected at the same time of day after treatment with vehicle, ACCi or ACCi + fenofibrate. (D) mRNA expression of SREBP1c and PLIN2 (a canonical PPARα target) in the livers of mice treated as in (C). Data are presented as mean ± SEM. In panels (B-C), *P ≤ 0.05 compared to vehicle control by one-way ANOVA with Dunnett correction for multiple comparisons. In panels (D-E), *P ≤ 0.05 by unpaired student's t-test. BID: twice daily; QD: once daily.
PPARα agonists reverse the plasma triglyceride increase induced by ACCi
In a liver-specific ACC1 and 2 double knockout (ACC DKO) mouse (15), the PPARα agonist, WY14643, was shown to reduce elevated plasma triglycerides. Moreover, in NASH patients who developed hypertriglyceridemia during treatment with GS-0976 in a Phase 2 study, co-administration with fenofibrate led to reductions in plasma triglycerides (13). Therefore, we tested the ability of fenofibrate dosed in chow concomitantly with ACCi to ameliorate the increase in plasma triglycerides observed with ACCi alone in an established murine model of NASH with enhanced DNL (Figure S8A).
Mice were fed a diet high in fat, cholesterol, and fructose (the fast-food diet, FFD) for 5 months, and treated with vehicle, ACCi (10 mg/kg QD), or ACCi and fenofibrate (0.1% in chow) for 14 days. Substantial steatosis (∼20% steatotic area, with mean triglycerides > 160 μmol/g or 35 mg/g) were observed in this long-term FFD model (Figure 7A-B). In agreement with the data generated in HFSD-fed rats, 28 days of ACCi treatment in FFD-fed mice significantly increased plasma triglycerides, significantly lowered hepatic triglycerides, cholesterol, and PUFAs and reduced expression of a PPARα signature and increased SREBP1c and its target genes (Figures 7A–B, S8B, and S9A-B). After two weeks of dosing ACCi in this model, plasma triglycerides were significantly increased by 15% relative to vehicle-treated animals, whereas coadministration of fenofibrate in chow with ACCi significantly reduced plasma triglycerides relative to both the ACCi-treated group and the vehicle control group (Figure 7C–D). Of note, these changes were associated with reversed alterations in PPARα/LXRα/SREBP1 gene expression (Figures 7E and S10A–C), suggesting that LXRα/SREBP1 activation and PPARα reduction are required for ACCi-mediated hypertriglyceridemia. The addition of fenofibrate did not impact the ACCi-induced reduction of liver triglycerides, but significantly further decreased liver cholesterol (Figure S11A–B). In contrast to the 21 day treatment in rats fed a HFSD, plasma βOHB was significantly increased after 14 days of ACCi treatment in this mouse model (Figure S11C). Moreover, addition of fenofibrate significantly increased βOHB relative to ACCi alone (Figure S11C).
Discussion
Multiple studies support pharmacologic inhibition of ACC1 and ACC2 for the treatment of NAFLD via simultaneous inhibition of DNL and stimulation of fatty acid oxidation (34). In particular, two clinical trials of the liver-directed ACC inhibitor GS-0976 have found statistically significant decreases in hepatic steatosis and the marker of fibrosis tissue inhibitor of metalloproteinase 1 (TIMP-1) in patients with NASH and fibrosis (12, 13). However, the effect of ACC inhibition on hepatic mitochondrial oxidation, gluconeogenesis, hepatic glucose production, and ketogenesis in vivo has not yet been explored. Moreover, liver-directed inhibition of ACC has been associated with elevated plasma triglycerides in some patients (15). To understand the mechanism for these observations, we examined the impact of Compound 1 on lipid and glucose metabolism in chow-fed rats and in rodent models of DIO. Specifically, we demonstrate that acute treatment with Compound 1 significantly reduces hepatic ACC activity and malonyl-CoA levels in chow-fed rats. These results are in agreement with previous studies and reflect the specific liver-targeted biodistribution of Compound 1 (Figure 3B) due to its preferential uptake into hepatocytes through the organic anion-transporting polypeptide (OATP) transporters (23, 35).
Malonyl-CoA is a substrate for DNL and inhibits the transport of fatty acids into the mitochondria through negative regulation of CPT1, leading to inhibition of fatty acid oxidation (9). Consistent with Compound 1's ability to lower hepatic malonyl-CoA levels, liver-directed inhibition of hepatic ACC significantly increased plasma βOHB and βOHB turnover in chow fed rats, indicative of increased fatty acid oxidation. Interestingly, the rise in plasma ketones was not detected after longer-term inhibition of ACC in overnight fasted HFSD-fed rats. Given that hepatic malonyl-CoA levels are lower in the fasting state then in the fed state (∼0.4 nmol/g vs. ∼2 nmol/g), it is not surprising that ACCi had no further effect on fatty acid oxidation. Indeed, we have previously demonstrated that ASO-induced reductions in ACC in high-fat fed rats only led to detectable increases in plasma ketones in the fed state (11). Accordingly, in FFD-fed mice, fasted for only 4–6 h prior to sacrifice, plasma βOHB was significantly increased by ACCi and addition of fenofibrate increased βOHB relative to ACCi alone.
An important contributor to lipid accumulation in rodent models of NAFLD and in patients with NAFLD is elevated rates of hepatic DNL (36). Here, liver-directed inhibition of ACC in rats led to a significant reduction in hepatic DNL and triglycerides after only one week of high-fructose feeding. Longer-term inhibition of hepatic ACC also markedly reduced hepatic triglycerides in rats fed a HFSD and mice fed a FFD independent of changes in body weight; however, this was likely a combined effect of ACCi-mediated reductions in DNL and increases in fatty acid oxidation. Nonetheless, this marked reduction in hepatic steatosis was associated with improved hepatic insulin sensitivity due to decreased hepatic DAGs and a reduction in PKCε activation in the liver. While other lipid intermediates have been shown to trigger diet-induced insulin resistance, ACCi treatment did not alter hepatic LCCoAs, acylcarnitines or the ceramide species previously implicated in resistance (C16 and C18 (24, 25)). Collectively, these data are consistent with previous studies reporting an association with reductions in DAG-mediated PKCε activation and improved hepatic insulin responsiveness at the level of IRK (2).
Despite the reduction in hepatic lipid accumulation and improved hepatic insulin sensitivity, as reflected by suppression of HGP during a hyperinsulinemic-euglycemic clamp, long-term ACC inhibition was associated with increases in rates of basal HGP in HFSD-fed rats. This increase was independent of changes in plasma insulin concentrations and likley reflect increases in hepatic acetyl-CoA content, which has previously been shown to increase hepatic gluconeogenesis through allosteric activation of pyruvate carboxylase (7). The opposing effects of increased hepatic insulin sensitivity and increased HGP may explain the lack of an effect on fasting plasma glucose observed in this study and in NASH patients treated for 12 weeks with GS-0976 (12, 13). The net effect of these changes on long term glycemic control in patients with metabolic dysfunction requires further clinical investigation.
In agreement with recent clinical data, allosteric inhibition of ACC by Compound 1 was also associated with a significant rise in plasma triglyceride levels, despite a marked reduction in hepatic steatosis (12, 13). These effects were mediated by a disequilibrium in nuclear receptor regulation due to a loss of hepatic malonyl-CoA, which is essential for the formation of PUFAs. n-3 and n-6 PUFAs act at the nuclear level to affect the expression of genes involved in lipogenesis and fatty acid oxidation (33). Specifically, reduced SREBP1 activation by PUFAs has been proposed to be mediated by the inhibition of LXRα activity (37, 38), a potent inducer of SREBP1 transcription, or by direct inhibition of SREBP1 proteolytic maturation (39). PUFAs also act as endogenous ligands for PPAR and have been shown to stimulate hepatic fatty acid catabolism through the activation of PPARα and CPT1. Here, we demonstrate that ACCi treatment significantly reduced PUFAs in the hepatic triglyceride compartment and induced the expression of multiple LXR/SREBP1 target genes in HFSD-fed rats and FFD-fed mice, which led to increased hepatic VLDL secretion (Figure S12). This was somewhat surprising given the marked reduction of hepatic triglycerides observed in ACCi-treated animals. However, previous studies have shown that a large percentage of VLDL-triglycerides are derived from peripheral fatty acids and that GPAT1 plays an important role in channeling fatty acids into triglycerides for VLDL secretion (36, 40). Indeed, Kim et al recently reported that increased hepatic expression of GPAT1 was necessary for the increased VLDL secretion and subsequent hypertriglyceridemia observed in liver-specific ACC DKO mice (15).
While our data are consistent with previous studies documenting increased plasma triglycerides following ACC inhibition in humans and rodents (15, 41), there are several differences that warrant discussion. Studies generated from the Wakil lab were the first to report increased plasma triglycerides due to decreases in ACC activity. Specifically, they demonstrated that whole-body ACC2 KO mice fed a chow-diet have a 30% increase in plasma triglyceride levels despite markedly lower hepatic triglycerides. While the mechanism remains to be determined, it was postulated that the hypertriglyceridemia observed in ACC2 KO mice was due to the mobilization of triglycerides and fatty acids from peripheral organs to provide substrates for oxidation (41). Given that Compound 1 is liver-directed, it is unlikely that the rise in plasma triglycerides is due to mobilization of lipids from the periphery due to reduced malonyl-CoA; instead the hypertriglyceridemia most likely results from a disequilibrium in hepatic nuclear receptor signaling pathways in an attempt to compensate for markedly low levels of hepatic DNL. Similar to observations by Kim et al, we demonstrate that ACC inhibition by Compound 1 results in a ∼15% increase in VLDL secretion. However, we also report that liver-directed allosteric inhibition of ACC results in a 20% reduction in plasma LPL activity and corresponding decrease in triglyceride clearance. This is most likely a result of secreted factors that inhibit LPL activity, including PPARα-regulated APOC3. While Kim et al, did not assess lipid clearance in their liver-directed ACC DKO mice, it is possible that reduced triglyceride catabolism also contributes to the rise in plasma triglycerides observed in their model (15).
The degree of ACC inhibition could ultimately determine the mechanism(s) that drive hypertriglyceridemia in rodents and humans. In particular, a recent study reported that allosteric inhibition of ACC markedly reduced hepatic steatosis and increased insulin sensitivity, without altering plasma triglycerides in rat models of DIO (23). In contrast, and consistent with our data, MK-4074 (a liver-directed inhibitor of ACC) was recently shown to increase plasma triglycerides in humans and mouse models of DIO (15). It is worth mentioning, however, that ACC was completely inhibited by MK-4074 and in the liver-directed ACC DKO mice described by Kim et al. (15). Here, we used a lower dose of Compound 1 that better approximated the dose of GS-0976 (20 mg once daily) being assessed in human clinical trials. This may explain the different magnitudes of aberrant nuclear receptor signaling pathways and suggests that the mechanism for ACCi-mediated hypertriglyceridemia proposed in this study may be more relevant to ACC inhibitors that are currently under evaluation in clinical trials. Nonetheless, Kim et al. elegantly showed that PUFA supplementation or the use of a PPARα agonist (WY14643) normalizes ACCi-mediated increases in plasma triglycerides (15). Here, we also demonstrate that co-administration of fenofibrate could ameliorate hypertriglyceridemia and stimulate fatty acid oxidation in FFD-fed mice.
In summary, the present study demonstrates that liver-directed allosteric inhibition of ACC significantly reduces hepatic malonyl-CoA levels, % DNL, and hepatic triglycerides. Using stable isotope tracers, we also report for the first time that acute ACCi treatment significantly increases whole-body βOHB turnover in chow-fed rats, without altering whole-body glucose turnover or rates of hepatic mitochondrial citrate synthase flux (VCS). Importantly, ACCi treatment markedly reduced HFSD-induced hepatic steatosis, which correlated with a significant reduction in hepatic DAGs, PKCε translocation, and improved hepatic insulin signaling at the level of the insulin receptor kinase. Steatosis reduction was also observed in mice on a FFD for 5 months. In rats, 21-day inhibition of ACC increased hepatic acetyl-CoA levels and HGP. Because this may serve to offset improvements in hepatic insulin sensitivity, it remains to be determined whether allosteric inhibition of hepatic ACC would have therapeutic benefit in treating fasting hyperglycemia in patients with NAFLD and T2DM. Moreover, ACCi treatment led to a significant rise in plasma triglycerides in HFSD-fed rats and FFD-fed mice. These effects were mediated by a reduction in hepatic PUFA levels and disequilibrium in nuclear hormone receptor expression that, in rats, resulted in increased hepatic VLDL secretion and reduced systemic triglyceride clearance. While increased hypertriglyceridemia has been observed across species and in the clinic, preclinical and clinical data suggest that co-administration with a PPARα agonist can ameliorate hypertriglyceridemia induced by ACC inhibition.
Supplementary Material
Acknowledgments
We thank Jianying Dong, Mario Khan, Xian-Man Zhang, Sylvie Dufour, Irina Smolgovsky, Gina Butrico, John Stack, Yongliang Wang, Yuichi Nozaki, Codruta Todeasa, Maria Batsu, and the Yale Diabetes Research Core Facility for their expert technical assistance. We thank G. Mani Subramanian and Rob P. Myers for helpful discussions.
Financial Support: This study was supported by grants from the NIH (P30 DK059635, R01 DK-040936, R01 DK113984 to GIS and F32 DK114954 to LG) and an investigator-initiated award from Gilead.
List of Abbreviations
- ABCA1
ATP binding cassette transporter A1
- ACC
acetyl-CoA carboxylase
- ALT
alanine aminotransferase activity
- ANGPTL3
angiopoietin-like protein 3
- ASO
antisense oligonucleotide
- AST
aspartate aminotransferase
- BC
biotin carboxylase
- BID
twice daily
- CPT1
carnitine palmitoyltransferase 1
- DAG
diacylglycerol
- DGAT2
diacylglycerol o-acyltransferase 2
- DIO
diet-induced obesity
- DKO
double knockout
- DNL
de novo lipogenesis
- EGP
endogenous glucose production
- eWAT
epididymal white adipose tissue
- FASN
fatty acid synthase
- GPAT1
glycerol-3-phosphate acyltransferase, mitochondrial
- HDL
high-density lipoprotein
- HFSD
high-fat sucrose diet
- HGP
hepatic glucose production
- LCCoA
long-chain acyl-CoA
- LDL
low-density lipoprotein
- LPL
lipoprotein lipase
- LXR
liver X receptor
- MTTP
microsomal triglyceride transfer protein
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NEFA
non-esterified fatty acid
- PC
pyruvate carboxylase
- PKC
protein kinase C
- PNPLA3
patatin-like phospholipase domain containing 3
- PPAR
peroxisome proliferator-activated receptor
- PUFA
polyunsaturated fatty acid
- QD
once daily
- qPCR
quantitative polymerase chain reaction
- SCD1
stearoyl-CoA desaturase 1
- SD
Sprague-Dawley
- SREBP1
sterol response element-binding protein
- TIMP-1
metalloproteinase 1
- T2D
type 2 diabetes
- VCS
citrase synthase flux
- VLDL
very low-density lipoprotein
- VPC
pyruvate carboxylase flux
Footnotes
Conflicts of interest: JB, TW, MS, and ASR are employed by and may own stock in Gilead.
Author Contributions: This study was designed by LG, JB, ASR and GIS. Experiments were performed by LG, DV, JB, RJP, TW, MWE and DZ. Data was analyzed and interpreted by LG, DV, JB, RR, LL, RJP, TW, MS, ASR and GIS. The manuscript was written by LG, JB, AR and GIS, with input from all authors.
References
- 1.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 2.Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014;371:1131–1141. doi: 10.1056/NEJMra1011035. [DOI] [PubMed] [Google Scholar]
- 3.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Birkenfeld AL, Shulman GI. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology. 2014;59:713–723. doi: 10.1002/hep.26672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farese RV, Jr, Zechner R, Newgard CB, Walther TC. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab. 2012;15:570–573. doi: 10.1016/j.cmet.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13:572–587. doi: 10.1038/nrendo.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ, Jurczak MJ, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015;160:745–758. doi: 10.1016/j.cell.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brownsey RW, Zhande R, Boone AN. Isoforms of acetyl-CoA carboxylase: structures, regulatory properties and metabolic functions. Biochem Soc Trans. 1997;25:1232–1238. doi: 10.1042/bst0251232. [DOI] [PubMed] [Google Scholar]
- 9.McGarry JD, Mannaerts GP, Foster DW. A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J Clin Invest. 1977;60:265–270. doi: 10.1172/JCI108764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong L, Harwood HJ., Jr Acetyl-coenzyme A carboxylases: versatile targets for drug discovery. J Cell Biochem. 2006;99:1476–1488. doi: 10.1002/jcb.21077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savage DB, Choi CS, Samuel VT, Liu ZX, Zhang D, Wang A, Zhang XM, et al. Reversal of diet-induced hepatic steatosis and hepatic insulin resistance by antisense oligonucleotide inhibitors of acetyl-CoA carboxylases 1 and 2. J Clin Invest. 2006;116:817–824. doi: 10.1172/JCI27300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawitz EJ, Poordad F, Coste A, Loo N, Djedjos CS, McColgan B, Jia C, et al. Acetyl-CoA carboxylase (ACC) inhibitor GS-0976 leads to suppression of hepatic de novo lipogenesis and significant improvements in MRI-PDFF, MRE, and markers of fibrosis after 12 weeks of therapy in patients with NASH. Journal of Hepatology. 2017;66:S34–S34. [Google Scholar]
- 13.Late-Breaking Abstracts - Presented at the 68th Annual Meeting of the American Association for the Study of Liver Diseases: The Liver Meeting 2017. Hepatology. 2017;66:1260A–1261A. [Google Scholar]
- 14.Stiede K, Miao W, Blanchette HS, Beysen C, Harriman G, Harwood HJ, Jr, Kelley H, et al. Acetyl-coenzyme A carboxylase inhibition reduces de novo lipogenesis in overweight male subjects: A randomized, double-blind, crossover study. Hepatology. 2017;66:324–334. doi: 10.1002/hep.29246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, Burgess SC, et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017;26:576. doi: 10.1016/j.cmet.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 16.Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011;301:G825–834. doi: 10.1152/ajpgi.00145.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perry RJ, Peng L, Cline GW, Petersen KF, Shulman GI. A Non-invasive Method to Assess Hepatic Acetyl-CoA In Vivo. Cell Metab. 2017;25:749–756. doi: 10.1016/j.cmet.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vatner DF, Majumdar SK, Kumashiro N, Petersen MC, Rahimi Y, Gattu AK, Bears M, et al. Insulin-independent regulation of hepatic triglyceride synthesis by fatty acids. Proc Natl Acad Sci U S A. 2015;112:1143–1148. doi: 10.1073/pnas.1423952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goedeke L, Rotllan N, Canfran-Duque A, Aranda JF, Ramirez CM, Araldi E, Lin CS, et al. MicroRNA-148a regulates LDL receptor and ABCA1 expression to control circulating lipoprotein levels. Nat Med. 2015;21:1280–1289. doi: 10.1038/nm.3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perry RJ, Peng L, Cline GW, Butrico GM, Wang Y, Zhang XM, Rothman DL, et al. Non-invasive assessment of hepatic mitochondrial metabolism by positional isotopomer NMR tracer analysis (PINTA) Nat Commun. 2017;8:798. doi: 10.1038/s41467-017-01143-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camporez JP, Kanda S, Petersen MC, Jornayvaz FR, Samuel VT, Bhanot S, Petersen KF, et al. ApoA5 knockdown improves whole-body insulin sensitivity in high-fat-fed mice by reducing ectopic lipid content. J Lipid Res. 2015;56:526–536. doi: 10.1194/jlr.M054080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, Marcucci MJ, et al. Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest. 2016;126:4361–4371. doi: 10.1172/JCI86013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harriman G, Greenwood J, Bhat S, Huang X, Wang R, Paul D, Tong L, et al. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc Natl Acad Sci U S A. 2016;113:E1796–1805. doi: 10.1073/pnas.1520686113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blachnio-Zabielska AU, Chacinska M, Vendelbo MH, Zabielski P. The Crucial Role of C18-Cer in Fat-Induced Skeletal Muscle Insulin Resistance. Cell Physiol Biochem. 2016;40:1207–1220. doi: 10.1159/000453174. [DOI] [PubMed] [Google Scholar]
- 25.Hla T, Kolesnick R. C16:0-ceramide signals insulin resistance. Cell Metab. 2014;20:703–705. doi: 10.1016/j.cmet.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yuan G, Al-Shali KZ, Hegele RA. Hypertriglyceridemia: its etiology, effects and treatment. CMAJ. 2007;176:1113–1120. doi: 10.1503/cmaj.060963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Millar JS, Cromley DA, McCoy MG, Rader DJ, Billheimer JT. Determining hepatic triglyceride production in mice: comparison of poloxamer 407 with Triton WR-1339. J Lipid Res. 2005;46:2023–2028. doi: 10.1194/jlr.D500019-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Merkel M, Eckel RH, Goldberg IJ. Lipoprotein lipase: genetics, lipid uptake, and regulation. J Lipid Res. 2002;43:1997–2006. doi: 10.1194/jlr.r200015-jlr200. [DOI] [PubMed] [Google Scholar]
- 29.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hertz R, Bishara-Shieban J, Bar-Tana J. Mode of action of peroxisome proliferators as hypolipidemic drugs. Suppression of apolipoprotein C-III. J Biol Chem. 1995;270:13470–13475. doi: 10.1074/jbc.270.22.13470. [DOI] [PubMed] [Google Scholar]
- 31.Inaba T, Matsuda M, Shimamura M, Takei N, Terasaka N, Ando Y, Yasumo H, et al. Angiopoietin-like protein 3 mediates hypertriglyceridemia induced by the liver X receptor. J Biol Chem. 2003;278:21344–21351. doi: 10.1074/jbc.M213202200. [DOI] [PubMed] [Google Scholar]
- 32.Lee JY, Parks JS. ATP-binding cassette transporter AI and its role in HDL formation. Curr Opin Lipidol. 2005;16:19–25. doi: 10.1097/00041433-200502000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Georgiadi A, Kersten S. Mechanisms of gene regulation by fatty acids. Adv Nutr. 2012;3:127–134. doi: 10.3945/an.111.001602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bourbeau MP, Bartberger MD. Recent advances in the development of acetyl-CoA carboxylase (ACC) inhibitors for the treatment of metabolic disease. J Med Chem. 2015;58:525–536. doi: 10.1021/jm500695e. [DOI] [PubMed] [Google Scholar]
- 35.Svensson RU, Parker SJ, Eichner LJ, Kolar MJ, Wallace M, Brun SN, Lombardo PS, et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat Med. 2016;22:1108–1119. doi: 10.1038/nm.4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ou J, Tu H, Shan B, Luk A, DeBose-Boyd RA, Bashmakov Y, Goldstein JL, et al. Unsaturated fatty acids inhibit transcription of the sterol regulatory element-binding protein-1c (SREBP-1c) gene by antagonizing ligand-dependent activation of the LXR. Proc Natl Acad Sci U S A. 2001;98:6027–6032. doi: 10.1073/pnas.111138698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshikawa T, Shimano H, Yahagi N, Ide T, Amemiya-Kudo M, Matsuzaka T, Nakakuki M, et al. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J Biol Chem. 2002;277:1705–1711. doi: 10.1074/jbc.M105711200. [DOI] [PubMed] [Google Scholar]
- 39.Hannah VC, Ou J, Luong A, Goldstein JL, Brown MS. Unsaturated fatty acids down-regulate srebp isoforms 1a and 1c by two mechanisms in HEK-293 cells. J Biol Chem. 2001;276:4365–4372. doi: 10.1074/jbc.M007273200. [DOI] [PubMed] [Google Scholar]
- 40.Moon YA, Liang G, Xie X, Frank-Kamenetsky M, Fitzgerald K, Koteliansky V, Brown MS, et al. The Scap/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012;15:240–246. doi: 10.1016/j.cmet.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science. 2001;291:2613–2616. doi: 10.1126/science.1056843. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.