

Abstract
Eight cembrane-type diterpenoids, namely, (+)-(6R)-6-hydroxyisosarcophytoxide (1), (+)-(6R)-6-acetoxyisosarcophytoxide (2), (+)-17-hydroxyisosarcophytoxide (3), sarcomililatins A–D (4–7), and sarcomililatol (8), were isolated from the soft coral Sarcophyton mililatensis collected from Weizhou Island, Guangxi Autonomous Region, together with 2 known related analogues, (+)-isosarcophytoxide (9) and (+)-isosarcophine (10). The structures of these compounds were elucidated by a combination of detailed spectroscopic analyses, chemical methods, and comparison with reported data. The absolute configuration of compound 1 was established by the modified Mosher׳s method, while the absolute configurations of compounds 4 and 5 were assigned by electronic circular dichroism (ECD) spectroscopy and that of compound 8 was established by time-dependent density functional theory electronic circular dichroism (TD-DFT ECD) calculation. In in vitro bioassays, compound 9 displayed significant cytotoxicity against the human cancer cell lines human promyelocytic leukemia cells (HL-60) and human lung adenocarcinoma cells (A-549) with IC50 values of 0.78±0.21 and 1.26±0.80 μmol/L, respectively. Compounds 4 and 9 also showed moderate inhibitory effects on the TNFα-induced Nuclear factor kappa B (NF-κB, a therapeutical target in cancer) activation, showing IC50 values of 35.23±12.42 and 22.52±4.44 μmol/L, respectively.
Keywords: Soft coral, Sarcophyton, Sarcophyton mililatensis, Cembrane-type diterpe-noids, Modified Mosher׳s method, ECD calculation, Cytotoxicity, NF-κB inhibitory activity
Graphical abstract
Eight new cembrane-type diterpenoids (1–8) were isolated from the soft coral Sarcophyton mililatensis. Compound 9 displayed significant cytotoxicity, and compounds 4 and 9 showed inhibitory effects on the TNFα-induced NF-κB.

1. Introduction
Literature reports concerning the natural products chemistry of soft corals of the cosmopolitan genus Sarcophyton (Alcyonacea, Alcyoniidae) indicate that they are well-known to be a rich source of specialised metabolites, particularly diterpenoids of the cembrane-type1, 2. To date, more than 220 cembranes have been discovered, besides undefined species, from approximately 18 species of this genus. Moreover, some of them have been reported to be responsible for a diverse range of significant bioactivities, especially cytotoxic and anti-inflammatory effects2, 3, 4. Their excellent bioactivities have for a long time attracted great interest from synthetic organic chemists as challenging targets for total synthesis5, 6.
Sarcophyton species are prolific in the South China Sea. In the course of our ongoing search for bioactive secondary metabolites from the South China Sea marine invertebrates7, 8, 9, we collected the soft coral S. mililatensis from Weizhou Island, Guangxi Autonomous Region, China. Notably, only 2 prior phytochemical studies have been performed on this species collected from Baycanh Island, Vietnam, resulting in the isolation of one 9,11-secosteroid and 6 cembranes10, 11. The present investigation of the Et2O-soluble fraction from the acetone extract of S. mililatensis has now led to the discovery of eight previously undescribed cembrane-type diterpenoids, namely, (+)-(6 R)-6-hydroxyisosarcophytoxide (1), (+)-(6 R)-6-acetoxyisosarcophytoxide (2), (+)-17-hydroxyisosarcophytoxide (3), sarcomililatins A–D (4–7), and sarcomililatol (8), along with 2 known structural analogues, (+)-isosarcophytoxide (9) and (+)-isosarcophine (10) as shown in Fig. 1. The absolute configuration of compound 1 was established by the modified Mosher׳s method. The absolute configurations of compounds 4 and 5 were assigned by ECD spectroscopy, while for compound 8 TD-DFT ECD calculation was used.
Figure 1.

Chemical structures of compounds 1–10.
Cancer is a group of diseases characterized by uncontrolled cell growth, which has become the major public health concern over the last several decades12, 13, 14, 15. NF-κB, as a family of inducible transcription factors in all cells discovered by Sen and Baltimore16 in 1986, has become one of the major targets for drug development17. In particular, the aberrant activation of NF-κB has been frequently observed in various types of human cancers, and suppression of NF-κB can limit the proliferation of cancer cells18. Hence, we focus on testing cytotoxic activity and the NF-κB inhibitory effects. According to the results, compound 9 exhibited moderate cytotoxic activity, both compounds 4 and 9 showed moderate NF-κB inhibitory effects on the TNFα-induced NF-κB. Reported herein are the isolation and structural elucidation of these compounds as well as their biological properties.
2. Results and discussion
Compound 1 was isolated as a colorless oil, and had a molecular formula of C20H30O3 as established by (+)-HR-ESI-MS ion peak at m/z 341.2096 [M + Na]+ (Calcd. for C20H30O3Na, 341.2087) and 13C NMR data (Table 1), implying 6 degrees of unsaturation. Its IR spectrum showed the presence of a hydroxyl group (3363 cm−1). The 1H NMR spectrum (Table 1) displayed signals due to 3 vinyl methyls at δH 1.83 (3 H, s, H3-19), 1.66 (3 H, s, H3-17), and 1.60 (3 H, s, H3-18), a tertiary methyl at δH 1.29 (3 H, s, H3-20), and 2 olefinic protons appearing as doublets at δH 5.22 (1 H, d, J = 9.2 Hz, H-7) and 5.09 (1 H, d, J = 10.0 Hz, H-3), which were attributed to 2 trisubstituted double bonds. In addition, proton signals were also observed for one oxymethylene at δH 4.52 (1 H, dd, J = 12.0, 4.0 Hz, H-16a) and 4.47 (1 H, dd, J = 12.0, 3.2 Hz, H-16b) and 2 oxymethines at δH 5.38 (1 H, ddd, J = 10.0, 4.0, 3.2 Hz, H-2) and 2.39 (1 H, dd, J = 11.2, 2.8 Hz, H-11) in the 1H NMR spectrum. The 13C NMR spectrum indicated the presence of 20 signals which were attributed by DEPT and HSQC experiments to 4 methyls, 6 methylenes, 5 methines, and 5 quaternary carbons. Of these carbons, 5 were bonded to oxygen and 6 were olefinic (2 were trisubstituted). These data suggested that 1 was a cembrane-type diterpenoid.
Table 1.
1H NMR and 13C NMR spectroscopic data for compounds 1–4 in CDCl3a.
| Position |
1b |
2c |
3c |
4c |
||||
|---|---|---|---|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
| 1 | 132.1, C | 131.9, C | 136.8, C | 161.3, C | ||||
| 2 | 83.0, CH | 5.38, ddd (10.0, 4.0, 3.2) | 82.9, CH | 5.37, br d(10.2) | 83.8, CH | 5.45, ddd (10.2, 4.8, 3.6) | 79.1, CH | 5.42, d (10.2) |
| 3 | 128.4, CH | 5.09, d (10.0) | 128.9, CH | 5.11, d (10.2) | 125.8, CH | 5.09, d (10.2) | 120.4, CH | 4.91, d (10.2) |
| 4 | 136.9, C | 136.0, C | 141.0, C | 143.4, C | ||||
| 5 | 48.4, CH2 | 2.16, dd (12.4, 10.8) | 44.9, CH2 | 2.22, dd (12.6, 10.8) | 39.0, CH2 | 2.18, m | 42.1, CH2 | 2.80, dd (13.8, 7.2) |
| 2.70, dd (12.4, 5.2) | 2.67, dd (12.6, 4.8) | 2.31, m | 2.86, dd (13.8, 7.2) | |||||
| 6 | 65.4, CH | 4.65, ddd (10.8, 9.2, 5.2) | 67.9, CH | 5.78, ddd (10.8, 10.2, 4.8) | 22.8, CH2 | 1.09, m | 129.6, CH | 5.97, dt (15.6, 7.2) |
| 2.42, m | ||||||||
| 7 | 128.5, CH | 5.22, d (9.2) | 124.3, CH | 5.16, d (9.6) | 125.7, CH | 5.00, br d (9.0) | 135.7, CH | 5.62, d (15.6) |
| 8 | 139.5, C | 141.8, C | 133.4, C | 84.3, C | ||||
| 9 | 36.8, CH2 | 2.02, m | 36.6, CH2 | 2.01, m | 36.8, CH2 | 1.97, m | 35.4, CH2 | 1.81, m |
| 2.35, m | 2.36, m | 2.29, m | 1.85, m | |||||
| 10 | 23.7, CH2 | 1.28, m | 23.7, CH2 | 1.28, m | 24.3, CH2 | 1.25, m | 24.0, CH2 | 1.66, m |
| 2.13, m | 2.15, m | 1.69, m | 1.76, m | |||||
| 11 | 62.1, CH | 2.39, dd (11.2, 2.8) | 61.8, CH | 2.38, dd (10.2, 1.8) | 62.5, CH | 2.51, dd (10.8, 3.0) | 61.7, CH | 2.69, dd (7.8, 4.8) |
| 12 | 61.7, C | 61.5, C | 61.5, C | 61.0, C | ||||
| 13 | 37.5, CH2 | 0.92, m | 37.3, CH2 | 0.92, m | 38.0, CH2 | 0.97, m | 35.8, CH2 | 1.30, m |
| 1.85, m | 1.86, m | 1.84, m | 1.87, m | |||||
| 14 | 22.5, CH2 | 1.71, m | 22.3, CH2 | 1.68, m | 23.8, CH2 | 1.84, m | 23.4, CH2 | 2.18, m |
| 2.33, m | 2.33, m | 2.15, m | 2.35, dt (13.2, 4.8) | |||||
| 15 | 128.8, C | 128.7, C | 132.0, C | 123.8, C | ||||
| 16 | 78.5, CH2 | 4.47, dd (12.0, 3.2) | 78.4, CH2 | 4.47, br d (11.4) | 75.9, CH2 | 4.66, dd (12.0, 3.6) | 174.7, C | |
| 4.52, dd (12.0, 4.0) | 4.52, br d (11.4) | 4.76, dd (12.0, 4.8) | ||||||
| 17 | 10.1, CH3 | 1.66, s | 10.0, CH3 | 1.66, s | 57.0, CH2 | 4.26, d (12.6) | 9.1, CH3 | 1.85, br s |
| 4.32, d (12.6) | ||||||||
| 18 | 15.5, CH3 | 1.60, s | 15.1, CH3 | 1.64, s | 14.8, CH3 | 1.61, s | 17.1, CH3 | 1.83, br s |
| 19 | 14.9, CH3 | 1.83, s | 15.3, CH3 | 1.83, s | 14.9, CH3 | 1.66, s | 21.2, CH3 | 1.47, s |
| 20 | 15.8, CH3 | 1.29, s | 15.7, CH3 | 1.29, s | 15.9, CH3 | 1.28, s | 16.7, CH3 | 1.30, s |
| 6-OAc | 170.2, C | 2.03, s | ||||||
| 21.4, CH3 | ||||||||
| 8-OOH | 7.35, s | |||||||
δ in ppm, assignments made by DEPT, COSY, HSQC, HMBC, and NOESY experiments.
At 400 MHz for 1H and 100 MHz for 13C NMR experiments.
At 600 MHz for 1H and 150 MHz for 13C NMR experiments.
A comparison of the NMR data of 1 with those of the co-occurring known cembrane diterpenoid, (+)-isosarcophytoxide (9)19, 20, revealed that they were structural analogues, with the only difference being the presence of an additional hydroxyl group at C-6 in 1, in agreement with the mass data. The hydroxyl group was connected to C-6, as evidenced by the observation of the downfield chemical shift of C-6 from δC 24.5 to δC 65.4 in 1. This assignment was further established by the 1H–1H COSY cross-peaks (Fig. 2) of H-6 (δH 4.65) with both H2-5 (δH 2.70 and 2.16) and H-7 (δH 5.22) and the HMBC correlation (Fig. 2) from H-7 to C-6. The geometries of the double bonds at Δ3 and Δ7 were assigned to be both E by the shielded carbon resonances of the 2 vinyl methyls at δC 15.5 (C-18) and 14.9 (C-19)21, which was further supported by the NOESY correlations (Fig. 3) of H3-18 (δH 1.60)/H-2 (δH 5.38) and of H3-19 (δH 1.83)/H-6 (δH 4.65). The relative configurations of C-2, C-11, and C-12 in 1 were proven to be the same as those of 9 due to the diagnostic NOESY correlations of H3-20 (δH 1.29)/H2-10 (δH 2.13 and 1.28) and H2-14 (δH 2.33 and 1.71); H-11 (δH 2.39)/H-7 (δH 5.22) and H-13a (δH 0.92); and H-2/H-13a and H3-18. Moreover, the NOESY correlations of H3-18/H-6 and H-2 suggested that 6-OH was β-oriented.
Figure 2.

Selected 1H–1H COSY and HMBC correlations of 1, 4, 6, and 8.
Figure 3.

Key NOESY correlations for compounds 1, 4, 6, and 8.
The absolute configuration at C-6 of 1 was assigned via a modified Mosher׳s method. Esterification of 1 with (R)- and (S)-MTPA chloride occurred at the C-6 hydroxyl group to give the (S)- and (R)-MTPA ester derivatives, 1s and 1r, respectively. The observed ∆δH(S-R) value distribution pattern (Fig. 4) established the 6R-configuration for 1. Therefore, the structure of 1 was elucidated as (+)-(6 R)-6-hydroxyisosarcophytoxide.
Figure 4.
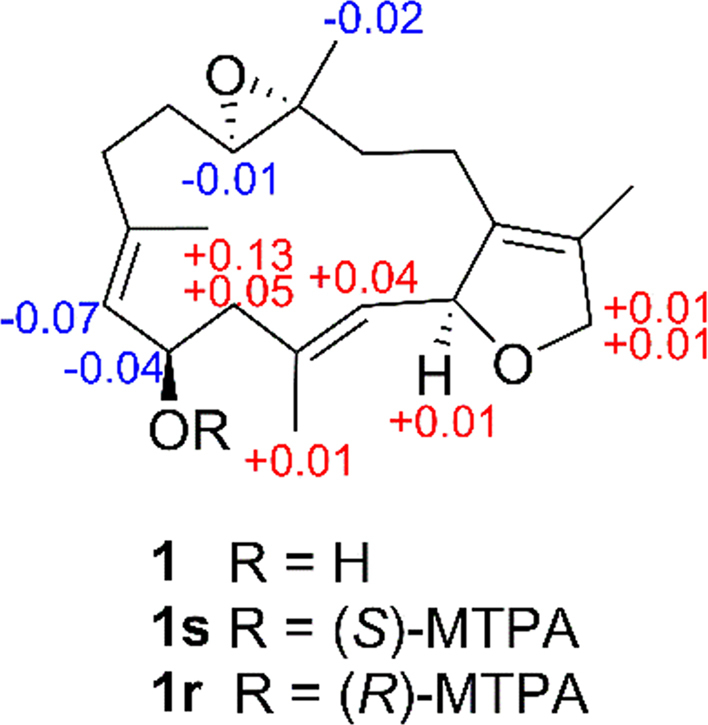
∆δH values [∆δ (in ppm) = δS–δR] obtained for (S)- and (R)-MTPA esters of compound 1 in pyridine-d5.
Compound 2, which was obtained as a colorless oil, gave the molecular formula C22H32O4 on the basis of its (+)-HR-ESI-MS ion peak at m/z 383.2197 [M + Na]+ (Calcd. for C22H32O4Na, 383.2193), requiring 7 degrees of unsaturation. The IR spectrum displayed a strong absorption at 1732 cm—1, consistent with the presence of a saturated ester carbonyl group. The 1H and 13C NMR spectra (Table 1) of 2 were virtually identical to those of 1, with the exception of an acetoxy moiety in 2 instead of the C-6 hydroxyl group in 1. This replacement caused the 13C NMR resonance of C-6 to be shifted downfield (from δC 65.4 to δC 67.9). The position of the acetoxy group at C-6 was further secured by the HMBC correlation (Fig. 2) from H-6 (δH 5.78) and the ester carbonyl carbon (δC 170.2). The similar NOESY correlation (Fig. 3) patterns of 2 and 1 indicated that they have the same relative configuration. Finally, the absolute configuration of 2 was assigned as 2S,6R,11R,12R, the same as those of 1. This is because acetylation of 1 yielded 2, which gave optical rotations { +51 (c 0.09, CH3OH); +65 (c 0.09, CHCl3)}, compared to those { +53 (c 0.5, CH3OH); +63 (c 0.24, CHCl3)} observed for the natural sample of 2. The structure of 2 was thereby proposed as (+)-(6 R)-6-acetoxyisosarcophytoxide.
A literature search revealed that the assigned structure of 2 was the same as that of sarcophytonoxide A, a known cembrane diterpenoid isolated previously from the soft coral Sarcophyton ehrenbergi22. Furthermore, the 1H and 13C NMR data of 2 were also the same as those of sarcophytonoxide A. However, when the optical rotation of 2{dextrorotatory, +53 (c 0.5, CH3OH)}, recorded in the same conditions, was compared with that reported for sarcophytonoxide A {levorotatory, –36.8 (c 0.5, CH3OH)}22, it appeared quite equal in value but opposite in sign. This result indicated that the 2 compounds are enantiomers and that the absolute configuration of sarcophytonoxide A should be 2R,6S,11S,12S.
Compound 3 was isolated as a colorless oil. Its molecular formula of C20H30O3, the same as that of 1, was deduced from the (+)-HR-ESI-MS ion peak at m/z 341.2077 [M + Na]+ (Calcd. for C20H30O3Na, 341.2087). A comparison of the 1H and 13C NMR data (Table 1) of 3 and 1 indicated similarities between them. In fact, the structure of 3 differed from that of 1 only by the location of the hydroxyl group from C-6 to C-17 in 3. This deduction was based on the chemical shift observed for C-6 (δC 22.8) and C-17 (δC 57.0), which was further confirmed by the 1H–1H COSY cross-peaks (Fig. 2) of H2-5 (δH 2.31 and 2.18)/H2-6 (δH 2.42 and 1.09)/H-7 (δH 5.00) and the HMBC correlations (Fig. 2) from H2-17 to C-1 (δC 136.8), C-15 (δC 132.0), and C-16 (δC 75.9). The relative configurations of all the asymmetric centers were determined to be the same as those of 1 on the basis of the NOESY experiment (Fig. 3). According to the previous findings2, 19, the absolute configuration at C-2 of 3 was proposed as S from the large dextrorotatory optical rotation { +98 (c 0.1, CHCl3)}. The absolute configuration of 3 was tentatively assigned as 2S,11R,12R. Hence, the structure of 3 was shown to be (+)-17-hydroxyisosarcophytoxide.
Compound 4 was obtained as a colorless oil with the molecular formula of C20H28O5 on the basis of (+)-HR-ESI-MS ion peak at m/z 371.1822 [M + Na]+ (Calcd. for C20H28O5Na, 371.1829) and 13C NMR data (Table 1), suggesting that 4 possessed 7 degrees of unsaturation. Its IR spectrum exhibited a broad absorption at 3356 cm−1 (OH) and strong absorptions at 1751 and 1678 cm−1, consistent with the presence of an α,β-unsaturated γ-lactone moiety. This were supported by the 13C NMR signals at δC 174.7 (C-16), 161.3 (C-1), 123.8 (C-15), and 79.1 (C-2) and UV absorption maxima at 246 and 275 nm23. The third oxygen atom was determined to be part of a trisubstituted epoxy ring, which was confirmed by the appearance of signals at δH 2.69 (1 H, dd, J = 7.8, 4.8 Hz, H-11) and δC 61.7 (C-11) and 61.0 (C-12). The NMR signals of an oxygenated quaternary carbon and an exchangeable proton were observed at δC 84.3 (C-8) and δH 7.35 (1 H, s, 8-OOH), respectively, strongly implying that the remaining 2 oxygen atoms were involved in a hydroperoxy group24. This conclusion was also supported by the significant downfield shift of the resonance for C-8 in 4 with respect to that of the corresponding carbon (δC 72.6) for mayolide B, a known cembranoid with the same 14-membered ring substituted with a hydroxy group at C-8 previously isolated the soft coral Sinularia mayi25. Further analysis of the 13C NMR and DEPT spectra of 4 displayed 20 signals for 4 methyls, 5 methylenes, 5 methines (3 olefinic and 2 oxygenated), 5 quaternary carbons (3 olefinic and 2 oxygenated), and one conjugated ester carbonyl carbon (C-16). The planar structure of 4 was extensively elucidated by 1H–1H COSY and HMBC spectra (Fig. 2). The 1H–1H COSY cross-peaks readily determined the presence of 4 spin systems from H-2 to H-3; H2-5 to H-7; H2-9 to H-11; and H2-13 to H2-14. The significant HMBC correlations from Me-17 to C-1, C-15, and C-16; Me-18 to C-3, C-4, and C-5; Me-19 to C-7, C-8, and C-9; Me-20 to C-11, C-12, and C-13;H-2 to C-1, C-15, and C-16; and H2-14 to C-1 and C-15 constructed the cembrane skeleton as depicted in Fig. 1. The hydroperoxy group and the epoxy ring in 4 were placed at C-8 and C-11/C-12, respectively, based on the strong HMBC correlations from Me-19 to C-8 and from Me-20 to C-11 and C-12.
The relative configuration of 4 was deduced from interpretation of the coupling constants and a NOESY experiment (Fig. 3). The large coupling constants (J6,7 = 15.6 Hz) and the chemical shift of the C-18 methyl group (δC 17.1) established the E geometries of the Δ3 and Δ6 double bonds, and this was further supported by the strong NOESY cross-peaks of H-6 (δH 5.97)/Me-18 (δH 1.83) and Me-19 (δH 1.47) and of H-5a (δH 2.86)/H-3 (δH 4.91) and H-7 (δH 5.62). The observed correlations in the NOESY spectrum as shown in Fig. 3 assigned H-11, Me-19, and Me-20 as β-oriented, while H-2 was the α-orientation. Furthermore, the absolute configuration at C-2 in 4 was defined by an ECD experiment. The ECD spectrum of 4 (Fig. 5) showed a negative Cotton effect at 249 nm (Δɛ = –3.1) and a positive Cotton effect at 221 nm (Δɛ = +33.4), indicative of a 2S-configuration26, 27, 28. Accordingly, the structure of 4 was characterized as shown, and the compound was designated with the trivial name sarcomililatin A.
Figure 5.

ECD spectra for compounds 4 (0.0014 mol/L, CH3CN, cell length 2 cm) and 5 (0.0014 mol/L, CH3CN, cell length 2 cm).
Compound 5 was isolated as a colorless oil, possessing the same molecular formula of C20H28O5 as 4 by (+)-HR-ESI-MS ion peak at m/z 371.1830 [M + Na]+ (Calcd. for C20H28O5Na, 371.1829). The IR, UV, and 1D NMR data (Table 2) of 5 were similar to those of 4, except for the migration of the Δ6 double bonds in 4 to Δ8(19) [δH 5.24 (1 H, br s, H-19a) and 5.18 (1 H, br s, H-19b); δC 148.4 (s, C-8) and 113.8 (t, C-19)] and the position of the hydroperoxy group at C-8 in 4 to C-7 [δH 7.71 (1 H, s, 7-OOH) and 4.22 (1 H, dd, J = 7.6, 5.6 Hz, H-7); δC 83.3 (d, C-7)]. However, a literature survey revealed that 5 showed exactly the same 1H and 13C NMR data as trocheliolide A, a known cembrane diterpenoid isolated previously from the soft coral Sarcophyton trocheliophorum29. This observation readily prompted us to assign the structure of 5 as trocheliolide A. Nevertheless, the optical rotations {dextrorotatory, +34 (c 0.1, CHCl3) and +23 (c 0.1, CH3OH)} sign of 5 as opposite to that of trocheliolide A {levorotatory, –76 (c 0.3, CHCl3)}29. These results suggested that 5 is the enantiomer of trocheliolide A. Moreover, the absolute configuration at C-2 in 5 was deduced to the same (S) as in 4 on the basis of an ECD spectrum with ɛ values (–1.5 at 249 nm and +11.3 at 223 nm). Thus, the structure of 5 (the trivial name sarcomililatin B) was assigned as depicted.
Table 2.
1H and 13C NMR spectroscopic data for compounds 5–8 in CDCl3a.
| Position |
5c |
6b |
7c |
8b |
||||
|---|---|---|---|---|---|---|---|---|
| δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | δC, type | δH (J in Hz) | |
| 1 | 161.2, C | 151.0, C | 151.3, C | 149.0, C | ||||
| 2 | 78.9, CH | 5.49, dd (10.0, 1.6) | 147.8, C | 147.4, C | 119.0, CH | 6.02, d (10.8) | ||
| 3 | 122.5, CH | 5.05, dt (10.0, 1.2) | 115.6, CH | 5.31, s | 116.4, CH | 5.50, s | 119.0, CH | 5.93, d (10.8) |
| 4 | 142.8, C | 73.7, C | 72.9, C | 137.7, C | ||||
| 5 | 35.8, CH2 | 2.22, m | 47.0, CH2 | 2.53, m | 42.7, CH2 | 1.82, ddd (13.6, 9.6, 2.4) | 38.1, CH2 | 2.20, m |
| 2.47, ddd (13.2, 9.6, 6.8) | 1.97, ddd (13.6, 5.2, 2.0) | |||||||
| 6 | 27.5, CH2 | 1.81, m | 126.8, CH | 5.81, dt (15.6, 7.2) | 23.2, CH2 | 2.22, m | 25.0, CH2 | 2.21, m |
| 2.41, m | 2.30, m | |||||||
| 7 | 83.3, CH | 4.22, dd (7.6, 5.6) | 136.1, CH | 5.63, d (15.6) | 127.4, CH | 5.26, t (6.8) | 127.3, CH | 5.15, dd (6.0, 4.2) |
| 8 | 148.4, C | 84.3, C | 134.0, C | 133.3, C | ||||
| 9 | 31.5, CH2 | 2.22, m | 35.0, CH2 | 1.50, m | 36.5, CH2 | 2.07, m | 36.9, CH2 | 1.09, m |
| 2.35, m | 1.97, m | 2.26, m | 2.24, m | |||||
| 10 | 30.3, CH2 | 1.54, m | 22.7, CH2 | 1.45, m | 24.6, CH2 | 1.53, m | 24.5, CH2 | 1.70, m |
| 2.08, m | 1.73, m | 1.85, m | 1.78, m | |||||
| 11 | 62.9, CH | 2.61, dd (10.0, 2.8) | 62.5, CH | 2.70, dd (7.2, 4.8) | 60.6, CH | 2.71, dd (7.2, 5.2) | 62.4, CH | 2.68, dd (7.2, 3.0) |
| 12 | 61.2, C | 60.3, C | 60.4, C | 61.6, C | ||||
| 13 | 35.3, CH2 | 1.35, td (13.2, 4.0) | 35.9, CH2 | 1.60, m | 35.3, CH2 | 1.64, m | 46.0, CH2 | 1.28, dd (14.4, 1.8) |
| 2.03, m | 2.19, m | 2.17, ddd (14.4, 8.4, 6.8) | 2.31, dd (14.4, 7.8) | |||||
| 14 | 22.8, CH2 | 2.26, m | 20.1, CH2 | 2.40, ddd (15.0, 10.2, 4.8) | 19.8, CH2 | 2.28, m | 69.4, CH | 4.86, dd (7.8, 1.8) |
| 2.38, m | 2.55, m | 2.42, m | ||||||
| 15 | 124.0, C | 124.4, C | 123.8, C | 27.6, CH | 2.55, septet (6.6) | |||
| 16 | 174.7, C | 169.7, C | 169.7, C | 24.5, CH3 | 1.07, d (6.6) | |||
| 17 | 9.1, CH3 | 1.85. dd (1.6, 1.2) | 9.4, CH3 | 1.93, s | 9.3, CH3 | 1.94, s | 25.2, CH3 | 1.09, d (6.6) |
| 18 | 16.0, CH3 | 1.84, br s | 29.6, CH3 | 1.55, s | 30.1, CH3 | 1.41, s | 17.6, CH3 | 1.71, br s |
| 19 | 113.8, CH2 | 5.18, br s | 23.2, CH3 | 1.43, s | 15.5, CH3 | 1.66, br s | 15.3, CH3 | 1.65, br s |
| 5.24, br s | ||||||||
| 20 | 17.0, CH3 | 1.29, s | 17.4, CH3 | 1.24, s | 17.6, CH3 | 1.30, s | 18.2, CH3 | 1.40, s |
| 7-OOH | 7.71, s | |||||||
| 8-OOH | 7.40, s | |||||||
δ in ppm, assignments made by DEPT, COSY, HSQC, HMBC, and NOESY experiments.
At 600 MHz for 1H and 150 MHz for 13C NMR experiments.
At 400 MHz for 1H and 100 MHz for 13C NMR experiments.
Compound 6 was obtained as a colorless oil. The molecular formula of 6 was established as C20H28O6 based on the [M – H]– ion peak at m/z 363.1813 (Calcd. for C20H27O6, 363.1813) in its (—)-HR-ESI-MS, which was 16 mass units more than that of 4, appropriate for 7 degrees of unsaturation. The IR spectrum of 6 closely resembled that of 4, showing similar functionalities in the molecule. Analysis of the 1H and 13C NMR spectra (Table 2) of 6 also revealed similarities to 4, except for the location of one of the double bond from the Δ3 to Δ2 accompanied by hydroxylation occurring at C-3 in 6. These observations were supported by the HMBC correlations (Fig. 2) from the olefinic proton H-3 resonating at δH 5.31 to C-1 (δC 151.0), C-2 (δC 147.8), C-4 (δC 73.7), C-5 (δC 47.0), and C-18 (δC 29.6) and from the methyl proton singlet H3-18 (δH 1.55) to C-3 (δC 115.6), C-4, and C-5. Its relative configurations at C-8, C-11, and C-12 were proven the same as those of 4 on the basis of a NOESY experiment (Fig. 3). The diagnostic NOESY cross-peaks of H3-19/H-7 and of H3-18/one of the H2-5 protons (δH 2.53) and H-6 (δH 5.81) suggested that Me-18 was α-oriented. Finally, from a biogenetic point of view, the absolute configurations at C-8, C-11, and C-12 of 6 were suggested to be the same as those of 4. The absolute configuration of 6 was tentatively defined as 4S,8R,11R,12R. Accordingly, the structure of 6 (the trivial name sarcomililatin C) was characterized as depicted.
Compound 7 was isolated as a colorless oil. The HR-ESI-MS of 7 showed a fragment ion peak at m/z 315.1957 [M – H2O + H]+ (Calcd. for C20H27O3, 315.1955) corresponding to the loss of water from 7, suggesting a molecular formula of C20H28O4 with 7 degrees of unsaturation. The 1H and 13C NMR spectra (Table 2) of 7 showed similarities to those of 6. However, the signal resonating at δH 7.40 (1 H, s) for the hydroperoxy group at C-8 in 6 was absent. Also, the 2 mutually coupled olefinic proton signals at δH 5.81 (1 H, dt, J = 15.6, 7.2 Hz, H-6) and 5.63 (1 H, d, J = 15.6 Hz, H-7) in 6 were replaced by an olefinic proton triplet at δH 5.26 (1 H, t, J = 6.8 Hz, H-7) in 7. The above observations, combined with the 1H–1H COSY cross-peaks (Fig. 2) of H2-6 (δH 2.41 and 2.22) with H-7 and the HMBC correlations from H3-19 (δH 1.66) to C-7 (δC 127.4) and C-8 (δC 134.0), indicated the loss of the hydroperoxy group in 6 accompanied by the isomerization of the olefin from the Δ6 to Δ7. The E geometry of the Δ7 double bond in 7 was suggested by the chemical shift of the signal for the C-19 methyl group (δC 15.5), which was further confirmed by the NOESY cross-peaks (Fig. 3) of H3-19/H2-6 and of H-7/H2-9 (δH 2.26 and 2.07). The relative configurations of all the asymmetric centers in 7 remained intact, with respect to those of 6, which was supported by a NOESY experiment. Analogously, the absolute configurations at C-11 and C-12 of 7 were suggested to be the same as those of 4. The absolute configuration of 7 was tentatively defined as 4S,11R,12R. Therefore, the structure of 7 (the trivial name sarcomililatin D) was proposed as depicted.
Compound 8, assigned the trivial name sarcomililatol, was isolated as a colorless oil and exhibited the molecular formula C20H32O2 as determined by its HR-EI-MS peak at m/z 304.2409 [M]+ (Calcd. for C20H32O2, 304.2402), suggestive of 5 degrees of unsaturation, of which one was accounted for by a 14-membered macrocyclic ring, 3 by 2 olefinic bonds, and one by an ether linkage. The intense IR absorption at 3362 cm−1 was indicative of the presence of a hydroxyl group. The 1H NMR spectrum (Table 2) showed typical signals for a cembrane nucleus with 2 geminal methyls at δH 1.09 (3 H, d, J = 6.6 Hz, H3-17) and 1.07 (3 H, d, J = 6.6 Hz, H3-16), a tertiary methyl at δH 1.40 (3 H, s, H3-20), and 2 vinyl methyls at δH 1.71 (3 H, br s, H3-18) and 1.65 (3 H, br s, H3-19). The 13C NMR spectrum (Table 2) showed the presence of 20 carbon signals which were classified with the aid of DEPT and HSQC experiments as 5 methyl, 5 methylene, 6 methine (3 sp2 hybridized and 2 oxygenated), and 4 quaternary (2 sp2 hybridized and one oxygenated) carbons. The aforementioned data of 8 revealed that it should be a stereoisomer of (+)-11,12-epoxysarcophytol A, which was first isolated from the soft coral Lobophytum sp.30. When comparing their 13CNMR spectra, the signals of C-11and C-12 were shown to be markedly different [δC 62.4 and 61.6 for 8; δC 58.4 and 59.8 for (+)-11,12-epoxysarcophytol A, respectively], indicating that the structural difference between them resided in the different configuration of the 11,12-ether linkage. Detailed analysis of its 1H–1H COSY, HSQC, and HMBC spectra (Fig. 2) allowed the assignment for all proton and carbon resonances of 8.
The relative configuration of 8 was deduced from interpretation of a NOESY experiment (Fig. 3).The NOESY cross-peaks of H-14 (δH 4.86)/H-11(δH 2.68) and Me-20 implied that these protons were cofacial and were assigned a β-orientation. The geometries of the Δ1(2), Δ3, and Δ7 double bonds were assigned as Z, E, and E, respectively, on the basis of the carbon chemical shifts of Me-18 (δC 17.6) and Me-19 (δC 15.3), and this was supported by the key NOESY cross-peaks of H-2 (δH 6.02)/H3-16, H3-17, and H3-18; H-3 (δH 5.93)/H2-5 (δH 2.20) and H-14; H-7 (δH 5.15)/H2-9 (δH 2.24 and 1.09); and H3-19/H2-6 (δH 2.30 and 2.21). Thus, the relative configuration of 8 was determined as 1Z,3E,7E,11R*,12R*,14S*. However, the absolute configuration of the chiral center at C-14 in 8 was not determined by a modified Mosher׳s method due to the limited quantity of sample.
In order to determine the absolute configuration of 8, the TD-DFT ECD calculation method was applied, which has proven to be a powerful and reliable method for the elucidation of the absolute configurations of natural products9. As shown in Fig. 6, the ECD spectrum (CH3CN) of 8 displayed a positive π−π* Cotton effect at 242 nm. The conformational searches of (11R,12R,14S)-8 were carried out using the torsional sampling (MCMM) method and OPLS_2005 force field. Conformers above 1% population were re-optimized at the B3LYP/6–311 G(d,p) level with IEFPCM (Polarizable Continuum Model using the Integral Equation Formalism variant) solvent model for acetonitrile (Supplementary Information Fig. S1 and Table S1). For the resulting geometries, ECD spectra were obtained by TD-DFT calculations performed with the same functional basis set and solvent model as the energy optimization. Finally, the Boltzmann-averaged ECD spectrum of (11 R,12 R,14 S)-8 highly matched to the experimental ECD spectrum of 8, while the enantiomer showed completely opposite curve. Consequently, the absolute configuration of 8 was determined to be 11R,12R,14S.
Figure 6.

Experimental ECD spectrum of 8 [0.0016 mol/L, CH3CN, cell length 2 cm] (black) compared with the calculated ECD spectra of 8 (red) and its enantiomer (blue).
The known diterpenoids were characterized as (+)-isosarcophytoxide (9)19, 20 and (+)-isosarcophine (10)31 by comparing their observed and reported spectroscopic data. Their HR-MS spectra are also supplied in the Supplementary Information Fig. S72 and Fig. S73, respectively.
Despite numerous cembrane-type diterpenoids isolated from soft corals of the genus Sarcophyton, the investigation of the Et2O-soluble portion of the acetone extract of the soft coral S. mililatensis led to the identification of eight new cembrane diterpenoids (1–8), of which sarcomililatins A–C (4–6) possess a rare hydroperoxy group at C-7 or C-8 in the family of cembrane diterpenoids. To explain the biogenetic origin of compounds 4–6, putative biosynthetic pathways are proposed as shown in Scheme 1. The 3 metabolites can be considered to derive from a common precursor, the co-occurring known cembrane (+)-isosarcophytoxide (9).
Scheme 1.

Putative biosynthetic pathways toward the formation of compounds 4–6.
The growth inhibitory potential towards various cancer cell lines of numerous cembrane-type diterpenoids has been documented widely. Accordingly, the cytotoxicities of all the isolates were evaluated in vitro against HL-60 and A-549 by using the MTT and SRB methods, respectively. However, all of the tested compound, except for the known compound (+)-isosarcophytoxide (9), were inactive (IC50>10 μmol/L). Compound 9 exhibited strong cytotoxic activities, with IC50 values of 0.78±0.21 and 1.26±0.80 μmol/L against HL-60 and A-549 cells, respectively, comparable to the positive control (adriamycin, IC50 0.07 μmol/L for HL-60 and 0.01 μmol/L for A-549). In addition, all of the isolated compounds were also subjected to testing in vitro for their inhibitory activities against the tumor necrosis factor (TNF)-α-induced nuclear factor (NF)-κB, a transcription factor that plays a critically important role in regulation of cell cycle as well as influencing cell death pathways and has been used as a key target for the treatment of inflammatory diseases and cancer32, 33. The results indicated that sarcomilitatin A (4) and (+)-isosarcophytoxide (9) showed moderate inhibitory activities, showing IC50 values of 35.23±12.42 and 22.52±4.44 μmol/L, respectively, compared with the positive control bortezomib (IC50, 0.03 μmol/L), whereas the other compounds were inactive (% inhibition < 50% at 20 μg/mL).
3. Conclusions
A large amount of scientific research has been reported on the specialised metabolite chemistry of soft corals of the genus Sarcophyton, whereas related reports on S. mililatensis are relatively rare. In the present study, eight new cembrane-type diterpenoids, (+)-(6 R)-6-hydroxyisosarcophytoxide (1), (+)-(6 R)-6-acetoxyisosarcophytoxide (2), (+)-17-hydroxyisosarcophytoxide (3), sarcomililatins A–D (4–7), and sarcomililatol (8), were isolated and characterized from the soft coral S. mililatensis, along with 2 known ones (9 and 10). The absolute configurations of compounds 4 and 5 were elucidated by ECD spectroscopy, while the absolute configurations of compounds 1 and 8 were established by the modified Mosher׳s method and TD-DFT ECD calculation, respectively. Among these isolates, compounds 4 and 9 showed inhibitory effects on the TNFα-induced NF-κB activation, while compound 9 also exhibited promising cytotoxicity.
4. Experimental
4.1. General experimental procedures
Optical rotations were measured on a Perkin-Elmer 241MC polarimeter (PerkinElmer, Fremont, CA, USA). UV spectroscopic spectra were recorded in chromatographic grade CH3OH on a Varian Cary 300 UV−Vis spectrophotometer (Varian, Palo Alto, CA, USA); peak wavelengths are reported in nm. ECD spectra were recorded on a Jasco J-810 spectropolarimeter (JASCO, Japan) at ambient temperature using chromatographic grade CH3OH and acetonitrile as solvent. IR spectra were recorded on a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA, USA); peaks are reported in cm–1. The NMR spectra were measured at 300 K on Bruker DRX 400 and Avance 600 MHz NMR spectrometers (Bruker Biospin AG, Fallanden, Germany). Chemical shifts are reported in parts per million (δ) in CDCl3 (δH reported referred to CHCl3 at 7.26 ppm; δC reported referred to CDCl3 at 77.16 ppm) and coupling constants (J) in Hz; assignments were supported by 1H–1H COSY, HSQC, HMBC, and NOESY experiments. EIMS and HREIMS spectra (70 eV) were recorded on a Finnigan-MAT-95 mass spectrometer (ThermoFisher Scientific, Waltham, USA). ESI-MS and HR-ESI-MS were carried out on a Bruker Daltonics Esquire 3000 plus instrument (Bruker Daltonics K. K., Kanagawa, Japan) and a Waters Q-TOF Ultima mass spectrometer (Waters, MA, USA), respectively. Semi-preparative HPLC was performed on an Agilent-1260 system equipped with a DAD G1315D detector using ODS-HG-5 (250 mm × 9.4 mm, 5 µm) by eluting with CH3OH–H2O or CH3CN–H2O system at 3 mL/min. Commercial silica gel (200−300 and 400−500 mesh; Qingdao, China) was used for column chromatography (CC). Precoated SiO2 plates (HSGF-254; Yantai, China) were used for analytical TLC. Spots were detected on TLC under UV light or by heating after spraying with anisaldehyde H2SO4 reagent. Sephadex LH-20 (Amersham Biosciences) was also used for CC. All solvents used for extraction and isolation were of analytical grade.
4.2. Biological material
Specimens of Sarcophyton mililatensis, identified by Prof. Hui Huang from South China Sea Institute of Oceanology, Chinese Academy Sciences, were collected along the coast of Weizhou Island (21 °0′58″ N, 109 °6′50″ E), Beihai, Guangxi Autonomous Region, China, in May 2007, at a depth of —20 m, and were frozen immediately after collection. A voucher specimen is available for inspection at Shanghai Institute of Materia Medica, SIBS-CAS (No. WZ82).
4.3. Extraction and isolation
The frozen animals (170 g, dry weight) were cut into pieces and ultrasonically extracted with acetone at room temperature (1 L × 3). The combined acetone extracts were filtered and concentrated in vacuo, affording a brown residue, which was suspended in H2O (4 L) and then partitioned with Et2O (3 times with 2 L each). The Et2O-soluble portion (5.0 g) was concentrated in vacuo, and then fractionated by silica gel CC (column: 40 cm×8 cm) eluting with petroleum ether (PE, 60–90 °C)–acetone (98:2–0:100) to afford nine fractions (A–I), which were combined on the basis of the analysis of TLC.
Fraction D (1.2 g), eluted with PE–acetone (9:1), was initially chromatographed over Sephadex LH-20 (column: 150 cm×2 cm), using PE (60–90 °C)–CH2Cl2–CH3OH (2:1:1) as the mobile phase, to give 5 subfractions (D1–D5). Subfraction D3 (219 mg) was further separated by silica gel CC (column: 20 cm×2 cm) eluting with PE (60–90 °C)–CH2Cl2 (8:2–3:7), and successively purified by reversed-phase semi-preparative HPLC (column: ODS-HG-5, 250 mm×9.4 mm, 5 µm; mobile phase: CH3OH–H2O, 87:13; flow rate: 3 mL/min) to afford compounds 8 (1.5 mg, tR = 14.3 min) and 9 (51.0 mg, tR = 19.8 min).
Fraction E (251 mg), eluted with PE–acetone (8.5:1.5), was initially fractionated by Sephadex LH-20 CC (column: 150cm×2cm) eluting with PE (60–90 °C)–CH2Cl2–CH3OH (2:1:1) to give 4 subfractions (E1–E4). Purification of subfraction E2 (79.7 mg) by silica gel CC (column: 20 cm×2 cm) eluting with PE (60–90 °C)–Et2O (7.5:2.5) to afford compound 2 (4.2 mg). Subfraction E3 (56.9 mg) was further chromatographed over silica gel (column: 20 cm×2 cm) eluting with PE (60–90 °C)–Et2O (8:2) followed by semi-preparative RP-HPLC (CH3CN–H2O, 55:45), affording compound 10 (35.7 mg, tR = 22.8 min).
Fraction F (280 mg), eluted with PE–acetone (8:2), was initially chromatographed over silica gel (column: 20 cm×3 cm) eluting with CH2Cl2–acetone (10:0–9:1), affording 3 subfractions (F1–F3). Subfraction F2 (70.1 mg) was further separated by Sephadex LH-20 CC (column: 150 cm×2 cm) eluting with PE (60–90 °C)–CH2Cl2–CH3OH (2:1:1), and then purified by semi-preparative RP-HPLC (CH3CN–H2O, 60:40) to afford compounds 4 (3.1 mg, tR = 5.8 min) and 5 (6.1 mg, tR = 4.9 min).
Fraction G (1.3 g), eluted with PE–acetone (7:3–6:4), was initially fractionated by Sephadex LH-20 CC (column: 150 cm × 2 cm) eluting with PE (60–90 °C)–CH2Cl2–CH3OH (2:1:1) to give 6 subfractions (G1–G6). Subfraction G4 (91.3 mg) was further subjected to silica gel CC (column: 25 cm×2 cm) eluting with PE (60–90 °C)–acetone (1:1), yielding compound 7 (2.1 mg) and 3 subfractions (G4-1–G4-3). Purification of subfraction G4-2 (18.5 mg) by semi-preparative RP-HPLC (CH3CN–H2O, 55:45) yielded compound 6 (2.0 mg, tR = 7.6 min). Purification of subfraction G6 (170.7 mg) through silica gel CC (column: 20 ×2 cm) eluting with PE (60–90 °C)–Et2O (1:1–3:7) followed by semi-preparative RP-HPLC (CH3CN–H2O, 50:50) yielded compounds 1 (6.0 mg, tR = 6.7 min) and 3 (3.0 mg, tR = 11.7 min).
4.3.1. (+)-(6R)-6-Hydroxyisosarcophytoxide (1)
Colorless oil; +100 (c 0.25, CHCl3); IR (KBr) νmax 3363, 2961, 2925, 2855, 1755, 1261, 1077, 1033 cm–1; For 1H and 13C NMR spectroscopic data, see Table 1; (+)-HR-ESI-MS m/z 341.2096 [M + Na]+ (Calcd. for C20H30O3Na, 341.2087).
4.3.2. (+)-(6R)-6-Acetoxyisosarcophytoxide (2)
Colorless oil; +63 (c 0.24, CHCl3); +53 (c 0.5, MeOH); IR (KBr) νmax 2921, 2850, 1732, 1240, 1195, 1131, 1077 cm—1; For 1H and 13C NMR spectroscopic data, see Table 1; (+)-HR-ESI-MS m/z383.2197 [M + Na]+ (Calcd. for C22H32O4Na, 383.2193).
4.3.3. (+)-17-Hydroxyisosarcophytoxide (3)
Colorless oil; +98 (c 0.1, CHCl3); IR (KBr) νmax 3358, 2921, 2851, 1661, 1180, 1131, 1077 cm—1; For 1H and 13C NMR spectroscopic data, see Table 1; (+)-HR-ESI-MS m/z 341.2077 [M + Na]+ (Calcd. for C20H30O3Na, 341.2087).
4.3.4. Sarcomilitatin A (4)
Colorless oil; +43 (c 0.5, CHCl3); ECD {CH3CN, λ[nm] (Δɛ), c 0.0014 M}: 249 (–3.1), 221 (+33.4); UV (MeOH) λmax (logɛ) 246 (2.86), 275 (2.68) nm; IR (KBr) νmax 3356, 2924, 2853, 1751, 1678, 1450, 1180, 1132, 1077 cm—1; For 1H and 13C NMR spectroscopic data, see Table 1; (+)-HR-ESI-MS m/z 371.1822 [M + Na]+ (Calcd. for C20H28O5Na, 371.1829).
4.3.5. Sarcomilitatin B (5)
Colorless oil; +34 (c 0.1, CHCl3); +23 (c 0.1, MeOH); ECD { CH3CN, λ[nm](Δɛ), c 0.0014 M}: 249 (–1.5), 223 (+11.3); UV (MeOH) λmax (logɛ) 247 (2.87), 275 (2.69) nm; IR(KBr) νmax 3358, 2921, 2851, 1659, 1468, 1180, 1132, 1077 cm—1; For 1H and 13C NMR spectroscopic data, see Table 2; (+)-HR-ESI-MS m/z 371.1830 [M + Na]+ (Calcd. for C20H28O5Na, 371.1829).
4.3.6. Sarcomilitatin C (6)
Colorless oil; +3 (c 0.2, CHCl3); UV (MeOH) λmax (logɛ) 247 (2.72), 288 (2.52), 295 (2.40) nm; IR (KBr) νmax 3357, 2920, 2850, 1762, 1662, 1463, 1180, 1132, 1077 cm—1; For 1H and 13C NMR spectroscopic data, see Table 2; (—)-HR-ESI-MS m/z 363.1813 [M – H]– (Calcd. for C20H27O6, 363.1813).
4.3.7. Sarcomilitatin D (7)
Colorless oil; +35 (c 0.1, CHCl3); UV (MeOH) λmax (logɛ) 247 (2.73), 286 (2.50), 297 (2.39) nm; IR (KBr) νmax 3359, 2921, 2851, 1763, 1659, 1632, 1468, 1180, 1132, 1077 cm—1; For 1H and 13C NMR spectroscopic data, see Table 2; (+)-HR-ESI-MS m/z 315.1957 [M – H2O +H]+ (Calcd. for C20H27O3, 315.1955).
4.3.8. Sarcomililatol (8)
Colorless oil; +24 (c 0.1, CHCl3); ECD { CH3CN, λ[nm] (Δɛ), c 0.0016 M}: 242 (+13.7); UV (MeOH) λmax (logɛ) 246 (3.87) nm; IR (KBr) νmax 3362, 2922, 2852, 1195, 1132, 1077 cm—1; For 1H and 13C NMR spectroscopic data, see Table 2; EI-MS m/z 304 [M]+ (5), 289 (4), 261 (5), 243 (6), 151 (29), 137 (71), 135 (23), 133 (25), 123 (39), 121 (37), 109 (100), 107 (49); HR-EI-MS m/z 304.2409 [M]+ (Calcd. for C20H32O2, 304.2402).
4.3.9. (+)-Isosarcophytoxide (9)
Colorless oil; +111 (c 0.6, CHCl3); lit.: +160 (c 0.22, CHCl3)20; (+)-HR-EI-MS m/z 302.2240 [M]+ (Calcd. for C20H30O2, 302.2246).
4.3.10. (+)-Isosarcophine (10)
Colorless oil; +181 (c 0.2, CHCl3); lit.: +170 (c 1.01, CHCl3)20; (+)-HR-ESI-MS m/z 317.2111 [M + H]+ (Calcd. for C20H29O3, 317.2111).
4.4. Preparation of the (S)- and (R)-MTPA ester derivatives of compound 1
To 0.92 mg of compound 1 was added 450 μL of pyridine-d5, and the solution was transferred into an NMR tube. To initiate the reaction, 15 μL of (S)-MTPA-Cl was added with careful shaking and then monitored immediately by 1H NMR at the following time points: 0, 5, 10, 15, and 20 min. The reaction was found to be complete in 20 min, yielding the mono (R)-MTPA ester derivative (1r) of 1.
In an analogous manner, 0.85 mg of compound 1 dissolved in 450 μL of pyridine-d5 was reacted in a second NMR tube with 15 μL of (R)-MTPA-Cl for 20 min, to afford the mono (S)-MTPA ester (1s).
4.4.1. (R)-MTPA ester (1r) of 1
1H NMR data of 1r (400 MHz, pyridine-d5) δH 7.570−7.351 (5 H, m, Ar-H), 6.147 (1H, ddd, J = 10.8, 9.2, 5.6 Hz, H-6), 5.513 (1H, m, H-2), 5.256 (1H, d, J = 9.2 Hz, H-7), 5.157 (1 H, d, J = 9.6 Hz, H-3), 4.581 (1H, dd, J = 12.0, 4.0 Hz, H-16a), 4.497 (1H, dd, J = 12.0, 3.2 Hz, H-16b), 3.627 (3H, s, OCH3-MPTA), 2.754 (1H, dd, J = 12.4, 5.2 Hz, H-5a), 2.441 (1H, dd, J = 11.2, 2.8 Hz, H-11), 2.316 (1H, dd, J = 12.4, 10.8 Hz, H-5b), 1.851 (3H, s, H3–19), 1.608 (3H, s, H3–17), 1.523 (3H, s, H3–18), 1.301 (3H, s, H3–20).
4.4.2. (S)-MTPA ester (1s) of 1
1H NMR data of 1s (400 MHz, pyridine-d5) δH 7.628−7.351 (5H, m, Ar-H), 6.112 (1H, ddd, J = 10.8, 9.2, 5.6 Hz, H-6), 5.518 (1 H, m, H-2), 5.193 (1H, d, J = 9.2 Hz, H-7), 5.120 (1H, d, J = 9.6 Hz, H-3), 4.586 (1H, dd, J = 12.0, 4.0 Hz, H-16a), 4.502 (1H, dd, J = 12.0, 3.2 Hz, H-16b), 3.632 (3H, s, OCH3-MPTA), 2.801 (1H, dd, J = 12.4, 5.2 Hz, H-5a), 2.431 (1H, dd, J = 11.2, 2.8 Hz, H-11), 2.444 (1H, dd, J = 12.4, 10.8 Hz, H-5b), 1.850 (3H, s, H3–19), 1.608 (3H, s, H3–17), 1.532 (3H, s, H3–18), 1.279 (3H, s, H3–20).
4.5. Acetylation of compound 1
Compound 1 (1.2 mg) was dissolved in pyridine (0.5 mL) and Ac2O (0.5 mL), and the reaction was left to stir at room temperature overnight. MeOH (5 mL) was added to the reaction mixture to remove excess pyridine and Ac2O in vacuo, yielding a brown oil (3.4 mg). The crude product was purified by silica gel CC eluting with PE (60–90 °C)–Et2O (8:2–7:3) to afford a colorless oil {0.8 mg, +65 (c 0.09, CHCl3); +51 (c 0.09, MeOH)}, which was identical to the natural sample of 2 in all respects.
4.6. ECD calculation of compound 8
Torsional sampling (MCMM) conformational searches using OPLS_2005 force field were carried out by means of the conformational search module in the Macro model 9.9.223 software (Schrödinger; http://www.schrodinger.com/MacroModel) applying an energy window of 21 kJ/mol, which afforded 74 conformers for 8. The Boltzmann populations of the conformers were obtained based on the potential energy provided by the OPLS_2005 force field, which afforded 13 conformers for 8 above 1% population for re-optimization. The re-optimization and the following TDDFT calculations of the re-optimized geometries were all performed with Gaussian 0934 at the B3LYP/6–311 G(d,p) level with IEFPCM solvent model for acetonitrile. Frequency analysis was performed as well to confirm that the re-optimized geometries were at the energy minima. Finally, the SpecDis1.62 software35 was used to obtain the Boltzmann-averaged ECD spectrum of the compound and visualize the results as shown (Fig. 6).
4.7. Cytotoxicity bioassays
The cytotoxicities of compounds 1–10 against human promyelocytic leukemia cells (HL-60) and human lung adenocarcinoma cells (A-549) was evaluated by using the MTT and SRB methods, respectively, according to the protocols described in previous literature36, 37. The half-maximal inhibition (IC50) was calculated with Graphpad Prism 5.0. IC50>10 μmol/L was considered inactive. Adriamycin was used as the positive control, with IC50 values of 0.07 μmol/L for the HL-60 cell line and 0.01 μmol/L for the A-549 cell line, respectively.
4.8. NF-κB signaling pathway inhibitory activity bioassays
NF-κB signaling pathway inhibitory activity was evaluated according to the previously reported protocol38. Stable HEK293/NF-κB cells were plated into 96 well plates at a concentration of approximate 10,000 cells per well. After culturing overnight, compounds were added to the medium at a final concentration of 10 ng/mL. HEK293/NF-κB cells were seeded into 96 well cell culture plates (Corning, NY, USA) and allowed to grow for 24 h. The cells were then treated with compounds, followed by stimulation with TNF-α. Four hours later, cell title blue was added to each well. 2 hours later, the luciferase substrate was added to each well, and the released luciferin signal was detected using an EnVision microplate reader. The IC50 was calculated with Prism 4 software (Graphpad, CA, USA) from the nonlinear curve fitting of the percentage of inhibition (% inhibition) versus the inhibitor concentration [I] by using the Eq. (1):
| (1) |
where k is the Hill coefficient. Bortezomib was used as a positive control with an IC50 value of 0.03 μmol/L.
Acknowledgments
This work was financially supported by the Natural Science Foundation of China (Nos. 41506187, 81520108028, 21672230 and 81603022), NSFC-Shandong Joint Fund for Marine Science Research Centers (No. U1606403), SCTSM Project (No. 15431901000), the SKLDR/SIMM Projects (SIMM 1705ZZ-01). We thank Prof. Hui Huang from South China Sea Institute of Oceanology, Chinese Academy of Sciences, for the identification of the soft coral material.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association
Supplementary data associated with this article can be found in the online version at https://doi.org/10.1016/j.apsb.2018.06.004.
Contributor Information
Shuichun Mao, Email: maoshuichun@ncu.edu.cn.
Yuewei Guo, Email: ywguo@simm.ac.cn.
Appendix A. Supplementary material
Supplementary material
.
References
- 1.Yang B., Zhou X.F., Lin X.P., Liu J., Peng Y., Yang X.W. Cembrane diterpenes chemistry and biological properties. Curr Org Chem. 2012;16:1512–1539. [Google Scholar]
- 2.Gross H., Kehraus S., Nett M., König G.M., Beil W., Wright A.D. New cytotoxic cembrane based diterpenes from the soft corals Sarcophyton cherbonnieri and Nephthea sp. Org Biomol Chem. 2003;1:944–949. doi: 10.1039/b210039h. [DOI] [PubMed] [Google Scholar]
- 3.Lin W.Y., Su J.H., Lu Y., Wen Z.H., Dai C.F., Kuo Y.H. Cytotoxic and anti-inflammatory cembranoids from the Dongsha Atoll soft coral Sarcophyton crassocaule. Bioorg Med Chem. 2010;18:1936–1941. doi: 10.1016/j.bmc.2010.01.036. [DOI] [PubMed] [Google Scholar]
- 4.Lin W.Y., Lu Y., Su J.H., Wen Z.H., Dai C.F., Kuo Y.H. Bioactive cembranoids from the dongsha atoll soft coral Sarcophyton crassocaule. Mar Drugs. 2011;9:994–1006. doi: 10.3390/md9060994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saitman A., Sullivan S.D., Theodorakis E.A. A strategy toward the synthesis of C13-oxidized cembrenolides. Tetrahedron Lett. 2013;54:1612–1615. doi: 10.1016/j.tetlet.2013.01.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lan J., Liu Z., Yuan H., Peng L., Li W.D., Li Y. First total synthesis and absolute configuration of marine cembrane diterpenoid (+)-11,12-epoxysarcophytol A. Tetrahedron Lett. 2000;41:2181–2184. [Google Scholar]
- 7.Li X.W., Chen S.H., Ye F., Mollo E., Zhu W.L., Liu H.L. Axiriabilines A—D, uncommon nitrogenous eudesmane-type sesquiterpenes from the Hainan sponge Axinyssa variabilis. Tetrahedron. 2017;73:5239–5243. [Google Scholar]
- 8.Xue D.Q., Liu H.L., Chen S.H., Mollo E., Gavagnin M., Li J. 5-Alkylpyrrole-2-carboxaldehyde derivatives from the Chinese sponge Mycale lissochela and their PTP1B inhibitory activities. Chin Chem Lett. 2017;28:1190–1193. [Google Scholar]
- 9.Ye F., Zhu Z.D., Chen J.S., Li J., Gu Y.C., Zhu W.L. Xishacorenes A—C, diterpenes with bicyclo[3.3.1]nonane nucleus from the Xisha soft coral Sinularia polydactyla. Org Lett. 2017;19:4183–4186. doi: 10.1021/acs.orglett.7b01716. [DOI] [PubMed] [Google Scholar]
- 10.Cuong N.X., Tuan T.A., Kiem P.V., Minh C.V., Choi E.M., Kim Y.H. New cembranoid diterpenes from the Vietnamese soft coral Sarcophyton mililatensis stimulate osteoblastic differentiation in MC3T3-E1 cells. Chem Pharm Bull. 2008;56:988–992. doi: 10.1248/cpb.56.988. [DOI] [PubMed] [Google Scholar]
- 11.Van Minh C., Cuong N.X., Tuan T.A., Choi E.M., Kim Y.H., Van Kiem P. A new 9,11-secosterol from the Vietnamese sea soft coral, Sarcophyton mililatensis, increases the function of osteoblastic MC3T3-E1 cells. Nat Prod Commun. 2007;2:1095–1100. [Google Scholar]
- 12.Khan Z., Bisen P.S. Oncoapoptotic signaling and deregulated target genes in cancers: special reference to oral cancer. Biochim Biophys Acta. 2013;1836:123–145. doi: 10.1016/j.bbcan.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Cui C.C., Merritt R., Fu L.W., Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017;7:3–17. doi: 10.1016/j.apsb.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng J. How to unleash mitochondrial apoptotic blockades to kill cancers? Acta Pharm Sin B. 2017;7:18–26. doi: 10.1016/j.apsb.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu T.C., Geng J., Guo W.J., Gao J., Zhu X.X. Asiatic acid inhibits lung cancer cell growth in vitro and in vivo by destroying mitochondria. Acta Pharm Sin B. 2017;7:65–72. doi: 10.1016/j.apsb.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sen R., Baltimore D. Inducibility of Kappa immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell. 1986;47:921–928. doi: 10.1016/0092-8674(86)90807-x. [DOI] [PubMed] [Google Scholar]
- 17.Peng S., Hang N., Liu W., Guo W.J., Jiang C.H., Yang X.L. Andrographolide sulfonate ameliorates lipopolysaccharide-induced acute lung injury in mice by down-regulating MAPK and NF-κB pathways. Acta Pharm Sin B. 2016;6:205–211. doi: 10.1016/j.apsb.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garg A., Aggarwal B.B. Nuclear transcription factor-κB as a target for cancer drug development. Leukemia. 2002;16:1053–1068. doi: 10.1038/sj.leu.2402482. [DOI] [PubMed] [Google Scholar]
- 19.Bowden B.F., Coll J.C., Heaton A., König G., Bruck M.A., Cramer R.E. The structures of four isomeric dihydrofuran-containing cembranoid diterpenes from several species of soft coral. J Nat Prod. 1987;50:650–659. [Google Scholar]
- 20.Wu Y.C., Hsieh P.W., Duh C.Y., Wang S.K., Soong K., Fang L.S. Studies on the Formosan soft corals I-Cytotoxic cembrane diterpenes from Sarcophyton trocheliophorum. J Chin Chem Soc. 1992;39:355–357. [Google Scholar]
- 21.Breitmaier E,Voelter W. Carbon-13 NMR spectroscopy. 3rd ed. New York: VCH; 1987.
- 22.Tang G.H., Sun Z.H., Zou Y.H., Yin S. New cembrane-type diterpenoids from the South China Sea soft coral Sarcophyton ehrenbergi. Molecules. 2016;21:E587. doi: 10.3390/molecules21050587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El Sayed K.A., Hamann M.T., Waddling C.A., Jensen C., Lee S.K., Dunstan C.A. Structurally novel bioconversion products of the marine natural product sarcophine effectively inhibit JB6 cell transformation. J Org Chem. 1998;63:7449–7455. doi: 10.1021/jo9813134. [DOI] [PubMed] [Google Scholar]
- 24.Su J.H., Ahmed A.F., Sung P.J., Chao C.H., Kuo Y.H., Kuo Y.H. Manaarenolides A—I, diterpenoids from the soft coral Sinularia manaarensis. J Nat Prod. 2006;69:1134–1139. doi: 10.1021/np050483q. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi M. Marine terpenes and terpenoids. IV. Isolation of new cembranoid and secocembranoid lactones from the soft coral Sinularia mayi. Chem Pharm Bull. 1988;36:488–494. doi: 10.1248/cpb.36.2331. [DOI] [PubMed] [Google Scholar]
- 26.Kamel H.N., Ferreira D., Garcia-Fernandez L.F., Slattery M. Cytotoxic diterpenoids from the hybrid soft coral Sinularia maxima × Sinularia polydactyla. J Nat Prod. 2007;70:1223–1227. doi: 10.1021/np070074p. [DOI] [PubMed] [Google Scholar]
- 27.Gawronski J.K., van Oeveren A., van der Deen H., Leung C.W., Feringa B.L. Simple circular dichroic method for the determination of absolute configuration of 5-substituted 2(5H)-furanones. J Org Chem. 1996;61:1513–1515. [Google Scholar]
- 28.Bernstein J., Shmeuli U., Zadock E., Kashman Y., Néeman I. Sarcophine, a new epoxy cembranolide from marine origin. Tetrahedron. 1974;30:2817–2824. [Google Scholar]
- 29.Liu K.M., Cheng C.H., Chen W.F., Lu M.C., Fang L.S., Wen Z.H. Trocheliolide A, a hydroperoxycembranoidal diterpene from the octocoral Sarcophyton trocheliophorum. Nat Prod Commun. 2015;10:1163–1165. [PubMed] [Google Scholar]
- 30.Bowden B.F., Coll J.C., Tapiolas D.M. Studies of Australian soft corals. XXXIII. New cembranoid diterpenes from a Lobophytum species. Aust J Chem. 1983;36:2289–2295. [Google Scholar]
- 31.Bowden B.F., Coll J.C., Hicks W., Kazlauskas R., Mitchell S.J. Studies of Australian soft corals. X. The isolation of epoxyisoneocembrene-A from Sinularia grayi and isoneocembrene-A from Sarcophyton ehrenbergi. Aust J Chem. 1978;31:2707–2712. [Google Scholar]
- 32.Pikarsky E., Porat R.M., Stein I., Abramovitch R., Amit S., Kasem S. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature. 2004;431:461–466. doi: 10.1038/nature02924. [DOI] [PubMed] [Google Scholar]
- 33.Heiss E., Herhaus C., Klimo K., Bartsch H., Gerhäuser C. Nuclear factor κB is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem. 2001;276:32008–32015. doi: 10.1074/jbc.M104794200. [DOI] [PubMed] [Google Scholar]
- 34.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R. Gaussian, Inc; Wallingford CT: 2010. Gaussian 09, Revision B.01. [Google Scholar]
- 35.Bruhn T., Schaumlöffel A., Hemberger Y., Bringmann G. SpecDis: quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality. 2013;25:243–249. doi: 10.1002/chir.22138. [DOI] [PubMed] [Google Scholar]
- 36.Yu X.Q., He W.F., Liu D.Q., Feng M.T., Fang Y., Wang B. A seco-laurane sesquiterpene and related laurane derivatives from the red alga Laurencia okamurai Yamada. Phytochemistry. 2014;103:162–170. doi: 10.1016/j.phytochem.2014.03.021. [DOI] [PubMed] [Google Scholar]
- 37.Yu X.Q., Jiang C.S., Zhang Y., Sun P., Kurtán T., Mándi A. Compositacins A—K: bioactive chamigrane-type halosesquiterpenoids from the red alga Laurencia composita Yamada. Phytochemistry. 2017;136:81–93. doi: 10.1016/j.phytochem.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 38.Huang R.Y., Chen W.T., Kurtán T., Mándi A., Ding J., Li J. Bioactive isoquinolinequinone alkaloids from the South China Sea nudibranch Jorunna funebris and its sponge-prey Xestospongia sp. Future Med Chem. 2016;8:17–27. doi: 10.4155/fmc.15.169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material