

Abstract
N6-Methyladenosine (m6A) modification is the most pervasive modification of human mRNA molecules. It is reversible via regulation of m6A modification methyltransferase, demethylase and proteins that preferentially recognize m6A modification as “writers”, “erasers” and “readers”, respectively. Altered expression levels of the m6A modification key regulators substantially affect their function, leading to significant phenotype changes in the cell and organism. Recent studies have proved that the m6A modification plays significant roles in regulation of metabolism, stem cell self-renewal, and metastasis in a variety of human cancers. In this review, we describe the potential roles of m6A modification in human cancers and summarize their underlying molecular mechanisms. Moreover, we will highlight potential therapeutic approaches by targeting the key m6A modification regulators for cancer drug development.
Key words: N6-Methyladenosine, Human cancer, Pharmacological target, m6A modification regulator, Drug development
Graphical abstract
N6-Methyladenosine (m6A) modification is the most pervasive internal modification in mRNA and plays an important role in cancers via affecting proliferation, invasion, drug resistance and immunosuppression. This review summarizes the pathogenesis and development of cancer that are mainly affected by m6A modification, and demonstrates the mechanisms of m6A modification inhibitors in various cancers. The key regulators for m6A modification may act as potential therapeutic targets for anti-cancer drug development.

1. Introduction
As the delivery vehicle of genome-encoded information to the functional protein, message RNA (mRNA) plays an important role in life processes in eukaryotes. A bulk of evidence suggested that abnormal transcription or expression of mRNA was closely related to various human diseases1, 2, 3. A high volume of drugs has been designed to target mRNA maturation, transportation, location and expression in the past decades4, 5, 6. In recent years, with development of gene examination technology, more than 100 types of chemical modifications of mRNA have been explored successively7, 8, such as m1A9, 10, m5C11, m6A12, 13, 5hmC14, pseudouridine, Ψ15, 16, 17, 2′-O-methylation18, etc. Those modifications join in regulating RNA splicing, translation and stability, thus effecting gene expression in diverse physiological processes12, 19, 20, 21. Studies on mRNA modification have identified the pathology, development and prognosis of various diseases, and provided relative therapeutics and numerous new targets for drug development. N6-methyladenosine (m6A) is the most pervasive internal modification of mRNA in the human cell, which is the methylation of the adenosine base at the nitrogen-6 position of mRNA (Fig. 1). m6A modification is widely distributed (on average, approximately 3–5 m6A modification sites per mRNA molecule22, 23, 24, 25) with high abundance (>25%) in the transcripts in human cells12, 13. m6A modification was first reported in poly (A) RNA fractions in 1970s26, 27. Due to the lack of specific technique for detecting the m6A modification sites in mRNAs, the research on m6A modification had been stagnant for decades until the gene FTO (the first m6A modification demethylase) was found to revive the field of RNA methylation. This discovery suggested that RNA modification, analogous to the well-studied reversible DNA and histone modifications, might also impact biological regulation28. N6-Methyladenosine often locates in the 5′ untranslated regions (5′-UTRs), near the stop codons, in the 3′-UTRs and within internal long exons22, 29, 30. Unlike other mRNA modification, the m6A modification is dynamic and versatile, which can be regulated by its formation as well as its removal. The m6A modification modulators include the m6A modification methyltransferase, demethylase and proteins that preferentially recognized m6A modification as “writers”, “erasers” and “readers”, respectively.
Figure 1.

m6A modification of mRNA occurs in methyl group at nitrogen atoms of adenosine.
1.1. m6A modification writers: a complex of METTL3, METTL14, and WTAP
The m6A modification “writers” is a methyltransferase complex comprising of methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14) and Wilms׳ tumor 1-associating protein (WTAP). METTL3 is the primary component of this complex. Knockout of METTL3 resulted in almost complete loss of the m6A modification activity in polyadenylated RNAs21, 31, 32. METTL14, another important component in the m6A modification methylation complex, forms a stable heterodimer complex with METTL3 in a stoichiometric ratio of 1:121. METTL14 is not a methyltransferase, but an adaptor required for enhancing the METTL3 activity by binding substrate RNAs and positioning the methyl group towards adenosine33, 34, 35. As a binding partner of the methyltransferase, WTAP is essential for RNA methylation. WTAP facilitates the METTL3-METTL14 complex to locate into nuclear speckles36. Genetic depletion of WTAP causes the dislocation of METTL3 and METTL14 from these speckles and affecting m6A modification formation in mRNA37.
1.2. m6A modification erasers: mRNA demethylation by FTO and ALKBH5
Fat mass and obesity-associated protein (FTO) and alkylated DNA repair protein AlkB homolog 5 (ALKBH5), referred as m6A modification “erasers”, are both the members of the AlkB family38, 39. FTO was the first m6A modification demethylase found in cells and it is able to demethylate m6A modification in mRNA both in vitro and in vivo28. The demethylation of m6A modification in nucleic acids by FTO relies on oxidative function of FTO in a Fe(II)- and α-KG-dependent manner. ALKBH5, another m6A modification demethylase found in the nucleus, regulates export and metabolism of mRNA by m6A modification demethylation39. Deletion of ALKBH5 affects 9% of total m6A modification sites, indicating that ALKBH5 may target specific m6A modification sites in mRNAs39.
1.3. m6A modification readers: proteins that recognize m6A modification containing mRNA
Functions of m6A-containing RNAs were achieved by recruiting m6A-binding proteins (termed as m6A modification readers) that can preferentially recognize m6A modification sites in methylated RNA and facilitate downstream processes. The “m6A readers” and the “m6A switch readers” include YTH domain family (YT521-B homology), HNRNP (Heterogeneous nuclear ribonucleoproteins) and insulin-like growth factor-2 mRNA-binding proteins 1, 2 and 3 (IGF2BP1—3). Mammalian genomes contain five YTH domain-containing proteins: YTHDF1—3 and YTHDC1—2. YTHDF1—3, the m6A modification readers in cytoplasm, prefer to bind methylated RNA with concentration ranging from 180 to 520 nmol/L40. Previous study suggested that YTHDF1 promoted the translation of m6A-containing transcripts and YTHDF2 mediated mRNA decay41. YTHDC1, another member of the YTH domain family, was identified as the major readers of nuclear m6A modification42. It was reported that YTHDC1 took part in the m6A-regulated splicing43. Originally, HNRNPA2B1 was shown to bind RG m6A C-containing sites on nuclear RNAs in vivo and in vitro, which regulated the alternative splicing of exons in a set of transcripts, and facilitated the processing of pri-miRNAs44. Subsequent structure analysis showed that HNRNPA2B1 functioned as the “m6A switch readers” instead of the “m6A readers”45. IGF2BP1–3 were the latest m6A readers as recently determined. Their K homology (KH) domains recognize m6A-containing RNAs selectively and promote their translation and stability46 (Fig. 2).
Figure 2.

The processes of mRNA N6-methyladenosine modification regulated by m6A modification “writers”, “erasers” and “readers”. The m6A modification methylation and demethylation occur in the nucleus. The m6A-containing RNAs are recognized by the m6A readers, such as HNRNPA2B1, YTHDC1 and HNRNPC, which affect the splicing and export of these mRNAs. In the cytoplasm, the m6A-“imprinting” mRNA is recognized by YTHF1–3 and IGF2BP1–3, which affecting the mRNA stability, translation and decay.
1.4. m6A modification involves in human cancers
Recently, N6-methyladenosine modification represents one of the hot spots of life sciences and attracts attention of a vast amount of studies worldwide. Studies have proved that m6A modification of mRNA can effect RNA stability40, 47, 48, RNA translation efficiency41, 49, RNA secondary structure50, RNA subcellular localization39, RNA alternative polyadenylation, and RNA splicing43. Furthermore, alterations of the expression of m6A modification key regulators will affect their functions and lead to significant biological changes. The abnormality of m6A modification bioprocess has been found to be associated with various diseases, including cancer. Cancer was one of the leading causes of morbidity and mortality worldwide, killing 8.8 million people every year and leading to 1 in 6 deaths globally. More than 14 million new cases developed per year and this was set to rise to over 21 million by 2030. The cutting-edge approaches of pharmaceutical research on human cancers mainly include as follows: seeking novel targets for cancer treatment and developing molecular target-based cancer therapy51, 52; screening inhibitors of proteins that contribute to chemotherapy resistance53; designing drugs that induce a powerful immune response to cancers54; enhancing the delivery of medicine to patients and overcoming obstacles in existing treatments55. While N6-methyladenosine modification acts as the most pervasive internal modification in mRNA and plays an important role in cancers via affecting tumor cell proliferation, invasion, drug resistance and immunosuppression, the key regulators for m6A modification are potentially important molecular targets for cancer therapy. In this review, we mainly summarized the pathogenesis and development of cancers that are mainly affected by m6A modification, demonstrated the mechanisms of the m6A-associated cancer process and highlighted potential pharmaceutical targets for anti-tumor drugs. Lastly, we summed up the present m6A modification regulators as the candidates for new cancer drug discovery.
2. m6A modification are potential targets for cancer therapy
The foundation of drug development was to identify and prove the association between specific pharmacological target and disease. Drugs were designed to recover the body through regulating the diseases-relevant molecular targets56. N6-Methyladenosine modification has been found to play significant roles in cancers and has provided a series of new pharmacological targets, such as m6A modification “writers”, “erasers” or “readers” for drug development. Their mechanisms referring as the molecular target of cancers will be summarized as follows.
2.1. The function of m6A modification methyltransferase in human cancers
METTL3 was the major RNA N6-adenosine methyltransferase, which was reported to be closely associated with the genesis and development of cancers. Chen et al.57 showed that METTL3 was significantly upregulated in human hepatocellular carcinoma (HCC) and multiple solid tumors. Clinically, overexpression of METTL3 was associated with poor prognosis of HCC patients. Knockdown or knockout of METTL3 would drastically reduce HCC cell proliferation, migration, colony formation in vitro and suppress HCC progression and lung metastasis in vivo in a mechanism of augmenting tumor suppressor gene SOCS2 expression post-transcriptionally57. In acute myeloid leukemia (AML), METTL3 mRNA and protein were highly expressed. METTL3 played as an essential gene for growth of AML cells. Downregulation of METTL3 resulted in cell cycle arrest, differentiation of leukemic cells and failure to establish leukemia in immune-deficient mice. In this regard, METTL3 could be recruited by the CAATT-box binding protein CEBPZ to the transcriptional start sites and initiate transcription of some target genes. METTL3 bound to the promoter and induced m6A modification within the coding region of the associated mRNA transcript, enhancing its translation by relieving ribosome stalling58, 59. Likewise, METTL3 expression was elevated in lung adenocarcinoma. Loss- and gain-of function studies showed that METTL3 promotes growth, survival, and invasion of human lung cancer cells. Mechanistically, METTL3 could enhance translation of certain mRNAs including epidermal growth factor receptor (EGFR) and the Hippo pathway effector TAZ though interacting with the translation initiation machinery, which was independent of methyltransferase activity and downstream m6A modification readers proteins60. Mammalian hepatitis B X-interacting protein (HBXIP) was originally discovered for its binding to the C terminus of the hepatitis B virus X protein61, which was documented as an oncoprotein with high expression in breast cancer62, 63, 64. Cai et al.65 showed that expression of METTL3 was positively related to that of HBXIP in clinical breast cancer tissues. HBXIP could up-regulate METTL3 by inhibiting miRNA let-7g which down-regulated the expression of METTL3 by targeting its 3′ UTR. As a feedback, METTL3 promoted the expression of HBXIP through m6A modification, forming a positive feedback loop of HBXIP/let-7g/METTL3/HBXIP and leading to acceleration of cell proliferation in breast cancer65. Similarly, Weng et al.66 showed that METTL14 was highly expressed in acute myeloid leukemia (AML) cells. Silencing of METTL14 promoted terminal myeloid differentiation of normal HSPCs and AML cells, and thus inhibiting AML cell survival and proliferation. Mechanistically, METTL14 exerted its oncogenic role by regulating its mRNA targets (MYB and MYC) through m6A modification, while the protein itself was negatively regulated by SPI166.
While some studies have shown that METTL3 and METTL14 may play oncogenic roles in cancers, which were essential for growth of tumors. Controversially, Cui et al.67 found that m6A modification functioned as a tumor suppressor for glioblastoma stem cell (GSC) self-renewal and tumorigenesis. Knockdown of METTL3 or METTL14 would dramatically upregulate expression of some oncogenes, such as ADAM19, EPHA3 and KLF4, which promote human GSC growth, self-renewal and tumorigenesis67. Similarly, Li et al.68 found that METTL3 might have a suppressive role in cell proliferation, migration, invasion and cell cycle of renal cell carcinoma (RCC). They found METTL3 mRNA and protein expression were downregulated in RCC samples and RCC cell lines. Up-regulation of METTL3 could obviously inhibit RCC cell proliferation, migration and invasion, and induce G0/G1 arrest thus significantly suppressed tumor growth in vivo68. In cervical cancer tissues, low expression levels of the m6A modification methyltransferases (METTL3 and METTL14) were found to be associated with tumor size, differentiation, lymph invasion and recurrence69. In pancreatic cancer, the fourth leading cause of cancer deaths70, METTL3 was confirmed as a potent target for enhancing therapeutic efficacy in patients with pancreatic cancer71. In pancreatic cancer patients with METTL3-depleted, they showed higher sensitivity to anticancer reagents, such as gemcitabine, 5-fluorouracil, cisplatin and irradiation compared to those without METTL3-depleted71.
Apart from the roles in regulating tumors growth, METTL3 plays an important role in the process of T cell homeostasis72. Deletion of METTL3 in mouse T cells resulted in the loss of the m6A modification marker and in turn reduced RNA decay and elevated SOCS family activity, which consequently inhibited IL-7-mediated STAT5 activation and T cell homeostatic proliferation and differentiation72. This interesting finding suggested that T cell-specific delivery of m6A-modifying agents might be a significant indicator for cancer immunotherapy. Consequently, METTL3 and METTL14 are closely associated with the occurrence and development of cancers via m6A modification process. They play diverse roles either as oncogenes or as tumor suppressor genes in specific tumors, and provide clues to the development of cancer drugs that are associated with m6A modification methyltransferases.
2.2. Oncogenic role of m6A modification demethylase in human cancers
As the first identified RNA demethylase that regulates demethylation of target mRNAs, FTO was reported to play critical roles in cancer development and progression. Previously, Li et al.73 showed that FTO expression was increased in acute myeloid leukemia (AML). Overexpression of FTO could promote cell proliferation and viability in two AML cell lines, MONOMAC-6 and MV4–11, while knockdown of FTO expression led to the opposite effects. Mechanically, FTO exerted its oncogenic role via targeting and suppressing expression of a set of critical transcripts, such as ASB2 and RARA73. Specifically, FTO decreased stability of ASB2 and RARA mRNA transcripts upon FTO-mediated demethylation of the m6A modification level in their mRNA transcripts73.
FTO was proved to be a direct target of R-2-hydroxyglutarate (R-2HG)74. R-2HG was reported to exhibit growth-suppressive activity and glycolysis-inhibitory function in gliomas75, 76. A recent study demonstrated that R-2HG showed growth-suppressive activity in leukemia and significantly inhibited progression of sensitive AMLs in vivo77. While FTO could inhibit accumulation of m6A modification on MYC transcripts, leading to the enhancement of MYC mRNA stability and upregulation of MYC signaling and contributing to tumor progression in many cancers74, R-2HG exerted its anti-tumor effect largely through inhibiting the enzymatic activity of FTO. Additionally, Cui et al.67 showed that FTO inhibitor MA2, the ethyl ester form of meclofenamic acid (MA), could increase mRNA m6A modification levels in glioblastoma stem cell (GSCs) and suppress GSC growth. Moreover, treatment of GSCs with the FTO inhibitor MA2 suppressed GSC-initiated tumorigenesis and prolonged the lifespan of GSC-engrafted mice. In cervical squamous cell carcinoma (CSCC), FTO was found to be elevated in CSCC tissues and promote chemo-radiotherapy resistance of CSCC in vitro and in vivo by decreasing m6A modification and promoting stability of β-catenin (an EMT maker) mRNA78. Collectively, these studies suggested a key role of FTO in suppressing stability of the critical factors in cancers by reducing m6A modification process, and further affecting development and prognosis of cancers. It indicates that FTO might be a potential molecular target for cancer therapy and drug development.
Similarly, another m6A modification demethylase ALKBH5 played a critical role in tumor growth. A study conducted on glioblastoma stem-like cells (GSCs) showed that ALKBH5 was highly expressed in GSCs79. Silencing ALKBH5 could suppress proliferation and tumorigenesis of patient-derived GSCs. Mechanistically, ALKBH5 demethylated the nascent transcripts of transcription factor FOXM1 that was identified as the central molecular mediator of GSC proliferation and enhanced the expression of mature RNA or protein80. Knockdown of ALKBH5 would cause 40% reduction of FOXM1 precursor mRNA expression80. Further, a long non-coding RNA antisense to FOXM1 (FOXM1-AS) was detected to promote the interaction between ALKBH5 and FOXM1 nascent transcripts81. The FOXM1 axis in GSC could be disrupted by depletion of FOXM1-AS. ALKBH5 could exert its oncogenic function under hypoxia as well, and promote EMT in a vary of aggressive cancers, thus causing resistance to cancer therapy81. Zhang et al.82 reported that exposure of breast cancer cells to hypoxia would stimulate a significant increase of hypoxia inducible factor (HIF)-1α- and HIF-2α dependent ALKBH5 expression. Moreover, the hypoxia induced ALKBH5 expression in HIF-dependent manner could enhance NANOG mRNA stability by catalyzing m6A modification demethylation82. Previous studies demonstrated that pluripotency factor NANOG played a critical role in the maintenance and specification of cancer stem cells, which is required for primary tumor formation and metastasis83. Therefore, ALKBH5 was proved to enhance BCSC enrichment in the hypoxic tumor microenvironment, and it was verified in immune deficient mice. Despite few studies work on the relationship between ALKBH5 and tumors, m6A modification demethylase ALKBH5 was certainly proved to involve in mechanism of tumor initiation and progression, holding the potential therapeutic role for anti-tumor drugs.
2.3. YTHDF2 links RNA metabolism to cancer progression
The role of m6A modification binding proteins in human cancers was poorly documented. YTHDF2 was found to promote tumorigenesis and cell proliferation in hepatocellular carcinoma (HCC)84. It showed that YTHDF2 was closely associated with malignance of HCC. Mechanistically, miR-145 was found to directly target YTHDF2 mRNA in 3′ UTR and down-regulate YTHDF2 expression level in HepG2 cells, consequently increasing the m6A modification levels of mRNAs and decreasing proliferation of HepG2 cells84.
In all, the impact of m6A modification in human cancers was mainly through three ways. Firstly, m6A modification regulates stabilities of various oncogene mRNAs. In this scenario, methylation would promote the mature mRNA decay, and inhibit cancer procession, which was evidenced in many studies. Therefore, the m6A modification erasers and readers play oncogenic roles in cancers and could be therapeutic targets accordingly. Secondly, the m6A modification readers METTL3 was found to bind to the transcriptional start site of some genes that are essential for cancer cell surviving. Thus, inducing m6A modification by upregulating METLL3 could increase mRNA transcripts to promote cancer growth. Lastly, it was notable that m6A modification could influence cancers by regulating immune system, providing clues to the link between m6A modification and cancer immunotherapy. Targeting such m6A modification could facilitate patient׳s own immune system to fight against the progressive cancers85. Collectively, these findings suggested that the m6A modification writers, erasers and readers could play significant roles in regulation of RNA metabolism, stem cell self-renewal, and metastasis in various cancers, and it indicates that m6A modification could be targeted for prevention and treatment of human cancers. Therefore, the key regulators of m6A modification could be theoretically served as the pharmacological targets for anti-cancer drug development.
3. Targeting m6A modification regulators in human cancers
As we know, the m6A modification is involved in cancer initiation, progression and prognosis. The key regulator genes of m6A modification become crucial to regulating the downstream targets. Targeting m6A modification regulators by small molecules has been proposed as a potential treatment for human cancers. Here, we focus on those small molecules, and discuss their potential applications in cancer treatment.
3.1. Inhibitors of 2-oxoglutarate (2OG) and iron-dependent oxygenases via suppressing m6A modification demethylation
2-Oxoglutarate and iron-dependent oxygenases (2OGX) are widely distributed in human beings86, and their function relies on Fe(II) as a co-factor, 2OG and molecular oxygen as co-substrates to catalyze a broad range of biochemical reactions87. ALKBH5 and FTO belong to 2OGX-dependent nucleic acid oxygenase (NAOX) family that catalyzed demethylation of N6-methyladenine in RNA88, 89. The existing inhibitors of 2OGX could be served as unspecific inhibitors of m6A modification demethylations90, including 2OG competitor (such as N-oxalylglycine and its cell-penetrating derivative dimethyl oxalylglycine, succinate, fumarate, 2-hydroxyglutarate, etc.), metal chelators (hydroxamic acids, flavonoids), divalent transition metal ions and endogenous׳ inhibitors that regulate the activity of 2OGX (succinate dehydrogenase, fumarate hydrataseand isocitrate dehydrogenase). We have summarized those inhibitors that were examined in ALKB family (Table 1). Later on, based on FTO and ALKBH5 domains, more and more 2OGX inhibitors have been developed to inhibit the demethylations in m6A modification. There are 3 main different 2OGX inhibitor types, and they are 2OG competitor, substrate competitor or substrate and 2OG competitor90 (Fig. 3). In this scenario, Aik et al.91 screened a set of 2OG analogues and related compounds using differential scanning fluorometry- and liquid chromatography-based assays. Sets of both cyclic and acyclic 2OG analogues had been identified as FTO inhibitors, including the well-characterized 2OG oxygenase inhibitors N-oxalylglycine and pyridine-2,4-dicarboxylate, as well as hydroxyquinoline-, pyridyl-, and isoquinoline-based compounds. Crystal structure analysis further showed that two compounds (compound 1 and 2) were able to closely bind with active site of FTO and show comparatively good inhibitory effects, with the IC50 at 3.3±1.1 and 2.8±0.9 µmol/L respectively. In a subsequent study, Zheng et al.92 had designed a new class of compounds to mimic ascorbic acid and inhibit 2-oxoglutarate-dependent hydroxylases. The compound 3 and 4 have been shown to inhibit the 2-oxoglutarate dependent hydroxylase FTO with IC50 of 4.9 and 8.7 µmol/L, respectively. To assess their cellular effect of FTO inhibition, the level of m6A modification in a cell-based model was examined and quantified. For example, treatment with compound 4 with a concentration of 25 µmol/L could result in a 9.3% increase in m6A modification in cells. Additionally, this compound 4 showed anticonvulsant activity in vivo and modulated various microRNAs92. By adopting dynamic combinatorial mass spectrometry, Woon et al.93 had identified the N-oxalyl-l-cysteine derivatives compound 5, 6 and 7 as potent inhibitors of AlkB. The researchers used a capillary electrophoresis-based assay to measure IC50 values of the compounds against AlkB94, 95, and they showed the IC50s of compounds 5, 6 and 7 were 0.5, 5.2 and 5.4 μmol/L, respectively. Further, 2 physiologically important human 2OG oxygenases PHD2 and PHF8 had been tested to be inhibited by these 3 compounds and found that their IC50 were all >1 mmol/L for both PHD232 and PHF896, representing significant selectivity towards AlkB. IOX3 was a known inhibitor of hypoxia inducible factor prolyl-hydroxylases (PHDs), and was proved to bind at the active site of both FTO and PHDs97, 98. IOX3 was able to occupy both the 2OG and the nucleotide binding sites91. McMurray et al.99 proved that the IC50 value of IOX3 for FTO was 2.8 μmol/L and it could decrease the protein expression of FTO, PHDs and other 2OG oxygenases in C2C12 mouse muscle myoblast cells in vitro. This in vivo experiment suggested that IOX3 might fail to alter FTO protein level of mice at the dose of 60 mg/kg, but it could significantly reduce bone mineral density and content, and alter adipose tissue distribution, which indicating IOX3 might function via affecting the enzyme activity of FTO. Nonetheless, these inhibitors (Table 2) were not selective and they could suppress all the Fe (II)- and 2OG-dependent oxygenases. Therefore, endogenous 2OG or substrate might compete with them and weaken their inhibitory effects. More highly selective and potent inhibitors of m6A modification demethylase were required (Table 3).
Table 1.
Table of available inhibitors of 2-oxoglutarate (2OG) oxygenases for ALKB family. IC50s are shown.
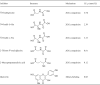 |
Figure 3.

The biochemical processes of the m6A modification demethylation. ALKBH5 and FTO are 2-oxoglutarate (2OG) and ferrous iron dependent nucleic acid oxygenase (NAOX). Inhibitors of the m6A modification demethylation could be designed as the 2OG and substrate competitors.
Table 2.
m6A modification demethylase inhibitors focusing on the 2OG and ferrous iron dependent oxidation.
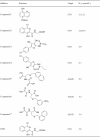 |
Table 3.
Crystal structure based m6A modification demethylase inhibitors.
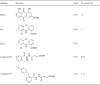 |
3.2. Crystal structures based inhibitors for m6A modification demethylase
Crystal structures of FTO100 and ALKBH5101 proteins have been studied, which provide a basis for understanding FTO and ALKBH5 substrate-specificity, and facilitate the rational design of FTO and ALKBH5 inhibitors. Following this strategy, structure-based FTO and ALKBH5 specific inhibitors had been developed extensively. For instance, Xu et al.101 have presented the crystal structures of the ALKBH5 catalytic domain. A citrate molecule was observed in the active site of ALKBH5 instead of 2OG and Mn2+ (Fig. 4A and B). Most of the residues involved in the citrate binding are involved in binding 2OG and Mn(II), that participated in regulation of the enzyme activity. Studies have showed that the IC50 of citrate for ALKBH5 was at 488 μmol/L, which is comparable to that for human FTO (300 μmol/L). Rhein was the first potent FTO m6A modification demethylase inhibitor, which was neither a structural mimic of 2-oxoglutarate nor a chelator of metal ion102. It was shown that Rhein reversibly bound to FTO catalytic domain and competitively prevented the recognition of m6A modification substrates. The details of interaction between FTO and Rhein were shown in Fig. 4C. The IC50 value of Rhein against FTO was 21 μmol/L, and its off-target selectivity analyses has proved that Rhein did not show inhibitory activity against other 2OG-dependent hydroxylases such as prolyl-4-hydroxylase, HDAC3 histone deacetylase and APOBEC3 DNA deaminases, which belong to transition metal-dependent histone and nucleic acid modifying enzymes. Additionally, Rhein was found with low cytotoxicity and was capable of increasing the modification level of m6A modification in mRNA in cells. In another study, meclofenamic acid (MA) was identified as a potent inhibitor of FTO, and belonged to a non-steroidal anti-inflammatory drug103. In this study, MA was shown to selectively and efficiently inhibit FTO demethylation in a dose dependent manner by competition on m6A-containing substrate binding in HeLa cells. It was notable that a β-hairpin motif, a part of the FTO nucleotide recognition lid (NRL), provides hydrophobic interactions between FTO and MA. However, ALKBH5 lacks such region of the part of NRL and causes leakage when binding to MA, making MA a selective inhibition of FTO over ALKBH5 (Fig. 4D). MA2 was the ester form of MA, which completely lost its inhibitory activity in vitro. The ester modification could facilitate penetration of the inhibitors in cells and further be hydrolyzed to yield active MA. Treatment with MA2 had led to the elevated levels of m6A modification in mRNA in HeLa cells. Previous study has shown that the substrate specificity of the AlkB enzymes could partly arise from structural differences within their nucleotide-binding sites104. Glu234FTO was likely a key residue that determined the affinity and specificity of FTO for its substrates. In this thread, Toh et al.104 had identified compound 8 (Fig. 4E) as a potent and subfamily-selective inhibitor of FTO that could selectively interacted with Glu234FTO. Further cell-based assays were shown that compound 9 was able to inhibit m6A modification demethylase activity in cells (Fig. 4F).
Figure 4.

Structure of m6A modification demethylase complexed with inhibitors. (A) Interaction between ALKBH5 and citrate. (B) Interaction between FTO and citrate. (C) Interaction between FTO and rhein. (D) Interaction between FTO and MA. (E) Interaction between FTO and compound 8. (F) Interactions between FTO and compound 9.
Collectively, emerging studies have worked on m6A modification in various diseases. Targeting m6A modification regulators have become a hot spot in drug design and development. As FTO was the first and most robust obesity-risk gene discovered in genome-wide association studies105, 106, 107, its inhibitors are currently the focus among other m6A modification regulators. Though many kinds of inhibitors targeting m6A modification demethylases were successfully identified, their pharmaceutical effects in vivo were rarely verified yet. Therefore, discovery of potent and selective inhibitors for m6A modification modulators is as important as design of pharmacological experiments for those identified inhibitors in clinical research.
4. Conclusions and perspectives
A large body of researches has confirmed that N6-methyladenosine modification was involved broadly in multiple types of human cancers. The m6A modification “writers” “erasers” and “readers” are certainly set at the important position of many biological pathways involved in cell metabolism, growth, proliferation and stem cell self-renewal. The m6A modification methyltransferases METTL3 and METTL14 are known to play diverse roles in specific tumors by affecting pre- or post- transcription of oncogenes, while the m6A modification demethylases and m6A modification recognition proteins sustain tumorigenicity of various cancers. Theoretically, these m6A modification regulators can be recognized as the bona fide targets in diagnosis and drug discovery of human cancers.
For the oncogenic roles of FTO and ALKBH5 being identified, small molecule inhibitors are served as the candidates for anti- cancer drug development. Up to now, FTO and ALKBH5 inhibitors are divided into two kinds of categories including broadly 2-oxoglutarate (2OG) and ferrous iron depressors, such as N-oxalylglycine and pyridine-2, 4-dicarboxylate, and structure-based selective inhibitors like Rhein, MA and IOX3. All these inhibitors had been proved to inhibit tumor growth through depressing the m6A modification levels in cancer cells. Although these inhibitors have not yet been verified in vivo and in clinical trials, it provides clues to development of m6A-specific regulators and paves the way for the treatment of human cancers, providing novel pharmacological targets for anti-cancer drug development.
Acknowledgments
This work was supported in part by a grant from the National Natural Science Foundation of China (No. 31701114) and the Fundamental Research Funds for University-Key Cultivation Project of Young Teacher in Sun Yat-Sen University (No. 17ykzd11).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Singh R.K., Cooper T.A. Pre-mRNA splicing in disease and therapeutics. Trends Mol Med. 2012;18:472–482. doi: 10.1016/j.molmed.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frischmeyer P.A., Dietz H.C. Nonsense-mediated mRNA decay in health and disease. Human Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 3.Carter B.Z., Malter J.S. Regulation of mRNA stability and its relevance to disease. Lab Investig. 1991;65:610–621. [PubMed] [Google Scholar]
- 4.Lightfoot H.L., Hall J. Target mRNA inhibition by oligonucleotide drugs in man. Nucleic Acids Res. 2012;40:10585–10595. doi: 10.1093/nar/gks861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shuman S. The mRNA capping apparatus as drug target and guide to eukaryotic phylogeny. Cold Spring Harb Symp Quant Biol. 2001;66:301–312. doi: 10.1101/sqb.2001.66.301. [DOI] [PubMed] [Google Scholar]
- 6.Masoud G.N., Wei L. HIF-1α pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B. 2015;5:378–389. doi: 10.1016/j.apsb.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Motorin Y., Helm M. RNA nucleotide methylation. Wiley Interdiscip Rev RNA. 2011;2:611–631. doi: 10.1002/wrna.79. [DOI] [PubMed] [Google Scholar]
- 8.Machnicka M.A., Milanowska K., Oglou O.O., Purta E., Kurkowska M., Olchowik A. MODOMICS: a database of RNA modification pathways—2013 update. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dominissini D., Nachtergaele S., Moshitch-Moshkovitz S., Peer E., Kol N., Ben-Haim M.S. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530:441–446. doi: 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li X., Xiong X., Wang K., Wang L., Shu X., Ma S. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat Chem Biol. 2016;12:311–316. doi: 10.1038/nchembio.2040. [DOI] [PubMed] [Google Scholar]
- 11.Edelheit S., Schwartz S., Mumbach M.R., Wurtzel O., Sorek R. Transcriptome-wide mapping of 5-methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet. 2013;9:e1003602. doi: 10.1371/journal.pgen.1003602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 13.Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delatte B., Wang F., Ngoc L.V., Collignon E., Bonvin E., Deplus R. RNA biochemistry. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science. 2016;351:282–285. doi: 10.1126/science.aac5253. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz S., Bernstein D.A., Mumbach M.R., Jovanovic M., Herbst R.H., León-Ricardo B.X. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014;159:148–162. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovejoy A.F., Riordan D.P., Brown P.O. Transcriptome-wide mapping of pseudouridines: pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLoS One. 2014;9:e110799. doi: 10.1371/journal.pone.0110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carlile T.M., Rojas-Duran M.F., Zinshteyn B., Shin H., Bartoli K.M., Gilbert W.V. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–146. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shatkin A.J. Capping of eucaryotic mRNAs. Cell. 1976;9:645–653. doi: 10.1016/0092-8674(76)90128-8. [DOI] [PubMed] [Google Scholar]
- 19.Izaurralde E., Lewis J., McGuigan C., Jankowska M., Darzynkiewicz E., Mattaj I.W. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–668. doi: 10.1016/0092-8674(94)90530-4. [DOI] [PubMed] [Google Scholar]
- 20.Li X., Zhu P., Ma S., Song J., Bai J., Sun F. Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome. Nat Chem Biol. 2015;11:592–597. doi: 10.1038/nchembio.1836. [DOI] [PubMed] [Google Scholar]
- 21.Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A.A., Kol N., Salmon-Divon M. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347:1002–1006. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- 22.Wei C.M., Gershowitz A., Moss B. Methylated nucleotides block 5′ terminus of HeLa cell messenger RNA. Cell. 1975;4:379–386. doi: 10.1016/0092-8674(75)90158-0. [DOI] [PubMed] [Google Scholar]
- 23.Narayan P., Rottman F.M. An in vitro system for accurate methylation of internal adenosine residues in messenger RNA. Science. 1988;242:1159–1162. doi: 10.1126/science.3187541. [DOI] [PubMed] [Google Scholar]
- 24.Horowitz S., Horowitz A., Nilsen T.W., Munns T.W., Rottman F.M. Mapping of N6-methyladenosine residues in bovine prolactin mRNA. Proc Natl Acad Sci U S A. 1984;81:5667–5671. doi: 10.1073/pnas.81.18.5667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harper J.E., Miceli S.M., Roberts R.J., Manley J.L. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 1990;18:5735–5741. doi: 10.1093/nar/18.19.5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desrosiers R., Friderici K., Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–3975. doi: 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perry R.P., Kelley D.E. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1:37–42. [Google Scholar]
- 28.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ke S., Alemu E.A., Mertens C., Gantman E.C., Fak J.J., Mele A. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015;29:2037–2053. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batista P.J., Molinie B., Wang J., Qu K., Zhang J., Li L. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–719. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agarwala S.D., Blitzblau H.G., Hochwagen A., Fink G.R. RNA methylation by the MIS complex regulates a cell fate decision in yeast. PLoS Genet. 2012;8:e1002732. doi: 10.1371/journal.pgen.1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong S., Li H., Bodi Z., Button J., Vespa L., Herzog M. MTA is an arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20:1278–1288. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang P., Doxtader K.A., Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63:306–317. doi: 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Śledź P., Jinek M. Structural insights into the molecular mechanism of the m6A writer complex. eLife. 2016;5:e18434. doi: 10.7554/eLife.18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X., Feng J., Xue Y., Guan Z., Zhang D., Liu Z. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575–578. doi: 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- 36.Ping X.L., Sun B.F., Wang L., Xiao W., Yang X., Wang W.J. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerken T., Girard C.A., Tung Y.C.L., Webby C.J., Saudek V., Hewitson K.S. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318:1469–1472. doi: 10.1126/science.1151710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C.M., Li C.J. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D. N6-Methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H. N6-Methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu C., Wang X., Liu K., Roundtree I.A., Tempel W., Li Y. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10:927–929. doi: 10.1038/nchembio.1654. [DOI] [PubMed] [Google Scholar]
- 43.Xiao W., Adhikari S., Dahal U., Chen Y.S., Hao Y.J., Sun B.F. Nuclear m6A readers YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–519. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 44.Alarcón C.R., Goodarzi H., Lee H., Liu X., Tavazoie S., Tavazoie S.F. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell. 2015;162:1299–1308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu B., Su S., Patil D.P., Liu H., Gan J., Jaffrey S.R. Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nat Commun. 2018;9:420. doi: 10.1038/s41467-017-02770-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou K.I., Pan T. An additional class of m6A readerss. Nat Cell Biol. 2018;20:230–232. doi: 10.1038/s41556-018-0046-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y., Li Y., Toth J.I., Petroski M.D., Zhang Z., Zhao J.C. N6-Methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang H., Weng H., Sun W., Qin X., Shi H., Wu H. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–295. doi: 10.1038/s41556-018-0045-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyer K.D., Patil D.P., Zhou J., Zinoviev A., Skabkin M.A., Elemento O. 5′ UTR m6A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T. N6-Methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cui C., Merritt R., Fu L., Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017;7:3–17. doi: 10.1016/j.apsb.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin M., Tang S., Zhang C., Chen H., Huang W., Liu Y. Euphorbia factor L2 induces apoptosis in A549 cells through the mitochondrial pathway. Acta Pharm Sin B. 2017;7:59–64. doi: 10.1016/j.apsb.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiong H., Ni Z., He J., Jiang S., Li X., He J. LncRNA HULC triggers autophagy via stabilizing Sirt1 and attenuates the chemosensitivity of HCC cells. Oncogene. 2017;36:3528–3540. doi: 10.1038/onc.2016.521. [DOI] [PubMed] [Google Scholar]
- 54.Wu M.Z., Cheng W.C., Chen S.F., Nieh S., O׳Connor C., Liu C.L. miR-25/93 mediates hypoxia-induced immunosuppression by repressing cGAS. Nat Cell Biol. 2017;19:1286–1296. doi: 10.1038/ncb3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu A.Y., Hou X.S., Ding Y. Advances of antineoplastic agents based on mitochondrial targeting mechanism. Acta Pharm Sin. 2017;52:879–887. [Google Scholar]
- 56.Pollio G., Roncarati R., Seredenina T., Terstappen G.C., Caricasole A. A reporter assay for target validation in primary neuronal cultures. J Neurosci Methods. 2008;172:34–37. doi: 10.1016/j.jneumeth.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 57.Chen M., Wei L., Law C.T., Tsang F.H., Shen J., Cheng C.L. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2017 doi: 10.1002/hep.29683. [DOI] [PubMed] [Google Scholar]
- 58.Vu L.P., Pickering B.F., Cheng Y., Zaccara S., Nguyen D., Minuesa G. The N6-methyladenosine (m6A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23:1369–1379. doi: 10.1038/nm.4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barbieri I., Tzelepis K., Pandolfini L., Shi J., Millán-Zambrano G., Robson S.C. Promoter-bound METTL3 maintains myeloid leukaemia by m6A-dependent translation control. Nature. 2017;552:126–131. doi: 10.1038/nature24678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lin S., Choe J., Du P., Triboulet R., Gregory R.I. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62:335–345. doi: 10.1016/j.molcel.2016.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Melegari M., Scaglioni P.P., Wands J.R. Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J Virol. 1998;72:1737–1743. doi: 10.1128/jvi.72.3.1737-1743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yue L., Li L., Liu F., Hu N., Zhang W., Bai X. The oncoprotein HBXIP activates transcriptional coregulatory protein LMO4 via Sp1 to promote proliferation of breast cancer cells. Carcinogenesis. 2013;34:927–935. doi: 10.1093/carcin/bgs399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu F., You X., Wang Y., Liu Q., Liu Y., Zhang S. The oncoprotein HBXIP enhances angiogenesis and growth of breast cancer through modulating FGF8 and VEGF. Carcinogenesis. 2014;35:1144–1153. doi: 10.1093/carcin/bgu021. [DOI] [PubMed] [Google Scholar]
- 64.Liu Q., Bai X., Li H., Zhang Y., Zhao Y., Zhang X. The oncoprotein HBXIP upregulates Lin28B via activating TF II D to promote proliferation of breast cancer cells. Int J Cancer. 2013;133:1310–1322. doi: 10.1002/ijc.28154. [DOI] [PubMed] [Google Scholar]
- 65.Cai X., Wang X., Cao C., Gao Y., Zhang S., Yang Z. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–19. doi: 10.1016/j.canlet.2017.11.018. [DOI] [PubMed] [Google Scholar]
- 66.Weng H., Huang H., Wu H., Qin X., Zhao B.S., Dong L. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m6A modification. Cell Stem Cell. 2018;22:191–205. doi: 10.1016/j.stem.2017.11.016. .e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cui Q., Shi H., Ye P., Li L., Qu Q., Sun G. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–2634. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li X., Tang J., Huang W., Wang F., Li P., Qin C. The m6A methyltransferase METTL3: acting as a tumor suppressor in renal cell carcinoma. Oncotarget. 2017;8:96103–96116. doi: 10.18632/oncotarget.21726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang X., Li Z., Kong B., Song C., Cong J., Hou J. Reduced m6A mRNA methylation is correlated with the progression of human cervical cancer. Oncotarget. 2017;8:98918–98930. doi: 10.18632/oncotarget.22041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vincent A., Herman J., Schulick R., Hruban R.H., Goggins M. Pancreatic cancer. Lancet. 2011;378:607–620. doi: 10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taketo K., Konno M., Asai A., Koseki J., Toratani M., Satoh T. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52:621–629. doi: 10.3892/ijo.2017.4219. [DOI] [PubMed] [Google Scholar]
- 72.Li H.B., Tong J., Zhu S., Batista P.J., Duffy E.E., Zhao J. m6A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338–342. doi: 10.1038/nature23450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li Z., Weng H., Su R., Weng X., Zuo Z., Li C. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell. 2016;31:127–141. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dang C.V. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fu X., Chin R.M., Vergnes L., Hwang H., Gang D., Xing Y. 2-hydroxyglutarate inhibits ATP synthase and mTOR signaling. Cell Metab. 2015;22:508–515. doi: 10.1016/j.cmet.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bralten L.B., Kloosterhof N.K., Balvers R., Sacchetti A., Lapre L., Lamfers M. IDH1 R132H decreases proliferation of glioma cell lines in vitro and in vivo. Ann Neurol. 2011;69:455–463. doi: 10.1002/ana.22390. [DOI] [PubMed] [Google Scholar]
- 77.Su R., Dong L., Li C., Nachtergaele S., Wunderlich M., Qing Y. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell. 2018;172:90. doi: 10.1016/j.cell.2017.11.031. [105.e23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou S., Bai Z.L., Xia D., Zhao Z.J., Zhao R., Wang Y.Y. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β-catenin through mRNA demethylation. Mol Carcinog. 2018;57:590–597. doi: 10.1002/mc.22782. [DOI] [PubMed] [Google Scholar]
- 79.Zhang S., Zhao B.S., Zhou A., Lin K., Zheng S., Lu Z. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591. doi: 10.1016/j.ccell.2017.02.013. [606.e6] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Schonberg D.L., Miller T.E., Wu Q., Flavahan W.A., Das N.K., Hale J.S. Preferential iron trafficking characterizes glioblastoma stem-like cells. Cancer Cell. 2015;28:441–455. doi: 10.1016/j.ccell.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vaupel P., Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metast Rev. 2007;26:225–239. doi: 10.1007/s10555-007-9055-1. [DOI] [PubMed] [Google Scholar]
- 82.Zhang C., Samanta D., Lu H., Bullen J.W., Zhang H., Chen I. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–E2056. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jeter C.R., Liu B., Liu X., Chen X., Liu C., Calhoun-Davis T. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene. 2011;30:3833–3845. doi: 10.1038/onc.2011.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yang Z., Li J., Feng G., Gao S., Wang Y., Zhang S. MicroRNA-145 Modulates N6-methyladenosine levels by targeting the 3′-untranslated mRNA region of the N6-methyladenosine binding YTH domain family 2 protein. J Biol Chem. 2017;292:3614–3623. doi: 10.1074/jbc.M116.749689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Couzin-Frankel J. Cancer immunotherapy. Science. 2013;342:1432–1433. doi: 10.1126/science.342.6165.1432. [DOI] [PubMed] [Google Scholar]
- 86.Loenarz C., Schofield C.J. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem Sci. 2011;36:7–18. doi: 10.1016/j.tibs.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 87.Kershaw N.J., Caines M.E., Sleeman M.C., Schofield C.J. The enzymology of clavam and carbapenem biosynthesis. Chem Commun. 2005;2005:4251–4263. doi: 10.1039/b505964j. [DOI] [PubMed] [Google Scholar]
- 88.Sancar A. DNA repair in humans. Ann Rev Genet. 1995;29:69–105. doi: 10.1146/annurev.ge.29.120195.000441. [DOI] [PubMed] [Google Scholar]
- 89.Tsujikawa K., Koike K., Kitae K., Shinkawa A., Arima H., Suzuki T. Expression and sub-cellular localization of human ABH family molecules. J Cell Mol Med. 2007;11:1105–1116. doi: 10.1111/j.1582-4934.2007.00094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Islam M.S., Leissing T.M., Chowdhury R., Hopkinson R.J., Schofield C.J. 2-oxoglutarate-dependent oxygenases. Ann Rev Biochem. 2018 doi: 10.1146/annurev-biochem-061516-044724. [DOI] [PubMed] [Google Scholar]
- 91.Aik W., Demetriades M., Hamdan M.K., Bagg E.A., Yeoh K.K., Lejeune C. Structural basis for inhibition of the fat mass and obesity associated protein (FTO) J Med Chem. 2013;56:3680–3688. doi: 10.1021/jm400193d. [DOI] [PubMed] [Google Scholar]
- 92.Zheng G., Cox T., Tribbey L., Wang G.Z., Iacoban P., Booher M.E. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem Neurosci. 2014;5:658–665. doi: 10.1021/cn500042t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Woon E.C., Demetriades M., Bagg E.A., Aik W., Krylova S.M., Ma J.H. Dynamic combinatorial mass spectrometry leads to inhibitors of a 2-oxoglutarate-dependent nucleic acid demethylase. J Med Chem. 2012;55:2173–2184. doi: 10.1021/jm201417e. [DOI] [PubMed] [Google Scholar]
- 94.Karkhanina A.A., Mecinović J., Musheev M.U., Krylova S.M., Petrov A.P., Hewitson K.S. Direct analysis of enzyme-catalyzed DNA demethylation. Anal Chem. 2009;81:5871–5875. doi: 10.1021/ac9010556. [DOI] [PubMed] [Google Scholar]
- 95.Krylova S.M., Karkhanina A.A., Musheev M.U., Bagg E.A.L., Schofield C.J., Krylov S.N. DNA aptamers for as analytical tools for the quantitative analysis of DNA-dealkylating enzymes. Anal Biochem. 2011;414:261–265. doi: 10.1016/j.ab.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 96.Loenarz C., Ge W., Coleman M.L., Rose N.R., Cooper C.D., Klose R.J. PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an Nε-dimethyl lysine demethylase. Human Mol Genet. 2010;19:217–222. doi: 10.1093/hmg/ddp480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chowdhury R., Candelalena J.I., Chan M.C., Greenald D.J., Yeoh K.K., Tian Y.M. Selective small molecule probes for the hypoxia inducible factor (HIF) prolyl hydroxylases. ACS Chem Biol. 2013;8:1488–1496. doi: 10.1021/cb400088q. [DOI] [PubMed] [Google Scholar]
- 98.Rose N.R., McDonough M.A., King O.N., Kawamura A., Schofield C.J. Inhibition of 2-oxoglutarate dependent oxygenases. Chem Soc Rev. 2011;40:4364–4397. doi: 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- 99.McMurray F., Demetriades M., Aik W., Merkestein M., Kramer H., Andrew D.S. Pharmacological inhibition of FTO. PLoS One. 2015;10:e0121829. doi: 10.1371/journal.pone.0121829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Han Z., Niu T., Chang J., Lei X., Zhao M., Wang Q. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature. 2010;464:1205–1209. doi: 10.1038/nature08921. [DOI] [PubMed] [Google Scholar]
- 101.Xu C., Liu K., Tempel W., Demetriades M., Aik W., Schofield C.J. Structures of human ALKBH5 demethylase reveal a unique binding mode for specific single-stranded N6-methyladenosine RNA demethylation. J Biol Chem. 2014;289:17299–17311. doi: 10.1074/jbc.M114.550350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chen B., Ye F., Yu L., Jia G., Huang X., Zhang X. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134:17963–17971. doi: 10.1021/ja3064149. [DOI] [PubMed] [Google Scholar]
- 103.Huang Y., Yan J., Li Q., Li J., Gong S., Zhou H. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–384. doi: 10.1093/nar/gku1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Toh Joel D.W., Sun L., Lau Lisa Z.M., Tan J., Low Joanne J.A., Tang C.W. A strategy based on nucleotide specificity leads to a subfamily-selective and cell-active inhibitor of N6-methyladenosine demethylase FTO. Chem Sci. 2015;6:112–122. doi: 10.1039/c4sc02554g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Frayling T.M., Timpson N.J., Weedon M.N., Zeggini E., Freathy R.M., Lindgren C.M. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Legry V., Cottel D., Ferrières J., Arveiler D., Andrieux N., Bingham A. Effect of an FTO polymorphism on fat mass, obesity, and type 2 diabetes mellitus in the French MONICA Study. Metabolism. 2009;58:971–975. doi: 10.1016/j.metabol.2009.02.019. [DOI] [PubMed] [Google Scholar]
- 107.Yajnik C.S., Janipalli C.S., Bhaskar S., Kulkarni S.R., Freathy R.M., Prakash S. FTO gene variants are strongly associated with type 2 diabetes in South Asian Indians. Diabetologia. 2009;52:247–252. doi: 10.1007/s00125-008-1186-6. [DOI] [PMC free article] [PubMed] [Google Scholar]