

Abstract
Vaccines and immunotherapies that elicit specific types of immune responses offer transformative potential to tackle disease. The mechanisms governing the processing of immune signals – events that determine the type of response generated – are incredibly complex. Understanding these processes would inform more rational vaccine design by linking carrier properties, processing mechanisms, and relevant timescales to specific impacts on immune response. We pursued this goal using nanostructured materials – termed immune polyelectrolyte multilayers – built entirely from antigens and stimulatory toll-like receptors agonists (TLRas). This simplicity allowed isolation and quantification of the rates and mechanisms of intracellular signal processing, and the link to activation of distinct immune pathways. Each vaccine component is internalized in a co-localized manner through energy-dependent caveolae-mediated endocytosis. This process results in trafficking through endosome/lysosome pathways and stimulation of TLRs expressed on endosomes/lysosomes. The maximum rates for these events occur within four hours, but are detectable in minutes, ultimately driving downstream pro-immune functions. Interestingly, these uptake, processing, and activation kinetics are significantly faster for TLRas in particulate form compared with free TLRa. Our findings provide insight into specific mechanisms by which particulate vaccines enhance initiation of immune response, and highlight quantitative strategies to assess other carrier technologies.
Keywords: immunotherapy, vaccine, nanotechnology, rational design, self-assembly
Graphical Abstract
Nanostructured materials composed entirely of immune signals allow isolation and quantification of the interactions between immune cells and new carriers. These electrostatically-assembled complexes of antigens and adjuvants were used to measure the routes and rates that signals reach distinct intracellular receptors, and the link to activation of downstream immune pathways.
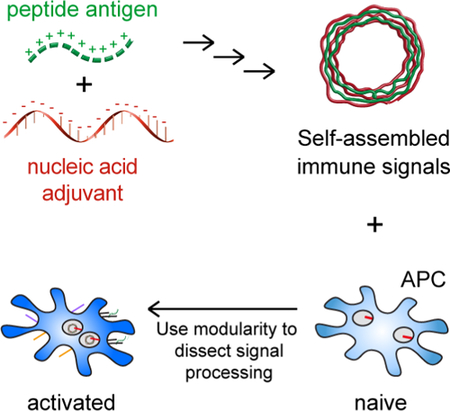
1. Introduction
Vaccines have reduced the public health threat of many infectious diseases, including polio, measles, mumps, and small pox. [1] However, exploiting the immune system to combat emerging pathogens such as Zika virus, persistent challenges such as HIV, and the heterogeneous features of cancer require more elaborate control over the specific features of immune responses elicited. Biomaterials offer the potential to enable this control, and are being intensely studied to improve the efficacy of vaccines and immunotherapies. We recently described immune polyelectrolyte multilayers (iPEMs) composed entirely of disease-relevant antigens and stimulatory molecular adjuvants. iPEMs juxtapose these vaccine components at high densities, enhancing immune responses and antitumor immunity. [2, 3] Here, we use iPEMs as simple tools to isolate the timescales and mechanisms by which nanostructured materials can interact with and be processed by antigen presenting cells (APCs), insight to support more rational design and assessment of technologies for vaccines and immunotherapy.
Despite the clinical success of vaccines, the complexity of many new and existing diseases requires greater control over the design and composition of vaccines and immunotherapies. One of the major limitations in this area is the lack of simple, modular approaches to vaccine design. For example, vaccines are generally developed through trial-and-error approach that requires significant resources and time compared with future methodologies that might allow de novo selection of the immune signal combinations and formulation properties needed for a given vaccine or immunotherapy. Additionally, most vaccines being developed, and those already in the clinic, are complex formulations consisting of antigens, adjuvants, stabilizers, carriers, attenuated pathogens, or other components required to increase potency. [4] The complexity of these mixtures further hinders development because of the increased difficulty of vaccine characterization, stability testing, and elucidation of mechanism of action. [5]
A potential route to tackle the inefficiencies above is development of well-defined adjuvants that target specific immune pathways. Once such set of pathways are toll like receptors (TLRs), which detect pathogen-associated molecular patterns (PAMPs) common in viruses and bacteria, but not in humans. [6, 7] TLR signaling pathways are activated when their respective ligand binds to the TLR. [8, 9] In the past few decades, more than a dozen TLRs have been identified and characterized. TLRs are found both on the cell surface and on intracellular membranes. [10] Generally, surface TLRs recognize PAMPs associated with bacteria, while intracellular TLRs recognize PAMPs associated with viruses; thus, this spatially-restricted distribution of receptors allows for recognition of multiple classes of pathogens. Intracellular TLRs are primarily found on endoplasmic reticulum and endosomal membranes where the TLRs can detect nucleic acid ligands. Some of the important intracellular TLRs include TLR3, TLR7/8, and TLR9. TLR3, for example, recognizes double stranded RNA such as polyinosinic-polycytidylic acid (polyIC), while TLR9 recognizes unmethylated CpG motifs. These moities each have molecular features uncommon in humans.
Due to their well-defined structure and selective pathway activation, TLR agonists are being intensely investigated as molecular adjuvants to improve vaccine efficacy and control. [6, 11–13] Importantly, different patterns of TLR signaling can result in different types of immune responses against an antigen. [14] TLR3as are already being investigated as a means of generating cytotoxic T cells and enhancing antigen cross presentation [6, 11] TLR3 agonists, such as polyIC, have shown potential as adjuvants in many mouse models of disease, inclduing influenza and cancer. [15–17] However, polyIC has seen limit success as an adjuvant in non-human primates and humans thus far due to degradation by serum nucleases and safety concerns during clinical trials. [6] Meanwhile, CpG – a TLR9a – has enhanced antigen-specific immune responses in vaccine trials for malaria, hepatitis B, influenza, and anthrax. [6] Taken together, TLRas are promising as adjuvants because of both the well defined nature, and these recent promising clinical results. TLRas could help address the broader need mentioned above for technologies that elicit specific types of immune responses, but this advance will require better understanding and control of the kinetics, trafficking, and processing of TLR-based adjuvants.
Biomaterials are being investigated to improve vaccine design because of advantages such as co-delivery, tunable loading, control over release kinetics, and protection of cargo from enzymatic degradation. [18] More specifically, for delivery of TLRas, biomaterials offer the opportunity to target these signals to specific receptor domains, which, as just described, can directly control the type of response generated against an antigen of interest. For example, a biomaterial matrix-based vaccine system has been developed that can deliver nanoparticles of CpG and drives strong cytotoxic T lymphocyte responses (CTL) responses in a mouse model of melanoma; this technology is now in human clinical trials for melanoma. [19] Another biomaterial approach has used polymer particles to co-deliver TLRas and antigen for induction of protective immunity against influenza in rhesus macaques. [20] Conversely, biomaterials have also been used to deliver regulatory TLR ligands that target intracellular TLRs signaling to promote tolerance in mouse models of multiple sclerosis. [21, 22] Recently, biomaterials are also being used to deliver TLRa and other signals for engineering or reprogramming immune tissues. [23] However, as with traditional vaccines, the incorporation of biomaterials for all of these applications adds complexity that can slow or even hinder vaccine design. Further, recent studies show that many common polymers exhibit intrinsic materials properties that can activate, suppress, or alter immune signaling. These phenomena can further complicate vaccine design because the carrier component might change the response to the antigen or other vaccine components. [24–29] Some key examples driving these intrinsic immunogenic features include physicochemical characteristics such as shape, [25] hydrophobicity, [26] and molecular weights [27, 28].
We recently developed a platform to self-assemble immune signals into nanostructured capsules. The materials mimic many of the attractive features of biomaterial carriers, but simplify vaccine design and remove complicating immunogenic effects because they are built entirely from immune signals. These capsules are assembled by electrostatic interaction of cationic peptide antigen and polyIC to form immune polyelectrolyte multilayers (PEMs). [2] While PEMs comprised of synthetic polymers have been used for sensing, drug delivery, catalysis [30–32] – and more recently, vaccine delivery [33–41] – iPEMs are unique in that they are built only from immune signals. This feature ensures a high density of signals, juxtaposition of antigen and TLRa adjuvant, and also eliminates the need for complicated carriers and excipients. [2, 3, 42]
Understanding how nanoparticulate materials – such as iPEMs, polymer particles, and liposomes – interact with distinct steps involved in initiation of adaptive immunity is critical, particularly for technologies that deliver TLRas, which are spatially restricted to distinct regions of APCs. [7] Thus, while our past studies with iPEMs focused on in vivo performance, here we use iPEMs as simple tools to probe and quantify interactions involved in antigen and adjuvant uptake, trafficking to target intracellular domains, activation of specific innate signaling pathways, and ultimately, initiation of downstream APC processes that directly drive adaptive immune responses in T cells. We show iPEMs promote co-delivery of antigen and TLRa into APCs through time and energy-dependent caveolae-mediated endocytosis uptake. This route leads to trafficking through endosomes/lysosomes, triggering of TLRs spatially-restricted to these sites, and downstream activation of APCs to present T cell co-stimulatory signals. Importantly, the rates most of these process occur at are significantly faster compared with equivalent doses of free TLRas. These findings are significant for iPEMs and other nano- and micro-particulate vaccine candidates targeting TLR signaling because understanding the mechanisms and timescale by which TLRs are activated could enable design of technologies to tailor immune responses by directing TLR signaling.
2. Results and Discussion
2.1. iPEM assembly results in well-defined capsules composed from antigen and TLRa
iPEMs were assembled on sacrificial calcium carbonate templates through electrostatic interaction of SIINFEKLR9 (SIINR9) – a cationic peptide antigen, and anionic TLR3a polyIC, an immunostimulatory double stranded RNA. After assembly of 3 bilayers, the core was removed using a chelator to create hollow capsules, (SIINR9/polyIC)3. We used these structures to quantify the kinetics and uptake mechanisms of processing, as well as the link to activation of TLR signaling and immune function (Figure 1a). Fluorescence microscopy was used to visualize the colocalization of each iPEM component after assembly, which were labeled with distinct fluorophores (Figure 1b). iPEMs exhibited diameters of 5.01 ± 0.84 μm prior to core removal, and 2.32 ± 0.68 μm after the core was removed to create the capsules (Figure S1a, Supporting Information). Capsules were composed of 57.7 ± 4.1 % SIINR9 and 42.3 ± 4.1 % polyIC by weight percent (Figure S1b, Supporting Information). Loading of antigen and adjuvant in this study varied from the previous studies with these cargos because the synthesis was performed entirely in water, which created a more facile and consistent synthesis result. [2, 3] Biomaterials offer many advantages to improve vaccine design such as co-delivery and cargo protection but have also been shown to activate an immune response. [18, 24] iPEMs are a promising alternative that offer many of the same advantages of biomaterials, such as co-delivery of signals, while eliminating the complicating intrinsic immune effects exhibited by many synthetic polymers. Further, due to the synthesis approach, iPEMs are composed 100 % of cargo, in this case, SIINR9 and polyIC. This is in contrast to traditional carriers involving encapsulation or adsorption of vaccine or immunotherapy components in or on polymer matrices and particles. iPEMs are a modular platform that can in principle be extended to any peptide antigen; antigens that are cationic do not need to be modified, while the epitope sequence of other antigens can be preserved by appending arginine or other cationic amino acids. In our past in vivo studies we have not observed the appended amino acids to hinder antigen-specific responses using either CD4 and CD8 dominant epitopes, however, it is possible such modifications could change antigen processing. [2, 3, 21, 41, 42]. For example, if a different (non-functional) subsequence of a an antigen appended with charged amino acids were loaded into MHC, the selectivity of response would be altered. Future molecular docking experiments might allow this possibility to be quantified as a function of peptide modification.
Figure 1.

iPEMs can control immune cell activation by delivering multiple immune signals. A) Schematic of iPEM synthesis and immune activation following treatment with iPEMs. Starting at the top left, iPEMs are synthesized through the electrostatic interactions between cationic peptide antigen and anionic nucleic acid-based TLR3a agonist. iPEMs can be used to control the kinetics of APC uptake and activation by delivering signals over time to target receptors in endosomes. B) Fluorescent microcopy images of iPEMs composed of model antigen SIINFEKLR9 (green) and TLR3a, polyIC (red). iPEMs colocalize both signals (yellow) in carrier-free capsules. Scale bar is 10 μm.
2.2. iPEM components are co-localized in DCs and internalized at similar rates
We synthesized iPEMs to deliver immune signals intracellularly where spatially-restricted intracellular TLRs reside. To investigate the rates at which iPEMs are internalized, and the density of iPEMs per cell, we incubated iPEMs with primary CD11c+ dendritic cells (DCs) for defined intervals. Fluorescence microscopy (Figure 2a) and automated image analysis (Figure 2b) revealed an increase in iPEMs in DCs over time, with a mean density of 2 iPEM events/cell after 15 minutes, and a mean density of 12 iPEM events/cell after 1 hour. These results were further confirmed by flow cytometry, which revealed an increase in the iPEM cargo signals over 1 hour, and continuing for 4 hours (Figure 2c). We note that the absolute MFI values across fluorophores is not indicative of the amounts of each cargo relative to one another since the labeling density and excitation/emission efficiency are different. However, analysis of the frequency of cells positive for each cargo revealed a significant increase in cargo uptake between 15 minutes and 4 hours (Figure 2d). Quantifying uptake kinetics over longer durations of ~1 day revealed a maximum in the antigen and adjuvant signal in cells after 2 to 4 hours incubation (Figure 2e). Interestingly, the maximums for each iPEM component (i.e., antigen and adjuvant) occurred at approximately the same interval, suggesting the components were co-delivered and entered cells at similar rates, as expected for juxtaposed cargos.
Figure 2.

Time dependent uptake of iPEMs by primary DCs. A) Fluorescent microscopy images of primary CD11c+ DCs incubated with iPEMs for 15 to 60 minutes reveals an increase in iPEMs taken up over time. Scale bar is 10 μm. B) Image analysis shows quantitatively an increase in the number of iPEMs per cell between 15 and 60 minutes. C) Flow cytometry of DCs incubated with iPEMs for 0 to 4 hours, showed an increase in iPEM cargo fluorescent intensity. D) Flow cytometry analysis of cells positive for iPEM cargo show an increase in iPEM cargo uptake (red and green bars) over 4 hours. Free polyIC was also taken up over 4 hours but to lesser extent. E) Flow cytometry analysis of DCs incubated with iPEMs for 24 hours shows an increase in iPEM signals over the first 4 hours followed by a decrease over the course of the next 20 hours. DCs incubated with dose-matched soluble polyIC show only a modest increase in polyIC intensity after 24 hours. Error bars are standard deviation. Statistical analysis performed by 2 way ANOVA; **** = P≤0.0001.
Initiation of immune response requires a combination of antigen and co-stimulatory molecules. Activation of the TLR signaling cascade leads to an upregulation of these co-stimulatory molecules. [10] Thus, co-delivery of antigen and TLRa to APCs provides the necessary components for immune activation in one structure. In contrast, cells treated with a dose-matched amount of soluble polyIC (“Free polyIC”) did not take up polyIC as rapidly or to the same extent as cells treated with iPEMs containing equivalent levels of polyIC. For example, at 4 hours 16 % of cells were positive for free polyIC compared to over 72 % of cells positive for antigen and polyIC delivered by iPEMs, (Figure 2d). This result indicates iPEMs deliver TLRas more efficiently and more quickly to APCs compared with free TLRa. The clinical efficacy of TLRas is limited by rapid clearance and degradation following soluble delivery in vivo. [6, 11–13] Thus, several biomaterial strategies – which allow sustained delivery and targeting – are being tested in the clinic. [18] iPEMs may create unique advantages in these contexts, for example, Figure 2 suggests iPEMs might allow more rapid or concentrated delivery of TLR over a given interval (e.g., before being cleared or degraded) relative to free cargo. Further, this more concentrated cell delivery might improve vaccine efficiency and limit off-target effects by reducing the total injected dose. However, these possibilities will need to be validated in vivo and compared with other particulate-based delivery systems. Overall, the data in Figure 2 reveals iPEMs increase the rate and magnitude that TLRas are delivered to immune cells, which peaks between 2 and 4 hours.
We next directly quantified the extent of cargo co-localization in DCs as a function of time. Flow cytometry revealed the change in the frequency of cells positive for both iPEM components (i.e., SIIN-R9+/polyIC+ cells) over the course of 24 hours (Figure 3a). At all assessment times, the fraction of cells positive for a single component was small, and generally decreased as time increased (Figure 3a). As expected from Figure 2, the maximum for value for cells positive for both iPEM components occurred after just 4 hours, then began to wane by 24 hours (Figure 3b). Since the rate of change (i.e., slope) in the frequency of cells double positive for both components was greatest at the earliest time points during flow cytometry studies (Figure 3b), we next used fluorescence microscopy and automated image analysis to measure the spatial co-localization of iPEM components within cells over one hour. In these experiments, each image pixel was plotted according to fluorescence intensity in the polyIC (x-axis) and SIIN-R9 (y-axis) channels for that same pixel position (Figure 3c). These scatterplots allowed calculation of the fraction of pixels positive for SIIN-R9 that were also positive for polyIC (Figure 3d). Intriguingly, while the frequency of cells positive for both iPEMs component increased with time (Figure 3b), the fraction of antigen (SIIN-R9) associated with adjuvant (polyIC) remained constant at all time points (Figure 3d). This result indicates components used to build iPEMs are internalized into DCs together. This co-delivery characteristic is important because generating adaptive immune response requires both an antigen (e.g., SIIN-R9) and inflammatory signal (e.g., polyIC) to be processed and presented by APCs. [10] Past mechanistic studies have elucidated specific mechanistic benefits associated with co-delivering immune signals to improve vaccine efficacy. [19] Further, many biomaterials strategies achieve co-delivery of signals to provide synergistic effects in pre-clinical models. [43, 44] Taken together then, our studies show that iPEMs provide a simpler, alternative platform to effectively co-deliver immune signals to APCs without need for additional components or carriers that complicated characterization and design, or that introduce intrinsic immunogenic carrier properties.
Figure 3.

Co-delivery of immune signals to primary DCs with iPEMs. A) Flow cytometry analysis of primary DCs incubated with iPEMs shows that in roughly 40–80% of cells, iPEMs deliver both antigen and adjuvant. B) Flow cytometry analysis of cells positive for both antigen and adjuvant reveals an increase in the frequency of cells over the first 4 hours which was sustained for up to 18 hours. C) Image analysis of fluorescent microscopy images of iPEM uptake shows an increase in pixels positive for iPEM signals over time. Each pixel is plotted by the fluorescent intensity of both the FITC signal resulting from antigen and Cy5 signal from polyIC with the highest intensity of both signals in the top right corner. D) Quantifying the fraction of SIIN-R9 signal co-localized with polyIC signal from the image analysis plots in B, shows that over 80% of immune signals are colocalized, which is sustained over 1 hour. Error bars are standard deviation.
2.3. Uptake of iPEMs is energy-dependent
Since iPEMs were internalized in a time dependent manner, we next tested the hypothesis that this uptake was energy dependent. iPEMs were incubated with DCs at either 4 °C – to inhibit energy-dependent uptake – or a standard culture temperature of 37 °C. [45] Fluorescence microscopy revealed iPEMs were nearly absent in DCs after incubation at 4 °C, while iPEMs were ubiquitous in cells for the same interval at 37 °C (Figure 4a). Quantification by image analysis confirmed a significant increase in iPEM density following incubation at 37 °C compared to incubation at 4 °C (Figure 4b). This was further confirmed using flow cytometry, which also revealed a significant increase in mean signal intensity and percent of iPEM positive cells comparing incubation at 37 °C versus 4 °C (Figure 4c and 4d). We initially hypothesized that the R9 cationic anchor on SIIN may serve as a cell-penetrating moiety. [46–48] The energy dependence of uptake in Figure 4 suggests otherwise, since this alternate mechanism would be expected to occur even at low temperature. However, a fraction of cells contained low levels of iPEMs following incubation at 4 °C, so cell-penetrating mechanisms may still be a contributing factor. In future studies the cationic anchor (i.e., R9) will be altered using different lengths and compositions of amino acids to test if these parameters increase cell-penetration. The findings in Figure 4 added greater evidence that iPEMs are taken up by energy-dependent endocytosis processes, motivating studies to identify specific mechanisms of internalization by DCs.
Figure 4.

Energy dependent uptake of iPEMs by primary DCs. A) Fluorescent microcopy images of primary DCs incubated with iPEMs at 4°C and 37 °C shows that DCs require energy to take up iPEMs. Scale bar is 10μm. B) Images analysis quantitatively reveals a decrease in the number of iPEMs per cell when energy dependent processes are inhibited by incubating at 4°C. C) Representative flow cytometry plots and D) analysis of the percentage of cells positive for iPEM signal FITC from SIIN-R9 confirm a statistically significant decrease in iPEM uptake when energy dependent processes are inhibited. Error bars are standard deviation. Statistical analysis performed with one-way ANOVA with a Tukey post-test to correct for multiple comparison. **** = P≤0.0001.
2.4. iPEMs are internalized by caveolae-mediated endocytosis
To dissect the specific pathways by which iPEMs are internalized and processed through, we cultured DCs with iPEMs and either a total inhibitor of endocytosis (2-deoxy-D-glucose, 2DG), or a selective inhibitor of caveolae-mediated endocytosis (Filipin). We first confirmed the inhibitors had no impact on the viability of DCs relative to untreated cells at several concentrations (Figure 5a). Next we measured the impact of the inhibitors on iPEM uptake by DCs using flow cytometry (Figure 5b). These studies revealed that 80 mM 2DG inhibited 60 % of iPEM uptake. This result indicates that uptake of iPEMs is dependent on endocytosis. Further, 10 μg mL−1 Filipin inhibited nearly 50 % of iPEM uptake by DCs, indicating more specifically that iPEMs are internalized to a significant extent by caveolae-mediated endocytosis. Caveolae are smooth invaginations of the plasma membrane. Endocytosis involving these structures relies on the presence of cholesterol and dynamin function. During caveolae-mediated endocytosis, caveolae buds containing endocytosed cargo are pinched off from the cell membrane to form caveolar vesicles that ultimately move to the endosomal pathway. [49] Caveolae-mediated endocytosis is known to be involved in cellular uptake of large bacteria and viruses [50] and has been shown to play a part in the uptake of microspheres as well. [51] Thus, although particles size and chemistry can impact the uptake route, it is not surprising that iPEMs are at least in part internalized by caveolae-mediated endocytosis. The uptake experiments in Figure 3 confirmed iPEM components are taken up together and co-delivered to APCs, suggesting iPEMs are still likely in capsule form at the onset of uptake. In light of this possibility, it is possible that the carrier-free, capsular nature of iPEMs provides a degree of flexibility during uptake compared to solid particulate delivery systems. Measuring the mechanical properties of iPEM capsules directly might reveal if such features exist, and help further explain the link between material properties and the caveolae-mediated endocytosis route. Downstream, with respect to stimulating immune response, iPEMs must be trafficked through endosomal pathways to deliver TLR3a to the TLR3 located in endosomes. [10] Caveolae-mediated endocytosis uptake aligns with this trafficking route, so we investigated the next processing step along this route.
Figure 5.

Inhibitor dependent uptake of iPEMs by primary DCs. A) Flow cytometry analysis of primary DCs incubated with iPEMs show no change in viability relative to untreated when incubated with a total inhibitor of endocytosis, 2-deoxy-D-glucose (2DG), or caveolae-mediated endocytosis inhibitor, Filipin. B) Analysis of the percentage of cells positive for the FITC signal from iPEMs relative to no inhibitor shows a reduction in uptake following incubation with either inhibitors. Error bars are standard deviation. Statistical analysis was performed with one-way ANOVA against the group without inhibitors using a Tukey post-test to correct for multiple comparisons. **** = P≤0.0001, n.s. = not significant.
2.5. iPEMs are trafficked through lysosomes
Because iPEMs are taken up at least in part by caveolae-mediated endocytosis, we investigated if these capsules are processed through lysosomes. Fluorescence microscopy was used to track colocalization of iPEMs within lysosomal compartments of DCs over time (Figure 6a). These experiments revealed an immediate increase in lysosome signal after iPEM treatment, which was expected since a known outcome of antigen internalization by DCs is an increase in lysosomal acidification. [52] To determine if iPEMs were co-localized with lysosomes, we quantified the colocalization of signal from each of these sources over time using pixel intensity plots (Figure 6b). Within 30 minutes, iPEM signal began to co-localize with lysosome signal, reaching a mean value of approximately 40 % that persisted for the remaining time points (Figure 6c); after 1 hr, for example, roughly 44 % of iPEMs were colocalized with lysosomes. Analysis of the frequency of lysosomes colocalized with iPEMs also revealed an increase by 30 minutes, but the stabilization point was at a much higher value of approximately 85 % (Figure 6d). This indicates that while most lysosomes contain iPEMs, a fraction of iPEMs in cells are not co-localized with lysosomes. One explanation is that iPEMs entering the DCs are in structures not yet exhibiting the characteristics of endosomes. Some early evidence supporting this possible explanation is the observation that after 15 min., a very small fraction of iPEMs are in lysosomes (Figure 6c), whereas at the same time, a significant fraction of lysosomes contain iPEMs (Figure 6d). However, there are several other considerations around this discovery. As referenced earlier, it is possible that a certain number of iPEMs are entering the cell using cell-penetration, which would not result in endosomal delivery to lysosomes. Another possibility is degradation of iPEMs occurs during uptake, leading to some components escaping the endosome/lysosome pathway. These possibilities can be tested by observing iPEM uptake with both signals labeled in live cells and staining for early endosomes in addition to lysosomes. This study would also reveal whether improved uptake and activation kinetics of iPEMs are preferentially associated with early or late endosomal stages. As already mentioned, the TLR for polyIC is located within endosomes and lysosomes of DCs. [53, 54] Thus, the ability of iPEMs to traffic through lysosomes is critical to fulfilling their immunostimulatory function in the context of vaccination and immunotherapy.
Figure 6.

Trafficking of iPEMs through primary DCs. A) Fluorescent microscopy images of primary DCs incubated with iPEMs shows iPEMs co-localizing with lysosomes over time. B) Image analysis of fluorescent microscopy images of iPEM uptake shows an increase in pixels positive for iPEM signal FITC and lysosomes over time. Each pixel is plotted by the fluorescent intensity of both the FITC signal resulting from antigen and Texas-red signal from lysotracker red with the highest intensity of both signals in the top right corner. C and D) quantifying the fraction of iPEMs and lysosomes colocalized from the image analysis plots in B, shows an increase in colocalization after 30 minutes that was sustained over 60 minutes. Error bars are standard deviation. Statistical analysis performed with a T test comparing each time point to the 15 min time point. * = P≤0.05, ** = P≤0.01, **** = P≤0.0001.
2.6. iPEMs activate DCs more efficiently than soluble TLRa
Uptake of iPEMs is time dependent (Figure 2) and involves processing through lysosomes (Figure 6). Thus, we hypothesized activation of endosomal/lysosomal TLRas such as TLR3 would be time dependent, and subsequently, lead to DC activation. To test these ideas we incubated DCs with either iPEMs or dose-matched free polyIC for defined times, then used flow cytometry to analyze cells for surface markers associated with DC activation, CD86 (Figure 7a) and CD40 (Figure 7b). iPEMs activated DCs much more quickly and to greater extents compared with free polyIC. This was true for all markers over the first 4 hours, and at even longer times for CD86. These data were particularly interesting since the dose of polyIC was fixed in all cases, irrespective of whether cells were treated with free polyIC or iPEMs. Based on the results in Figure 2, this rapid DC activation is likely because iPEMs are taken up at much greater levels than free polyIC, and further, quickly associate with lysosomes (Figure 6). To assess a further step in the cascade of activating antigen-specific T cells, we tested if antigen in iPEMs is displayed in the appropriate antigen presented complexes Major histocompatibility complex I (MHC-I) - at rates equivalent to controls of free antigen mixed with polyIC (“Free mix of polyIC & SIIN-R9”) (Figure 7c). After 2 hours, and continuing through 24 hours, treatment with iPEMs led to significant increases in antigen presentation (Figure 7c). The levels were similar to those measured in cells treated with simple mixtures of antigen and polyIC, though at some points the simple mixtures exhibited slight to modest increases. The results of these studies are important because they indicate that despite being tightly bound to polyIC, antigen is still displayed and processed in the appropriate machinery. Further, our data show iPEMs also lead to more rapid and potent activation. Thus, to specifically assess TLR signaling and confirm the enhanced activation driven by iPEMs results from improved uptake and trafficking – not from more efficient TLR activation, we treated TLR3 reporter cell line with iPEMs or polyIC and assessed TLR3 signaling over time (Figure 7d). Treatment with iPEMs and Free polyIC both drove high levels of TLR3 signaling relative to untreated control wells. The levels were generally similar, though iPEMs activated TLR3 signaling to slightly greater extents at early time points, while free polyIC activated TLR3 to slightly greater extents at the longest time points. These studies reveal that the kinetics of iPEM uptake and localization with endosomes and lysosomes – where TLR3 are expressed – are primary drivers of enhanced DC activation. Interestingly, although TLRa delivery peaked at 4 hours (Figure 2), DC activation persisted much longer, for at least 24 hours, illustrating the potential of using these types of analyses to dissect the specific roles and mechanisms involved in the immunogenicity of nanoparticulate vaccines and immunotherapies.
Figure 7.

DC activation with iPEMs analyzed by flow cytometry. Flow cytometry analysis of surface markers of DC activation A) CD86 and B) CD40 reveals an increase in DC activation following incubation of DCs with iPEMs over time, to a greater extent than dose-matched soluble delivery of polyIC, especially over the first 4 hours. C) Flow cytometry analysis of antigen presentation in MHCI shows an increase in antigen presentation over time following treatment with iPEMs comparable to treatment with dose-matched soluble components. D) TLR3 reporter cell activity shows iPEMs more efficiently activate the TLR3 signaling pathways than a dose-matched treatment with Free polyIC. Error bars are standard deviation. Statistical analysis performed with a T test comparing iPEMs to Free polyIC and SIINR9. * = P≤0.05, ** = P≤0.01, *** = P≤0.001, **** = P≤0.0001, n.s. = not significant.
3. Conclusion
In this work, we investigated the mechanism by which nanostructured assemblies of immune signals are taken up and processed to initiate adaptive immunity. Our data reveal iPEM uptake is time and energy dependent, involves caveolae-mediated endocytosis, and trafficking through lysosomes. This is significant because the adjuvant component in the iPEMs is a TLR3 agonist found on endocytic compartments of DCs. More significant is the discovery that iPEMs increase DC activation through TLR3 signaling because these structures are internalized more rapidly than free TLRas, and to a lesser extent, because of more efficient activation of TLRs. The ability to individually understand how antigens and adjuvants are processed in iPEMs – without confounding intrinsic immune activity of common synthetic polymers – creates a tool to better understand how specific features of nanostructures materials interact with antigen presenting cells. New insight such as this could help speed design and translation of biomaterial carriers being investigated for vaccination and immunotherapy.
4. Experimental Section
Synthesis of iPEMs:
SIINFEKLR9 (SIINR9) was synthesized by Genscript with >98 % purity, with a FITC label on the N-terminus. Low molecular weight polyinosinic-polycytidylic acid (polyIC) was purchased from Invivogen. iPEMs were synthesized as previously described with the exception that all cargo and wash solutions were in DI water. [2] After the final layer, the calcium carbonate core was removed by resuspending the iPEMs in 1 mL 0.1 M ethylenediaminetetraacetic acid (EDTA, Sigma-Aldrich), adjusted to pH 4 with 1 M HCl and 1 M NaOH solutions. Capsules were then centrifuged at 2000g for 2 min and washed with 100 μL 1X PBS. iPEMs were collected by centrifuging at 1000g for 1 min and then resuspended in 100 μL PBS. In some studies, iPEM capsules were formed from FITC SIINR9 and Cy5 labeled polyIC following the same protocol as above. Label-IT nucleic acid labeling kits (Cy5) were purchased from Mirus Bio LLC. PolyIC was labeled according to the Mirus Bio instructions.
Characterization of iPEMs:
iPEMs composed of FITC SIINR9 and Cy5 labeled polyIC were used for characterization studies. iPEMs were imaged using a 60X objective on a fluorescent microscope (Olympus IX-83) and analyzed using ImageJ. 1,825 iPEMs were sized by manually thresholding the capsules and using the analyze particle plugin to get the area of each iPEM. The diameter was calculated using the areas of each iPEM by assuming iPEMs are circles. PolyIC loading was determined by incubating iPEMs in 200μL trypsin (0.05 %) at 37 °C for 1 hr, then measuring the Cy5 signal by fluorimetry using a Gemini XPS fluorescence microplate reader (Molecular Devices). Loading the SIINR9 was measured as previously described using a MicroBCA Protein Assay Kit (Thermofisher Scientific). [2]
DC isolation.
Splenic DCs were isolated from C57BL/6 mice purchased from the Jackson Laboratory with CD11c positive magnetic isolation kit (Miltenyi Biotec) following manufacturer’s protocol. Following isolation, cells were resuspended in DC medium (RPMI1640, 10 % FBS, 0.5 % pen/strep, 50 μM 2-mercaptoethanol). The care of animals from which cells were isolated was approved and overseen by the institutional animal care and use committee (IACUC) at University of Maryland, College Park.
Time and Temperature dependent uptake:
To image cell internalization, DCs (1×106 cells in 2mL glass bottom dish) were incubated overnight with 10 μg CpG ODN at 37 °C. The following day, 50 μL iPEMs, composed of FITC SIINR9 and Cy5 labeled polyIC, were incubated with DCs for varying times at 4 °C or 37 °C. After incubation, cells were washed with PBS and fixed with 4 % paraformaldehyde, then stained as previously described. [3] Fluorescence microscopy (Olympus IX-83) was used to visualize both fluorescently tagged SIINR9 (FITC) and polyIC (Cy5) in DCs. iPEM uptake was quantified using the FITC SIINR9, Texas Red cell membrane signal, and ImageJ. At least 18 cells were analyzed for number of iPEMs for each time point and temperature. Cells were outlined and defined as regions of interests and iPEMs were thresholded and counted using the particle analyzer function in ImageJ within each region of interest. To calculate the co-localization of each iPEM signal, 3 replicates were used for each time point. Each iPEM signal was thresholded manually on ImageJ and then the JACoP plugin was used to determine the fraction of one signal co-localized with the other.
Quantifying iPEM internalization by DCs was performed as previously described. [3] Briefly, DCs were incubated with 20 μL iPEMs composed of FITC SIINR9 and polyIC for varying times at 4 °C or 37 °C. After incubation, cells were washed with FACs buffer (1 % BSA in PBS) and resuspended in 100 μL FACs buffer. 50 μL Trypan Blue Solution (ThermoFisher Scientific) was mixed in by pipetting to quench extracellular FITC signal from iPEMs. Samples were analyzed by flow cytometry (CantoII, BD) and Flowjo. iPEM positive cells were quantified using the FITC SIIN-R9 signal under a lymphocyte gate, manually determined using the forward and side scatter plots.
Inhibitor-dependent uptake:
Inhibitor experiments were performed by incubating DCs (1×105 cells per well in 24 well plate) with endocytosis inhibitors (2-deoxy-D-glucose (2DG, total inhibitor), Filipin (caveolae-mediated endocytosis)) for 1 hr at 37 °C. After 1 hr, 20 μL iPEMs composed of FITC SIIN-R9 and polyIC were added and cells were incubated for an additional 1 hr at 37 °C. Untreated, and iPEMs with no inhibitor were used as controls. Following incubation, cells were washed with FACs buffer and resuspended in 100 μL of either viability dye (DAPI, Invitrogen) diluted 1000× in FACs buffer or 100 μL FACs buffer with 50 μL Trypan Blue mixed in and analyzed by flow cytometry and Flowjo. iPEM uptake was quantified using the FITC SIIN-R9 signal in the lymphocyte population manually gated using the forward and side scatter plot.
Intracellular trafficking:
Intracellular trafficking studies were performed by incubating DCs with 2 μM LysoTracker Red DND-99 (ThermoFisher Scientific) and iPEMs composed of FITC SIIN-R9 and polyIC at 37 °C for 2 hrs. After incubation, cells were washed with PBS and fixed with 4 % paraformaldehyde, then stained with Hoescht and imaged using fluorescence microscopy. iPEM colocalization with lysosomes was determined using the JACoP plugin on ImageJ. 3 replicated were used for each time point. Both the FITC signal from iPEMs and LysoTracker Red signal were first thresholded manually on ImageJ before running the JACoP plugin to determine the fraction of one signal co-localized with the other.
DC activation and antigen presentation:
DC activation was quantified as previously described. [3] Briefly, DCs were incubated with 20 μL iPEMs composed of FITC SIIN-R9 and Cy5 polyIC for 15 mins to 24 hrs. Untreated, and soluble dose-matched Cy5 polyIC (20 μg) were used as controls. After incubation, cells were collected, washed, and stained as previously described. [3] Antibody stained samples were analyzed by flow cytometry and Flowjo. Cells to analyze for activation were selected by gating for lymphocytes and then excluding DAPI positive cells as dead cells. For antigen presentation studies, DCs were treated similarly with 20 μL iPEMs composed of FITC SIIN-R9 and polyIC for 1 hr to 24 hrs. Untreated, soluble dose-matched SIIN-R9, soluble dose-matched polyIC, and soluble dose-matched SIIN-R9 + polyIC were used as controls. After incubation, cells were collected, washed, and stained to assess antigen presentation as previously described. [3] Cells to analyze for activation were selected by gating for lymphocytes and then excluding DAPI positive cells as dead cells.
TLR3 signaling assay:
To assess the kinetics of TLR3-specific signaling, HEK-Blue mTLR3 reporter cells (InvivoGen) were plated at 5×105 cells/well in HEK-Blue Detection medium (InvivoGen) as previously described. [2] Cells were treated with Free polyIC (20 μg) or iPEMs for 15 minutes to 24 hours and the absorbance at 620 nm was measured using a Gemini XPS fluorescence microplate reader (Molecular Devices) per manufacturer’s instructions. To verify that increase in signaling was TLR3 specific, soluble CpG (1 μg), a TLR9a, soluble SIIN-R9 (20 μg), and media only were used as controls. Results are shown relative to media only absorbance.
Statistical Analysis:
Error bars were calculated using standard deviation. Statistical analysis was performed by either a one-way ANOVA with a Tukey post-test to correct for multiple comparisons or a T test. A P value less than or equal to 0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
This work was supported in part by the United States Department of Veteran Affairs # 1I01BX003690, the Damon Runyon Foundation # DRR3415, NSF CAREER Award # 1351688, and Alliance for Cancer Gene Therapy (#15051543). M.L.B. is a trainee of the NIH T32 Host-Pathogen Interaction Fellowship (# AI089621). K.L.H. is a SMART Graduate Fellow funded by ASD/R&E, Defense – Wide/PE0601120D8Z National Defense Education Program (NDEP)/BA-1, Basic Research. C.M.J. is a Young Investigator of the Melanoma Research Alliance (# 348963).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Disclosures
C.M.J. is an employee of the Maryland Veterans Affairs (VA) Health Care System at the Baltimore VA Medical Center. The views reported in this paper do not reflect the views of the Department of Veterans Affairs or the United States Government. C.M.J. holds an equity position in Cellth LLC.
References
- 1.D’Amelio E; Salemi S; D’Amelio R, International Reviews of Immunology 2016, 35 (3), 260–290. DOI 10.3109/08830185.2015.1082177. [DOI] [PubMed] [Google Scholar]
- 2.Chiu Y-C; Gammon JM; Andorko JI; Tostanoski LH; Jewell CM, ACS biomaterials science & engineering 2015, 1 (12), 1200–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiu Y-C; Gammon JM; Andorko JI; Tostanoski LH; Jewell CM, ACS applied materials & interfaces 2016, 8 (29), 18722–18731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gellin BG; Salisbury DM, Vaccine 2015, 33, B44–B46. DOI https://doi.org/10.1016/j.vaccine.2015.02.073. [DOI] [PubMed] [Google Scholar]
- 5.Wu TY-H; Singh M; Miller AT; De Gregorio E; Doro F; D’Oro U; Skibinski DA; Mbow ML; Bufali S; Herman AE, Science translational medicine 2014, 6 (263), 263ra160–263ra160. [DOI] [PubMed] [Google Scholar]
- 6.Steinhagen F; Kinjo T; Bode C; Klinman DM, Vaccine 2011, 29 (17), 3341–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeda K; Akira S, Current protocols in immunology 2015, 109 (1), 14.12. 1–14.12. 10. [DOI] [PubMed] [Google Scholar]
- 8.O’neill LA; Golenbock D; Bowie AG, Nature Reviews Immunology 2013, 13 (6), 453. [DOI] [PubMed] [Google Scholar]
- 9.De Nardo D, Cytokine 2015, 74 (2), 181–189. DOI https://doi.org/10.1016/j.cyto.2015.02.025. [DOI] [PubMed] [Google Scholar]
- 10.Abbas AK; Lichtman AH; Pillai S, Cellular and molecular immunology E-book. Elsevier Health Sciences: 2014. [Google Scholar]
- 11.Duthie MS; Windish HP; Fox CB; Reed SG, Immunological reviews 2011, 239 (1), 178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reed SG; Orr MT; Fox CB, Nature medicine 2013, 19 (12), 1597–1608. [DOI] [PubMed] [Google Scholar]
- 13.Shirota H; Klinman DM, Expert review of vaccines 2014, 13 (2), 299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Q; Ding JL, Immunology And Cell Biology 2016, 94, 538 DOI 10.1038/icb.2016.18. [DOI] [PubMed] [Google Scholar]
- 15.Currie AJ; van der Most RG; Broomfield SA; Prosser AC; Tovey MG; Robinson BWS, The Journal of Immunology 2008, 180 (3), 1535–1544. DOI 10.4049/jimmunol.180.3.1535. [DOI] [PubMed] [Google Scholar]
- 16.Pulko V; Liu X; Krco CJ; Harris KJ; Frigola X; Kwon ED; Dong H, The Journal of Immunology 2009, 183 (6), 3634–3641. DOI 10.4049/jimmunol.0900974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ichinohe T; Watanabe I; Ito S; Fujii H; Moriyama M; Tamura S.-i.; Takahashi H; Sawa H; Chiba J; Kurata T, Journal of virology 2005, 79 (5), 2910–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bookstaver ML; Tsai SJ; Bromberg JS; Jewell CM, Trends in immunology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ali OA; Huebsch N; Cao L; Dranoff G; Mooney DJ, Nature materials 2009, 8 (2), 151–8. DOI 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kasturi SP; Skountzou I; Albrecht RA; Koutsonanos D; Hua T; Nakaya H; Ravindran R; Stewart S; Alam M; Kwissa M; Villinger F; Murthy N; Steel J; Jacob J; Hogan RJ; García-Sastre A; Compans R; Pulendran B, Nature 2011, 470 (7335), 543–547. DOI 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tostanoski LH; Chiu Y-C; Andorko JI; Guo M; Zeng X; Zhang P; Royal W III; Jewell CM, ACS nano 2016, 10 (10), 9334–9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hess KL; Andorko JI; Tostanoski LH; Jewell CM, Biomaterials 2017, 118, 51–62. DOI https://doi.org/10.1016/j.biomaterials.2016.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gosselin EA; Eppler HB; Bromberg JS; Jewell CM, Nature Materials 2018, 17 (6), 484–498. DOI 10.1038/s41563-018-0077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharp FA; Ruane D; Claass B; Creagh E; Harris J; Malyala P; Singh M; O’Hagan DT; Pétrilli V; Tschopp J, Proceedings of the National Academy of Sciences 2009, 106 (3), 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sunshine JC; Perica K; Schneck JP; Green JJ, Biomaterials 2014, 35 (1), 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moyano DF; Goldsmith M; Solfiell DJ; Landesman-Milo D; Miranda OR; Peer D; Rotello VM, Journal of the American Chemical Society 2012, 134 (9), 3965–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andorko JI; Hess KL; Pineault KG; Jewell CM, Acta biomaterialia 2016, 32, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andorko JI; Pineault KG; Jewell CM, Journal of Biomedical Materials Research Part A 2017, 105 (4), 1219–1229. [DOI] [PubMed] [Google Scholar]
- 29.Dold NM; Zeng Q; Zeng X; Jewell CM, Acta biomaterialia 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jewell CM; Lynn DM, Advanced drug delivery reviews 2008, 60 (9), 979–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wong SY; Moskowitz JS; Veselinovic J; Rosario RA; Timachova K; Blaisse MR; Fuller RC; Klibanov AM; Hammond PT, Journal of the American Chemical Society 2010, 132 (50), 17840–17848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stein EW; Volodkin DV; McShane MJ; Sukhorukov GB, Biomacromolecules 2006, 7 (3), 710–719. [DOI] [PubMed] [Google Scholar]
- 33.De Koker S; De Geest BG; Singh SK; De Rycke R; Naessens T; Van Kooyk Y; Demeester J; De Smedt SC; Grooten J, Angewandte Chemie 2009, 121 (45), 8637–8641. [DOI] [PubMed] [Google Scholar]
- 34.De Geest BG; Willart MA; Lambrecht BN; Pollard C; Vervaet C; Remon JP; Grooten J; De Koker S, Angewandte Chemie International Edition 2012, 51 (16), 3862–3866. [DOI] [PubMed] [Google Scholar]
- 35.De Koker S; Hoogenboom R; De Geest BG, Chemical Society Reviews 2012, 41 (7), 2867–2884. [DOI] [PubMed] [Google Scholar]
- 36.De Geest BG; Willart MA; Hammad H; Lambrecht BN; Pollard C; Bogaert P; De Filette M; Saelens X; Vervaet C; Remon JP, ACS nano 2012, 6 (3), 2136–2149. [DOI] [PubMed] [Google Scholar]
- 37.Yan Y; Björnmalm M; Caruso F, Chemistry of Materials 2013, 26 (1), 452–460. [Google Scholar]
- 38.DeMuth PC; Min Y; Huang B; Kramer JA; Miller AD; Barouch DH; Hammond PT; Irvine DJ, Nature materials 2013, 12 (4), 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.De Koker S; Fierens K; Dierendonck M; De Rycke R; Lambrecht BN; Grooten J; Remon JP; De Geest BG, Journal of Controlled Release 2014, 195, 99–109. [DOI] [PubMed] [Google Scholar]
- 40.Hsu BB; Park M-H; Hagerman SR; Hammond PT, Proceedings of the National Academy of Sciences 2014, 111 (33), 12175–12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng Q; Gammon JM; Tostanoski LH; Chiu Y-C; Jewell CM, ACS Biomaterials Science & Engineering 2016, 3 (2), 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang P; Chiu Y-C; Tostanoski LH; Jewell CM, Acs Nano 2015, 9 (6), 6465–6477. [DOI] [PubMed] [Google Scholar]
- 43.Moon JJ; Suh H; Bershteyn A; Stephan MT; Liu H; Huang B; Sohail M; Luo S; Um SH; Khant H, Nature materials 2011, 10 (3), 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldinger SM; Dummer R; Baumgaertner P; Mihic‐Probst D; Schwarz K; Hammann‐Haenni A; Willers J; Geldhof C; Prior JO; Kündig TM, European journal of immunology 2012, 42 (11), 3049–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vácha R; Martinez-Veracoechea FJ; Frenkel D, Nano letters 2011, 11 (12), 5391–5395. [DOI] [PubMed] [Google Scholar]
- 46.Deshayes S; Morris M; Divita G; Heitz F, Cellular and molecular life sciences 2005, 62 (16), 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walrant A; Correia I; Jiao C-Y; Lequin O; Bent EH; Goasdoué N; Lacombe C; Chassaing G; Sagan S; Alves ID, Biochimica et Biophysica Acta (BBA)-Biomembranes 2011, 1808 (1), 382–393. [DOI] [PubMed] [Google Scholar]
- 48.Copolovici DM; Langel K; Eriste E; Langel U, ACS nano 2014, 8 (3), 1972–1994. [DOI] [PubMed] [Google Scholar]
- 49.Sandvig K; Kavaliauskiene S; Skotland T, Histochemistry and cell biology 2018, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rejman J; Oberle V; Zuhorn IS; Hoekstra D, Biochemical Journal 2004, 377 (1), 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JS; Hur W, International Journal of Nanomedicine 2016, 11, 2069–2079. DOI 10.2147/IJN.S103830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trombetta ES; Ebersold M; Garrett W; Pypaert M; Mellman I, Science 2003, 299 (5611), 1400–1403. [DOI] [PubMed] [Google Scholar]
- 53.Matsumoto M; Funami K; Tanabe M; Oshiumi H; Shingai M; Seto Y; Yamamoto A; Seya T, The Journal of Immunology 2003, 171 (6), 3154–3162. [DOI] [PubMed] [Google Scholar]
- 54.Muzio M; Bosisio D; Polentarutti N; D’amico G; Stoppacciaro A; Mancinelli R; van’t Veer C; Penton-Rol G; Ruco LP; Allavena P, The Journal of Immunology 2000, 164 (11), 5998–6004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.