

Abstract
Estrogen receptor alpha (ERα), a key driver of breast cancer, normally requires estrogen for activation. Mutations that constitutively activate ERα without the need for hormone are frequently found in endocrine therapy-resistant breast cancer metastases and are associated with poor patient outcomes. The location of these mutations in the ER ligand-binding domain and their impact on receptor conformation suggest that they subvert distinct mechanisms that normally maintain the low basal state of wild-type ERα in the absence of hormone. Such mutations provide opportunities to probe fundamental issues underlying ligand-mediated control of ERα activity. Instructive contrasts between these ER mutations and those that arise in androgen receptor (AR) during antiandrogen treatment of prostate cancer highlight differences in how activating functions in ER vs. AR control receptor activity, how hormonal pressures (deprivation vs. antagonism) drive the selection of phenotypically different mutants, and how altered protein conformations can reduce antagonist potency and altered ligand-receptor contacts can invert the response that a receptor has to an agonist vs. an antagonist. A deeper understanding of how ligand regulation of receptor conformation is linked to receptor function offers a conceptual framework for developing new antiestrogens that might be more effective in preventing and treating breast cancer.
[INTRODUCTION]
Understanding how protein structure relates to protein activity is a problem of fundamental importance in biology that is being studied from many directions. Members of the nuclear hormone receptor superfamily provide compelling examples of how molecular biology, structural biology, biochemistry and modeling can combine to provide a progressively refined, molecular-level understanding of how this class of transcription factors work and, in particular, how many of them are regulated by ligands. The estrogen receptor α (ERα), in particular, has led the way in defining the roles played by the different domains of these nuclear hormone receptors in their interaction with agonist and antagonist ligands, and how these interactions translate into the regulation of transcription (Box 1).
BOX 1. Abbreviations and Glossary of Terms.
AE – antiestrogen Antiestrogens are ligands for the estrogen receptor used as one form of endocrine therapy for breast cancer. They bind to the estrogen receptor but alter its conformation so that it is unable to stimulate the proliferation and progression of breast cancer cells.
AI – aromatase inhibitor Aromatase inhibitors are another form of endocrine therapy for breast cancer. They work by blocking the production of estrogens produced by the ovaries, by other tissues such as the adrenal, and by the tumor itself
Apo – a binding protein in an unliganded state
AR – androgen receptor A transcription factor that is a member of the nuclear hormone receptor superfamily. It is the principal mediator of the biological effects of androgens and a major driver of the proliferation and progression of prostate cancer.
ERα – estrogen receptor α A transcription factor that is a member of the nuclear hormone receptor superfamily. It is the principal mediator of the biological effects of estrogens and a major driver of the proliferation and progression of breast cancer. ERα is distinguished from another ER subtype, ERβ, which has very different biological activities, largely unrelated to driving breast cancer progression
E2 – estradiol A steroid with an aromatic A-ring that is the principal endogenous estrogenic hormone that drives the proliferation and progression of breast cancer cells.
h – helix A characteristic motif of protein secondary structure consisting of a right-handed helix of amino acids in a peptide chain, stabilized by internal hydrogen bonds between carbonyl groups and N-H groups.
HSP – heat shock proteins A family of proteins that selectively bind other proteins that are intrinsically or aberrantly unfolded. HSP90 is the major protein to which WT apo-ERα binds, although other HSPs likely also participate in this binding.
LBD – ligand binding domain A domain of the estrogen receptor responsible for binding estrogens and antiestrogens. It is domain E out of the domains A-F, and stretches from ca. amino acid 304 to 554 out of a total of 595 amino acids, accounting for about 40% of the overall length of ERα. It is constituted of some 12 α-helices and a few β-strand elements of secondary structure.
LBP – ligand binding pocket An interior region of the LBD within which both agonist and antagonist ligand bind, with occasional portions of the ligand extending beyond the confines of the pocket.
MD – molecular dynamics A computationally intensive method for exploring the conformation and dynamic features of proteins by providing alternating inputs of velocity on individual atoms and relaxation within the energy force field confines of the protein.
NR – nuclear receptors A superfamily of proteins of which ERα and AR are members. Most members of the superfamily function largely as transcription factors, many of which are regulated by the binding of ligands, which can be endogenous metabolites (hormones) exogenous ligands (pharmaceuticals, xenobiotics, etc.).
SERD – selective estrogen receptor downregulator A class of ligands for ERα such as fulvestrant that cause a reduction in the levels of the ERα protein; they also function as ERs antagonists and are used in breast cancer endocrine therapies.
SERM – selective estrogen receptor modulator A class of ligands for ERα that can have tissue-selective pharmacological effects, acting as agonists in some tissues (such as bone and vasculature) and antagonists in others (breast and uterus). SERMs such as tamoxifen are used in breast cancer endocrine therapy; other SERMs such as raloxifene are used in hormone replacement therapies to protect bone in post-menopausal women.
WT – wild type The naturally occurring form of a protein, as distinguished from various mutant forms.
ERα, a member of a large superfamily of nuclear receptors (NRs), is the main transcription factor regulating the biology and properties of over 70% of human breast cancers1–3. The ERα, like the androgen receptor (AR) and other members of the NR superfamily, has a DNA-binding domain C (DBD), a ligand-binding domain E (LBD), and activating functions (AF1 in the N-terminal A/B domain and AF2 in the LBD) that control transcription and hormone-dependent gene expression (Figure 1A). The LBD of ERα (ca. amino acids 304–554) consists of 12 α-helices (h1-h12) linked mostly by loop regions. In the absence of a bound ligand, this domain is inactive, likely partially disordered, bound by heat shock proteins (largely HSP90) and likely a monomer (Figure 1B)4,5. When an agonist like estradiol (E2) binds, the LBD sheds the HSPs, dimerizes, and becomes stabilized in a conformation in which the last helix (h12) folds over the ligand binding pocket (LBP), forming a hydrophobic groove into which coactivators bind (Figure 1C). In contrast, when an antiestrogen (AE) like tamoxifen binds to the LBD, its side chain prevents h12 from forming an active AF-2 conformation; so, h12 docks in the AF-2 hydrophobic groove and blocks coactivator binding (Figure 1D).
Figure 1. Overview of nuclear receptor domain structure, activating functions, ligand-induced conformations, and dimer formation.
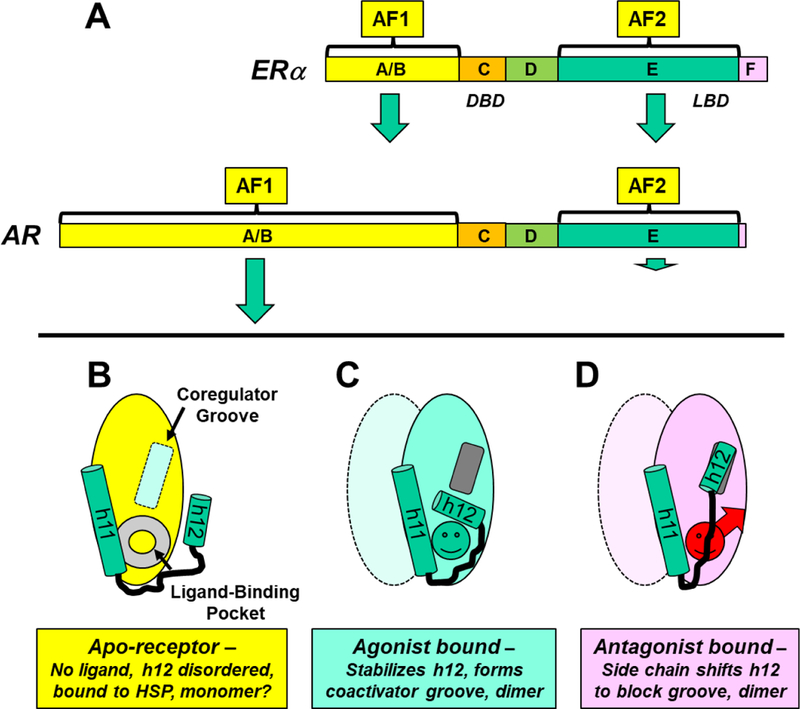
A. Domain structure of two nuclear hormone receptors, ERα and AR. The domains, shown to scale, are labeled A/B through F. The size of the green arrows associated with the activating functions (AF1 and AF2) illustrate their relative contribution to the transcriptional activity of ER or AR. The D domain is called the hinge, and the most C-terminal F domain is much more prominent in ERα than in AR. B-D. Schematic representation of three conformational states of the LBD of hormone-regulated nuclear receptors, highlighting the relative positions of the two carboxy-terminal helices, h11 and h12, and dimer states. B. In the unliganded or apo-receptor LBD, the LBP is empty (gray doughnut) and the coactivator groove (light blue rectangle) is incomplete and empty. C. With a bound agonist (green circle), h12 folds back, covering the LBP and generating the coactivator binding groove (dark gray rectangle). D. With a bound AE (red circle with arrow for a side chain), h12 moves to block the coactivator binding groove. The liganded LBDs (C and D) are represented as dimers; the second monomer is shown in lighter color, and conformational details are omitted. Although uncertain, the apo-LBD (B) is shown as a monomer.
When activated by an estrogen, ERα increases proliferation and progression of ERα-positive breast cancers that comprise largely the luminal A and B breast cancer subtypes6,7. Many of these breast cancers are effectively treated with aromatase inhibitors (AIs) such as letrozole or with antiestrogen ligands of either the selective ER modulator (SERM) or selective ER downregulator (SERD) type, the mainstays of endocrine therapies. Recently, deep DNA sequencing has revealed activating mutations in the ligand binding domain (LBD) of ERα in ca. 40% of recurrent, ER-positive breast cancers8–30. Most of these mutations convey constitutive activity to levels approximating those achieved by hormone stimulation12–15,18,24,31–33. It is no surprise then that these mutations are strongly associated with reduced efficacy of estrogen-deprivation therapies such as AIs9,10,24. Moreover, these mutations alter the efficacy of some ER antagonists such as tamoxifen14–16,18,24,29. Thus, these mutations clearly pose a challenge to the continued effectiveness of endocrine therapies and highlight the clinical need for developing more effective endocrine-therapy agents or treatment combinations3,24,34. These mutations also provide important vehicles for greatly expanding our understanding of how the structure of ERα, most notably the ligand-induced conformation of its LBD, is related to the activity of this major transcription factor.
In this review, we bring together various threads, some from decades ago, but most from recent X-ray crystallography, biophysical and biochemical assays, and molecular dynamics modeling studies, to illuminate the carefully refined nature of the LBD of wild type (WT) ERα that enables its nuanced regulation by structurally diverse hormonal ligands. By first appreciating that the LBD of unliganded WT ERα (WT apo-ERα) is, of necessity, in an “off state”, we can better understand how this off-state, which is intrinsically disordered, is subverted in varying ways by the different activating mutations that convey resistance to estrogen-deprivation therapy by engendering folding into an agonistic conformation without a bound estrogen. We then examine two modes by which a constitutively active ERα has reduced sensitivity to ER antagonists. Instructive contrasts are then made between the activating ERα mutations found in breast cancer and mutations in androgen receptor (AR) that arise in castration-resistant prostate cancer: This comparison highlights differences in the two activating functions (AF1 and AF2) in ER vs. AR, the types of mutations that provide constitutive activity, the different hormonal pressures that control selection of the antiandrogen resistance, and nature of mutational changes that reduce antagonist potency and can even invert ligand activity from antagonist to agonist. This AR vs. ERα mutant comparison raises a cautionary note regarding new types of ERα mutations that might be encountered as endocrine therapies in breast cancer evolve. A deeper understanding of how ligand regulation of receptor conformation is linked to receptor function provides a framework to guide development of new antiestrogens that might be more effective in preventing and treating breast cancer.
ERα Activating Mutations
Constitutively active mutant ERα’s were first reported in the 1990’s through structure-function studies that employed random mutagenesis and phenotypic selection in the absence of estradiol or the presence of antiestrogens32,35, and site-directed mutagenesis31. The first report of an activating ERα mutation in a breast cancer metastasis also appeared in the 1990’s33, but the clinical prevalence of these single nucleotide polymorphisms became evident only more recently as a result of technological advances that facilitated deep DNA sequencing of tumors8,36. The prevalent, activating mutations in ERα are all located in the ER LBD and found in three distinct zones (Figure 2, Supplemental Movie S1). (A compendium of ER mutations can be found in a database of cancer mutations from more than 10,000 samples: http://www.cbioportal.org/study?id=msk_impact_2017#summary.)
Figure 2. Activating Mutations in the ERα LBD, Pharmacological Phenotypes, and Mechanisms.
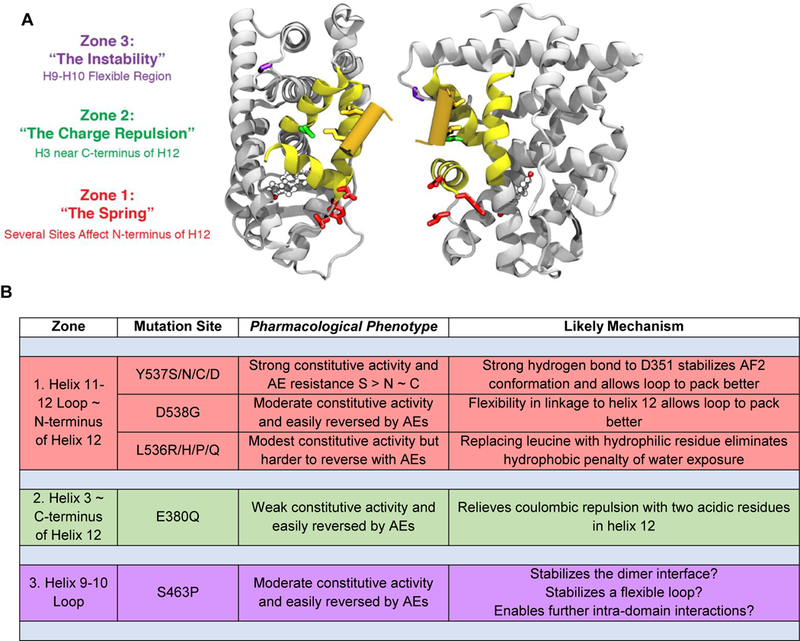
A. Ribbon diagrams of two perspectives illustrating the three zones for the principal activating mutations in the ERα ligand-binding domain (LBD). The color codes (red, green, and purple) match the zone designations with the residues in each zone. All of these mutations alter residues that are far from the bound ligand, estradiol (ball and stick model). The region shown in yellow constitutes the coactivator binding groove and the orange cylinder, the coactivator LxxLL helical domain. (A movie showing a rotating 3D version of this structure is available – Supplemental Movie S1.) B. Tabular listing by zone of the principal activating mutations, with their pharmacological phenotype and likely functional mechanism. Constitutive activity and reversal here refers principally to the transcriptional activation functions of the receptor.
“The Importance of Being Off”
The activating mutations in the LBD occur at sites outside the LBP and therefore involve residues that are not directly in contact with bound ligand. Hence, it is tempting to ask “How can mutations distributed at these three very different sites, all remote from the ligand, result in receptors that are active without ligand and resistant to antagonism?” It makes more sense to reverse the question, “How is it that the WT ERα is ‘Off’ when it is not bound to agonist ligand?” Posed in this way, the question recognizes a simple axiom: A ligand-regulated transcription factor needs to be off in the absence of agonist binding, so that its activity can be increased upon binding of an agonist. By recognizing the “Importance of Being Off” (a phrase adapted from the 1895 Oscar Wilde Play “The Importance of Being Earnest”), we can better examine how each of these mutations subverts several distinct mechanisms that enforce the off-state of WT apo-ERα.
We use the term “Off” to characterize the inability of unliganded WT ERα to enhance breast cancer proliferation and progression. In the absence of ligand, much of WT ERα, as well as that of other NR superfamily members, are bound by heat shock proteins,4 an interaction that protects them from proteolysis and from which they can be released in an active form by ligand binding. Even without ligand, however, WT ERα may still regulate some genes and cellular functions, often through post-transcriptional modifications37,38 that respond to other growth-regulating signaling systems, such as epidermal or insulin-like growth factors39–42. Also, the estrogen receptor does not work in isolation, and alterations in the levels and nature of associated and interacting factors—coregulators, other transcription factors, modifiers of the epigenome, post-translational modifications, cell-signaling pathways, the ERβ subtype43—can all modify how breast cancer will respond to hormone deprivation and antiestrogen treatments and lead to ligand-independent activation. The constitutively active ERα mutations we discuss, however, are responsible for a significant proportion of endocrine resistance found clinically in women with breast cancer.
Zones of Activating Mutations
In the sections below, we examine how mutations undermine the off state of WT apo-ERα and lead to constitutive activity. The prevalent, activating mutations in ERα are all located in the ER LBD, and they are found in three distinct zones (Figure 2, Supplemental Movie S1). Notably, their activating effects are directed at distinct mechanisms by which the off-state of WT ERα—in the absence of a bound agonist—is being enforced. To keep track of these mechanisms, we have designated the three zones of mutant interactions with “functional terms”: “The Spring” (Zone 1), “The Charge Repulsion” (Zone 2), and “The Instability” (Zone 3) (Figure 2).
Zone 1 – The “Spring”
Loops in protein structures are typically thought of as flexible, often unstructured turn regions with no function beyond that needed to connect the ends of nearby secondary structural elements. The h11–12 loop in WT ERα, by contrast, appears to provide a critical function to keep WT apo-ERα off. Several lines of evidence (X-ray crystallography, MD Modeling, and deuterium exchange kinetics) suggest that when h12 bends over the LBP to adopt the active conformation (Figure 1C), the h11–12 loop experiences an inherent spring-like strain due to the aqueous exposure of consecutive hydrophobic residues in this sequence, V533, V534, P535, and L53616. WT ERα requires the energy from binding an agonist ligand to bend this spring into the agonist conformation, and in the absence of a bound ligand, the spring-like nature of the h11–12 loop plays a key role in keeping WT-ERα off. It also ensures that this domain retains some degree of disorder (Figure 1B)44, which we will later see serves other important functions. This intrinsic disorder is likely the reason for the lack of success in crystallizing the WT LBD unless it has bound a ligand 45, for the binding of WT apo-ERα to heat shock proteins4, and also for the great ease with which proteases can make clips in the h11–12 loop46; these are all markedly changed by mutations in this loop16,45.
Y537S/N/C – “Latching the h11–12 spring” with a stronger hydrogen bond.
Among the ERα mutants identified in endocrine therapy-resistant disease, the Y537 site is the one most frequently mutated9,22,24. Cell activity assays show that mutation from Y to S at this 537 site engenders high, nearly full constitutive activity and reduces antiestrogen potencies; the penetrance of these characteristics is reduced somewhat when mutation is to N and particularly to C (Figure 2B)24. Recently identified in one patient, Y537D is a constitutively active mutant found at this site as well24. Other residues introduced at this site by mutagenesis, Y537A, E, F, or K, have some, generally lower, levels of constitutive activity, but of these, only Y537F could occur through a single nucleotide change32,47,48, and thus far has not been reported clinically.
The growing corpus of crystal structures obtained for the Y537S mutant clearly shows that h12 is in the agonist conformation16,45,49,50, even without bound ligand45. The hydrogen bonding partner of S537 in the Y537S mutant is D351 on h3, and this tight interaction appears to be “latching the h11–12 spring” in the agonist conformation, turning on constitutive activity. This S537-D351 interaction in the mutant ERα appears more highly-optimized than the Y537-N548 interaction in WT ERα, predicted from MD modeling16. In addition to its strong latching hydrogen bond with D351, the Y537S mutation also allows for a more optimal packing of the three hydrophobic side chains16.
D538G – “Lengthening the h11–12 spring” by unwinding helix 12.
The D538G mutation, observed in ~20% of patients with AI-treated metastatic breast cancers9,12,20,22, in the h11–12 loop of the ERα LBD, has constitutive activity comparable to or somewhat less than that of Y537S (Figure 2B)24,27,28. Structural data shows that the charged D538 residue in WT apo-ERα introduces a kink in the protein backbone driven by its strong preference for solvent exposure and electrostatic repulsion from other nearby acidic residues (e.g., D351), and it initiates the helical character at the start of h1216. The glycine mutation at this position eliminates the electrostatic components, and coupled with the glycine “helix-breaking” backbone conformational preferences, results in a change in loop conformation that erodes the beginning of h12 from 538 to 539. This “lengthening of the h11–12 spring” in D538G ERα allows better side chain packing for the hydrophobic residues. Curiously, these changes also disrupt the canonical hydrogen bond presumed to form between Y537 and N348 in the WT LBD16. MD simulations that predicted the loss of this interaction in the mutant and indicated that Y537 adopts multiple orientations of its side chain were later confirmed by x-ray crystal structures16. None of the other synthetic mutations explored at this site (A, N, and V) were reported to have constitutive activity18,48,51, suggesting that the unique flexibility afforded by G is essential for lengthening the spring.
L536R/H/P/Q – “Softening the h11–12 spring” by reducing hydrophobicity.
Of the h11–12 mutations, changes at 536 are found less often (~1% of patients with AI-treated metastatic breast cancers)9,24, and they convey relatively modest constitutive activity (Figure 2B). Nevertheless, in cell-based assays the L536R mutation is quite difficult to fully suppress with antiestrogens, suggesting that it may be overcoming a major contribution to the h11–12 loop strain in the WT sequence. Although no X-ray structures yet available for mutations at this site, L536 is observed in a solvent-exposed conformation in the majority of wild-type ERα LBD structures (21 out of 26 monomers in WT ER LBD crystal structures have L536 exposed: PDB codes 1ERE, 3ERD, 1QKU, 1GWR, 1L2I, 1G50, 1PCG, 1X7R, 1X7E, 2YJA, 4DMA). This observation, when put into the context of structural changes observed to the h11–12 loop for the mutations described above, suggests that changing residue 536 from strongly hydrophobic to charged (R), polar (H and Q), or less hydrophobic (P), allows for a more energetically optimal arrangement of the remaining hydrophobic side chains (i.e., amino acids 533–535), in essence “softening the h11–12 spring”.
Nature has provided an “experiment” that supports our proposed mechanism for softening the h11–12 spring by replacing L536 with less hydrophobic residues52. Fortuitously, the h11–12 loops in both ERα and ERβ have identical sequences, except in place of L536 in ERα, ERβ has at the corresponding 487 site the less hydrophobic valine residue (Supplemental Figure S1A), and from a number of cell-based and cell-free assays, ERβ is known to have higher constitutive activity than ERα52,53. In addition, using FRET assays 52, we could not detect any binding of the steroid receptor coactivator 3 (SRC3) NR interaction domain to WT apo-ERα (Supplemental Figure S1B), consistent with its lack of constitutive activity, whereas there was good binding to WT apo-ERβ (Supplemental Figure S1C), consistent with its significant basal activity52, and comparable to SRC3 binding found in some constitutively active ERα mutants16,32,35. While the two ER subtypes differ in sequence elsewhere, this specific comparison is consonant with our hypothesis that exposure of a hydrophobic residue at the 536 position in ERα is a major contributor to the strength of the h11–12 spring keeping WT ERα off in the absence of agonist ligand binding, with the L536 in ERα giving a stronger “turn-off spring” than the corresponding V487 in ERβ. A number of other mutations synthetically at 536 (A, I, E, G, K, and N) are also less hydrophobic than L and most have some constitutive activity48,54, but of these, only I could occur through a single nucleotide change.
Zone 2 – The “Charge Repulsion”
Among the ERα mutants identified in endocrine therapy-resistant disease, E380 is the third most frequently mutated residue (~5% of patients with AI-treated metastatic breast cancers). In cell studies its constitutive activity and resistance to antiestrogens are relatively modest (Figure 2B)31, although clinical resistance from E380Q can be considerable.55 Despite the lack of structural information for this mutant, one can formulate a plausible mechanism by which ERα activity might be overcome by a change from a negatively charged to a neutral residue.
E380Q – “Neutralizing the Charge Repulsion”.
Unlike the Zone 1 mutations, which are immediately downstream in sequence from h12 and thus clearly in position to affect its orientation directly, the E380 residue is in h5, very far in sequence from the start of h12 (Supplemental Figure S2). Nevertheless, E380 is close in space to the C-terminal portion of h12, and from this position there is likely a repulsion between the negative charge of E380 and the two acidic—negatively charged—residues E542 and D545 in the middle of h12, disfavoring h12 positioning in the agonist conformation. The E to Q mutation at 380 would “eliminate this charge repulsion interaction”, enabling the active conformation to be achieved without the energy of agonist ligand binding. Supporting this hypothesis is a report that the synthetic E542K mutation, which would make this interaction attractive, has constitutive activity.48 In addition, an E542G mutation, which would also reduce coulombic repulsion, was found in a breast cancer patient with recurrent disease (Chandarlapaty, unpublished)56.
Zone 3 – The “Instability”
The S463P mutation, observed in ~2% patients with AI-treated MBC9, is curious in two respects: It has relatively low constitutive activity in reporter gene assays or resistance to antagonists, yet it drives estrogen independent tumor growth as well as or better than the other mutations18,24, and of all the most well characterized mutations, it is most remote both in sequence and in space from h12 (Figure 2A, Supplemental Movie S1). As with E380Q, there is no published structural information on S463P ERα; so, at this point we are left to speculate on the source of its activity.
S463P mutation – various modes for stabilizing the LBD.
The 463 site is found in a loop between h9 and h10 (Figure 2A). In many crystal structures of WT ERα LBD, this loop is absent, suggesting that it is unstructured and consistent with the avidity with which this region is cleaved by proteases46. If the intrinsic disorder of this loop were one of the factors keeping WT apo-ERα bound through binding to HSPs, then replacement of serine with the structurally more constrained proline residue might activate the LBD by favoring its release from HSP binding. This loop is also close to the dimer interface (Figure 1C and D); so the S463P mutation could affect the stability of ERα LBD dimers. Although it is not clear how this might be operating, there are intriguing mechanisms by which the S463P mutation could affect the structure and function of the ERα LBD and alter the dimer interface (Box 2).
BOX 2. A Role for the Mysterious F-Domain.
ER is unique among the members of the NR superfamily in having a substantial F-domain (Figure 1A). The structure of the 40 C-terminal residues (556–595) that constitute the F-domain in full-length ERα has been difficult to study because ERα LBD crystal structures end abruptly around R555, which is another site of active protease cleavage considered to be the end of h12 and the E-domain46,110. Nevertheless, the function of the F-domain is clearly substantial, as F-domain mutations and truncations alter the agonist/antagonist balance of different ligands and increase the stability of ER dimers111–114. In crystal structures of ERα LBD agonist complexes, the end of h12 is aimed at the dimer interface and appears to interfere with dimer stability (Figure 1C) because ERα LBD agonist dimers are less stable than antagonist dimers, in which the end of h12 is directed away from the dimer interface (Figure 1D)115.
Some idea of what might be happening to the dimer interface can be gleaned from AR and other members of the glucocorticoid receptor subfamily; these members lack a comparable F-domain, but have some residues that extend beyond the site corresponding to R555 in ERα, which are visible in crystal structures (See Supplemental Movies S2 and S3, and legends). These extended sequences form a β-sheet structure with the h9–10 loop and eventually interact with residues in the usual dimerization zone, blocking or altering dimerization of the AR LBD116. The dimerization of full length WT ERα might be similarly weakened by sequences at the start of the F-domain through β-sheet interaction with the intrinsically disordered h9–10 loop. Substitution of serine with the more structured proline in the S463P mutant might disfavor this β-sheet formation; by reducing interference with the dimer interface, it could stabilize and activate the mutant ER by minimizing its interaction with heat shock proteins.
While this review is focused on the prevalent mutations detailed above, the identification of additional mutations in the ERα LBD from breast tumor biopsy samples is an ongoing activity. Some more recently identified mutations are: (a) V422del – Seen more often than S463P and weakly activating24; located at the start of h8, close to but pointing away from the LBP. (b) S432L – Located in the middle of h8, but far from the LBP; found in tamoxifen-treated patients; lacks activity in absence of E224. (c) G442R – Found at low abundance and has constitutive activity; located at the start of h9, far from the ligand24. (d) L469V – A conservative mutation; has considerable constitutive activity; at the start of h10, far from ligand but close to the dimer interface24.
Mutations and AE Resistance
We have detailed how WT ERα is able to maintain an off state by relying on several built-in forces that oppose LBD folding into the agonist conformation when no ligand is bound. Intriguingly, this unfolded character of the WT ERα LBD also enables it to bind both agonist and antagonist ligands with particularly high affinities. The activating mutations that defeat these forces, however, also reduce the binding affinities of agonist and antagonist ligands that leads to AE resistance through interesting mechanisms, discussed below.
Intrinsic Disorder and Binding
Intrinsic disorder of binding domains affords two components of ligand binding energy.
From biophysical studies probing the dynamics of the ERα-LBD with fluorescent probes, we concluded that the LBD in WT-ERα could readily adopt a molten globule or intrinsically disordered state44. Intrinsic disorder is a well-recognized, functional feature of binding proteins in search of their binding partners57, such as NRs searching for their ligands. Intrinsically disordered domains can open and close spontaneously, enabling them to search for their binding partners efficiently, an activity aptly termed “fly casting”58,59. In addition, when intrinsically disordered protein domains interact with their binding partners they gain two components of energy: from new protein-ligand contacts as well as from new protein-protein contacts that form in the more fully folded protein-ligand complex60. In the case of NRs like ERα, in which agonist ligands become completely engulfed by the protein but ligand-protein contacts are relatively sparse61,62, the binding energy contribution from new protein-protein contacts must be considerable.
Prefolding bias of ERα activating mutants reduces protein-protein binding energy.
We have noted that the activating mutations—in various ways—enable the ERα LBD to pre-fold in the agonist conformation without ligand. One can presume that this mutation-induced pre-folding will eliminate much of the second component of ligand-binding energy, the one from new protein-protein contacts that develop from folding the intrinsically disordered WT ERα LBD around the ligand. Loss of this folding energy will reduce the binding affinity of agonist ligands like estradiol, but antiestrogens will experience a greater reduction in binding affinity: the bias of the mutant ERα’s to pre-fold in the agonist conformation means that additional energy is required by these mutants to access the antagonist conformations needed to bind the antiestrogens. The factors at play are illustrated schematically in Figure 3A by the relative magnitude of the arrows for E2 interaction with WT vs. mutant forms. This figure also illustrates that prior to ligand binding, WT ERα LBD is in an unfolded state, associated with HSPs, whereas the mutant ERα is agonist pre-folded; the agonist (E2) and antagonist (AE) complexes are folded in their respective conformations, whether mutant or wild type.
Figure 3. Schematic overview of estrogen agonist (E2) and antiestrogen (AE) binding to WT and mutant ERα LBDs and binding affinities of various antiestrogens to WT ERα and five mutant ERs.
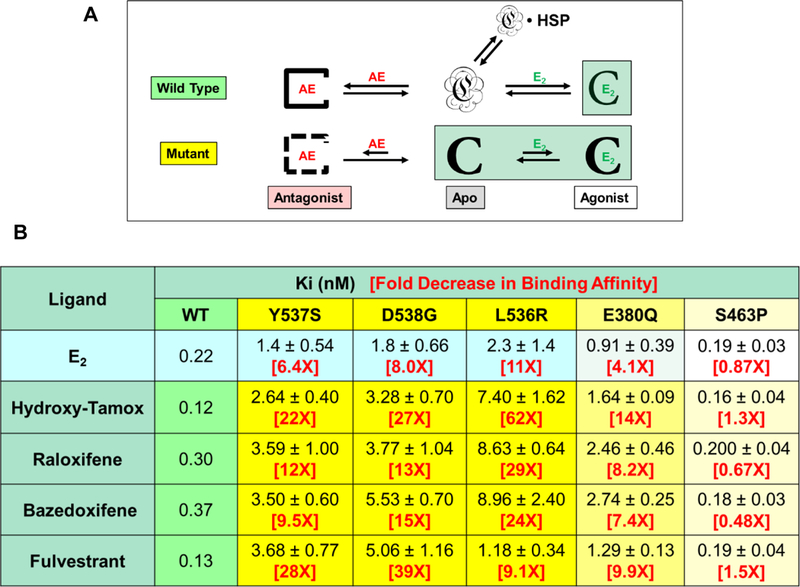
A. The WT ERα LBD, which is unfolded in the absence of ligands and largely bound by heat shock proteins (HSP), gains extra energy upon binding of either E2 or AEs from formation of additional protein-protein contacts in the stably folded ligand complexes. Mutant ER LBDs are pre-folded in the agonist conformation, which reduces the binding affinity of both E2 and AEs, but AE binding is reduced to a greater extent because the agonist folding bias has to be overcome to access the antagonist conformation. Active species are highlighted in light green. B. Binding affinities for E2 and AEs to WT and constitutively active ERα mutants. The level of constitutive activity is indicated by the intensity of the shading (blue for WT ERα and yellow for the mutants). Estradiol binding is determined by a direct assay with [3H]estradiol by Scatchard analysis16,29. Antiestrogen binding was determined by a competitive binding assay using [3H]estradiol as a tracer and LBDs of WT and ERα mutants29. Because the IC50 values from the competition assay are affected by the estradiol binding affinities, they have been converted to Ki values using the Cheng-Prusoff relationship117. (Data on WT, Y537S and D538G ERα LBDs for estradiol, hydroxy-tamoxifen, and fulvestrant are updated values with more replicates from our published work16,63; the values on the other ERα mutants and for raloxifene and bazedoxifene were determined in our laboratories using the same published methodology16,29,63.)
ERα activating mutants have reduced affinity for E2 and especially for AEs.
We and others have measured the binding of estradiol and several SERMs and SERDs to ERα’s with activating mutations by competitive radiometric and other binding assays16,29,34,63, and results from our studies are summarized in Figure 3B. (Affinities are expressed as Ki rather than IC50 or relative binding affinity values; see notes in Figure 3B.) Relative to WT ERα, the binding of both estradiol and antagonists to the mutants is substantially reduced in most cases, with antiestrogen affinities being reduced to a greater extent (up to 60 fold) than those of estradiol (up to 11 fold)16. Notably, the reduction in binding affinity parallels the level of mutant constitutive activity: Y537S ~ D538G ~ L536R > E380Q24, which can be considered a rough measure of the extent of agonist conformation pre-folding. The first 4 mutations also have a general or coordinate effect on the binding of all the antiestrogens rather than affecting only one of them (in marked contrast to endocrine therapy-resistant mutations found in AR, see below). The markedly reduced binding affinity of antagonists to these ERα mutants is likely a major factor underlying their resistance to antiestrogens.
Contrasting AR and ERα Mutations
While we have thus far focused on how mutations in ERα can undermine the therapeutic benefit from AIs and antiestrogens in breast cancer, instructive comparisons can be made between these ERα mutations and those that arise in the AR when prostate cancer becomes resistant to antiandrogens. The sequence of endocrine therapies for prostate cancer roughly parallels that for breast cancer: When the disease becomes resistant to androgen-deprivation therapy through suppression of gonadal androgen biosynthesis and progresses to the castration-resistant stage, therapy then shifts to complete AR blockade, which involves the additional use of antiandrogens such as flutamide bicalutamide, or enzalutamide, or systemic inhibitors of androgen biosynthesis such as abiraterone64. Despite this more intensive antiandrogen therapy, further resistance can develop as a result of mutations in the AR LBD64–66. The nature of the resistance mutations in AR and ERα, however, are very different and reflect inherent differences between the activating function strengths of the two receptors, the selection conditions under which the mutations arise, the location of the mutations, and their pharmacological phenotype. These contrasts (summarized in Table 1) are very informative and raise a cautionary note because they suggest ways in which a different class of therapy-resistant mutations might arise in ERα if the nature and sequence of endocrine therapies used in breast cancer patients were to be changed in ways that expose the cancers initially and/or predominantly to AEs rather than AIs.
Table 1.
Comparative characteristics of endocrine therapy-resistance mutations in the estrogen receptor and androgen receptor
| Characteristic of Mutation | Estrogen Receptor | Androgen Receptor |
|---|---|---|
| 1. Phenotype | Constitutive activity and antiestrogen resistance from LBD mutations | Constitutive activity with C-terminal truncation; antiandrogen resistance from LBD mutations |
| 2. Selective pressure | Estrogen deprivation through aromatase inhibition | Androgen deprivation favors AR C-terminal truncation; antiandrogens favor LBP mutations |
| 3. Location | Outside ligand binding pocket, far from contact with ligand | Either C-terminal truncation or within ligand binding pocket in close contact with ligand |
| 4. Specificity of antagonist resistance | Resistance is consequence of preferential agonist folding that coordinately affects current antiestrogens | Resistance is characteristic for each antiandrogen and converts it from an antagonist to an agonist with increased potency |
| 5. Structure and mechanism of hormone antagonists | Current antiestrogens appear to act largely as “direct antagonists” | Current antiandrogens are presumed to function largely as “indirect antagonists” |
C-terminal truncated AR mutants are constitutively active.
AR can become constitutively active simply by C-terminal truncations, whereas ER requires mutations in the LBD to become constitutively active. This reflects contrasting ways by which these receptors use their two activating functions, AF1 in the N-terminal A/B domain and AF2 in the LBD (Figure 1A). The large AF1 in AR has intrinsic activity sufficient to drive proliferation in prostate cancer without the need for agonist binding; however, in the absence of ligands, AF2 in AR blocks AF1 activity. Hence, C-terminal truncation of the AR LBD removes the inhibitory effect of AF2 and produces a constitutively active AR mutant67. By contrast, AF1 in ER is smaller and requires contributions from AF2 activated by agonist binding to drive proliferation in breast cancer. Consequently, constitutive activity in ER arises from mutations in the LBD that activate AF2 in the absence of ligand.
Locations of AR vs. ER mutations reflect selection and phenotype.
The activating mutations in ERα are all outside of the ligand binding pocket, and in contact with ligands. By contrast, AR mutations resistant to antiandrogens are all inside the ligand pocket (Supplement Movies S2 and S3) in contact with the antagonist ligands (Figure 4A). The ER mutations arise predominantly under endocrine-deprivation selection conditions (AIs) and are constitutively active; their resistance to AEs results in lowered antagonist affinity and potency that affects multiple antiestrogens generally, rather than being specific for individual ones (Figure 3B). The AR mutations arise under androgen-antagonist selective conditions, and affect different antagonist ligands in distinct ways (Table 1 and Figure 4B).
Figure 4. Comparative view of the locations of activating mutations in ER and in AR relative to the position of the ligand, and the relationship of AR mutations to specific AR antagonists.
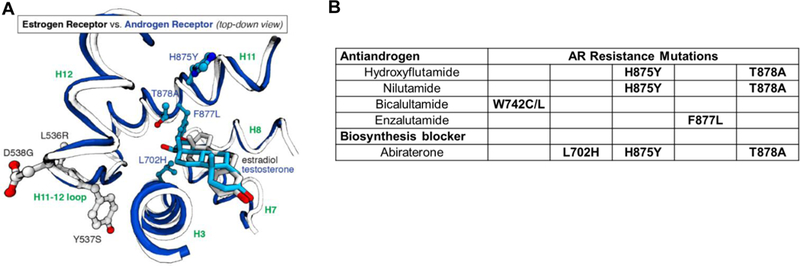
A. A structural overlay of a portion of the LBDs of ER and estradiol (in gray) and testosterone (in blue). The sites of mutation in AR (702, 875, 877, and 878, blue residues) are within the ligand-binding pocket, close to the ligand, whereas the mutation sites in ER (536, 537, and 538, standard atom colored residues) are outside of the pocket, far from ligand contact. (Two rotating 3D movies are available: Supplemental Movie S2 shows that the resistance mutations in the AR LBD are within the LBP. Supplemental Movie S3 shows an overlap comparison of the two LBDs, contrasting the locations of the mutations relative to the ligand.) B. Specific AR mutations are associated with antiandrogens with different structures.64–66 (Because hydroxyflutamide and nilutamide have similar structures, they have similar sites of mutation.) While nominally a blocker of androgen biosynthesis from adrenal precursors, Abiraterone, and particularly its oxidized metabolite D4A, are also direct AR antagonist ligands.118,119 Abiraterone therapy also elevates levels of progestational ligands and suppresses corticosteroid production, necessitating corticosteroid supplementation. These three mutations reduce AR binding specificity and are activated by progestins and corticosteroids.120
AR activity-inversion mutants
The AR LBD mutations prevent antiandrogens from inducing an antagonist conformation. They are located at characteristic sites where certain residues interact strongly with a portion of an individual antiandrogen (Figure 4A), and the mutational change reduces the size or increases the flexibility of these contact residues. This reduces the steric strain through which the antiandrogen is thought to distort the AR LBD into an antagonist conformation (Figure 4B), with the net result that that the specific antiandrogen becomes an agonist. In fact, increasing the size of the antiandrogen substituent juxtaposed to the smaller-sized residue in the mutant AR appears to be a viable strategy for designing analogs having restored antiandrogen activity against these specific AR mutant proteins68,69.
Detailed analyses of antagonist ligand binding to AR are limited because, unlike the ERα LBD, there are no crystal structures available for AR LBD ligand complexes having an antagonist conformation; so, structures illustrating complexes with antiandrogens (Figure 4A and Supplemental Movies S2 and S3) come from modeling based on agonist conformations. Nevertheless, the resistance mutations in AR can be classified as “ligand activity-inversion mutations” because they change the receptor’s interpretation of the ligand from an antagonist to an agonist.
ERα activity-inversion mutants
The constitutively active mutations in ERα that arose under conditions of estrogen deprivation also have reduced sensitivity to antiestrogens, because of the agonist pre-folded conformational preference of these mutations (Figure 3), but they do not convert antiestrogens into estrogens. Could a different type of ERα mutant—with ligand activity-inversion character like in AR—arise in breast cancer patients treated solely with antiestrogens? An informative prospect is L540Q ERα, which was identified in early ERα mutagenesis studies when selection was done in the presence of antiestrogens51,70–73. The L540Q mutant is substantially activated by the antiestrogens hydroxytamoxifen, ICI164,384, and RU54,876 (akin to the antiandrogen activation of the AR mutations), but L540Q ERα also functions as a dominant negative: It is neither active without ligand nor in the presence of estradiol, functioning under these conditions as a potent suppressor of WT ERα activity70–72.
Remarkably, a careful search of available clinical databases identified one patient who presented with a metastasis harboring this L540Q mutation after 5 years of tamoxifen-only therapy (Chandarlapaty, unpublished). While the L540Q mutation has not yet been reported elsewhere, there has been limited reporting of deep sequencing of the ERα gene in recurrent disease after exposure to ER antagonists alone. This mutation would not arise under estrogen deprivation by AIs because its dominant negative activity would inhibit WT ERα function and actually suppress, not stimulate, proliferation. If an L540Q mutation were to arise in a patient being treated with an antiestrogen, withdrawal of the drug might cause marked regression because of the dominant negative effect of the unoccupied L540Q ER70–72.
The L540Q mutation in ERα resembles the mutations in AR by having ligand activity-inversion character, but differs from them by not being specific for individual structurally diverse antagonists. The L540Q change places a polar residue in the middle of h12, which probably prevents its adopting either the agonist or the antagonist conformation (Figure 1C and D). Of interest, synthetic mutations of other hydrophobic residues in h12 also lead to this type of ligand activity-inversion character74. Hence, the molecular mechanisms by which the L540Q ERα and related h12 mutations function certainly deserve careful study.
ERα LBD mutations at two other sites convey an increased agonist response to SERMs. The first mutation, D351Y, was identified in one MCF-7 xenograft grown in the presence of tamoxifen75. Like L540Q, a variety of SERMs have substantial partial agonist activity on D351Y ERα in transcription assays; however, unlike L540Q ER, the SERD fulvestrant remains a full antagonist, and estradiol, a full agonist76–78. D351 is an acidic residue is in the middle of h3, close to the side chains of antiestrogens, and appears to have an important attractive interaction with the basic side chains of certain SERMs (e.g., tamoxifen and raloxifene)61,62 and a repulsive interaction with the acidic side chain of other ER antagonists (e.g., GW-5638 and AZD-9496)78–81. Both of these interactions would be abrogated by replacement of the negatively charged aspartate with the uncharged tyrosine. The second mutation, G400V, occurred during the initial cloning of ERα82; in cells, it too conveys a more agonistic response to some SERMs83 but not to a SERD84–86. G400 is located far from the ligand, at the start of the β-sheet region following h6; so, there is no clear molecular basis for these behaviors. Unlike L540Q ER, neither of these ERα mutations has yet been found in breast cancer sequencing databases.
CONCLUSIONS
In this review we have presented our current understanding of how activating mutations in the ERα LBD contribute to endocrine-therapy resistance in breast cancer, highlighting the molecular mechanisms by which they undermine the varied structural features of WT ERα that ensure it is off in the absence of estrogens. Selection under conditions of estrogen deprivation (AI therapy) leads to distinct sets of ligand-remote mutations in the ERα LBD that enable it to access an active/agonist conformation without agonist binding; this gives constitutive activity and resistance to AIs, but this preference for pre-folding in the agonist conformation also leads to a general decrease in binding and sensitivity to antiestrogens. By contrast, endocrine therapy-resistant AR mutations in prostate cancer are quite different, with C-terminal truncation being sufficient to give constitutive activity and resistance to androgen deprivation, but with antiandrogen therapy selecting for distinct AR LBD ligand-contact mutations that invert the activity of specific androgen antagonists to agonists. From this perspective, one can advance some general thoughts about how endocrine therapies for breast cancer might be improved. One can also appreciate the critical importance of sharing the lessons learned from ER mutations in breast cancer and AR mutations in prostate cancer in formulating the most beneficial and durable endocrine therapy strategies for each of these cancers.
Improved Design of Antiestrogens
Even though the ERs with activating mutations have reduced sensitivity to ER antagonists, the proliferative drive of ERs bearing activating mutations can still be overcome with higher concentrations of current antiestrogens, although the dose required depends on the nature of the mutation24,63. Thus, a strategy already underway is the improvement of pharmacokinetic properties to increase the internal exposure of tumors to highly potent antagonists. Some emerging orally active antiestrogens, such as AZD-9496, GDC-0810, as well as others, appear to provide just such encouraging behavior34,63,87–95 although further studies are needed regarding side-effect profiles and prevention of disease recurrence.
While most antiestrogens appear to block ER activity largely by a direct mechanism, through which the antiestrogen side chain repositions h12 from the agonist conformation (Figure 1D), surprisingly few structural strategies have been explored to accomplish this, with most antiestrogens having either a basic amine as in tamoxifen and raloxifene96,97, an acrylic acid side chain as in AZD-9496 and GDC-081034,80,88,98, or a long, largely extended largely hydrophobic chain as in ICI 182,780 (fulvestrant)99 and RU 58,668100. Other side chain design strategies could be evaluated, as could the optimal matching between antiestrogen side chains and core structural elements, with the goal of optimizing affinity, potency, and antiproliferative efficacy, while also seeking the best pharmacokinetic behavior. There are even alternative approaches to disrupting the ERα agonist conformation by indirect mechanisms using ligands with expanded core elements (even ones without side chains) that distort the positioning of regions within the ligand-binding pocket that are needed to support the agonist conformation of helix 1250,101–103. Finally, it will also be important to clarify the necessity—or even the desirability—of coupling ERα antagonism with ERα degradation (SERD activity), and determine whether ERα levels of both WT and mutant ERs can be lowered sufficiently to afford broad suppression of breast cancer progression.
Resistance Mutation Screening
Although the mutant ERα proteins appear to be uniformly less sensitive to multiple antiestrogens, it is possible that a broader exploration of modes of antiestrogen action might lead to antagonists that are active on specific mutations. In any case, the clinical exploration of structurally novel antiestrogens or new endocrine therapy strategies should be coupled with forward-looking mutagenesis studies to explore new ERα alterations by which resistance might develop. By getting ahead of potential limitations to the durability of their clinical efficacy, drug developers could explore structural modifications that might overcome the resistance due to these specific new mutations in advance of their appearance in the clinic. Thus far, this has been done more extensively with new antiandrogens68,104, but the generation and clinical observation of the L540Q ligand-activity inversion mutation in ERα after exposure to tamoxifen and the finding of other ERα activity-inversion mutations suggest that resistance to any structurally new antiestrogens through ERα LBD mutation should be screened for proactively. The general trend to combine antiestrogen endocrine therapies with other targeted-therapy agents, such as CDK4/6 inhibitors as well as other agents, may minimize the risk from different modes of therapy resistance105–109, including those due to mutations in ER.
We have described how the structural features of the LBD of WT ERα are optimized for it to be off in the absence of estradiol. Through this, we can also better understand how the receptor functions, appreciate the diverse ways by which mutations can undermine or even reverse these functions, and consider how the activation of ERα by oncological means (i.e., by mutation) vs. physiological means (i.e., by ligands) are both similar and different. Metastatic, therapy-resistant breast cancers due to ERα mutations are a significant medical issue and the cause of many deaths22; so, a deeper understanding of the molecular mechanisms by which these mutant ERα proteins generate hormone-independent, constitutive and antagonist-resistant activities should facilitate the development of a toolbox of antiestrogens to overcome this “on-state” of the receptor so as to improve endocrine therapies of breast cancer for patients, now and in the future.
Supplementary Material
REFERENCES
- 1.Katzenellenbogen BS & Frasor J Therapeutic targeting in the estrogen receptor hormonal pathway. Semin. Oncol 31, 28–38, (2004). [DOI] [PubMed] [Google Scholar]
- 2.Rugo HS et al. Endocrine Therapy for Hormone Receptor-Positive Metastatic Breast Cancer: American Society of Clinical Oncology Guideline. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 34, 3069–3103, (2016). [DOI] [PubMed] [Google Scholar]
- 3.Tryfonidis K, Zardavas D, Katzenellenbogen BS & Piccart M Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer treatment reviews 50, 68–81, (2016). [DOI] [PubMed] [Google Scholar]
- 4.Smith DF & Toft DO Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol. Endocrinol 22, 2229–2240, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pratt WB & Toft DO Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood) 228, 111–133, (2003). [DOI] [PubMed] [Google Scholar]
- 6.Ciriello G et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weigelt B et al. Molecular portraits and 70-gene prognosis signature are preserved throughout the metastatic process of breast cancer. Cancer Res 65, 9155–9158, (2005). [DOI] [PubMed] [Google Scholar]
- 8.Chu D et al. ESR1 Mutations in Circulating Plasma Tumor DNA from Metastatic Breast Cancer Patients. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 993–999, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fribbens C et al. Plasma ESR1 Mutations and the Treatment of Estrogen Receptor-Positive Advanced Breast Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 34, 2961–2968, (2016). [DOI] [PubMed] [Google Scholar]
- 10.Garcia-Murillas I et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci Transl Med 7, 302ra133, (2015). [DOI] [PubMed] [Google Scholar]
- 11.Jeselsohn R, Buchwalter G, De Angelis C, Brown M & Schiff R ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol 12, 573–583, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeselsohn R et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin. Cancer. Res 20, 1757–1767, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merenbakh-Lamin K et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer Res 73, 6856–6864, (2013). [DOI] [PubMed] [Google Scholar]
- 14.Li S et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell reports 4, 1116–1130, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson DR et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet 45, 1446–1451, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanning SW et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife 5, February 2;5 pii: e12792. doi: 12710.17554/eLife.12792., (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee AV, Oesterreich S & Davidson NE MCF-7 cells--changing the course of breast cancer research and care for 45 years. J. Natl. Cancer Inst 107, (2015). [DOI] [PubMed] [Google Scholar]
- 18.Toy W et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet 45, 1439–1445, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang P et al. Sensitive Detection of Mono- and Polyclonal ESR1 Mutations in Primary Tumors, Metastatic Lesions, and Cell-Free DNA of Breast Cancer Patients. Clinical cancer research : an official journal of the American Association for Cancer Research 22, 1130–1137, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spoerke JM et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun 7, 11579, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas C & Gustafsson JA Estrogen receptor mutations and functional consequences for breast cancer. Trends Endocrinol. Metab 26, 467–476, (2015). [DOI] [PubMed] [Google Scholar]
- 22.Chandarlapaty S et al. Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol 2, 1310–1315, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clatot F et al. Kinetics, prognostic and predictive values of ESR1 circulating mutations in metastatic breast cancer patients progressing on aromatase inhibitor. Oncotarget 7, 74448–74459, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Toy W et al. Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov 7, 277–287, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alluri PG, Speers C & Chinnaiyan AM Estrogen receptor mutations and their role in breast cancer progression. Breast Cancer Res 16, 494, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fuqua SA, Gu G & Rechoum Y Estrogen receptor (ER) alpha mutations in breast cancer: hidden in plain sight. Breast Cancer Res Treat 144, 11–19, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bahreini A et al. Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res 19, 60, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mao C, Livezey M, Kim JE & Shapiro DJ Antiestrogen Resistant Cell Lines Expressing Estrogen Receptor alpha Mutations Upregulate the Unfolded Protein Response and are Killed by BHPI. Scientific reports 6, 34753, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlson KE, Choi I, Gee A, Katzenellenbogen BS & Katzenellenbogen JA Altered ligand binding properties and enhanced stability of a constitutively active estrogen receptor: evidence that an open pocket conformation is required for ligand interaction. Biochemistry 36, 14897–14905, (1997). [DOI] [PubMed] [Google Scholar]
- 30.Jordan VC, Curpan R & Maximov PY Estrogen receptor mutations found in breast cancer metastases integrated with the molecular pharmacology of selective ER modulators. J. Natl. Cancer Inst 107, djv075, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pakdel F, Reese JC & Katzenellenbogen BS Identification of charged residues in an N-terminal portion of the hormone-binding domain of the human estrogen receptor important in transcriptional activity of the receptor. Mol. Endocrinol 7, 1408–1417, (1993). [DOI] [PubMed] [Google Scholar]
- 32.Weis KE, Ekena K, Thomas JA, Lazennec G & Katzenellenbogen BS Constitutively active human estrogen receptors containing amino acid substitutions for tyrosine 537 in the receptor protein. Mol. Endocrinol 10, 1388–1398, (1996). [DOI] [PubMed] [Google Scholar]
- 33.Zhang QX, Borg A, Wolf DM, Oesterreich S & Fuqua SA An estrogen receptor mutant with strong hormone-independent activity from a metastatic breast cancer. Cancer Res 57, 1244–1249, (1997). [PubMed] [Google Scholar]
- 34.Joseph JD et al. The selective estrogen receptor downregulator GDC-0810 is efficacious in diverse models of ER+ breast cancer. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lazennec G, Ediger TR, Petz LN, Nardulli AM & Katzenellenbogen BS Mechanistic aspects of estrogen receptor activation probed with constitutively active estrogen receptors: correlations with DNA and coregulator interactions and receptor conformational changes. Mol. Endocrinol 11, 1375–1386, (1997). [DOI] [PubMed] [Google Scholar]
- 36.Kircher M & Kelso J High-throughput DNA sequencing--concepts and limitations. Bioessays 32, 524–536, (2010). [DOI] [PubMed] [Google Scholar]
- 37.Trevino LS & Weigel NL Phosphorylation: a fundamental regulator of steroid receptor action. Trends Endocrinol. Metab 24, 515–524, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Likhite VS, Stossi F, Kim K, Katzenellenbogen BS & Katzenellenbogen JA Kinase-specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, deoxyribonucleic acid, and coregulators associated with alterations in estrogen and tamoxifen activity. Mol. Endocrinol 20, 3120–3132, (2006). [DOI] [PubMed] [Google Scholar]
- 39.Yee D & Lee AV Crosstalk between the insulin-like growth factors and estrogens in breast cancer. J Mammary Gland Biol Neoplasia 5, 107–115, (2000). [DOI] [PubMed] [Google Scholar]
- 40.Voudouri K, Berdiaki A, Tzardi M, Tzanakakis GN & Nikitovic D Insulin-like growth factor and epidermal growth factor signaling in breast cancer cell growth: focus on endocrine resistant disease. Anal Cell Pathol (Amst) 2015, 975495, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curtis SH & Korach KS Steroid receptor knockout models: phenotypes and responses illustrate interactions between receptor signaling pathways in vivo. Adv Pharmacol 47, 357–380, (2000). [DOI] [PubMed] [Google Scholar]
- 42.Stellato C et al. The “busy life” of unliganded estrogen receptors. Proteomics 16, 288–300, (2016). [DOI] [PubMed] [Google Scholar]
- 43.Iwase H Molecular action of the estrogen receptor and hormone dependency in breast cancer. Breast cancer (Tokyo, Japan) 10, 89–96, (2003). [DOI] [PubMed] [Google Scholar]
- 44.Gee AC & Katzenellenbogen JA Probing conformational changes in the estrogen receptor: evidence for a partially unfolded intermediate facilitating ligand binding and release. Mol. Endocrinol 15, 421–428, (2001). [DOI] [PubMed] [Google Scholar]
- 45.Nettles KW et al. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat. Chem. Biol 4, 241–247, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seielstad DA, Carlson KE, Kushner PJ, Greene GL & Katzenellenbogen JA Analysis of the structural core of the human estrogen receptor ligand binding domain by selective proteolysis/mass spectrometric analysis. Biochemistry 34, 12605–12615, (1995). [DOI] [PubMed] [Google Scholar]
- 47.White R, Sjoberg M, Kalkhoven E & Parker MG Ligand-independent activation of the oestrogen receptor by mutation of a conserved tyrosine. EMBO J 16, 1427–1435, (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herynk MH & Fuqua SA Estrogen receptor mutations in human disease. Endocr. Rev 25, 869–898, (2004). [DOI] [PubMed] [Google Scholar]
- 49.Nwachukwu JC et al. Systems Structural Biology Analysis of Ligand Effects on ERalpha Predicts Cellular Response to Environmental Estrogens and Anti-hormone Therapies. Cell chemical biology 24, 35–45, (2017). [DOI] [PubMed] [Google Scholar]
- 50.Nwachukwu JC et al. Predictive features of ligand-specific signaling through the estrogen receptor. Mol. Syst. Biol 12, 864, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wrenn CK & Katzenellenbogen BS Structure-function analysis of the hormone binding domain of the human estrogen receptor by region-specific mutagenesis and phenotypic screening in yeast. J. Biol. Chem 268, 24089–24098, (1993). [PubMed] [Google Scholar]
- 52.Jeyakumar M, Carlson KE, Gunther JR & Katzenellenbogen JA Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities. J. Biol. Chem 286, 12971–12982, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Madak-Erdogan Z et al. Integrative genomics of gene and metabolic regulation by estrogen receptors alpha and beta, and their coregulators. Mol. Syst. Biol 9, 676, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhao C et al. Mutation of Leu-536 in human estrogen receptor-alpha alters the coupling between ligand binding, transcription activation, and receptor conformation. J. Biol. Chem 278, 27278–27286, (2003). [DOI] [PubMed] [Google Scholar]
- 55.De Mattos-Arruda L et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann. Oncol 25, 1729–1735, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zehir A et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med 23, 703–713, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Staby L et al. Eukaryotic transcription factors: paradigms of protein intrinsic disorder. Biochem. J 474, 2509–2532, (2017). [DOI] [PubMed] [Google Scholar]
- 58.Jain VP & Tu RS Coupled folding and specific binding: fishing for amphiphilicity. Int J Mol Sci 12, 1431–1450, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trizac E, Levy Y & Wolynes PG Capillarity theory for the fly-casting mechanism. Proc Natl Acad Sci U S A 107, 2746–2750, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang Y & Liu Z Nonnative interactions in coupled folding and binding processes of intrinsically disordered proteins. PLoS One 5, e15375, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brzozowski AM et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389, 753–758, (1997). [DOI] [PubMed] [Google Scholar]
- 62.Shiau AK et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 95, 927–937, (1998). [DOI] [PubMed] [Google Scholar]
- 63.Zhao Y et al. Constitutively active mutant estrogen receptors show differential responsiveness to structurally novel antiestrogens in breast cancer cells and tumors. Cancer Research In Press XXX, XXX–XXX, (2017). [DOI] [PMC free article] [PubMed]
- 64.Watson PA, Arora VK & Sawyers CL Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lallous N et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol 17, 10, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Imamura Y & Sadar MD Androgen receptor targeted therapies in castration-resistant prostate cancer: Bench to clinic. Int J Urol 23, 654–665, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nyquist MD et al. TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer. Proc Natl Acad Sci U S A 110, 17492–17497, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Balbas MD et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife 2, e00499, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGinley PL & Koh JT Circumventing anti-androgen resistance by molecular design. J. Am. Chem. Soc 129, 3822–3823, (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ince BA, Zhuang Y, Wrenn CK, Shapiro DJ & Katzenellenbogen BS Powerful dominant negative mutants of the human estrogen receptor. J. Biol. Chem 268, 14026–14032, (1993). [PubMed] [Google Scholar]
- 71.Ince BA, Schodin DJ, Shapiro DJ & Katzenellenbogen BS Repression of endogenous estrogen receptor activity in MCF-7 human breast cancer cells by dominant negative estrogen receptors. Endocrinology 136, 3194–3199, (1995). [DOI] [PubMed] [Google Scholar]
- 72.Schodin DJ, Zhuang Y, Shapiro DJ & Katzenellenbogen BS Analysis of mechanisms that determine dominant negative estrogen receptor effectiveness. J. Biol. Chem 270, 31163–31171, (1995). [DOI] [PubMed] [Google Scholar]
- 73.Montano MM, Ekena K, Krueger KD, Keller AL & Katzenellenbogen BS Human estrogen receptor ligand activity inversion mutants: receptors that interpret antiestrogens as estrogens and estrogens as antiestrogens and discriminate among different antiestrogens. Mol. Endocrinol 10, 230–242, (1996). [DOI] [PubMed] [Google Scholar]
- 74.Mahfoudi A, Roulet E, Dauvois S, Parker MG & Wahli W Specific mutations in the estrogen receptor change the properties of antiestrogens to full agonists. Proc Natl Acad Sci U S A 92, 4206–4210, (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levenson AS, Catherino WH & Jordan VC Estrogenic activity is increased for an antiestrogen by a natural mutation of the estrogen receptor. J. Steroid Biochem. Mol. Biol 60, 261–268, (1997). [DOI] [PubMed] [Google Scholar]
- 76.Catherino WH, Wolf DM & Jordan VC A naturally occurring estrogen receptor mutation results in increased estrogenicity of a tamoxifen analog. Mol. Endocrinol 9, 1053–1063, (1995). [DOI] [PubMed] [Google Scholar]
- 77.Levenson AS, MacGregor Schafer JI, Bentrem DJ, Pease KM & Jordan VC Control of the estrogen-like actions of the tamoxifen-estrogen receptor complex by the surface amino acid at position 351. J. Steroid Biochem. Mol. Biol 76, 61–70, (2001). [DOI] [PubMed] [Google Scholar]
- 78.Bentrem D et al. Molecular mechanism of action at estrogen receptor alpha of a new clinically relevant antiestrogen (GW7604) related to tamoxifen. Endocrinology 142, 838–846, (2001). [DOI] [PubMed] [Google Scholar]
- 79.Wu YL et al. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol. Cell 18, 413–424, (2005). [DOI] [PubMed] [Google Scholar]
- 80.De Savi C et al. Optimization of a Novel Binding Motif to (E)-3-(3,5-Difluoro-4-((1R,3R)-2-(2-fluoro-2-methylpropyl)-3-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)phenyl)acrylic Acid (AZD9496), a Potent and Orally Bioavailable Selective Estrogen Receptor Downregulator and Antagonist. J. Med. Chem 58, 8128–8140, (2015). [DOI] [PubMed] [Google Scholar]
- 81.Liu H et al. Structure-function relationships of the raloxifene-estrogen receptor-alpha complex for regulating transforming growth factor-alpha expression in breast cancer cells. J. Biol. Chem 277, 9189–9198, (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tora L et al. The cloned human oestrogen receptor contains a mutation which alters its hormone binding properties. EMBO J 8, 1981–1986, (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Levenson AS & Jordan VC Transfection of human estrogen receptor (ER) cDNA into ER-negative mammalian cell lines. J. Steroid Biochem. Mol. Biol 51, 229–239, (1994). [DOI] [PubMed] [Google Scholar]
- 84.Levenson AS & Jordan VC The key to the antiestrogenic mechanism of raloxifene is amino acid 351 (aspartate) in the estrogen receptor. Cancer Res 58, 1872–1875, (1998). [PubMed] [Google Scholar]
- 85.Jiang SY, Parker CJ & Jordan VC A model to describe how a point mutation of the estrogen receptor alters the structure-function relationship of antiestrogens. Breast Cancer Res Treat 26, 139–147, (1993). [DOI] [PubMed] [Google Scholar]
- 86.Jiang SY, Langan-Fahey SM, Stella AL, McCague R & Jordan VC Point mutation of estrogen receptor (ER) in the ligand-binding domain changes the pharmacology of antiestrogens in ER-negative breast cancer cells stably expressing complementary DNAs for ER. Mol. Endocrinol 6, 2167–2174, (1992). [DOI] [PubMed] [Google Scholar]
- 87.Robertson JF et al. A good drug made better: the fulvestrant dose-response story. Clin Breast Cancer 14, 381–389, (2014). [DOI] [PubMed] [Google Scholar]
- 88.Weir HM et al. AZD9496: An Oral Estrogen Receptor Inhibitor That Blocks the Growth of ER-Positive and ESR1-Mutant Breast Tumors in Preclinical Models. Cancer Res 76, 3307–3318, (2016). [DOI] [PubMed] [Google Scholar]
- 89.Garner F, Shomali M, Paquin D, Lyttle CR & Hattersley G RAD1901: a novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs 26, 948–956, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wardell SE et al. Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 21, 5121–5130, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hattersley G, Harris AG, Simon JA & Constantine GD Clinical investigation of RAD1901, a novel estrogen receptor ligand, for the treatment of postmenopausal vasomotor symptoms: a phase 2 randomized, placebo-controlled, double-blind, dose-ranging, proof-of-concept trial. Menopause 24, 92–99, (2017). [DOI] [PubMed] [Google Scholar]
- 92.Min J et al. Adamantyl Antiestrogens with Novel Side Chains Reveal a Spectrum of Activities in Suppressing Estrogen Receptor (ER)-Mediated Activities in Breast Cancer Cells. J. Med. Chem, (2017). [DOI] [PMC free article] [PubMed]
- 93.Liu J et al. Rational Design of a Boron-Modified Triphenylethylene (GLL398) as an Oral Selective Estrogen Receptor Downregulator. ACS Med Chem Lett 8, 102–106, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu J et al. Fulvestrant-3 Boronic Acid (ZB716): An Orally Bioavailable Selective Estrogen Receptor Downregulator (SERD). J. Med. Chem 59, 8134–8140, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang Q, Zhong Q, Zhang Q, Zheng S & Wang G Boron-Based 4-Hydroxytamoxifen Bioisosteres for Treatment of de Novo Tamoxifen Resistant Breast Cancer. ACS Med Chem Lett 3, 392–396, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jordan VC Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 1. Receptor interactions. J. Med. Chem 46, 883–908, (2003). [DOI] [PubMed] [Google Scholar]
- 97.Jordan VC Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 2. Clinical considerations and new agents. J. Med. Chem 46, 1081–1111, (2003). [DOI] [PubMed] [Google Scholar]
- 98.Dickler M et al. in American Association for Cancer Research CT231 (Philadelphia PA, 2015). [Google Scholar]
- 99.Wakeling AE & Bowler J ICI 182,780, a new antioestrogen with clinical potential. J. Steroid Biochem. Mol. Biol 43, 173–177, (1992). [DOI] [PubMed] [Google Scholar]
- 100.Van de Velde P et al. RU 58,668, a new pure antiestrogen inducing a regression of human mammary carcinoma implanted in nude mice. J. Steroid Biochem. Mol. Biol 48, 187–196, (1994). [DOI] [PubMed] [Google Scholar]
- 101.Srinivasan S et al. Full antagonism of the estrogen receptor without a prototypical ligand side chain. Nat. Chem. Biol 13, 111–118, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhu M et al. Bicyclic core estrogens as full antagonists: synthesis, biological evaluation and structure-activity relationships of estrogen receptor ligands based on bridged oxabicyclic core arylsulfonamides. Org Biomol Chem 10, 8692–8700, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zheng Y et al. Development of selective estrogen receptor modulator (SERM)-like activity through an indirect mechanism of estrogen receptor antagonism: defining the binding mode of 7-oxabicyclo[2.2.1]hept-5-ene scaffold core ligands. ChemMedChem 7, 1094–1100, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Josan JS & Katzenellenbogen JA Designer antiandrogens join the race against drug resistance. Elife 2, e00692, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nardone A, De Angelis C, Trivedi MV, Osborne CK & Schiff R The changing role of ER in endocrine resistance. Breast 24 Suppl 2, S60–66, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Maurer C, Martel S, Zardavas D & Ignatiadis M New agents for endocrine resistance in breast cancer. Breast 34, 1–11, (2017). [DOI] [PubMed] [Google Scholar]
- 107.Augereau P et al. Hormonoresistance in advanced breast cancer: a new revolution in endocrine therapy. Ther Adv Med Oncol 9, 335–346, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.O’Sullivan CC Overcoming Endocrine Resistance in Hormone-Receptor Positive Advanced Breast Cancer-The Emerging Role of CDK4/6 Inhibitors. Int J Cancer Clin Res 2, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.O’Sullivan CC CDK4/6 inhibitors for the treatment of advanced hormone receptor positive breast cancer and beyond: 2016 update. Expert Opin Pharmacother 17, 1657–1667, (2016). [DOI] [PubMed] [Google Scholar]
- 110.Seielstad DA, Carlson KE, Katzenellenbogen JA, Kushner PJ & Greene GL Molecular characterization by mass spectrometry of the human estrogen receptor ligand-binding domain expressed in Escherichia coli. Mol. Endocrinol 9, 647–658, (1995). [DOI] [PubMed] [Google Scholar]
- 111.Montano MM, Müller V, Trobaugh A & Katzenellenbogen BS The carboxy-terminal F domain of the human estrogen receptor: role in the transcriptional activity of the receptor and the effectiveness of antiestrogens as estrogen antagonists. Mol. Endocrinol 9, 814–825, (1995). [DOI] [PubMed] [Google Scholar]
- 112.Patel SR & Skafar DF Modulation of nuclear receptor activity by the F domain. Mol. Cell. Endocrinol 418 Pt 3, 298–305, (2015). [DOI] [PubMed] [Google Scholar]
- 113.Yang J, Singleton DW, Shaughnessy EA & Khan SA The F-domain of estrogen receptor-alpha inhibits ligand induced receptor dimerization. Mol. Cell. Endocrinol 295, 94–100, (2008). [DOI] [PubMed] [Google Scholar]
- 114.Schwartz JA, Zhong L, Deighton-Collins S, Zhao C & Skafar DF Mutations targeted to a predicted helix in the extreme carboxyl-terminal region of the human estrogen receptor-alpha alter its response to estradiol and 4-hydroxytamoxifen. J. Biol. Chem 277, 13202–13209, (2002). [DOI] [PubMed] [Google Scholar]
- 115.Tamrazi A, Carlson KE, Daniels JR, Hurth KM & Katzenellenbogen JA Estrogen receptor dimerization: ligand binding regulates dimer affinity and dimer dissociation rate. Mol. Endocrinol 16, 2706–2719, (2002). [DOI] [PubMed] [Google Scholar]
- 116.Nadal M et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat Commun 8, 14388, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cheng Y & Prusoff WH Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol 22, 3099–3108, (1973). [DOI] [PubMed] [Google Scholar]
- 118.Li Z et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature 523, 347–351, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Norris JD et al. Androgen receptor antagonism drives cytochrome P450 17A1 inhibitor efficacy in prostate cancer. J. Clin. Invest 127, 2326–2338, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Krishnan AV et al. A glucocorticoid-responsive mutant androgen receptor exhibits unique ligand specificity: therapeutic implications for androgen-independent prostate cancer. Endocrinology 143, 1889–1900, (2002). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.