

Abstract
Pathogens and cancers are pervasive health risks in the human population. I argue that if we are to better understand disease and its treatment, then we need to take an ecological perspective of disease itself. I generalize and extend an emerging framework that views disease as an ecosystem and many of its components as interacting in a community. I develop the framework for biological etiological agents (BEAs) that multiply within humans—focusing on bacterial pathogens and cancers—but the framework could be extended to include other host and parasite species. I begin by describing why we need an ecosystem framework to understand disease, and the main components and interactions in bacterial and cancer disease ecosystems. Focus is then given to the BEA and how it may proceed through characteristic states, including emergence, growth, spread and regression. The framework is then applied to therapeutic interventions. Central to success is preventing BEA evasion, the best known being antibiotic resistance and chemotherapeutic resistance in cancers. With risks of evasion in mind, I propose six measures that either introduce new components into the disease ecosystem or manipulate existing ones. An ecosystem framework promises to enhance our understanding of disease, BEA and host (co)evolution, and how we can improve therapeutic outcomes.
Keywords: antibiotics, cancer, immune system, resistance, microbiota, pathogens
INTRODUCTION
Despite the pervasiveness of parasites and cancers across animal species and in humans in particular, our knowledge of the ecology and evolution of disease itself is surprisingly limited. When disease is studied in detail, it is often through descriptions of signs and symptoms, or the characterization of the biological etiological agent (BEA) and of diseased tissue ultrastructure. Other study—focusing on population-level interactions—usually abstracts disease into a single variable such as aggressiveness or pathogenicity. Although both approaches produce insights into BEA–host interactions, they oversimplify disease by omitting what may be its single most important driving force—ecology.
I advocate the perspective that when examined through an ecological lens, disease has a characteristic structure. Investigating this structure and variations on it is important not only for understanding ecology and evolution in a fundamental type of habitat—the organism—but also to develop approaches that lessen the burden of BEAs in animal husbandry, wildlife, on endangered and domesticated species, and in the human population—the latter being the focus of this review. The usual approach is to treat BEAs with drugs, but these can fail due to selection for resistance. The two most familiar types in humans are antibiotic resistance and chemotherapeutic resistance in cancers.
Early in the 20th century, Paul Ehrlich encouraged the search for the ‘Magic Bullet’—a single drug that targets and cures a disease with minimal residual toxicity for the patient. Ehrlich [1] also argued for the accepted approach of the day frapper fort et frapper vite (hit hard and without delay), based on the construct that drug dose must be commensurate with the severity of the disease and delaying drug delivery allows BEAs to multiply and cause irreparable damage and possibly death. Ehrlich and his contemporaries knew that some parasites could resist chemotherapy but were unaware of the underlying mechanisms. Despite clear arguments for why attempting to eradicate BEAs risks selecting for resistance [2, 3], frapper vite et frapper fort is still widely regarded as the surest way to cure disease.
The above two challenges—understanding disease and improving therapeutic outcomes—are interrelated because achieving the former will help address the latter, and because both natural, evolved processes that remediate disease and therapeutic interventions have ecology at their foundation. Here, I develop a framework for addressing these challenges. Focus is on humans because of the rich knowledge of some of their diseases and the growing importance of finding solutions to drug resistance and the improvement of therapeutic outcomes. In particular, I describe how disease can be understood as an ecosystem, the components and processes of which can be either manipulated separately or in combination toward a therapeutic objective. The disease ecosystem employs analogies from terrestrial, aquatic and agricultural ecosystems (hereafter ‘classic’ ecosystems), including predators, prey, competitors, detrivores, resources and the environmental surroundings. Thus, like classic ecosystems, disease ecosystems are intrinsically ecological: they are influenced by the environment and composed of feeding relationships (food webs) among ‘species’ in an interactive community.
I begin by justifying the need for an ecosystem framework of disease. I argue that in both healthy and diseased tissues there is a largely undiscovered world of ecosystem-like processes including resource fluxes, inter- and intraspecific competition, predation by immune systems and waste removal. I develop this concept as the central feature of the framework and then focus on how the state of the BEA population and associated disease provide reliable information about disease progression or remediation. Before applying the framework to therapies, I discuss what failures due to resistance typically resemble and present several other routes to therapeutic evasion that have received little attention. Key to applying the framework is how decisions are made about whether a treatment should attempt to eradicate or contain the BEA, or rather simply limit disease, and how strategies and tactics can be either used singly or in combination. Finally, I present some limitations to the framework and concluding thoughts.
The framework is centered on BEAs that replicate in the disease ecosystem such as microparasites and cancers, and not asymptomatic infections or diseases stemming from non-BEAs. The current framework should therefore be viewed as part of a larger picture. I also do not address the important question of how to measure ecological and evolutionary features of disease (see reviews in e.g. [4, 5]). Finally, I do not attempt a systematic overview of the huge diversity of human BEAs. Rather, I refer where appropriate to specific examples in several well-studied BEA groups, principally bacteria and cancers. Comparing and contrasting the basic features of these groups is the first step towards an understanding of disease ecosystems and how they can be used to devise successful therapies.
THE NEED FOR A GENERAL FRAMEWORK
Disease is a complex system of multiple species interacting at multiple biological and spatial scales. Whether and how a disease progresses, what are the implications for the BEA and the host, and how to design therapies can only be answered if we characterize the underlying components, their functions and interrelationships.
The limited accessibility and microscopic nature of disease makes it particularly challenging to study and to measure. As a consequence, in vivo work typically takes physiological and epidemiological approaches, describing signs and symptoms, changes in tissue ultrastructure, and consequences for morbidity and mortality. In contrast, in vitro study usually focuses on the BEA itself and abstracts-out the complexity characteristic of in vivo environments. This is problematic, for example, because in vitro observations can be misleading about in vivo behaviors [6]. The combined situation—the general ignorance of ecological and evolutionary mechanism in vivo, the oversimplification of real environments in vitro —limits our knowledge of how disease-generating processes actually work.
Beyond the importance of how both ecology and evolution influence disease, a general framework needs to have the flexibility to accommodate the effects of therapeutic interventions. Therapy can be viewed as an ecological perturbation intended to reset a disrupted system to its healthy state. As indicated above, the prevailing approach is frapper fort et frapper vite to eliminate BEAs and minimize the chances that (contagious) BEAs are transmitted to other hosts. However, a large body of work indicates that targeted antimicrobial and anticancer chemotherapies select for resistance [3, 7], and there is increasing awareness that drug therapies can have enduring disruptive effects on the microbiome [8] (but see [9]) and the host itself (e.g. contributing to other diseases, such as secondary cancers).
Despite the large body of evidence that treat-to-cure chemotherapy can lead to resistance and treatment failure, this approach has stood the test of time, largely due to its simplicity and common sense, but also because it often works [10]. Aggressive chemotherapy also continues largely unchallenged because scientific study is still in early days of evaluating alternatives, most of which are centered on preventing or limiting the evolution of resistance. Until relatively recently much of what was known about resistance and its management derived from pesticide use (Box 1). Although the lessons learned are foundational, they often abstract-out ecology, and are sometimes difficult to apply to disease due to differences in biology and ecology, habitat structure and habitat resilience to treatment.
Box 1: The lessons of pesticide use
Much of our knowledge about therapeutic resistance and how to manage it can be traced to lessons from the use of pesticides in agriculture [181, 182]. Pesticide treatments attempt to reduce damage below an economic threshold while not exceeding acceptable levels of residual toxicity for consumers and the environment (e.g. non-target species). By the very nature of pest status (i.e. large, dispersive populations with high potential growth rates) and high but nevertheless limited doses of chemicals (meaning that sufficiently resistant strains are likely to survive a given dose), the repeated, blanket application of the same ‘magic bullet’ over a population will eventually select for resistance. The usual course of action is to search for new compounds to replace the failing ones. But our ability to create new active substances is declining and the number of resistant pests is increasing [183]. Ecologically- and evolutionarily informed approaches have become promising alternatives. Primary among them is the prediction that resistance can only be contained by ‘conserving’ sensitive strains of the pest. For example, the evolution of resistance can be slowed if the competitive balance is shifted in some places and times in favor of sensitive strains [184]. There is evidence that this and other tactics increase the likelihood of managing resistance in pest control programs [185].
We therefore need a general framework that is firmly rooted in ecology and evolution both as a basis for fundamental understanding and to guide therapies [11]. There are a number of developments and frameworks that integrate ecology and evolution (parasites and pathogens: [12–15]; cancers: [5, 16–20]), but none extend to both infectious and non-infectious BEAs. My aims are to generalize the disease ecosystem concept and to use its insights to suggest several novel therapeutic interventions.
AN ECOSYSTEM FRAMEWORK OF DISEASE
The framework for understanding and treating disease (Figs 1 and 2) focuses on how the components and processes in the disease ecosystem interact directly and indirectly with the BEA. The system can be in one of four main states that reflect BEA emergence, population growth, spread and pathogenicity, and regression. Understanding the state of the BEA and associated disease will be the basis for applying the framework to therapeutic interventions, as described below.
Figure 1.

Healthy and disease ecosystems. (A) Organ- and tissue-level scale of the main compartments in host ecosystems. The boundaries of the ecosystem are determined by the interactions and events of importance and interest to the observer. Organ type, tissue architecture and local environmental conditions will play roles in the structure and dynamics of the ecosystem. In particular, interactions among cells (depicted as a network) and immune system flows into and out of the reference ecosystem will mediate homeostasis and/or dysbiosis in association with the BEA. (B) Basic structure of a healthy tissue ecosystem. This consists of the local vascular system, epithelial cells, extracellular matrix (ECM), microbiota (e.g. bacteriophages and bacteria) and elements of the immune response, including phagocytes, lymphocytes and antimicrobial peptides. Nutrients delivered through the vascular system feed nearby living cells. Foreign cells may be engulfed by phagocytes, and waste removed by both phagocytes and the vascular system (diffusible wastes, e.g. CO2). Finally, fibroblasts as part of the innate immune system contribute to maintaining tissue structure (ECM and vascular system) and initiating the immune response to injury or BEA invasion. See caption C for key to symbols. (C) Basic structure of the disease ecosystem. BEAs, microbiota, their natural enemies (e.g. viruses) and immune cells interact in a community. Healthy host cells are also part of the community since they compete locally with BEAs. All living cells in this disease ecosystem consume nutrients and produce waste and by-products, some of the latter two of which is recycled. Like the healthy tissue ecosystem, the habitat is supported by fibroblasts, ECM and the vascular system, but, notably in the case of tumor microenvironments, the structure of these are disrupted by the damage caused by the BEA and chronic inflammation (not shown). Arrows indicate a subset of the possible directions of influence. See main text for details
The organism ecosystem
The ecosystem concept is a general way of representing species interaction networks, non-living organic and inorganic matter pools, and the physical habitat. Key to the concept is the fluxes, balances and stocks in these different compartments, and how events (e.g. environmental disturbance, species invasions) can have consequences for individual compartments or the ecosystem as a whole.
Similar to classic ecosystems, the healthy organism ecosystem is composed of supportive and regulatory structures (Fig. 1A and B). Supportive structures include habitats (tissues), resource replenishment (circulatory system) and waste removal (phagocytosis, circulatory system). Regulatory structures include cooperative interactions (cell–cell signaling, hormonal control), predation (immune systems), and commensalism, competition and parasitism (microbiota). These structures will vary between tissue types [14], suggesting that ecosystem dynamics may contrast as well.
Although it is tempting to perfectly equate different components in organism ecosystems with analogs in classic ecosystems, they do have at least one important difference: whereas Darwinian selection acts on many component species in the latter, it is centered on a single species in the former (but see [21]). Thus, we would expect the core of the organism ecosystem (i.e. host cells) to evolve traits that promote habitat maintenance and remediation, meaning more coordination and less autonomy than species interactions in classic ecosystems. We would nevertheless also expect that similar processes are at work between the two system types, including selection acting on traits ultimately affecting the ecosystem, and co-evolution with mutualists, commensals and parasites. More research is needed to explore these observations and expectations.
The disease ecosystem
The disease ecosystem results when a healthy ecosystem is disrupted by a BEA and the host organism attempts remediation. This is in some ways analogous to environmental disturbances in classic ecosystems and more specifically to those stemming from invasive species (Box 2). Below I describe some of the main features of the disease ecosystem (Fig. 1C), and briefly review the most understood type—the ‘tumor microenvironment’—in Box 3.
Box 2: BEAs and invasive species
BEAs have commonalities with invasive species [186]. They colonize a spatially heterogeneous and potentially hostile environment, and may adapt to or transform the prevailing host ecosystem into one where they can grow, evade predation (or grow despite it), gain access to resources, and multiply and disperse. In so doing BEAs interact with the microbiome, host cells and tissues, and produce waste. Moreover, like invasive species, BEAs can be spatially structured. For example, Lloyd et al. [187] examined spatial models of tumor growth and found that cells toward the center of the tumor aggressively competed for resources and attained high densities, whereas cells toward the periphery grew faster and competed less. Similar to invasive species [188], BEAs may be associated with specific microbial communities (e.g. [14, 140, 189, 190]), but any causation between the two can be difficult to determine (see [191]). Also, BEA activity may lead to the damage of host cells, tissues and organ systems, and the impairment of waste removal, and there are parallels with invasive species in classic ecosystems [192].
Box 3: The tumor microenvironment
The most understood type of disease ecosystem is the ‘tumor microenvironment’. One principal difference between tumor cells and other BEAs is that the former are - themselves - diseased. Tumor cells divide, move, compete and may cooperate in a 3D semi-structured mass [193–196]. However, only those tumors successfully evading the immune response will progress to cause significant disease [65, 85]. Similar to healthy host cells or growing parasites, tumor cells sequester resources from the host. But in contrast to the first two, tumor cells dramatically increase their glucose uptake through aerobic glycolysis and produce waste in the form of lactate (i.e. the Warburg Effect [197]).
Spatial structure plays an important role in the disease ecosystem of solid tumors. Because of the disruption of otherwise healthy tissue, diffusible wastes increase and nutrients decrease with distance to the nearest capillaries. This is particularly important in cancer growth, where cells toward the interior of a tumor tend to be deprived of resources (glucose and oxygen) and exposed to high concentrations of various wastes (especially lactate). These conditions can lead to altered cellular behavior or cell death [198], and can even extend beyond stressed areas of a tumor: hypoxic cells may diffuse molecular signals that stimulate (i) angiogenesis thereby fostering cell survival and tumor expansion, and (ii) cell motility, which promotes metastasis [199].
Another characteristic of the cancer disease ecosystem is that massive tumor cell division and genomic instability of tumor cells generate considerable phenotypic variability (State 2, Fig. 2), some of which will increase adaptation to changing micro-environmental conditions. Adaptive traits include changes in cellular growth (r or K) strategies, escape from immune responses, movement away from inhospitable microsites and dispersal to other tissue ecosystems in the body [34, 194, 200–202]. Although the details differ, many of the above elements are analogous to interactions in classic ecosystems and in food webs [203].
When the BEA enters or emerges from within a healthy host ecosystem it is likely to be confronted by one or more of physical structures [22, 23], oxidative stress, chemical defenses [24] and innate immune responses [25]. Immune systems in particular are central to somatic maintenance, including the host’s ability to either limit the growth and spread of (or eradicate) a BEA [26], and to repair tissue damage [27]. However, some BEAs may evade the immune response, grow, spread and generate disease. BEA and host genotypes, the environment and tissue type are among the many factors influencing these processes and the associated severity and extent of disease [25, 28–31].
Many disease ecosystem interactions involve vying for resources. As the BEA grows it competes for space and/or nutrients and this may result in stressful local conditions (e.g. hypoxia, [32, 33]), and in certain cancers, favor disease progression [34]. Competition for space or nutrients can occur with the same or other BEA types [35–37], host cells [38] or commensal microbiota [39]. For example, Balmer and Tanner [37] argued that multi-strain infections could have far reaching consequences for the ecological and evolutionary dynamics of disease, including mutualistic interactions and direct and indirect competitive interactions (e.g. apparent competition). Nutrient levels have also been shown to influence BEAs indirectly via the immune system [40]. Despite the multitude of potential indirect interactions suggested by Fig. 1C—particularly with the microbiota—little study to date has investigated these in detail (e.g. [41]).
Although many of the events in disease ecosystems have analogs in classic ecosystems, the detailed functions often contrast. Thus, for example, whereas immune systems and predators ultimately kill their ‘prey’, unlike predators, immune cells do not appear to gain significant energy in the process. Rather, the latter derive energy from nutrients in situ, including in the disease ecosystem (for the tumor microenvironment, see [42]). Moreover, unlike predators, different immune cells cooperate in killing diseased/damaged cells and pathogens. (A notable exception to this in classic communities is cooperative hunting.) Thus, lymphocytes function as precursors for phagocyte activity, either marking or killing target cells, whereas phagocytes engulf and remove target cells and waste. Despite detailed knowledge about immune systems, we know very little about their functional and numerical responses (but see [43]) and how these may contrast with classic species communities.
States of BEA and associated disease
The BEA is at the center of the disease ecosystem framework. The BEA population is dynamic, yet proceeds through one or more of a characteristic series of states. These states can convey considerable information about the current and future growth of the BEA and disease. Torres et al. [44] proposed a ‘disease map’ that tracks pathogen load and patient health through the course of a disease. Below, I reinterpret this concept emphasizing the growth and evolvability of the BEA population and associated disease (Fig. 2).
Figure 2.
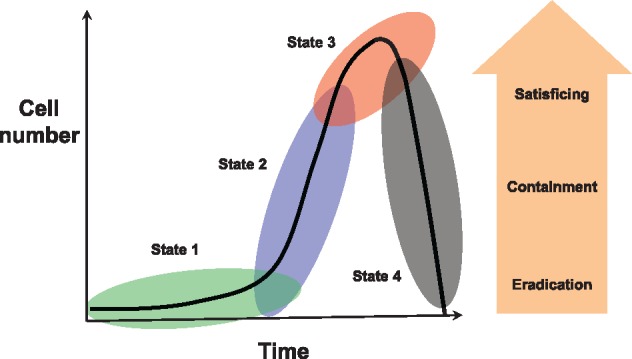
Four states in BEA and associated disease. This classification is based on growth in a novel and hostile environment (State 1), BEA population size (cumulative number of births) and therefore evolutionary potential (State 2), and spread and colonization of local and distant within-host habitats with correspondingly greater impact of disease on the host (State 3). Therapeutic objectives (eradication, containment, satisficing) change with progression through the three first states (it is assumed that natural remission (State 4) will not be subject to therapeutic intervention). In particular, although optimal protocols are feasible in States 1 and 2, treatment options may be limited to ‘satisficing’ in (late) State 3, for example due to reduced tolerance to drug toxicity. Should the immune response and any corresponding inflammatory response be sufficient, then the BEA and associated disease would regress to the healthy tissue state and homeostasis (State 4). This is shown for remediation from State 3, but it could also occur at States 2 or 1. See main text for further discussion
State 1. Establishment: small numbers, low evolutionary potential, negligible disease
As described above, during establishment, BEAs confront novel and possibly hostile environments [45, 46]. Establishment will depend on the BEA’s ability to evade immune responses and either already possess or plastically express virulence factors permitting resource acquisition and growth [47]. Should the immune response be sufficient, the BEA enters directly into State 4 (see below) and declines. Even should a small BEA population continue to grow, its total mass and the extent of associated disease is likely to be minute. State 1 continues until (i) cumulative cell turnover is on the order of the reciprocal of the beneficial mutation rate (entry into State 2), and/or (ii) the intensification of disease due to some combination of direct damage by the BEA and possible chronic inflammation (entry into State 3).
Detailed study of the initial phases of BEA population growth is uncommon. For example, Margolis and Levin [48] considered how commensal Haemophilus influenzae could become invasive from the nasal passage to the blood and then evolve. They showed that invasive infections have a significant stochastic component, are typically initiated by a single bacterial cell and found evidence for infrequent within-host evolution. Leggett et al. [49] looking across 43 different human pathogens found that lower inoculum numbers were required for infections generating pathogenesis at local as opposed to distant sites. The former tended to be more virulent than the latter. In contrast to parasitic BEAs, tumorigenesis requires key initiating mutations that compromise cooperative cellular functions [50], and together with subsequent tumor progression and metastasis (States 2 and 3), this can take years or even decades to achieve (e.g. [51]).
State 2. Growth: mutation, adaptation and resistance
The transition from a relatively slow growing, evolutionarily limited BEA in State 1 to an exponentially growing, evolving BEA in State 2 is associated with both population turnover (total births per unit time) and mutation rate [52, 53]. Population growth will be influenced by the host immune system and nutrient levels [40, 54]. The per-birth mutation rate can be highly variable between different BEA strains and types, with more rapid passage from State 1 to State 2 expected for hypermutating bacteria, RNA viruses such as HIV-1, and genomically unstable tumor cells. For example, Sottoriva et al. [55] studied the spatio-temporal dynamics of beneficial mutations in colorectal cancers. They found that the first dominant mutations emerged in tumors containing as few as ∼<10 000 cells, which can be explained in part by mutation rates as high as ∼10−5 (see also [56]).
BEA population growth and evolutionary potential have two notable consequences. First, growing BEA populations result in alteration of host cell behaviors and for some BEA types, greater tissue damage. Second, higher mutation rate favors anti-immune defenses [57], adaptations in nutrient use [58, 59] and resistance to therapeutic interventions [60, 61].
State 3. Spread: growth and dispersal magnify disease
Continued BEA growth eventually results in metabolic competition and—particularly in the tumor microenvironment—waste accumulation. One or both of these factors can promote local colonization and migratory behaviors [62, 63], which lead to increased pathogenicity. Although disease can be significant at low BEA densities (States 1 and 2), all else being equal, it usually worsens with time and as BEA numbers grow (see contrasting scenarios in [64]). Cancers are arguably the best examples of how growth, local tissue invasion and distant tissue colonization—together with chronic inflammation—compromise host condition and increase mortality. Local tissue invasion and metastatic spread are indicative of escape from immune control [65]. Similar patterns are observed in certain microparasites, where disease progression is associated with local tissue invasion and cell entry [23].
State 4: Regression: immune responses abate BEA and disease
Should the combined effects of the innate and adaptive immune systems prevail resulting in BEA population decline, then disease too will eventually regress [44]. Depending on immune and BEA dynamics, State 4 may obtain at any point during States 1 to 3 for infectious BEAs, whereas for cancers State 4 is most likely to obtain during State 1, before the tumor evolves to evade the immune response. BEAs in State 3 that do not enter State 4 either lead to chronic disease or result in host death.
The above classification gives a clear message for therapy. A disease is much more treatable in State 1, but either impossible or difficult to diagnose compared to State 2 and particularly State 3. In States 2 and 3, resistance is a concern, and special measures may be needed to treat the BEA. Finally, drug toxicity becomes an issue as the BEA population grows, disease spreads, and the person’s health is at risk or life is engaged, as would be observed (late) in State 3. Observation that the BEA and associated disease are in State 4 would often preclude a therapeutic intervention.
APPLYING THE FRAMEWORK
The population ecology of BEAs makes them particularly difficult to eradicate. Once disease is detected, BEAs typically will have attained large population sizes and generated mutant strains that result in opportunities for evolutionary responses. There are useful parallels between seeking to cure a disease and attempting to eradicate an invasive species. Perhaps, the most compelling is that eradication becomes more achievable as the population is smaller and more spatially circumscribed (States 1 and 2, Fig. 2). Thus, for example, invasive species are more difficult to eradicate on mainland (or interconnected habitats) than on islands (or isolated habitats) [66]. Both ecological dynamics (e.g. larger population sizes, refugia) and evolutionary dynamics (e.g. greater additive genetic variation) contribute to explaining this observation. The same basic principle applies to treating a BEA, where diversification (State 2) and spread (State 3) make eradication less probable, as is amply demonstrated by relapse due to chemotherapeutic resistance in metastatic cancers.
Below, I first review different ways in which therapies fail. I then present six eco-evolutionary-sensible measures that can be used singly or in combination to achieve therapeutic objectives.
Why do therapies fail?
Excepting technical issues of inappropriate drugs, the main probable cause for treatment failure is BEA resistance [10, 67]. Drug resistance due to high-dose therapy is a textbook example of ‘evolutionary rescue’ [68, 69], where the drug (if it were to be maintained) drives the sensitive population to extinction, but resistant variants already present or emerging during the treatment grow and repopulate the disease ecosystem. The actual sequence of mutational events leading to rescue has rarely been studied [70], and we are only beginning to learn about the underlying ecological processes (see [69] for contrasts between medicine, agriculture and conservation biology). For instance, although the evolution of resistance is often associated with high-dose chemotherapies, lower dosing (e.g. to regulate toxicity, especially in cancer chemotherapies) can also select for resistance [71, 72].
The most discussed mechanism leading to evolutionary rescue is ‘competitive release’, whereby reductions in the population of the sensitive strain open the niche for growth of otherwise competitively suppressed resistant strains [73, 74]. Qualifying all instances of evolutionary rescue as stemming from competitive release is an oversimplification, because there are notable contrasts in the mechanism depending on the absolute and relative fitnesses of sensitive and resistant strains, both in the presence and absence of a drug [74]. Nevertheless, there is some support for the general phenomenon of competitive release [75–77].
Much of what is known about treatment failure comes from studies of the evolution of drug resistance. But barring data actually demonstrating resistance, other less studied, non-mutually exclusive mechanisms may be involved in BEA resurgence or relapse.
Competitive release—although sometimes used for inter-strain competitive effects (see above)—is originally an ecological concept [78]. In ecological competitive release, the elimination of the target BEA results in the emergence of one or more otherwise competitively suppressed BEAs [79].
Therapeutic tolerance is the ability of BEAs to withstand the transient effects of a treatment. Examples include reduced receptor sensitivity or density-limited drug impact in cell sub-populations of certain cancers [80], and quiescent or dormant states in cancer stem cells [81] and bacterial persister cells [82]. Other forms of tolerance involve protective structures, such as bacterial biofilms against antibiotics [83], biofilms limiting host immune responses [84], and immune tolerance in the tumor microenvironment [85].
Escape mutants do not have specific resistance or tolerance mechanisms, but rather simply outgrow the suppressive effects of the therapy. This may occur, for example, due to insufficient drug dosing [86, 87].
Spatial and temporal refuges are similar to escape mutants, except that BEA survival is due to heterogeneity in therapeutic exposure. Examples include limited drug diffusion [88–90] and ‘drug holidays’ or patient non-compliance [86].
High mutation rates may favor escape by increasing the chances of resistance emerging in bacteria [91], HIV [92] and certain cancers [93]. Hypoxic stress in leading to higher mutation rates in tumors could explain certain origins of therapeutic resistance [33].
Therapeutic measures
Applying the framework involves first acquiring information about the BEA and disease ecosystem (Figs 1 and 2). A therapeutic objective is chosen based on this information and on actionable strategies and associated tactics. The choice considers failure risks due to BEA resistance or evasion. Typically, a single strategy and tactic will be deployed to achieve an objective, but as will be explained below, under some circumstances different strategies/tactics may complement one another and become a combination strategy. Below, I refer to strategies and tactics as ‘measures’. A treatment is deemed a success if it achieves its objective (see Supplementary Material for a simple criterion).
Importantly, a chosen measure need not do all the ‘work’ to achieve a therapeutic objective. Rather, it can be employed as an adjuvant either to other measures as part of a combination strategy or to processes already active in the disease ecosystem. For example, amoxicillin is a common antibiotic used for treating type A strep throat associated with Streptococcal pharyngitis. The antibiotic interferes with BEA cell wall synthesis and thus targets bacterial replication. Amoxicillin may not be 100% effective on its own (due to limited pharmacokinetics) and different components of the immune system ensure the final stages of the complete clearing of the infection [94].
Below, I present a series of six measures for treating disease (Fig. 3), starting with the most employed—drug-based targeted therapy (Measure 1). Measure 1 has three main variants depending on the objective: eradicate, contain or satisfice. A second approach is to focus on reducing damage, rather than target the BEA per se (Measure 2). A third approach is to target other components of the disease ecosystem (Measures 3–6). Finally, I describe the strategy of combining measures into more effective therapies.
Figure 3.
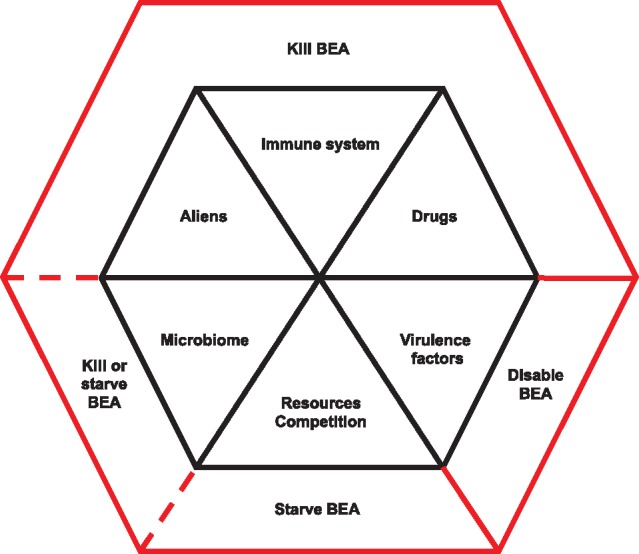
Six measures for treating disease. Measures have one or more of three proximate effects: kill (cytotoxic), disable (cytostatic) or starve the BEA. Killing the BEA is the most likely of the three to select for resistance. Combination therapies (two or more measures) are most likely to succeed if they do not interfere with one another in mode of action and do not select for resistance in the same or in linked genes. See main text for further discussion
Measure 1: Drugs
Alexander Fleming expressed a nuanced version of frapper fort et frapper vite [95]. His recommendation ‘if you use penicillin, use enough’ was based on the idea that sensitive strains were the origin of de novo resistant mutants, and the former needed to be eliminated quickly and decisively. Resistant mutants may however be present even before a therapy commences, either in the pathogen inoculum (or the first cells of an invasive carcinoma) or due to mutations in a growing BEA population (i.e. State 2, Fig. 2). The observation that the Ehrlich–Fleming approach applied to a large enough BEA population risks selecting for resistance has fueled theoretical research aimed at determining the drug doses and scheduling that will either contain or eradicate a BEA (e.g. [96–101]). These studies and others have contributed the following basic insights.
Eradicate
If the turnover of the BEA population and/or its genetic diversity are low (State 1), then resistance mutants are unlikely to be present [53] or may fail to emerge even under treatment (see also [102]). Eradication becomes an option. Depending on the information available about possible resistance mutants, the minimal dosing strategy is (in order of decreasing dose) either to apply the maximum tolerated dose (no information), attain the mutant prevention concentration [103] (information about resistance) or apply the dose necessary to clear sensitive strains (no resistance detected).
Contain
When the turnover of the BEA population and/or genetic diversity are high (States 2 and 3), then high-dose, targeted therapy is likely to be useless or even counterproductive. Examples where this may occur include clonal expansions [16], and genetically diverse infections [104]. Often the course of action taken when resistance is suspected or identified is to employ particularly aggressive drugs or—for some multi-drug-resistant pathogens—last resort drugs [105]. These treatment strategies, and indeed intermediate dose treatment strategies, are likely to fail [101, 106, 107]. Experimental and theoretical study indicates that the most sensible approach is to aim to contain the BEA through low but sufficient dose chemotherapies (e.g. [101, 108], and references therein). Containment requires that sensitive BEA strains be held in check by the therapy, but maintained at levels so as to competitively limit the populations of any drug-resistant strains when therapy is released. This is most readily achieved through managing the total BEA burden, whereby resistant types express a fitness cost relative to sensitives during periods of relaxed therapeutic selection (recent reviews in e.g. [98, 109]). More nuanced ‘adaptive’ approaches monitor therapeutic performance and eventually adjust drug dose or drug type so as to manage disease burden [110]. Recent experimental demonstrations of tumor containment show some promise (in vitro [111]; in vivo [77]), and the same approach should be applicable to certain microbial pathogens [112]. There are nevertheless situations where containment strategies are unlikely to succeed, such as when fitness-compensatory mutations have emerged in resistant strains during evolutionary rescue or have been transferred horizontally (e.g. [113–115]).
Satisfice
Simple dosing rules can be misleading when toxicity is an issue (late State 3 and no indication of entering State 4), as is often encountered in metastatic cancers. This is because the doses needed to optimally treat the (advanced) BEA are too toxic for the weakened patient. This situation becomes particularly acute when the patient’s life is engaged and rapid decisions need to be made [116]. In such scenarios aiming for ‘optimality’ is better replaced by ‘satisficing’, where success is equated with an acceptable improvement in signs and/or symptoms [117]. Much of what is known about containing a BEA while managing toxicity through scheduled dosing comes from theoretical study. For example, Foo et al. [118] examined pharmacokinetic models of the emergence of drug resistance in patients with epidermal growth factor receptor lung cancers. They found that the pulsed dosing (to control toxicity) could limit tumor growth, but long intervals risked the emergence of new drug-resistant clones.
Drug therapies are not ‘stand-alone’—particularly when targeting a microparasitic infection. Unless a patient is immunosuppressed, the immune system will act in conjunction with the treatment. This means that optimal dosing may depend on host condition (drug tolerance, ability to mount an immune response) and the disease ecosystem (the BEA population state). For example, Ankomah and Levin [119] studied a model of how the innate and adaptive immune systems could complement the effects of a drug to clear a bacterial infection. They showed that high drug doses led to the most successful outcomes (but see contrasting findings in [101]).
Despite empirical work showing that drug dose can influence treatment outcomes (for literature survey, see [101]), the necessary information required to make strategic decisions is often lacking. This could in part explain why eco-evolutionary reasoning is not being used to its full potential in deciding whether and how to employ aggressive treatment or containment strategies [98, 104, 107].
Measure 2: Virulence factors
The severity of disease will be influenced by the expression of ‘virulence factors’ that enable the BEA to construct its niche. Virulence factors may promote resource extraction, protect from host defenses, or attach to cell or tissue surfaces. They are most documented for microparasitic BEAs and include adhesive structures, motility enhancers such as Type IV pili, and toxins [120, 121]). Virulence may also have a social component, for example via quorum sensing in certain bacteria [122, 123].
Anti-virulence drugs have gained attention as possibly being ‘evolution proof’, because the traits actually targeted have little influence on fitness [120, 124]. Examples of drug targets include rendering bacteria more susceptible to immune clearance and increasing antibiotic efficacy [125], and cytostatic drugs that stall cancer cell division rather than targeting cell survival [126]. Other approaches seek to change BEA behaviors, such as increasing pH in cancer microenvironments that reduces tumor growth and metastasis [127]. Although in vitro studies of anti-virulence strategies appear promising (e.g. [128]), resistance is a concern, and there may be limitations in their efficacy in vivo [117, 120, 121].
Measure 3: Immune system
The immune system is the single most important mechanism responsible for preventing disease and limiting its spread. Immune systems can be remarkably complex, and their detailed description is beyond the scope of this review. Broadly speaking, immune responses may involve specialized molecules and/or cell types. Introducing molecules from the innate immune response—notably antimicrobial peptides—shows great therapeutic potential by both their stunning diversity and the lower likelihood of resistance evolution compared to antibiotics [129]. Immunotherapies that stimulate the production of specific immune cell motifs show promise in treating certain cancers [130] and bacterial diseases [131], but their use is still controversial due to risks of immunotoxicity and autoimmune disorders [132]. Related to this, Smyth et al. [133] recently argued for using information about components of the tumor microenvironment in decisions about immunotherapies, particularly as adjuvants to other therapies.
Measure 4: Resources—competition
BEAs need resources to grow and multiply. Resources include hospitable space and nutrients such as glucose, oxygen, carbon, nitrogen and phosphorus. Glucose use in particular differs fundamentally between healthy cells and tumor cells (with the latter consuming up to 18 times the former; [134]), as does phosphorus [135], and leveraging these may contribute to a strategy for tumor control. Resource deprivation by curtailing angiogenesis has proved successful in certain cancer treatments, but can come at a cost of limiting drug diffusion through the tumor [136]. In Plasmodium, Wale et al. [137] recently showed how resource limitation could differentially impact sensitive and resistant strains. Other tactics either limiting or excluding BEAs include boosting the competitiveness of healthy cells [138], or of commensals in the microbiome [139].
Measure 5: Microbiome
The microbiome is increasingly recognized as foundational to organism biology and condition. Some analogies are likely to apply between the functions of microbes in classic ecosystems and in healthy and diseased ecosystems [15]. For example, dysbiosis may be associated with certain BEA infections [121] and cancers [140], and there is accumulating evidence that the gut microbiota modulates the effectiveness of cancer therapies and associated toxic effects [141].
That microbiome disruption can influence disease suggests that re-normalizing it could prevent or ameliorate certain diseases. For example, lifestyle changes (e.g. avoiding high caloric intake and excessive hygiene) may indirectly prevent disease via their impacts on the microbiome, and more active interventions such as microbiome transplants could either be used preventively or therapeutically [140, 141].
Measure 6: Aliens
‘Living drugs’ such as bacteriophages (or ‘phages’) are akin to introducing a predator to control an agricultural pest. Phages self-amplify and adapt to their bacterial hosts, meaning that they can continually counter bacterial resistance [142]. In addition to their use as self-propagating, evolving antimicrobials, bacteriophage can be applied prophylactically to prevent bacterial pathogens from emerging [143], or engineered and introduced to disrupt antibiotic resistance [144]. Although less studied, replicating oncolytic viruses have clinical potential similar to baculoviruses [145], but with the added twist that under some circumstances they can induce anti-tumor immunity [146].
Similar to drug therapies, when submitted to phage predation, sufficiently large and diverse bacterial populations evolve resistance. Although a phage population will typically respond by evolving countermeasures, a more proactive, promising strategy is to evolutionarily ‘train’ phage before they are actually employed [147, 148]. Training involves selecting phage in vitro for traits that impact current bacteria at the infection site and prevent the future emergence of resistant strains. In vitro study indicates that passaging phage achieves the former objective, whereas coevolving them with the bacterium achieves the latter [148–150]. Training oncolytic viruses on tumor cells in vitro remains unexplored.
Combining measures
An important principle from integrated pest management is that attaining a control objective is more likely as multiple, complementary practices are put into place. This same idea—combining two or more variations of the same measure or two or more measures—is therapeutically sensible both ecologically and evolutionarily [151]. Combination therapies are most likely to succeed when each measure (i) reduces BEA numbers or turnover, thereby removing existing resistance mutations and lowering the probability of such mutations emerging de novo to other measures (e.g. therapy A kills single mutants with resistance to therapy B, and vice versa) and (ii) acts through a different, non-antagonistic mechanism in targeting the BEA. Both of these effects imply that different genes are involved in resisting different measures. Crucially, because in sufficiently small populations it is unlikely that multiple resistance factors will occur in the same BEA individual, combinations are less likely than single measures to select for resistance [86, 152, 153] (see [60, 154] for possible exceptions in tumors).
Combination therapies have become a mainstay for treating HIV, malaria and tuberculosis and are particularly relevant to cancers [133, 145, 155]. They are also effective against bacterial pathogens, for example, the use of phage cocktails [156, 157], multiple antibiotics [158, 159], certain aminoglycosides and metabolites against persister bacteria [160], antimicrobial peptide cocktails [161], phage–antibiotic combinations [157], and phage to select for increased sensitivity to antibiotics [162]. Like phage–antibiotic combinations, antibiotic combinations that target different critical bacterial functions may not only increase the chances of therapeutic success but also limit the evolution and spread of antibiotic resistance [163]. Due in part to their great diversity, combinations involving natural host-derived antimicrobial peptides are particularly promising in this regard [164].
Depending on the nature of their interactions, measures can be combined either simultaneously or sequentially. For instance, Roemhild et al. [165] demonstrated how the order of sub-lethal doses of antibiotics could have substantial effects on bacterial population sizes and resistance (see also [96]). Torres-Barceló et al. [166] showed how intermediate delays between phage and antibiotic application not only minimized numbers of Pseudomonas aeruginosa but also minimized resistance to both antibiotics and phage. Sequential applications have also shown promise in combinations of chemotherapy and immunotherapy [167], and polyADP ribose polymerase inhibitors capitalizing on synthetic lethality in certain breast and ovarian cancers [168]. Other more information-based combination strategies such as evolutionary steering [169] and resistance reversal [170] have shown proof of concept.
Combination therapies nevertheless have several notable drawbacks. First, they can fail if not carefully devised. Hegreness et al. [171] showed how synergistic antibiotic combinations could accelerate the evolution of resistance compared to (a priori, less preferred) antagonistic combinations (see also [96]). Second and similarly, the use of multiple tactics could mean that lower intensities of each are employed to limit toxicity; these lower doses are more likely to fall within the ‘mutant selection window’ should such resistant strains be present, and result in resistance evolution [101, 172]. Third, it is logistically more challenging to administer combinations than monotherapies. This extends both to the willingness of medical practitioners to employ unfamiliar novel therapies and for patients to follow protocols.
POTENTIAL LIMITATIONS
The disease ecosystem framework focuses on a well-studied host (humans) and a small number of their well-studied BEAs (most examples from bacteria and cancer). The remarkable diversity of host species and BEAs and their intra-population variation—but also the fact that many BEAs are associated with multiple disease ecosystems within the same host individual, or exploit multiple host species, or have complex life cycles with intermediate hosts—mean that the framework presented here is only an initial step towards a more refined general framework and more taxon-specific versions. This situation is no different from the challenges of understanding classic ecosystems, and much of the progress in identifying process and pattern in the latter should be useful in instructing approaches to understanding disease ecosystems.
Second and similarly, many BEA taxa do not replicate in all hosts (particularly for complex life-cycle parasites, such as helminth worms), meaning that in situ mutation is not a factor. These species are confronted with some of the same basic constraints as are parasites that can replicate and evolve within their host (resource levels, competition, immune responses), and consequently have evolved mechanisms to plastically adapt to the host ecosystem and in particular, regulate host immunity [173]. The present framework could be extended to accommodate these BEAs.
Third, the ecosystem framework notably ignores host-to-host transmission of BEAs and the spread of resistance between hosts. This concern is evidently not relevant to cancers but is an important factor for infectious diseases, where there is a potential conflict between the immediate interests of individual patients and future interests of the greater population (e.g. [7, 108, 163, 174, 175]).
A fourth limitation is the need for sufficiently rich information to employ certain therapeutic measures, particularly those aiming to contain the BEA. Some BEAs can be quantified either directly or through correlations with measurable signs of disease (e.g. biomarkers, scans). Quantifying ecosystem components such as the immune system (e.g. [176]) could show promise as performance indicators of natural or therapeutically influenced BEA control.
CONCLUDING REMARKS
The immense body of knowledge on classic ecosystems can contribute to our understanding of disease ecosystems. We will however need more than these analogies to achieve a predictive theory of disease ecosystem dynamics. The principal reason is their underlying complexity. Healthy organism ecosystems and their responses to disease have evolved to maintain homeostasis/performance and remediation to homeostasis, respectively. Although the former should be fairly predictable, the latter will be dynamic, context dependent and more challenging to forecast. The disease ecosystem framework represents an initial step toward understanding disease, predicting its course and designing eco-evolutionarily sensible therapies.
Disease ecosystems are complex and dynamic and have no a priori spatial limits. The ecosystem framework focuses on proximal interactions at the host cellular and tissue levels. These interactions will interlock with other tissue and organ systems in the host individual, and ultimately influence and be influenced by population and external environmental-scale systems. Maintaining generality even at the cellular and tissue scales necessarily means omitting considerable realistic detail (see e.g. Table 2 in [177]). Moreover, at aggregate levels, ecological and evolutionary dynamics are likely to be complex given the feedback structures of homeostatic regulation [27], interaction networks [14], and stochasticity in the mutation process and the host’s immune response [178, 179]. Despite this complexity, it is encouraging that across two pervasive BEA types in humans (bacteria and cancer) there is broad similarity in the basic interactions of the disease ecosystem (this study), and with regard to therapies, correspondences in drug resistance evolution [115]. This suggests generality in the framework presented here.
The ecosystem framework is a scaffolding. To be fully operative, it needs to be supplemented with quantitative descriptions of each component process. Although the framework on its own can advise specific types of therapeutic decisions (either ones where resistance is not an issue or satisficing is the only option—i.e. State 1 and late State 3), it will be limited without parameterized models to guide finer containment approaches such as adaptive therapies (States 2 and 3; see e.g. [111]). Such a model would link the most important components of the ecosystem to the variables of interest, typically BEA population size, frequency of resistance, virulence factors or aggressiveness, and disease signs. The model would then be used to evaluate how different candidate therapeutic interventions affect the likelihood that an objective will be achieved. Usually but not always, preventing or limiting BEA evasion (e.g. resistance) will be an integral part of which measure(s) is (are) finally employed.
We are only beginning to scratch the surface of how the disease ecosystem functions and its relevance as part of a larger network, including other ecosystems in the individual, the population and as part of wider (classic) ecosystems [180]. Understanding these processes, their interactions, and importance in creating pattern at different scales will be considerable challenges (Box 4), but promise to lead to novel insights about the ecology and evolution of symbioses, and new breakthroughs in conceptualizing and treating disease.
Box 4: Questions for future research
How do disease ecosystems change with host diet, sex and age?
To what extent do ecology and evolution in classic ecosystems produce patterns that resemble those in healthy and diseased host ecosystems?
Does the likelihood of BEA resistance differ between natural (e.g. boosting existing immune responses) and novel (e.g. synthetic drug) interventions?
How important is spatial heterogeneity (spatial distributions of immune cells, microbiota and BEAs) and indirect interactions (local nutrients mediating competition with microbiota or influencing the immune system) to BEAs and disease dynamics?
What characteristics of the disease ecosystem predict pathogenicity, within-host migration, and (for parasites) host to host transmission?
What are the common features and specificities in disease ecosystems found in different tissues and organs, host individuals within a population, closely related species and distantly related taxa?
The ecosystem framework should be applicable to both healthy and disrupted within-organism systems more generally (e.g. due to injury, aging, non-BEA associated diseases). Is there a useful common framework across organism–ecosystem types?
GLOSSARY
Disease. Changes to the structure and function of host cells, tissues or organ systems associated with interactions between the BEA and the host.
Signs. Objective observations of disease, typically of macroscopic features, such as swollen tonsils consistent with strep throat.
Symptoms. Subjective assessments of disease, such as pain or discomfort.
Pathogenicity. The propensity for disease to result in tissue damage, morbidity and mortality. Like disease, pathogenicity is a property of the interaction between the BEA and host.
Resistance. A BEA phenotype that protects from specific antagonisms such as immune responses or therapies.
Cure. Disease remediation to no detectable signs and symptoms.
Chemotherapy. The use of chemicals to treat disease, usually focused at killing the etiological agent, either microparasites or tumor cells.
Eradication. The complete elimination of the BEA.
Containment. Maintaining disease below an acceptable maximum threshold.
Microparasites. Microbial parasites that replicate within their host, including viruses, bacteria, fungi and protozoa.
Virulence factors. BEA traits that increase BEA fitness (growth, survival and reproduction) and intensification of disease (i.e. pathogenicity).
Inflammation. Alteration of the disease ecosystem that promotes the intensification of the immune response and remediation of disease damage. Chronic inflammation can result in tissue damage.
Satisficing. An acceptable improvement in disease signs and symptoms.
Supplementary Material
Acknowledgements
The author acknowledges participants of the ‘Aging and Adaptation in Infectious Diseases’ workshop at the Santa Fe Institute, three anonymous reviewers, and Sebastian Bonhoeffer, Troy Day, Andrea Graham, Andrew Read, Roland Regoes and Jens Rolff for discussions.
Funding
I acknowledge financial support from the McDonnell Foundation (Studying Complex Systems Research Award No. 220020294), ITMO Cancer AVIESAN (Program ‘HetColi’) and the Institut National du Cancer (2014-1-PL-BIO-12-IGR-1) for funding.
Conflict of interest: None declared.
REFERENCES
- 1. Ehrlich P. Address in pathology on chemotherapeutics: methods, and results. Lancet 1913; 182:445–51. [Google Scholar]
- 2. Read AF, Day T, Huijben S.. The evolution of drug resistance and the curious orthodoxy of aggressive chemotherapy. Proc Natl Acad Sci U S A 2011; 108:10871–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gillies RJ, Verduzco D, Gatenby RA.. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat Rev Cancer 2012; 12:487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haegeman B, Hamelin J, Moriarty J. et al. Robust estimation of microbial diversity in theory and in practice. ISME J 2013; 7:1092–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maley CC, Aktipis A, Graham TA. et al. Classifying the evolutionary and ecological features of neoplasms. Nat Rev Cancer 2017; 17:605–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cornforth DM, Dees JL, Ibberson CB. et al. Pseudomonas aeruginosa transcriptome during human infection. Proc Natl Acad Sci U S A 2018; 115:E5125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lipsitch M, Samore MH.. Antimicrobial use and antimicrobial resistance: a population perspective. Emerg Infect Dis 2002; 8:347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Becattini S, Taur Y, Pamer EG.. Antibiotic-induced changes in the intestinal microbiota and disease. Trends Mol Med 2016; 22:458–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Estrela S, Brown SP.. Community interactions and spatial structure shape selection on antibiotic resistant lineages. PLoS Comput Biol 2018; 14:e1006179.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Currie CJ, Berni E, Jenkins-Jones S. et al. Antibiotic treatment failure in four common infections in UK primary care 1991-2012: longitudinal analysis. BMJ 2014; 349:g5493. [DOI] [PubMed] [Google Scholar]
- 11. Courchamp F, Dunne JA, Le Maho Y. et al. Fundamental ecology is fundamental. Trends Ecol Evol 2015; 30:1–8. [DOI] [PubMed] [Google Scholar]
- 12. Conrad D, Haynes M, Salamon P. et al. Cystic fibrosis therapy: a community ecology perspective. Am J Respir Cell Mol Biol 2013; 48:150–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McNally L, Brown SP.. Building the microbiome in health and disease: niche construction and social conflict in bacteria. Philos Trans R Soc Lond B Biol Sci 2015; 370:20140298.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rynkiewicz EC, Pedersen AB, Fenton A.. An ecosystem approach to understanding and managing within-host parasite community dynamics. Trends Parasitol 2015; 31:1–10. [DOI] [PubMed] [Google Scholar]
- 15. Robinson CJ, Bohannan BJM, Young VB.. From structure to function: the ecology of host-associated microbial communities. Microbiol Mol Biol Rev 2010; 74:453–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Merlo LMF, Pepper JW, Reid BJ. et al. Cancer as an evolutionary and ecological process. Nat Rev Cancer 2006; 6:924–35. [DOI] [PubMed] [Google Scholar]
- 17. Pienta KJ, McGregor N, Axelrod R. et al. Ecological therapy for cancer: defining tumors using an ecosystem paradigm suggests new opportunities for novel cancer treatments. Transl Oncol 2008; 1:158–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Basanta D, Anderson ARA.. Exploiting ecological principles to better understand cancer progression and treatment. Interface Focus 2013; 3:20130020.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Amend SR, Roy S, Brown JS. et al. Ecological paradigms to understand the dynamics of metastasis. Cancer Lett 2016; 380:237–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Alfarouk KO, Ibrahim ME, Gatenby RA. et al. Riparian ecosystems in human cancers. Evol Appl 2013; 6:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Free A, Barton NH.. Do evolution and ecology need the Gaia hypothesis? Trends Ecol Evol 2007; 22:611–9. [DOI] [PubMed] [Google Scholar]
- 22. Doran KS, Banerjee A, Disson O. et al. Concepts and mechanisms: crossing host barriers. Cold Spring Harb Perspect Med 2013; 3:a010090.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ribet D, Cossart P.. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect 2015; 17:173–83. [DOI] [PubMed] [Google Scholar]
- 24. Levin BR, Antia R.. Why we don’t get sick: the within-host population dynamics of bacterial infections. Science 2001; 292:1112–5. [DOI] [PubMed] [Google Scholar]
- 25. Schmid-Hempel P. Immune defence, parasite evasion strategies and their relevance for “macroscopic phenomena” such as virulence. Philos Trans R Soc Lond B Biol Sci 2009; 364:85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brown EM, Sadarangani M, Finlay BB.. The role of the immune system in governing host-microbe interactions in the intestine. Nat Immunol 2013; 14:660–7. [DOI] [PubMed] [Google Scholar]
- 27. Rankin LC, Artis D.. Beyond host defense: emerging functions of the immune system in regulating complex tissue physiology. Cell 2018; 173:554–67. [DOI] [PubMed] [Google Scholar]
- 28. McDade TW, Georgiev AV, Kuzawa CW.. Trade-offs between acquired and innate immune defenses in humans. Evol Med Public Health 2016; 2016:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hochberg ME, Noble RJ.. A framework for how environment contributes to cancer risk. Ecol Lett 2017; 20:117–24. [DOI] [PubMed] [Google Scholar]
- 30. Schmid-Hempel P. Parasite immune evasion: a momentous molecular war. Trends Ecol Evol 2008; 23:318–26. [DOI] [PubMed] [Google Scholar]
- 31. Noble R, Kaltz O, Hochberg ME.. Peto’s paradox and human cancers. Philos Trans R Soc Lond B Biol Sci 2015; 370:20150104.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schaffer K, Taylor CT.. The impact of hypoxia on bacterial infection. FEBS J 2015; 282:2260–6. [DOI] [PubMed] [Google Scholar]
- 33. Verduzco D, Lloyd M, Xu L. et al. Intermittent hypoxia selects for genotypes and phenotypes that increase survival, invasion, and therapy resistance. PLoS One 2015; 10:e0120958.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chang C-H, Qiu J, O’Sullivan D. et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 2015; 162:1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Read AF, Taylor LH.. The ecology of genetically diverse infections. Science 2001; 292:1099–102. [DOI] [PubMed] [Google Scholar]
- 36. Mideo N. Parasite adaptations to within-host competition. Trends Parasitol 2009; 25:261–8. [DOI] [PubMed] [Google Scholar]
- 37. Balmer O, Tanner M.. Prevalence and implications of multiple-strain infections. Lancet Infect Dis 2011; 11:868–78. [DOI] [PubMed] [Google Scholar]
- 38. Smith VH, Holt RD.. Resource competition and within-host disease dynamics. Trends Ecol Evol 1996; 11:386–9. [DOI] [PubMed] [Google Scholar]
- 39. Bäumler AJ, Sperandio V.. Interactions between the microbiota and pathogenic bacteria in the gut. Nature 2016; 535:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cressler CE, Nelson WA, Day T. et al. Disentangling the interaction among host resources, the immune system and pathogens. Ecol Lett 2014; 17:284–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cobey S, Lipsitch M.. Pathogen diversity and hidden regimes of apparent competition. Am Nat 2013; 181:12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Buck MD, Sowell RT, Kaech SM. et al. Metabolic instruction of immunity. Cell 2017; 169:570–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fenton A, Perkins SE.. Applying predator-prey theory to modelling immune-mediated, within-host interspecific parasite interactions. Parasitology 2010; 137:1027–38. [DOI] [PubMed] [Google Scholar]
- 44. Torres BY, Oliveira JHM, Thomas Tate A. et al. Tracking resilience to infections by mapping disease space. PLoS Biol 2016; 14:e1002436.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rolfe MD, Rice CJ, Lucchini S. et al. Lag phase is a distinct growth phase that prepares bacteria for exponential growth and involves transient metal accumulation. J Bacteriol 2012; 194:686–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gillies RJ, Gatenby RA.. Metabolism and its sequelae in cancer evolution and therapy. Cancer J 2015; 21:88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Brown SP, Cornforth DM, Mideo N.. Evolution of virulence in opportunistic pathogens: generalism, plasticity, and control. Trends Microbiol 2012; 20:336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Margolis E, Levin BR.. Within-host evolution for the invasiveness of commensal bacteria: an experimental study of bacteremias resulting from Haemophilus influenzae nasal carriage. J Infect Dis 2007; 196:1068–75. [DOI] [PubMed] [Google Scholar]
- 49. Leggett HC, Cornwallis CK, West SA.. Mechanisms of pathogenesis, infective dose and virulence in human parasites. PLoS Pathog 2012; 8:e1002512.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Aktipis CA, Boddy AM, Jansen G. et al. Cancer across the tree of life: cooperation and cheating in multicellularity. Philos Trans R Soc Lond B Biol Sci 2015; 370:20140219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yachida S, Jones S, Bozic I. et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010; 467:1114–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wodarz D, Komarova N.. Can loss of apoptosis protect against cancer? Trends Genet 2007; 23:232–7. [DOI] [PubMed] [Google Scholar]
- 53. Iwasa Y, Nowak MA, Michor F.. Evolution of resistance during clonal expansion. Genetics 2006; 172:2557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. De Boer RJ, Perelson AS.. Target cell limited and immune control models of HIV infection: a comparison. J Theor Biol 1998; 190:201–14. [DOI] [PubMed] [Google Scholar]
- 55. Sottoriva A, Kang H, Ma Z. et al. A Big Bang model of human colorectal tumor growth. Nat Genet 2015; 47:209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Williams MJ, Werner B, Barnes CP. et al. Identification of neutral tumor evolution across cancer types. Nat Genet 2016; 48:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Caswell DR, Swanton C.. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med 2017; 15:133.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Olive AJ, Sassetti CM.. Metabolic crosstalk between host and pathogen: sensing, adapting and competing. Nat Rev Microbiol 2016; 14:221–34. [DOI] [PubMed] [Google Scholar]
- 59. Rohmer L, Hocquet D, Miller SI.. Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends Microbiol 2011; 19:341–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Komarova NL, Wodarz D.. Drug resistance in cancer: principles of emergence and prevention. Proc Natl Acad Sci U S A 2005; 102:9714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bozic I, Nowak MA.. Timing and heterogeneity of mutations associated with drug resistance in metastatic cancers. Proc Natl Acad Sci U S A 2014; 111:15964–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hibbing ME, Fuqua C, Parsek MR. et al. Bacterial competition: surviving and thriving in the microbial jungle. Nat Rev Microbiol 2010; 8:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Renner K, Singer K, Koehl GE. et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol 2017; 8:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Casadevall A, Pirofski L-A.. The damage-response framework of microbial pathogenesis. Nat Rev Microbiol 2003; 1:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Khong HT, Restifo NP.. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol 2002; 3:999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Veitch CR, Clout MN, Towns DR.. Island Invasives: Eradication and Management. Gland, Switzerland: IUCN, 2011. [Google Scholar]
- 67. Longley DB, Johnston PG.. Molecular mechanisms of drug resistance. J Pathol 2005; 205:275–92. [DOI] [PubMed] [Google Scholar]
- 68. Gonzalez A, Ronce O, Ferriere R. et al. Evolutionary rescue: an emerging focus at the intersection between ecology and evolution. Philos Trans R Soc Lond B Biol Sci 2013; 368:20120404.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bell G. Evolutionary rescue. Annu Rev Ecol Evol Syst 2017; 48:605–27. [Google Scholar]
- 70. Mwangi MM, Wu SW, Zhou Y. et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci U S A 2007; 104:9451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gullberg E, Cao S, Berg OG. et al. Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog 2011; 7:e1002158.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Andersson DI, Hughes D.. Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol 2014; 12:465–78. [DOI] [PubMed] [Google Scholar]
- 73. de Roode JC, Culleton R, Bell AS. et al. Competitive release of drug resistance following drug treatment of mixed Plasmodium chabaudi infections. Malar J 2004; 3:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Day T, Huijben S, Read AF.. Is selection relevant in the evolutionary emergence of drug resistance? Trends Microbiol 2015; 23:126–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wargo AR, Huijben S, de Roode JC. et al. Competitive release and facilitation of drug-resistant parasites after therapeutic chemotherapy in a rodent malaria model. Proc Natl Acad Sci U S A 2007; 104:19914–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Enriquez-Navas PM, Wojtkowiak JW, Gatenby RA.. Application of evolutionary principles to cancer therapy. Cancer Res 2015; 75:4675–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Zhang J, Cunningham JJ, Brown JS. et al. Integrating evolutionary dynamics into treatment of metastatic castrate-resistant prostate cancer. Nat Commun 2017; 8:1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Yeaton RI, Cody ML.. Competitive release in island Song Sparrow populations. Theor Popul Biol 1974; 5:42–58. [DOI] [PubMed] [Google Scholar]
- 79. Lloyd-Smith JO. Vacated niches, competitive release and the community ecology of pathogen eradication. Philos Trans R Soc Lond B Biol Sci 2013; 368:20120150.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sharma SV, Lee DY, Li B. et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010; 141:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med 2011; 17:313–9. [DOI] [PubMed] [Google Scholar]
- 82. Balaban NQ, Merrin J, Chait R. et al. Bacterial persistence as a phenotypic switch. Science 2004; 305:1622–5. [DOI] [PubMed] [Google Scholar]
- 83. Stewart PS. Mechanisms of antibiotic resistance in bacterial biofilms. Int J Med Microbiol 2002; 292:107–13. [DOI] [PubMed] [Google Scholar]
- 84. Bjarnsholt T, Alhede M, Alhede M. et al. The in vivo biofilm. Trends Microbiol 2013; 21:466–74. [DOI] [PubMed] [Google Scholar]
- 85. Vinay DS, Ryan EP, Pawelec G. et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol 2015; 35(Suppl): S185–98. [DOI] [PubMed] [Google Scholar]
- 86. Lipsitch M, Levin BR.. The population dynamics of antimicrobial chemotherapy. Antimicrob Agents Chemother 1997; 41:363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Akhmetzhanov AR, Hochberg ME.. Dynamics of preventive vs post-diagnostic cancer control using low-impact measures. Elife 2015; 4:e06266.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Trédan O, Galmarini CM, Patel K. et al. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst 2007; 99:1441–54. [DOI] [PubMed] [Google Scholar]
- 89. Negri MC, Lipsitch M, Blázquez J. et al. Concentration-dependent selection of small phenotypic differences in TEM beta-lactamase-mediated antibiotic resistance. Antimicrob Agents Chemother 2000; 44:2485–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fu F, Nowak MA, Bonhoeffer S.. Spatial heterogeneity in drug concentrations can facilitate the emergence of resistance to cancer therapy. PLoS Comput Biol 2015; 11:e1004142.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Oliver A, Levin BR, Juan C. et al. Hypermutation and the preexistence of antibiotic-resistant Pseudomonas aeruginosa mutants: implications for susceptibility testing and treatment of chronic infections. Antimicrob Agents Chemother 2004; 48:4226–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Cuevas JM, Geller R, Garijo R. et al. Extremely high mutation rate of HIV-1 in vivo. PLoS Biol 2015; 13:e1002251.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Martincorena I, Campbell PJ.. Somatic mutation in cancer and normal cells. Science 2015; 349:1483–9. [DOI] [PubMed] [Google Scholar]
- 94. Soderholm AT, Barnett TC, Sweet MJ. et al. Group A streptococcal pharyngitis: immune responses involved in bacterial clearance and GAS-associated immunopathologies. J Leukoc Biol 2018; 103:193–213. [DOI] [PubMed] [Google Scholar]
- 95. Fleming A. Penicillin. Nobel Lectures, Physiology or Medicine 1942–1962. Amsterdam, NL: Elsevier Publishing Company, 1964. [Google Scholar]
- 96. Pena-Miller R, Laehnemann D, Jansen G. et al. When the most potent combination of antibiotics selects for the greatest bacterial load: the Smile-Frown transition. PLoS Biol 2013; 11:e1001540.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Foo J, Michor F.. Evolution of acquired resistance to anti-cancer therapy. J Theor Biol 2014; 355:10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hansen E, Woods RJ, Read AF.. How to use a chemotherapeutic agent when resistance to it threatens the patient. PLoS Biol 2017; 15:e2001110.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Slater HC, Okell LC, Ghani AC.. Mathematical modelling to guide drug development for malaria elimination. Trends Parasitol 2017; 33:175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Geli P, Laxminarayan R, Dunne M. et al. “One-size-fits-all”? Optimizing treatment duration for bacterial infections. PLoS One 2012; 7:e29838.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Day T, Read AF.. Does high-dose antimicrobial chemotherapy prevent the evolution of resistance? PLoS Comput Biol 2016; 12:e1004689.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hartfield M, Alizon S.. Within-host stochastic emergence dynamics of immune-escape mutants. PLoS Comput Biol 2015; 11:e1004149.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Drlica K, Zhao X.. Mutant selection window hypothesis updated. Clin Infect Dis 2007; 44:681–8. [DOI] [PubMed] [Google Scholar]
- 104. Zur Wiesch PA, Kouyos R, Engelstädter J. et al. Population biological principles of drug-resistance evolution in infectious diseases. Lancet Infect Dis 2011; 11:236–47. [DOI] [PubMed] [Google Scholar]
- 105. Weiss E, Zahar J-R, Lesprit P. et al. Elaboration of a consensual definition of de-escalation allowing a ranking of β-lactams. Clin Microbiol Infect 2015; 21:649.e1–10. [DOI] [PubMed] [Google Scholar]
- 106. Tam VH, Louie A, Deziel MR. et al. The relationship between quinolone exposures and resistance amplification is characterized by an inverted U: a new paradigm for optimizing pharmacodynamics to counterselect resistance. Antimicrob Agents Chemother 2007; 51:744–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kouyos RD, Metcalf CJE, Birger R. et al. The path of least resistance: aggressive or moderate treatment. Proc R Soc B Biol Sci 2014; 281:20140566.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Huijben S, Bell AS, Sim DG. et al. Aggressive chemotherapy and the selection of drug resistant pathogens. PLoS Pathog 2013; 9:e1003578.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Michor F, Beal K.. Improving cancer treatment via mathematical modeling: surmounting the challenges is worth the effort. Cell 2015; 163:1059–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Gatenby RA, Silva AS, Gillies RJ. et al. Adaptive therapy. Cancer Res 2009; 69:4894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Bacevic K, Noble R, Soffar A. et al. Spatial competition constrains resistance to targeted cancer therapy. Nat Commun 2017; 8:1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Gjini E, Brito PH.. Integrating antimicrobial therapy with host immunity to fight drug-resistant infections: classical vs. adaptive treatment. PLoS Comput Biol 2016; 12:e1004857.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. MacLean RC, Hall AR, Perron GG. et al. The population genetics of antibiotic resistance: integrating molecular mechanisms and treatment contexts. Nat Rev Genet 2010; 11:405–14. [DOI] [PubMed] [Google Scholar]
- 114. Schulz zur Wiesch P, Engelstadter J, Bonhoeffer S.. Compensation of fitness costs and reversibility of antibiotic resistance mutations. Antimicrob Agents Chemother 2010; 54:2085–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Hughes D, Andersson DI.. Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet 2015; 16:459–71. [DOI] [PubMed] [Google Scholar]
- 116. Woods RJ, Read AF.. Clinical management of resistance evolution in a bacterial infection. Evol Med Public Health 2015; 2015:281–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Vale PF, Fenton A, Brown SP.. Limiting damage during infection: lessons from infection tolerance for novel therapeutics. PLoS Biol 2014; 12:e1001769.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Foo J, Chmielecki J, Pao W. et al. Effects of pharmacokinetic processes and varied dosing schedules on the dynamics of acquired resistance to erlotinib in EGFR-mutant lung cancer. J Thorac Oncol 2012; 7:1583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Ankomah P, Levin BR.. Exploring the collaboration between antibiotics and the immune response in the treatment of acute, self-limiting infections. Proc Natl Acad Sci U S A 2014; 111:8331–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Allen RC, Popat R, Diggle SP. et al. Targeting virulence: can we make evolution-proof drugs? Nat Rev Microbiol 2014; 12:300–8. [DOI] [PubMed] [Google Scholar]
- 121. Vale PF, McNally L, Doeschl-Wilson A. et al. Beyond killing: can we find new ways to manage infection? Evol Med Public Health 2016; 2016:148–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Rumbaugh KP, Diggle SP, Watters CM. et al. Quorum sensing and the social evolution of bacterial virulence. Curr Biol 2009; 19:341–5. [DOI] [PubMed] [Google Scholar]
- 123. Mellbye B, Schuster M.. The sociomicrobiology of antivirulence drug resistance: a proof of concept. MBio 2011; 2:00131-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ternent L, Dyson RJ, Krachler A-M. et al. Bacterial fitness shapes the population dynamics of antibiotic-resistant and -susceptible bacteria in a model of combined antibiotic and anti-virulence treatment. J Theor Biol 2015; 372:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Johnson BK, Abramovitch RB.. Small molecules that sabotage bacterial virulence. Trends Pharmacol Sci 2017; 38:339–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Lorz A, Perthame B, Lorenzi T. et al. Populational adaptive evolution, chemotherapeutic resistance and multiple anti-cancer therapies. Esaim Math Model Numer Anal 2013; 47:2013–5. [Google Scholar]
- 127. Robey IF, Baggett BK, Kirkpatrick ND. et al. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res 2009; 69:2260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Ross-Gillespie A, Weigert M, Brown SP. et al. Gallium-mediated siderophore quenching as an evolutionarily robust antibacterial treatment. Evol Med Public Health 2014; 2014:18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Yu G, Baeder DY, Regoes RR. et al. Predicting drug resistance evolution: insights from antimicrobial peptides and antibiotics. Proc Biol Sci 2018; 285:20172687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Childs RW, Carlsten M.. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat Rev Drug Discov 2015; 14:487–98. [DOI] [PubMed] [Google Scholar]
- 131. Zumla A, Rao M, Wallis RS. et al. Host-directed therapies for infectious diseases: current status, recent progress, and future prospects. Lancet Infect Dis 2016; 16:e47–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. June CH, Warshauer JT, Bluestone JA.. Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med 2017; 23:540–7. [DOI] [PubMed] [Google Scholar]
- 133. Smyth MJ, Ngiow SF, Ribas A. et al. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol 2016; 13:143–58. [DOI] [PubMed] [Google Scholar]
- 134. Vander Heiden MG, Cantley LC, Thompson CB.. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324:1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Elser JJ, Kyle MM, Smith MS. et al. Biological stoichiometry in human cancer. PLoS One 2007; 2:e1028.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Yonucu S, Yιlmaz D, Phipps C. et al. Quantifying the effects of antiangiogenic and chemotherapy drug combinations on drug delivery and treatment efficacy. PLoS Comput Biol 2017; 13:e1005724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Wale N, Sim DG, Jones MJ. et al. Resource limitation prevents the emergence of drug resistance by intensifying within-host competition. Proc Natl Acad Sci U S A 2017; 114:13774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Maley CC, Reid BJ, Forrest S.. Cancer prevention strategies that address the evolutionary dynamics of neoplastic cells: simulating benign cell boosters and selection for chemosensitivity. Cancer Epidemiol Biomarkers Prev 2004; 13:1375–84. [PubMed] [Google Scholar]
- 139. Libberton B, Horsburgh MJ, Brockhurst MA.. The effects of spatial structure, frequency dependence and resistance evolution on the dynamics of toxin-mediated microbial invasions. Evol Appl 2015; 8:738–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Bhatt AP, Redinbo MR, Bultman SJ.. The role of the microbiome in cancer development and therapy. CA Cancer J Clin 2017; 67:326–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Roy S, Trinchieri G.. Microbiota: a key orchestrator of cancer therapy. Nat Rev Cancer 2017; 17:271–85. [DOI] [PubMed] [Google Scholar]
- 142. Pirnay J-P, Verbeken G, Rose T. et al. Introducing yesterday’s phage therapy in today’s medicine. Future Virol 2012; 7:379–90. [Google Scholar]
- 143. Levin BR, Bull JJ.. Phage therapy revisited: the population biology of a bacterial infection and its treatment with bacteriophage and antibiotics. Am Nat 1996; 147:881–98. [Google Scholar]
- 144. Yosef I, Manor M, Kiro R. et al. Temperate and lytic bacteriophages programmed to sensitize and kill antibiotic-resistant bacteria. Proc Natl Acad Sci U S A 2015; 112:7267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Twumasi-Boateng K, Pettigrew JL, Kwok YYE. et al. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat Rev Cancer 2018; 18:419–32. [DOI] [PubMed] [Google Scholar]
- 146. Kaufman HL, Kohlhapp FJ, Zloza A.. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov 2015; 14:642–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Pirnay JP, De Vos D, Verbeken G. et al. The phage therapy paradigm: prêt-à-porter or sur-mesure? Pharm Res 2011; 28:934–7. [DOI] [PubMed] [Google Scholar]
- 148. Betts A, Vasse M, Kaltz O. et al. Back to the future: evolving bacteriophages to increase their effectiveness against the pathogen Pseudomonas aeruginosa PAO1. Evol Appl 2013; 6:1054–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Poullain V, Gandon S, Brockhurst MA. et al. The evolution of specificity in evolving and coevolving antagonistic interactions between a bacteria and its phage. Evolution 2008; 62:1–11. [DOI] [PubMed] [Google Scholar]
- 150. Betts A, Kaltz O, Hochberg ME.. Contrasted coevolutionary dynamics between a bacterial pathogen and its bacteriophages. Proc Natl Acad Sci U S A 2014; 111:11109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Fitzgerald JB, Schoeberl B, Nielsen UB. et al. Systems biology and combination therapy in the quest for clinical efficacy. Nat Chem Biol 2006; 2:458–66. [DOI] [PubMed] [Google Scholar]
- 152. Torres-Barceló C, Franzon B, Vasse M. et al. Long-term effects of single and combined introductions of antibiotics and bacteriophages on populations of Pseudomonas aeruginosa. Evol Appl 2016; 9:583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Goldie JH, Coldman AJ.. The genetic origin of drug resistance in neoplasms: implications for systemic therapy. Cancer Res 1984; 44:3643–53. [PubMed] [Google Scholar]
- 154. Bozic I, Reiter JG, Allen B. et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife 2013; 2:e00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Vanneman M, Dranoff G.. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer 2012; 12:237–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Chan BK, Abedon ST, Loc-Carrillo C.. Phage cocktails and the future of phage therapy. Future Microbiol 2013; 8:769–83. [DOI] [PubMed] [Google Scholar]
- 157. Torres-Barceló C, Hochberg ME.. Evolutionary rationale for phages as complements of antibiotics. Trends Microbiol 2016; 24:1–8. [DOI] [PubMed] [Google Scholar]
- 158. Tamma PD, Cosgrove SE, Maragakis LL.. Combination therapy for treatment of infections with gram-negative bacteria. Clin Microbiol Rev 2012; 25:450–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Barbosa C, Beardmore R, Schulenburg H. et al. Antibiotic combination efficacy (ACE) networks for a Pseudomonas aeruginosa model. PLoS Biol 2018; 16:e2004356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Allison KR, Brynildsen MP, Collins JJ.. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 2011; 473:216–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Zanchi C, Johnston PR, Rolff J.. Evolution of defence cocktails: antimicrobial peptide combinations reduce mortality and persistent infection. Mol Ecol 2017; 26:5334–43. [DOI] [PubMed] [Google Scholar]
- 162. Chan BK, Sistrom M, Wertz JE. et al. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci Rep 2016; 6:26717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Tepekule B, Uecker H, Derungs I. et al. Modeling antibiotic treatment in hospitals: a systematic approach shows benefits of combination therapy over cycling, mixing, and mono-drug therapies. PLoS Comput Biol 2017; 13:e1005745.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yu G, Baeder DY, Regoes RR. et al. Combination effects of antimicrobial peptides. Antimicrob Agents Chemother 2016; 60:1717–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Roemhild R, Barbosa C, Beardmore RE. et al. Temporal variation in antibiotic environments slows down resistance evolution in pathogenic Pseudomonas aeruginosa. Evol Appl 2015; 8:945–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Torres-Barceló C, Arias-Sánchez FI, Vasse M. et al. A window of opportunity to control the bacterial pathogen Pseudomonas aeruginosa combining antibiotics and phages. PLoS One 2014; 9:e106628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Antonia SJ, Mirza N, Fricke I. et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin Cancer Res 2006; 12:878–87. [DOI] [PubMed] [Google Scholar]
- 168. Rottenberg S, Jaspers JE, Kersbergen A. et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A 2008; 105:17079–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Nichol D, Jeavons P, Fletcher AG. et al. Steering evolution with sequential therapy to prevent the emergence of bacterial antibiotic resistance. PLoS Comput Biol 2015; 11:e1004493.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Baym M, Stone LK, Kishony R.. Multidrug evolutionary strategies to reverse antibiotic resistance. Science 2016; 351:aad3292.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Hegreness M, Shoresh N, Damian D. et al. Accelerated evolution of resistance in multidrug environments. Proc Natl Acad Sci U S A 2008; 105:13977–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Andersson DI, Hughes D.. Evolution of antibiotic resistance at non-lethal drug concentrations. Drug Resist Updat 2012; 15:162–72. [DOI] [PubMed] [Google Scholar]
- 173. McSorley HJ, Maizels RM.. Helminth infections and host immune regulation. Clin Microbiol Rev 2012; 25:585–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Baquero F, Campos J.. The tragedy of the commons in antimicrobial chemotherapy. Rev Esp Quimioter 2003; 16:11–3. [PubMed] [Google Scholar]
- 175. Uecker H, Bonhoeffer S.. Modeling antimicrobial cycling and mixing: differences arising from an individual-based versus a population-based perspective. Math Biosci 2017; 294:85–91. [DOI] [PubMed] [Google Scholar]
- 176. Wargo JA, Reddy SM, Reuben A. et al. Monitoring immune responses in the tumor microenvironment. Curr Opin Immunol 2016; 41:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Gonzalez-Angulo AM, Morales-Vasquez F, Hortobagyi GN.. Overview of resistance to systemic therapy in patients with breast cancer. Adv Exp Med Biol 2007; 608:1–22. [DOI] [PubMed] [Google Scholar]
- 178. Lipniacki T, Paszek P, Brasier AR. et al. Stochastic regulation in early immune response. Biophys J 2006; 90:725–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Bozic I, Antal T, Ohtsuki H. et al. Accumulation of driver and passenger mutations during tumor progression. Proc Natl Acad Sci U S A 2010; 107:18545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Johnson PTJ, de Roode JC, Fenton A.. Why infectious disease research needs community ecology. Science 2015; 349:1259504.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Gatenby RA. A change of strategy in the war on cancer. Nature 2009; 459:508–9. [DOI] [PubMed] [Google Scholar]
- 182. Greene SE, Reid A.. Moving targets: fighting the evolution of resistance in infections, pests, and cancer. Microbe Wash DC 2013; 8:279–85. [Google Scholar]
- 183. Sparks TC, Nauen R.. IRAC: mode of action classification and insecticide resistance management. Pestic Biochem Physiol 2015; 121:122–8. [DOI] [PubMed] [Google Scholar]
- 184. Vacher C, Bourguet D, Rousset F. et al. Modelling the spatial configuration of refuges for a sustainable control of pests: a case study of Bt cotton. J Evol Biol 2003; 16:378–87. [DOI] [PubMed] [Google Scholar]
- 185. Rex Consortium. Heterogeneity of selection and the evolution of resistance. Trends Ecol Evol 2013; 28:110–8. [DOI] [PubMed] [Google Scholar]
- 186. Costello EK, Stagaman K, Dethlefsen L. et al. The application of ecological theory toward an understanding of the human microbiome. Science 2012; 336:1255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Lloyd MC, Cunningham JJ, Bui MM. et al. Darwinian dynamics of intratumoral heterogeneity: not solely random mutations but also variable environmental selection forces. Cancer Res 2016; 76:3136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Vilà M, Espinar JL, Hejda M. et al. Ecological impacts of invasive alien plants: a meta-analysis of their effects on species, communities and ecosystems. Ecol Lett 2011; 14:702–8. [DOI] [PubMed] [Google Scholar]
- 189. Jacqueline C, Abbate JL, Sorci G. et al. The macroecology of cancer incidences in humans is associated with large-scale assemblages of endemic infections. Infect Genet Evol 2018; 61:189–96. [DOI] [PubMed] [Google Scholar]
- 190. Dillon SM, Frank DN, Wilson CC.. The gut microbiome and HIV-1 pathogenesis: a two-way street. AIDS 2016; 30:2737–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Thomas RM, Jobin C.. The microbiome and cancer: is the “oncobiome” mirage real? Trends Cancer Res 2015; 1:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Amend SR, Pienta KJ.. Ecology meets cancer biology: the cancer swamp promotes the lethal cancer phenotype. Oncotarget 2015; 6:9669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Waclaw B, Bozic I, Pittman ME. et al. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature 2015; 525:261–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Anderson ARA, Weaver AM, Cummings PT. et al. Tumor morphology and phenotypic evolution driven by selective pressure from the microenvironment. Cell 2006; 127:905–15. [DOI] [PubMed] [Google Scholar]
- 195. Axelrod R, Axelrod DE, Pienta KJ.. Evolution of cooperation among tumor cells. Proc Natl Acad Sci U S A 2006; 103:13474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lyssiotis CA, Kimmelman AC.. Metabolic interactions in the tumor microenvironment. Trends Cell Biol 2017; 27:863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Liberti MV, Locasale JW.. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 2016; 41:211–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Smallbone K, Gatenby RA, Gillies RJ. et al. Metabolic changes during carcinogenesis: potential impact on invasiveness. J Theor Biol 2007; 244:703–13. [DOI] [PubMed] [Google Scholar]
- 199. Folkman J. Angiogenesis. Annu Rev Med 2006; 57:1–18. [DOI] [PubMed] [Google Scholar]
- 200. Carmona-Fontaine C, Bucci V, Akkari L. et al. Emergence of spatial structure in the tumor microenvironment due to the Warburg effect. Proc Natl Acad Sci U S A 2013; 110:19402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Castillo SPP, Rebolledo R, Arim M. et al. The Network Structure of Cancer Ecosystems 2017. BioRxiv DOI: 10.1101/240796. [Google Scholar]
- 202. Aktipis CA, Boddy AM, Gatenby RA. et al. Life history trade-offs in cancer evolution. Nat Rev Cancer 2013; 13:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. David P, Thébault E, Anneville O. et al. Impacts of invasive species on food webs In: Bohan DA, Dumbrell AJ, Massol F (eds). Networks of Invasion: A Synthesis of Concepts, London: Academic Press, 2017, 1–60 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.