

Abstract
Metabolite identification is a crucial step in mass spectrometry (MS)-based metabolomics. However, it is still challenging to assess the confidence of assigned metabolites. In this study, we report a novel method for estimating false discovery rate (FDR) of metabolite assignment with a target-decoy strategy, in which the decoys are generated through violating the octet rule of chemistry by adding small odd numbers of hydrogen atoms. The target-decoy strategy was integrated into JUMPm, an automated metabolite identification pipeline for large-scale MS analysis, and was also evaluated with two other metabolomics tools, mzMatch and MZmine 2. The reliability of FDR calculation was examined by false datasets, which were simulated by altering MS1 or MS2 spectra. Finally, we used the JUMPm pipeline coupled with the target-decoy strategy to process unlabeled and stable-isotope labeled metabolomic datasets. The results demonstrate that the target-decoy strategy is a simple and effective method for evaluating the confidence of high-throughput metabolite identification.
Keywords: False positive discovery, target-decoy strategy, metabolite identification, database search, mass spectrometry, metabolome, metabolomics
Graphical Abstract
INTRODUCTION
Mass spectrometry (MS)-based metabolomics has been widely used to gain insights into the mechanisms of human diseases1, and drug and biomarker discovery2. In a high-throughput metabolomic experiment, thousands of MS features can be detected from biological samples. To identify these features, numerous computational tools have been developed over the past decade, most of which are implemented in R packages and windows graphical user interface (GUI), such as XCMS3,4, MS-DIAL5, mzMatch6, and MZmine7 for liquid chromatography–mass spectrometry (LC-MS), as well as AMDIS8, MET-IDEA9, and SpectConnect10 for gas chromatography–mass spectrometry (GC-MS). Four levels of identification confidence are proposed by the Metabolomics Standards Initiative (MSI)11 in 2007. At level 1, metabolites are assigned by the comparison with authentic chemical standards analyzed under identical conditions within the same laboratory. Common metabolite identification tools with public library information can, at best, provide putative metabolite annotation (level 2), including CFM-ID12, FingerID13, MetFrag14, MAGMa15, and MyCompoundID16. Level 3 is defined as the determination of tentative candidates of compound classes, and the related tools contain MI-PACK17, MetAssign18, ProbMetab19, and xMSannotator20. Level 4 may have metabolite assignments of molecular formulas without sufficient evidence of structures, for which several programs have been developed, such as CAMERA21, SIRIUS22, and MZedDB23. In these tools, especially for level 2, the identification of metabolites is inferred from assigning MS2 spectra with library spectra with a matching score. However, the score itself may not provide sufficient power to discriminate true from false matches. Validation of metabolite assignments mainly relies on manual inspection of the corresponding product ions, followed by comparative analyses with individual compound standards. This “manual” validation is apparently subjective and error-prone, and the introduction of compound standards is expensive and labor intensive. Therefore, it is important to develop a robust method to estimate false discovery rate (FDR) during the metabolite identification.
Target-decoy strategy has been successfully applied in MS-based large-scale proteomics studies24,25. In proteomics, there are several methods for generating decoy sequences, including protein sequence reversal25,26, shuffling27, and completely randomization28. All of these methods rely on a basic assumption that decoys do not exist, but are similar to targets in the database with respect to amino acid composition, peptide length, and mass range. The advantage of the target-decoy strategy is that it can be applied to any database search tools29. However, this concept cannot be readily applied to metabolomics, because metabolites are small molecules with diverse structural isomers. In theory, decoys can be created by simulation and removal of all known target components, but it is challenging because the target database is incomplete30. For example, a simulation model was proposed to make decoys for formula search31. In a recent paper32, decoys were introduced by randomly choosing implausible adducts for high-resolution imaging mass spectrometry. The implausible adducts are large, producing much heavier decoys than the targets. Thus the decoys could not fully mimic the actual targets in the database, resulting in biased FDR estimation. In addition, four FDR methods were proposed for spectral library-based searches33: (1) an empirical Bayes approach; (2) randomizing MS1 spectra; (3) randomizing MS2 spectra; (4) fragmentation tree-based method. But these methods cannot be directly applied to spectral library independent database search.
Here we introduce a novel target-decoy strategy to estimate the FDR for metabolite identification. The strategy is based on the violation of the octet rule, yielding invalid formulas and structures. The validity of the strategy is assessed by two simulated MS1 and MS2 datasets. The strategy is currently implemented in the JUMPm, a metabolite identification pipeline that we recently developed (http://www.stjuderesearch.org/site/lab/peng/jumpm), and is also evaluated with two other metabolomics tools, mzMatch and MZmine 2. By applying the strategy to both labeled and unlabeled LC-MS/MS datasets, we show that this strategy is a general method for estimating FDR during metabolite identification.
MATERIALS AND METHODS
LC-MS/MS Data Acquisition
Both labeled and unlabeled yeast LC-MS/MS datasets were generated with a previously described protocol34. Briefly, for a labeled yeast dataset, cells were grown in four different minimal media conditions. For 13C labeling, the media remained the same except 13C-6 glucose (Cambridge Isotope Laboratories) to replace standard glucose. Similarly, for 15N labeling, 15N-2 ammonium sulfate was substituted into the media. Each culture was maintained for ~30 generations in the labeled media before analysis. Both labeled and unlabeled yeast cells were collected and extracted. The lysate was transferred to a fresh vial to exclude the glass beads, and the supernatant was then dried under centrifugal vacuum and dissolved for LC-MS/MS analysis by an Orbitrap Elite (Thermo Scientific) coupled to an Easy nLC™ system.
Structure Databases and Generation of Decoy Formulas
Three open metabolome databases, including PubChem35, the Human Metabolome Database (HMDB)36, and the Yeast Metabolome Database (YMDB)37, were used for formula and structure databases. All three databases (PubChem, HMDB, and YMDB) in XML format were downloaded, and processed by an in-house script to extract metadata associated with each metabolite, including ID, formula, InChI key, InChI string, SMILES, IUPAC, and monoisotopic mass. Radicals and other unstable structures were discarded. The remaining list of metabolites was used to create the target formula and structure databases. The decoy formulas were generated by adding one or other small odd hydrogen atom(s) to each target formula.
Generation of Theoretical Product Ions of a Decoy Structure
Theoretical product ion pattern of a decoy structure was generated in two steps: (1) the decoy-corresponding target structure was used to generate an MS2 pattern by MetFrag38, and (2) the MS pattern was edited by adding the mass of hydrogen (+1.007825) to a randomly selected carbon atom in the structure and passing the additional mass to each product ion containing the selected carbon. The method is termed as “H2C”. In addition, “Randomized Peaks” method is also evaluated33, which randomly select peaks from a pre-built theoretical product ion library that is generated from all compounds in the HMDB.
For labeled data, if the MS2 spectrum derives from 15N or 13C instead of a 12C precursor, then the predicted MS2 ions were adjusted to include the heavy isotopes. JUMPm uses the same scoring algorithm (i.e. hypergeometric test) for both target and decoy structure matchings. A p-value from the hypergeometric test is generated for each metabolite-spectrum match. A Mscore is generated based on the p-value: .
Spectral Library and Processing
We downloaded a processed spectral library of the Global Natural Product Social Molecular Networking (GNPS)39 (https://bio.informatik.uni-jena.de/passatutto) for FDR estimation. This spectral library was filtered by the following steps as in the previous study33: (1) containing a SMILES or InChI key; (2) removing low-resolution reference data; (3) considering only positive ion mode; (4) accepting compounds below 1,000 Da; (5) requiring at least 5 ion peaks with relative intensity above 2% of the base peak. A total of 4,096 MS2 spectra (2,196 formulas and 2,889 InChI keys) were accepted. We further removed spectra with the InChI key not present in the HMDB database, resulting in a spectral library containing 1,742 MS2 spectra, 799 formulas, and 870 InChI keys for HMDB database. We also downloaded a false MS2 spectral library (https://bio.informatik.uni-jena.de/passatutto), which was generated by randomizing MS2 spectra but keeping the same MS1 mass. From the library, we used 1,742 false MS2 spectra that correspond to true MS2 spectra in the HMDB.
Data and Parameters for mzMatch and MZmine 2
In addition to the JUMPm, two other metabolite identification tools, mzMatch (version 2.0.6) and MZmine 2 (version 2.31), were also used for validating the target-decoy strategy. We used the same set of simulated LC-MS/MS data, and the same HMDB database (7,967 metabolites provided by the mzMatch tool). The mass tolerance used for database search was 100 ppm. The mzMatch was tested in the RStudio environment (v1.0.143, 2016 RStudio, Inc.).
RESULTS AND DISCUSSION
Theoretical Background of the FDR and Target-decoy Strategy
In a large-scale “omics” study (e.g. metabolomics and proteomics), a number of statistic tests are performed for evaluating significance with a probability value (i.e. p-value or a derived score). The p-value represents the probability for a given test when the null hypothesis is true. For example, the null hypothesis in metabolite identification is that a spectrum matches to metabolites in the database on a random basis.
The target-decoy strategy is commonly used to estimate the level of random matches under null hypothesis. A target-decoy composite database has a basic theoretical assumption that both decoys and targets have the same possibility to be randomly identified in the database. To ensure this assumption, the strategy usually follows two rules in practice: (1) the number of decoys is the same as the number of targets in the database; (2) decoys are false but adequately mimic targets in terms of physical properties. Thus, the FDR of the target assignments can be estimated by FDR = nd / nt, where nt is target matches and nd is decoy matches40. The search results are then filtered to reduce the FDR to a user-defined level.
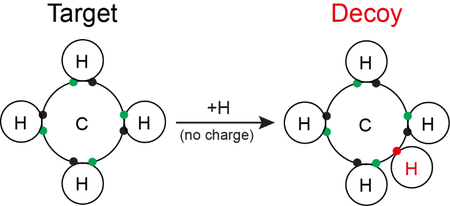
Here we design a target-decoy strategy for metabolite identification, which is based on the octet rule in Chemistry. The octet rule states that atoms combine in such a manner that each atom has eight electrons in its valence shell (Figure 1a, b). The rule is especially applicable to carbon, nitrogen, and oxygen. There are rare exceptions to the rule41,42 (e.g., radicals or expanded octets), but we found that all of the HMDB entries (n = 40,778) follow the octet rule after removing 18 radical exceptions. The strategy uses all formulas in the database (PubChem, HMDB, or YMDB) as targets, and creates decoy formulas by adding one hydrogen atom to each target formula without changing the charge state (Figure 1c). These decoys mimic mass distribution of the targets, but can only be assigned due to by-chance matches. For example, CH4 is a compound in the HMDB, and we create the decoy formula CH5 without charge in the decoy database. Similarly, we used target structures to generate decoy structures, which display product ions in MS2 spectra during database search (see Methods). In addition, the strategy can be expanded and generalized by adding any of small odd numbers (e.g., 3, 5, 7, and 9) of hydrogen atoms to formulas. Adding even numbers of hydrogen atoms, however, may convert unsaturated to saturated compounds, which cannot be applied to produce decoys.
Figure 1. Target-Decoy strategy.

(a) Common biological elements with characteristic number of electrons. (b) The valid Lewis structure for methane (CH4) shows shared electrons between hydrogen and carbon according to the octet rule. (c) Generation of decoy chemical formulas by computational addition of a hydrogen atom to each database formula, yielding an invalid structure without a change in the charge state. JUMPm treats all decoy formulas as neutral, ensuring that they are invalid. The impossible decoy structure for methane’s formula is shown.
Implementation of the Target-decoy Strategy in JUMPm
We have used the proposed target-decoy strategy in the JUMPm software, a tool that we have developed for metabolite identification from both unlabeled and stable-isotope labeled datasets. For an unlabeled dataset, JUMPm detects metabolite features from spectra, and searches for formulas and structures in the database. For a stable-isotope labeled dataset that contains unlabeled and fully labeled metabolite pairs, JUMPm can precisely determine the numbers of labeled atoms for unambiguously identifying formulas. The target-decoy strategy is implemented to estimate FDR in both formula and structure identification (Figure 2, see details in Supporting Information). The structure FDR is estimated at different levels of matching scores (i.e. Mscore). The Mscore is calculated by the hypergeometric test that compares theoretical (in silico) product ions with the observed MS2 peaks. At any given Mscore, the structure FDR can be estimated. With a defined FDR threshold (e.g. 0.05), JUMPm can filter the results with the related Mscore to produce a list of formulas and structures. The FDR threshold can be applied to different experimental LC-MS/MS runs since it is independent of experimental settings, and parameters of database search.
Figure 2. Workflow of the target-decoy implemented in JUMPm.

The FDR can be estimated at formula and structure levels.
For stable-isotope labeled datasets, a formula FDR is computed and linked with Pscore in JUMPm (see details in Supporting Information). The Pscore is used for assessing the reliability of identified formula, which is calculated by taking into consideration isotopic mass differences, relative ion intensity, and co-elution of the isotopic peaks. Analogous to the structure FDR, The formula FDR can be estimated at any given Pscore.
Validation of the Target-decoy Strategy by Null Datasets
We next examined the target-decoy strategy by simulated null MS1 and MS2 datasets, as a null spectrum has an equal probability of matching targets and decoys in the concatenated target-decoy database. We generated a falsified null dataset by shifting all precursor ion (i.e. MS1) mass by 4.5 Da in an unlabeled LC-MS/MS run (Figure 3a). When searched against the concatenated database, the target and decoy matches displayed an almost equal number (Figure 3b, left side), indicating that all of the target hits from the null dataset are due to random matching. Moreover, we tested the target-decoy strategy with a simulated stable-isotope labeled LC-MS/MS dataset. Similarly, after shifting precursor ion mass, it also showed identical numbers of targets and decoys (Figure 3b, right side), although the number of detected structures was low due to the stringent requirement in formula assignment of paired metabolites. We further assessed numerous target-decoy databases generated by alternative decoy methods (e.g. +3H, +5H, +7H, and +9H), all of which produced ~100% FDR with the simulated dataset (Figure 3c; Figure S1a in the Supporting Information). In addition, to approve that different mass shifts in the null dataset do not affect FDR estimation, we repeated the analysis with other mass shifts (i.e. 3.5, 5.5, 7.5, and 9.5 Da) and obtained the same result of an FDR of ~100% (Figure S1b in the Supporting Information).
Figure 3. Evaluation of the target-decoy strategy by null datasets.

(a) Diagram showing a simulation analysis by simulating MS1 precursor mass to assess the validity of the target-decoy strategy. The result shows that FDR of authentic labeled yeast data (4%) and null data (~100%). (b,c) The distribution of targets and decoys detected from both simulated unlabeled and labeled datasets by two decoy generating strategies (i.e. +H and +3H). (d) Evaluation of the target-decoy strategy by simulated MS2 spectra and PubChem database.
In addition, we validated the target-decoy strategy with mzMatch and MZmine 2, two widely used metabolite identification tools (see details in Supporting Information). The same set of the simulated null dataset by adding 4.5 Da to precursor ion mass was used. Similar to JUMPm, mzMatch and MZmine 2 showed 97% and 103% false discovery rates (Figure S2 in the Supporting Information), respectively, supporting that the target-decoy strategy is a general approach applicable to metabolite search programs.
We further evaluated the validity of the target-decoy strategy using completely simulated MS2 spectra. A total of 1,742 simulated MS2 spectra were generated by randomizing MS2 spectra approach33 based on the target spectra from the GNPS library (see Methods). By searching these simulated spectra with JUMPm, we found a similar Mscore distribution of targets and decoys (Figure 3d), indicative of random matching of the simulated MS2 spectra. In summary, the analyses of both simulated MS1 and MS2 spectra suggest that they are indistinguishable between target and decoy hits, demonstrating that our decoy database provides a representative model of the null hypothesis for FDR estimation in metabolite identification.
FDR Estimation for Experimental Metabolomic Analyses
To use the target-decoy strategy to estimate FDR for metabolomic analyses, we analyzed both unlabeled and stable-isotope labeled yeast datasets by the JUMPm algorithm. For the stable-isotope labeled dataset, the FDR can be estimated at formula and structure levels on the basis of Pscore and Mscore, respectively (Figure 2).
For any decoy structure, we first generated an MS2 product ion pattern from its corresponding target structure (Figure 4a), and then tested two structure decoy methods: “H2C” and “Randomized Peaks”. The “H2C” adds the small odd hydrogen(s) to a subset of product ions containing a selected carbon in the structure. Since the carbon is randomly selected, we evaluated the distribution of decoys in three replicates and obtained similar results (Figure 4b), indicating no obvious influence of carbon selection on decoy scoring. As the “H2C” only shifts a fraction of product ions, we further examined a method by adding the small odd hydrogen(s) to all product ions (“100% shift”, Figure 4c), yielding a decoy Mscore distribution indistinguishable from the “H2C” method. The result emphasizes that Mscore is contributed by matching both MS1 precursor ion and MS2 product ions, and the precursor ion mass has been already altered in the decoy database, reminiscent of the null strategy of changing the MS1 precusor ion mass (Figure 3a–c). The “Randomized Peaks” method randomly selects peaks from a pre-built theoretical product ion library derived from all HMDB compounds, leading to similar but slighly lower Mscores of the decoys than the “H2C” method (Figure 4c), implying that “Randomized Peaks” might result in more dramatic product ion pattern change than the “H2C” method. We then used the “H2C” method for the subsequent analysis of labeled and unlabeled datasets.
Figure 4. FDR estimation for metabolomic analyses.

(a) Step-wise search of a composite target-decoy database. (b) Distributions of target and decoy Mscores from a labeled dataset with the “H2C” method. (c) Comparison of “H2C”, 100% shift, and “Randomized Peaks” methods to generate decoy MS2 patterns using the same labeled dataset. (d) Distributions of target and decoy Pscores from the labeled dataset. (e) Distributions of target and decoy Mscores from an unlabeled dataset. (f) Histogram of targets and decoys with respect to mass error during JUMPm search. Target and decoy formulas are bins of 2 ppm across the mass error range (0.5 ppm within the grey rectangle); a zoomed-in range is also shown. (g) Two-dimensional distribution of target and decoy formulas by isotope mass defect (581 out of 613 within the green circle). Decoy formulas are randomly scattered in the plot while targets are tightly clustered around the expected mass defect values of 1.00335 for 13C and 0.99703 for 15N.
From a yeast labeled dataset, we detected 85 unique formulas at 5% FDR, and 91 unique structures at 5% FDR. Both Pscore and Mscore showed skewed distributions for target and decoy hits, with heavy right tails (Figure 4c,d). From an unlabeled dataset, we identified 265 metabolite-spectrum matches, corresponding to 151 unique formulas and unique 176 structures at the structure FDR of 5% filtered by the matching score (Mscore). Mscore also exhibited skewed distributions for targets and decoys towards high Mscores at the right side, but much more pronounced for targets (Figure 4e). In the low Mscore region, the numbers of targets and decoys were comparable, indicating that the FDR was exceedingly high for these poorly matched metabolites.
Finally, we further evaluated the target-decoy strategy by examining the distribution of assigned targets and decoys within different mass error ranges. In theory, when searching against a target-decoy database with a large mass error (e.g. 100 ppm), the true targets should be found in the range of defined mass error that matches the MS instrument setting (e.g. 2 ppm), whereas the decoys and false targets would be evenly distributed in the entire mass range due to by-chance matching. This concept was tested and confirmed in a previous proteomics target-decoy study43. To examine this concept in our metabolomics database search, we searched the labeled dataset with a mass error of 100 ppm. In the central window (± 2 ppm, see Figure 4f insert), we found the assignment of most targets and a few decoys, suggesting a low FDR in this range. In sharp contrast, outside of the central window (Figure 4f), the frequency of targets and decoys was almost equal, indicating ~100% FDR. The similar phenomenon should be observed when analyzing the target and decoy distributions with respect to the accuracy of mass defect of the labels (i.e., 1.00335 for 13C-12C mass difference, 0.99703 for 15N-14N mass difference). Indeed, only targets were identified within ± 0.001 Da of the theoretical isotope mass difference (see the square in Figure 4g), whereas decoys only appeared outside of the range of the square. These results further demonstrate that the proposed target-decoy strategy is a powerful tool for estimating FDR in metabolomics analysis.
CONCLUSIONS
In summary, we have introduced a novel target-decoy strategy by violating the octet rule to estimate the confidence of metabolite identifications at the levels of formula and structure assignments. The strategy can be expanded and generalized by adding one small odd number of hydrogen atoms to targets. It has been currently implemented in our recent developed metabolite identification tool (i.e. JUMPm), and can be applied to other metabolite identification tools. The validity of the strategy is strongly supported by the search results of simulated MS1 and MS2 datasets, as well as two unlabeled and stable isotope-labeled metabolomic datasets. Thus, the strategy is a simple and effective method for false discovery estimation of metabolite identification in high-throughput metabolomic studies.
Supplementary Material
Figure S1 - Evaluation of the target-decoy strategy with additional simulation analyses
Figure S2 - Evaluation of the target-decoy strategy with JUMPm, mzMatch, and MZmine 2.
Supplementary Text S1 - Introduction of JUMPm software tool.
Supplementary Text S2 - Detailed procedures for testing mzMatch.
Supplementary Text S3 - Detailed procedures for testing MZmine 2.
ACKNOWLEDGMENTS
The authors thank Z. Wu for laboratory assistance, A. High and V. Pagala for MS instrument guidance, and other lab and facility members for helpful discussion. This work was partially supported by National Institutes of Health grant R01AG047928, R01GM114260, R01AG053987, and ALSAC (American Lebanese Syrian Associated Charities). The MS analysis was performed in the St. Jude Children’s Research Hospital Proteomics Facility, partially supported by NIH Cancer Center Support Grant (P30CA021765).
Footnotes
CONFLICT OF INTEREST DISCLOSURE
The authors declare no competing financial interests.
REFERENCES
- (1).Johnson CH; Ivanisevic J; Siuzdak G Metabolomics: beyond biomarkers and towards mechanisms. Nat. Rev. Mol. Cell. Biol 2016, 17, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wishart DS Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov 2016, 15, 473–484. [DOI] [PubMed] [Google Scholar]
- (3).Benton HP; Wong DM; Trauger SA; Siuzdak G XCMS2: processing tandem mass spectrometry data for metabolite identification and structural characterization. Anal. Chem 2008, 80, 6382–6389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Smith CA; Want EJ; O’Maille G; Abagyan R; Siuzdak G XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem 2006, 78, 779–787. [DOI] [PubMed] [Google Scholar]
- (5).Tsugawa H; Cajka T; Kind T; Ma Y; Higgins B; Ikeda K; Kanazawa M; VanderGheynst J; Fiehn O; Arita M MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 2015, 12, 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Scheltema RA; Jankevics A; Jansen RC; Swertz MA; Breitling R PeakML/mzMatch: a file format, Java library, R library, and tool-chain for mass spectrometry data analysis. Anal. Chem 2011, 83, 2786–2793. [DOI] [PubMed] [Google Scholar]
- (7).Pluskal T; Castillo S; Villar-Briones A; Oresic M MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 2010, 11, 395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Meyer MR; Peters FT; Maurer HH Automated mass spectral deconvolution and identification system for GC-MS screening for drugs, poisons, and metabolites in urine. Clin. Chem 2010, 56, 575–584. [DOI] [PubMed] [Google Scholar]
- (9).Broeckling CD; Reddy IR; Duran AL; Zhao X; Sumner LW MET-IDEA: data extraction tool for mass spectrometry-based metabolomics. Anal. Chem 2006, 78, 4334–4341. [DOI] [PubMed] [Google Scholar]
- (10).Styczynski MP; Moxley JF; Tong LV; Walther JL; Jensen KL; Stephanopoulos GN Systematic identification of conserved metabolites in GC/MS data for metabolomics and biomarker discovery. Anal. Chem 2007, 79, 966–973. [DOI] [PubMed] [Google Scholar]
- (11).Sumner LW; Amberg A; Barrett D; Beale MH; Beger R; Daykin CA; Fan TW; Fiehn O; Goodacre R; Griffin JL; Hankemeier T; Hardy N; Harnly J; Higashi R; Kopka J; Lane AN; Lindon JC; Marriott P; Nicholls AW; Reily MD, et al. Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 2007, 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Allen F; Pon A; Wilson M; Greiner R; Wishart D CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42, W94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Heinonen M; Shen H; Zamboni N; Rousu J Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 2012, 28, 2333–2341. [DOI] [PubMed] [Google Scholar]
- (14).Ruttkies C; Schymanski EL; Wolf S; Hollender J; Neumann S MetFrag relaunched: incorporating strategies beyond in silico fragmentation. J. Cheminform 2016, 8, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ridder L; van der Hooft JJ; Verhoeven S; de Vos RC; Bino RJ; Vervoort J Automatic chemical structure annotation of an LC-MS(n) based metabolic profile from green tea. Anal. Chem 2013, 85, 6033–6040. [DOI] [PubMed] [Google Scholar]
- (16).Li L; Li R; Zhou J; Zuniga A; Stanislaus AE; Wu Y; Huan T; Zheng J; Shi Y; Wishart DS; Lin G MyCompoundID: using an evidence-based metabolome library for metabolite identification. Anal. Chem 2013, 85, 3401–3408. [DOI] [PubMed] [Google Scholar]
- (17).Weber RJ; Viant MR MI-Pack: Increased confidence of metabolite identification in mass spectra by integrating accurate masses and metabolic pathways. Chemom. Intell. Lab. Syst 2010, 104, 75–82. [Google Scholar]
- (18).Daly R; Rogers S; Wandy J; Jankevics A; Burgess KE; Breitling R MetAssign: probabilistic annotation of metabolites from LC-MS data using a Bayesian clustering approach. Bioinformatics 2014, 30, 2764–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Silva RR; Jourdan F; Salvanha DM; Letisse F; Jamin EL; Guidetti-Gonzalez S; Labate CA; Vencio RZ ProbMetab: an R package for Bayesian probabilistic annotation of LC-MS-based metabolomics. Bioinformatics 2014, 30, 1336–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Uppal K; Walker DI; Jones DP xMSannotator: An R Package for Network-Based Annotation of High-Resolution Metabolomics Data. Anal. Chem 2017, 89, 1063–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kuhl C; Tautenhahn R; Bottcher C; Larson TR; Neumann S CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem 2012, 84, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bocker S; Letzel MC; Liptak Z; Pervukhin A SIRIUS: decomposing isotope patterns for metabolite identification. Bioinformatics 2009, 25, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Draper J; Enot DP; Parker D; Beckmann M; Snowdon S; Lin W; Zubair H Metabolite signal identification in accurate mass metabolomics data with MZedDB, an interactive m/z annotation tool utilising predicted ionisation behaviour ‘rules’. BMC Bioinformatics 2009, 10, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Elias JE; Haas W; Faherty BK; Gygi SP Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat. Methods 2005, 2, 667–675. [DOI] [PubMed] [Google Scholar]
- (25).Peng J; Elias JE; Thoreen CC; Licklider LJ; Gygi SP Evaluation of multidimensional chromatography coupled with tandem mass spectrometry (LC/LC-MS/MS) for large-scale protein analysis: the yeast proteome. J. Proteome Res. 2003, 2, 43–50. [DOI] [PubMed] [Google Scholar]
- (26).Moore RE; Young MK; Lee TD Qscore: an algorithm for evaluating SEQUEST database search results. J. Am. Soc. Mass Spectrom 2002, 13, 378–386. [DOI] [PubMed] [Google Scholar]
- (27).Kall L; Storey JD; MacCoss MJ; Noble WS Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J. Proteome Res 2008, 7, 29–34. [DOI] [PubMed] [Google Scholar]
- (28).Colinge J; Masselot A; Giron M; Dessingy T; Magnin J OLAV: towards high-throughput tandem mass spectrometry data identification. Proteomics 2003, 3, 1454–1463. [DOI] [PubMed] [Google Scholar]
- (29).Balgley BM; Laudeman T; Yang L; Song T; Lee CS Comparative evaluation of tandem MS search algorithms using a target-decoy search strategy. Mol. Cell. Proteomics 2007, 6, 1599–1608. [DOI] [PubMed] [Google Scholar]
- (30).Matsuda F Rethinking Mass Spectrometry-Based Small Molecule Identification Strategies in Metabolomics. Mass Spectrom. (Tokyo) 2014, 3, S0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Matsuda F; Shinbo Y; Oikawa A; Hirai MY; Fiehn O; Kanaya S; Saito K Assessment of metabolome annotation quality: a method for evaluating the false discovery rate of elemental composition searches. PLoS One 2009, 4, e7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Palmer A; Phapale P; Chernyavsky I; Lavigne R; Fay D; Tarasov A; Kovalev V; Fuchser J; Nikolenko S; Pineau C; Becker M; Alexandrov T FDR-controlled metabolite annotation for high-resolution imaging mass spectrometry. Nat. Methods 2017, 14, 57–60. [DOI] [PubMed] [Google Scholar]
- (33).Scheubert K; Hufsky F; Petras D; Wang M; Nothias LF; Duhrkop K; Bandeira N; Dorrestein PC; Bocker S Significance estimation for large scale metabolomics annotations by spectral matching. Nat. Commun 2017, 8, 1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jones DR; Wu Z; Chauhan D; Anderson KC; Peng J A nano ultra-performance liquid chromatography-high resolution mass spectrometry approach for global metabolomic profiling and case study on drug-resistant multiple myeloma. Anal. Chem 2014, 86, 3667–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang Y; Xiao J; Suzek TO; Zhang J; Wang J; Bryant SH PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Wishart DS; Jewison T; Guo AC; Wilson M; Knox C; Liu Y; Djoumbou Y; Mandal R; Aziat F; Dong E; Bouatra S; Sinelnikov I; Arndt D; Xia J; Liu P; Yallou F; Bjorndahl T; Perez-Pineiro R; Eisner R; Allen F, et al. HMDB 3.0--The Human Metabolome Database in 2013. Nucleic Acids Res. 2013, 41, D801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ramirez-Gaona M; Marcu A; Pon A; Guo AC; Sajed T; Wishart NA; Karu N; Djoumbou Feunang Y; Arndt D; Wishart DS YMDB 2.0: a significantly expanded version of the yeast metabolome database. Nucleic Acids Res. 2017, 45, D440–D445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wolf S; Schmidt S; Muller-Hannemann M; Neumann S In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC bioinformatics 2010, 11, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan T; Porto C; Bouslimani A; Melnik AV; Meehan MJ; Liu WT; Crusemann M; Boudreau PD; Esquenazi E; Sandoval-Calderon M; Kersten RD, et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol 2016, 34, 828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kall L; Storey JD; MacCoss MJ; Noble WS Posterior error probabilities and false discovery rates: two sides of the same coin. J. Proteome Res 2008, 7, 40–44. [DOI] [PubMed] [Google Scholar]
- (41).Lewis GN The atom and the molecule. J. Am. Chem. Soc 1916, 38, 762–785. [Google Scholar]
- (42).Petrucci RHH, William S; Herring FG; Madura Jeffrey D. General Chemistry: Principles & Modern Applications, 9th Ed ed; Pearson Education, Inc: New Jersey, 2007. [Google Scholar]
- (43).Everley PA; Bakalarski CE; Elias JE; Waghorne CG; Beausoleil SA; Gerber SA; Faherty BK; Zetter BR; Gygi SP Enhanced analysis of metastatic prostate cancer using stable isotopes and high mass accuracy instrumentation. J. Proteome Res 2006, 5, 1224–1231. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 - Evaluation of the target-decoy strategy with additional simulation analyses
Figure S2 - Evaluation of the target-decoy strategy with JUMPm, mzMatch, and MZmine 2.
Supplementary Text S1 - Introduction of JUMPm software tool.
Supplementary Text S2 - Detailed procedures for testing mzMatch.
Supplementary Text S3 - Detailed procedures for testing MZmine 2.