

Abstract
The contribution of epithelial–mesenchymal transition (EMT) to human lung fibrogenesis is controversial. Here we provide evidence that ZEB1-mediated EMT in human alveolar epithelial type II (ATII) cells contributes to the development of lung fibrosis by paracrine signalling to underlying fibroblasts. Activation of EGFR–RAS–ERK signalling in ATII cells induced EMT via ZEB1. ATII cells had extremely low extracellular matrix gene expression even after induction of EMT, however conditioned media from ATII cells undergoing RAS-induced EMT augmented TGFβ-induced profibrogenic responses in lung fibroblasts. This epithelial–mesenchymal crosstalk was controlled by ZEB1 via the expression of tissue plasminogen activator (tPA). In human fibrotic lung tissue, nuclear ZEB1 expression was detected in alveolar epithelium adjacent to sites of extracellular matrix (ECM) deposition, suggesting that ZEB1-mediated paracrine signalling has the potential to contribute to early fibrotic changes in the lung interstitium. Targeting this novel ZEB1 regulatory axis may be a viable strategy for the treatment of pulmonary fibrosis.
Subject terms: Respiratory tract diseases, Predictive markers
Introduction
Epithelial–mesenchymal transition (EMT), a dynamic and reversible biological process by which epithelial cells lose their cell polarity and down-regulate cadherin-mediated cell–cell adhesion to gain migratory properties, is involved in embryonic development, wound healing, fibrosis and cancer metastasis [1]. EMT is executed in response to pleiotropic signalling factors, including the transforming growth factor β (TGFβ) superfamily, Sonic Hedgehog (Shh), Wnt/β-catenin, fibroblast growth factor (FGF) and epidermal growth factor (EGF). These factors regulate the expression of specific transcription factors (TFs) called EMT-TFs (e.g. Snail, ZEB, Twist and others) that promote repression of epithelial features and induction of mesenchymal characteristics [2, 3]. Unlike EMT in cancer, which is detrimental, wound-healing-driven EMT induced in response to injury is beneficial, but exaggerated healing responses can lead to fibrosis or tissue scarring.
Fibrosis is a hallmark of many chronic degenerative disorders and is associated with reduced organ function and eventual organ failure. Fibrotic disease is on the increase; for example, idiopathic pulmonary fibrosis (IPF), the most common type of idiopathic interstitial pneumonia, occurs with similar frequency to that of stomach, brain and testicular cancer [4]. IPF is now generally regarded as a consequence of multiple interacting genetic and environmental risk factors, with repetitive local micro-injuries to ageing alveolar epithelium playing a central role [5]. These micro-injuries initiate the progressive accumulation of extracellular matrix (ECM) deposited by myofibroblasts. The origin of these myofibroblasts has been debated for many years, with EMT being considered as a potential source by driving the transformation of epithelial cells into ECM producing myofibroblasts [6–10]. However, lineage tracing in transgenic mice indicates that the contribution of those cells to the population of myofibroblasts is negligible [11–14].
In this study, we identify a novel regulatory axis involved in lung fibrosis whereby EMT contributes to the fibrotic process via paracrine activation of fibroblasts. We demonstrate that epidermal growth factor receptor (EGFR)-RAS-extracellular signal-regulated kinase (ERK) signalling induces the transcription factor ZEB1, which not only controls EMT but also regulates the production of locally-acting mediators. Specifically, we identified tissue plasminogen activator (tPA) as a downstream effector of ZEB1 transcriptional activity that contributes to paracrine signalling by enhancing TGFβ-induced profibrogenic responses in fibroblasts. Consistent with this, increased ZEB1 nuclear expression was detected in alveolar epithelium adjacent to sites of ECM deposition in IPF lung tissue. Thus, rather than contributing directly to the mesenchymal population, our data suggest that ZEB1-dependent EMT of ATII cells contributes to fibrosis via epithelial–fibroblast crosstalk. The occurrence of ZEB1 activation at sites of local ECM deposition in IPF lung tissue is consistent with the concept that ZEB1-regulated paracrine signalling contributes to the development of a profibrogenic microenvironment leading to interstitial lung fibrosis.
Results
Activation of EGFR signalling induces EMT in alveolar epithelial cells
To investigate IPF associated signalling pathways, we analysed differentially expressed genes in IPF and control lung tissue from a publicly available microarray dataset (GSE24206) [15]. Using a false discovery rate (FDR) corrected P value of 0.05, we identified 7668 genes to be differentially expressed out of a total of 54,675 probe sets. Gene network analysis using the Consensus Pathways Database [16] identified a number of pathways. Of these the EGFR–ERK pathway was the top-ranked pathway with 150 of 458 pathway candidates being significantly (Q-value < 0.05) overrepresented in the dataset (Supplementary Fig. S1a).
Based on the transcriptomic data, we hypothesised an important role of EGFR signalling in IPF. Identification of pathological mechanisms of IPF has been challenging; however, dysregulation of alveolar type 2 (ATII) epithelial cells is thought to be central [5]. We therefore treated a human ATII cell line (ATIIER:KRASV12) [17, 18] with EGF (Fig. 1b–d; Supplementary Fig. S1b) or transforming growth factor α (TGFα) (Supplementary Fig. S1b) to activate EGFR signalling. The human ATII cell line grows in continuous culture and expresses the ATII cell marker, pro-surfactant protein C (ProSP-C) (Figs. 1a, 2f). Our results showed that treatment of ATIIER:KRASV12 cells with EGF for 24 h induced EMT, reflected by a change in their morphology from typical cuboidal epithelial cells to a more elongated mesenchymal cell phenotype with a reorganisation of the actin cytoskeleton as demonstrated using Phalloidin staining of filamentous actin (F-actin) (Fig. 1b). This phenotypic switch was accompanied by a significant increase in mRNA expression of ZEB1 and VIM (Vimentin), and a reduction in CDH1 (E-cadherin); mRNA levels of other EMT-TFs, such as SNAI1, SNAI2, TWIST and ZEB2 were not increased by activation of EGFR signalling (Fig. 1c). The changes in ZEB1 and E-cadherin were further confirmed by Western blot analysis (Fig. 1d; Supplementary Fig. S1b).
Fig. 1.

Activation of EGFR signalling induces EMT in alveolar epithelial cells. a Immunofluorescence staining of Pro-surfactant protein-C (Pro-SP-C) (green) in ATIIER:KRASV12 cells. DAPI (blue) was used to stain nuclei. Scale bars: 40 μm. b Immunofluorescence staining of F-actin (red) in ATIIER:KRASV12 cells cultured in the absence or presence of 100 ng/ml EGF for 24 h. Rhodamine-phalloidin was used to stain F-actin. DAPI (blue) was used to stain nuclei. Scale bars: 40 μm. c Fold change in mRNA levels of CDH1 (E-cadherin), VIM (Vimentin), SNAI1 (Snail1), SNAI2 (Snail2), TWIST, ZEB1 and ZEB2 in ATIIER:KRASV12 cells cultured in the absence or presence of 100 ng/ml EGF for 24 h. GAPDH-normalised mRNA levels in control cells were used to set the baseline value at unity. Data are mean ± s.d. n = 3 samples per group. **P < 0.01. ***P < 0.001. d Protein expression of E-cadherin, ZEB1 and phospho-ERK (p-ERK) in ATIIER:KRASV12 treated with 100 ng/ml EGF for 8 or 24 h. β-actin was used as a loading control. e Protein expression of E-cadherin, ZEB1, phospho-Smad2 (p-Smad2), phospho-ERK (p-ERK) in primary human ATII cells treated with 100 ng/ml EGF or 5 ng/ml TGFβ over 7 days. β-actin was used as a loading control
Fig. 2.

Activation of RAS signalling induces EMT in alveolar epithelial cells. a Protein expression of E-cadherin, ZEB1 and phospho-ERK (p-ERK) in ATIIER:KRASV12 treated with 100 ng/ml EGF or 250 nM 4-OHT for 24 h. β-tubulin was used as a loading control. b Fold change in mRNA levels of CDH1 (E-cadherin), VIM (Vimentin), SNAI1 (Snail1), SNAI2 (Snail2), TWIST, ZEB1 and ZEB2 in ATIIER:KRASV12 cells cultured in the absence or presence of 250 nM 4-OHT for 24 h. GAPDH-normalised mRNA levels in control cells were used to set the baseline value at unity. Data are mean ± s.d. n = 3 samples per group. ***P < 0.001. c Protein expression of E-cadherin, ZEB1, ZEB2, phospho-ERK (p-ERK) and phospho-AKT (p-AKT) in ATIIER:KRASV12 treated with 250 nM 4-OHT for the indicated period. β-actin was used as a loading control. d Immunofluorescence staining of ZEB1 (green) and F-actin (red) in ATIIER:KRASV12 cells cultured in the absence or presence of 250 nM 4-OHT for 24 h. Rhodamine-phalloidin was used to stain F-actin. DAPI (blue) was used to stain nuclei. Scale bars: 40 μm. e Representative 3D confocal images of ATIIER:KRASV12 cells cultured in Matrigel in the absence or presence of 250 nM 4-OHT for 48 h. Spheres were stained for F-actin with Rhodamine-phalloidin (red) and DAPI (blue). Scale bars: 40 μm. f Western blot analysis of lysates from 3D-cultured ATIIER:KRASV12 cells in Matrigel treated without or with 250 nM 4-OHT for 48 h showing effects on E-cadherin, ZEB1, Vimentin, phospho-ERK (p-ERK), phospho-AKT (p-AKT) and Pro-surfactant protein-C (Pro-SP-C). β-actin was used as a loading control
Similar results were obtained using primary human ATII cells treated with EGF where an increase in ZEB1 expression was associated with down-regulation of E-cadherin (Fig. 1e). Under the same conditions, however, TGFβ was not able to induce EMT in the primary human ATII cells (Fig. 1e). Together, these results demonstrate that activation of EGFR signalling is able to activate the EMT programme in ATII cells, which is supported by a morphology change, the induction of the EMT-TF ZEB1 and a mesenchymal marker Vimentin as well as a reduction in E-cadherin expression.
Activation of the RAS pathway drives EMT via ERK–ZEB1 in ATII cells
RAS signalling is one of the most important pathways downstream of EGFR activation and is involved in a variety of physiological and pathological responses, including EMT [19–21]. To investigate whether the RAS pathway is important for EMT in ATII cells, we utilised a RAS-inducible ATII cell model. KRASV12 (containing a single amino acid mutation in KRAS, glycine to valine at position 12) fused to the oestrogen receptor (ER) ligand-binding domain [22] was introduced into ATII cells to generate ATIIER:KRASV12, in which KRASV12 expression is induced by 4-hydroxytamoxifen (4-OHT) [17, 18]. Like EGF, direct activation of the RAS pathway in ATIIER:KRASV12 cells by treatment with 4-OHT induced EMT, reflected by a reduction in E-cadherin levels and an increase in ZEB1 and Vimentin expression (Fig. 2a, b). Time-course analysis further demonstrated that the induction of ZEB1 by RAS activation preceded the down-regulation of E-cadherin (Fig. 2c). Consistently, an EMT morphology change with an increase in ZEB1 expression was observed upon RAS activation (Fig. 2d). When grown on a thick layer of Matrigel, ATII cells form spheres (a 3D culture model) [23]. We adopted this experimental system and used ATIIER:KRASV12 cells to investigate whether RAS activation induces EMT in 3D cultures. Control ATIIER:KRASV12 cells formed single round spheres. Induction of oncogenic KRAS by 4-OHT resulted in spheres invading into the Matrigel with protrusions (Fig. 2e; Supplementary Fig. S2). We recovered these cells from the Matrigel, and examined the protein expression. We confirmed that RAS activation induced EMT in 3D cultures, demonstrated by a reduction in E-cadherin, and an increase in ZEB1 and Vimentin expression (Fig. 2f). These observations suggest that EGFR signalling and the downstream RAS pathway are able to induce EMT in ATII cells.
Since RAS activity regulates both the RAF–ERK and phosphoinositide 3-kinase (PI3K)–protein kinase B (AKT) signalling pathways, we next investigated which one is required for EMT in the ATII cells using inhibitors for these pathways. Treatment with the ERK inhibitor U0126 in ATIIER:KRASV12 cells was sufficient to inhibit RAS-induced ZEB1 and Vimentin expression, as well as to restore the expression of E-cadherin and the epithelial morphology; in contrast, the AKT inhibitor AKT VIII failed to do so (Fig. 3a, b; Supplementary Fig. S3a).
Fig. 3.

Activation of the RAS pathway drives EMT via ERK–ZEB1 in ATII cells. a Protein expression of E-cadherin, ZEB1, phospho-AKT (p-AKT) and phospho-ERK (p-ERK) in ATIIER:KRASV12 treated with 250 nM 4-OHT in the absence or presence of inhibitors AKT VIII (10 μM) or U0126 (10 μM) for 24 h. DMSO was used as a vehicle control and β-actin was used as a loading control. b Fold change in mRNA levels of CDH1 (E-cadherin), VIM (Vimentin) and ZEB1 in ATIIER:KRASV12 treated with 250 nM 4-OHT in the absence or presence of inhibitors AKT VIII (10 μM) or U0126 (10 μM) for 24 h. DMSO was used as a vehicle control. GAPDH-normalised mRNA levels in control cells were used to set the baseline value at unity. Data are mean ± s.d. n = 3 samples per group. **P < 0.01. ***P < 0.001. c Protein expression of E-cadherin, ZEB1, Snail1 and Snail2 in ATIIER:KRASV12 cells transfected with the indicated siRNA followed by treatment of 250 nM 4-OHT for 24 h. β-tubulin was used as a loading control. d Fold change in the mRNA level of CDH1 (E-cadherin) in ATIIER:KRASV12 cells transfected with the indicated siRNA followed by treatment of 250 nM 4-OHT for 24 h. GAPDH-normalised mRNA levels in control cells were used to set the baseline value at unity. Data are mean ± s.d. n = 3 samples per group. ***P < 0.001
We next investigated which EMT-TFs are important for RAS-induced EMT in ATII cells. ZEB1 RNA interference (RNAi), but not SNAI1 or SNAI2 RNAi, was able to restore E-cadherin expression and the epithelial morphology in 4-OHT-treated ATIIER:KRASV12 cells (Fig. 3c, d; Supplementary Fig. S3b), in line with the fact the ZEB1 was the only EMT-TF induced by EGFR–RAS signalling (Figs. 1, 2). Taken together, our results identify that RAS activation in human ATII cells drives EMT via ERK–ZEB1 pathway.
ZEB1 is highly expressed in IPF alveolar epithelium and is critical for transcriptional regulation of secreted factors that mediate crosstalk between ATII cells and fibroblasts
Given our in vitro findings, we compared ZEB1 expression in IPF and control lung tissue. In IPF tissue, we detected strong nuclear expression of ZEB1 not only in fibroblastic foci (Fig. 4a) but also in epithelial cells of thickened alveoli septae where collagen deposition in the interstitium was evident (Fig. 4b); in contrast, little ZEB1 staining or collagen deposition was observed in alveoli of control lung tissue (Fig. 4c). The presence of nuclear ZEB1 staining in alveolar epithelial cells within IPF lung tissue suggests that these cells are undergoing EMT; furthermore, the presence of ECM suggests induction of mesenchymal responses, either directly via the repairing epithelial cells undergoing EMT or by crosstalk with underlying fibroblasts.
Fig. 4.

ZEB1 is highly expressed in IPF fibroblastic foci and epithelial cells of thickened alveoli septae where collagen deposition in the interstitium is also evident. Serial sections of IPF (fibroblastic foci in a and epithelial cells of thickened alveoli septae in b) or control lung tissue (c) were stained for ZEB1 (left panel), with H/E (middle panel) or Masson’s trichrome stain (right panel, collagen shown in blue). n = 3. Arrows: representative positive ZEB1 staining. **—a fibroblastic focus. Scale bars: 50 μm
Comparison of the relative expression of ECM components in RAS-activated ATIIER:KRASV12 cells and fibroblasts highlights that ATII cells produce extremely low levels of ECM genes even after the induction of EMT (Supplementary Fig. S4a), suggesting that ECM production in fibrosis is more likely to be a consequence of fibroblast activation than direct deposition by epithelial cells undergoing EMT. Therefore we investigated whether ATII cells undergoing RAS-induced EMT produce paracrine factors that activate fibroblasts. For these experiments, we took advantage of the ability of 4-OHT to induce RAS pathway activation in ATIIER:KRASV12 cells, as this was not dependent on exogenous growth factors that might directly affect fibroblast responses. We treated the MRC5 or primary human parenchymal lung fibroblasts with conditioned media (CM) from control or 4-OHT-treated ATIIER:KRASV12 cells in the absence or presence of TGFβ, and evaluated the fibroblast responses by measuring the expression of α-smooth muscle actin (α-SMA, a myofibroblast marker) and other ECM genes, including COL1A1, COL3A1 and FN1. On its own, CM from RAS-activated ATIIER:KRASV12 cells (4-OHT-treated ATII CM) had little effect on the activation of fibroblasts (Fig. 5). However, 4-OHT-treated ATII CM together with TGFβ achieved a synergistic effect in activating fibroblasts, reflected by a larger increase in α-SMA (ACTA2), COL1A1 and FN1 levels (Fig. 5a, b). Of note, 4-OHT-treated ATII CM did not augment Smad2 phosphorylation suggesting a Smad2-independent response (Fig. 5a, c). Similar results were obtained using primary human lung fibroblasts from IPF patients (IPF fibroblasts, IPFFs) and control donors (normal human lung fibroblasts, NHLFs) (Fig. 5c; Supplementary Fig. S4b).
Fig. 5.
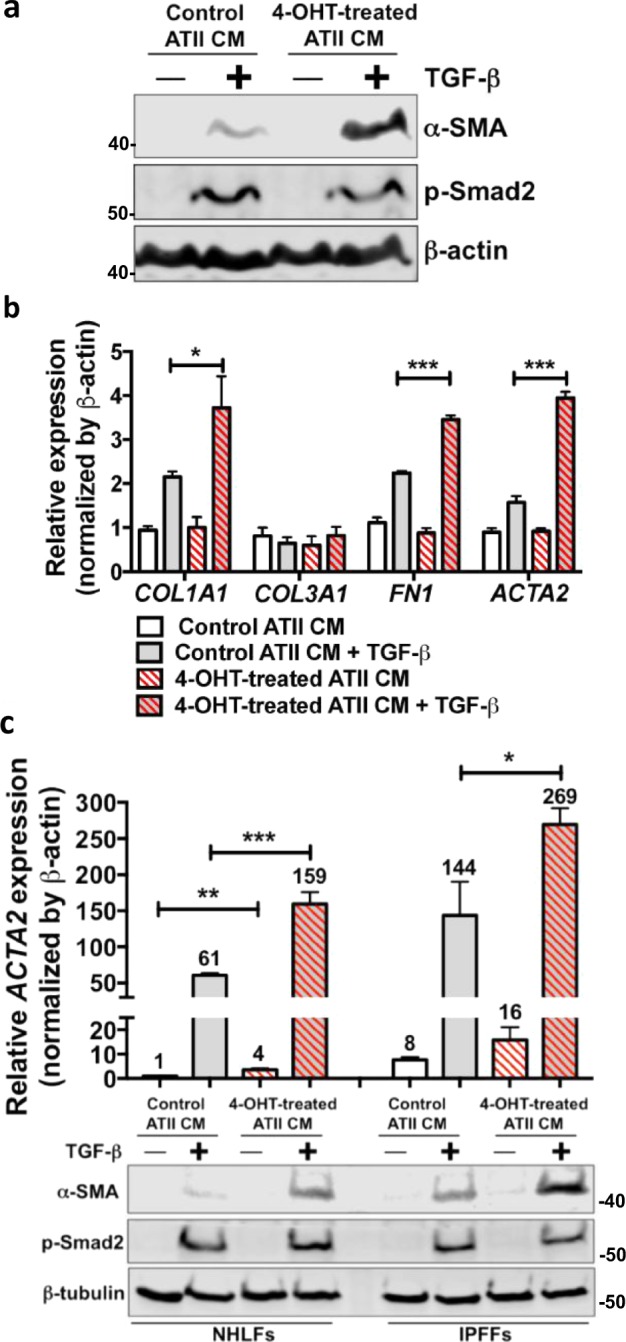
ATII cells undergoing RAS-induced EMT induce fibroblast activation via paracrine signalling. a Protein expression of α-SMA and phospho-Smad2 (p-Smad2) in MRC5 lung fibroblasts treated without or with 5 ng/ml TGFβ in the presence of conditioned media (CM) from control or 4-OHT-treated ATIIER:KRASV12 cells for 48 h. β-actin was used as a loading control. b Fold change in mRNA levels of COL1A1, COL3A1, FN1 and ACTA2 in MRC5 lung fibroblasts with indicated treatments. β-actin-normalised mRNA levels in control cells were used to set the baseline value at unity. Data are mean ± s.d. n = 3 samples per group. *P < 0.05. ***P < 0.001. c Protein expression of α-SMA and phospho-Smad2 (p-Smad2), and fold change in the mRNA level of ACTA2 (α-SMA) in primary human lung fibroblasts from IPF (IPFFs) or from normal healthy lung (NHLFs) with indicated treatments. β-tubulin was used as a loading control in Western blots. β-actin-normalised mRNA levels in control cells were used to set the baseline value at unity (indicated above bars). Data are mean ± s.d. n = 3 samples per group. *P < 0.05. **P < 0.01. ***P < 0.001
Given the important role of ZEB1 in mediating RAS-induced EMT and the fact that ZEB1 is highly expressed in the alveolar epithelium of IPF patients, we hypothesised that ZEB1 may determine the paracrine signalling produced by ATII cells undergoing RAS-induced EMT. ZEB1 RNAi (Fig. 6a) in ATII cells completely abolished the effects of CM from RAS-activated ATII cells on TGFβ-induced activation of fibroblasts (Fig. 6b; Supplementary Fig. S5), highlighting ZEB1 as a key regulator of EMT as well as the paracrine signalling between ATII cells and fibroblasts.
Fig. 6.
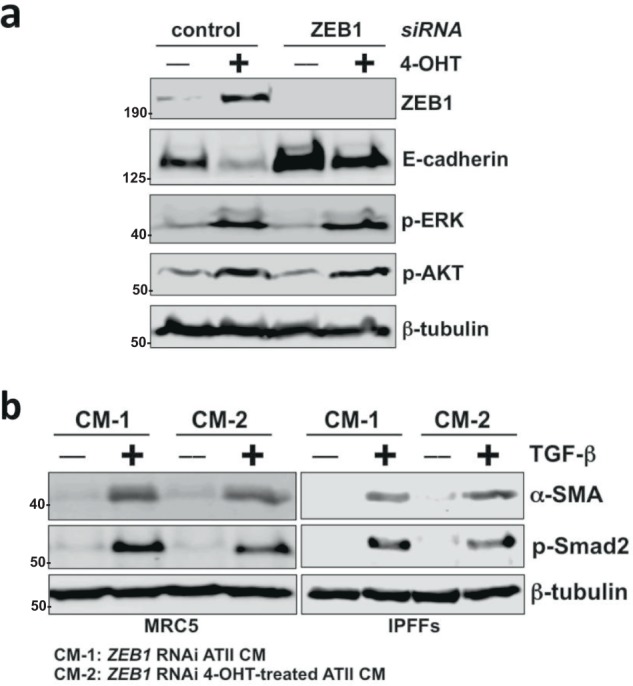
ZEB1 is a key regulator of the paracrine signalling between ATII cells and fibroblasts. a Protein expression of ZEB1, E-cadherin, phospho-ERK (p-ERK) and phospho-AKT (p-AKT) in ATIIER:KRASV12 cells with indicated treatments. β-tubulin was used as a loading control. b Protein expression of α-SMA and phospho-Smad2 (p-Smad2) in MRC5 lung fibroblasts or primary human lung fibroblasts from IPF (IPFFs) with indicated treatments. β-tubulin was used as a loading control
ZEB1 regulates the expression of tissue plasminogen activator, which acts as a paracrine regulator of TGFβ-induced fibroblast activation
By performing quantitative proteomic analysis of the CM from control or 4-OHT-treated ATIIER:KRASV12 cells, we identified ~430 secreted proteins whose levels changed during RAS-induced EMT. We then checked their expression in pulmonary epithelial cells from control and IPF lung tissue using a publicly available dataset [24], and identified a total number of 25 genes/proteins that were elevated in IPF lung epithelial cells as well as in CM from 4-OHT-treated ATIIER:KRASV12 cells (Supplementary Table S1). Of these, PLAT, which encodes tPA was most up-regulated in IPF epithelial cells (Fig. 7a; Supplementary Table S1) and we confirmed enhanced secretion of tPA in the CM from 4-OHT-treated ATIIER:KRASV12 cells by Western blotting (Fig. 7b). As we had identified ZEB1 as the key regulator of epithelial–mesenchymal crosstalk, we scanned the promoter of PLAT for the presence of ZEB1 binding motifs (5′-CANNTG-3′) and found a ZEB1 binding site −419 bp upstream of the transcriptional start site (TSS) (Supplementary Fig. S6a). Further experiments showed that the mRNA expression of PLAT was increased upon RAS-activation in ATII cells and this was repressed by ZEB1 RNAi (Fig. 7c).
Fig. 7.

ZEB1 regulates the expression of tissue plasminogen activator (tPA), which acts as a paracrine regulator of TGFβ-induced fibroblast activation. a Increased expression of PLAT (tPA) in IPF epithelial cells is shown by an online LGEA web portal (https://research.cchmc.org/pbge/lunggens/mainportal.html). b Quantitative secretome analysis identifies an increased level of tPA in the conditioned media (CM) from 4-OHT-treated ATIIER:KRASV12 cells and a representative tPA Western blot of CM from control or 4-OHT-treated ATIIER:KRASV12 cells. Data are individual values with mean and s.d. n = 3 samples per group. Values were normalised to total fmol of each sample multiplied by 10,000. c Fold change in mRNA levels of ZEB1 and PLAT (tPA) in ATIIER:KRASV12 cells with indicated treatments. β-actin-normalised mRNA levels in control cells were used to set the baseline value at unity. Data are mean ± s.d. n = 3 samples per group. ***P < 0.001. d ChIP assays of ZEB1's ability to bind the PLAT (tPA) promoter in ATIIER:KRASV12 cells with indicated treatments. The amplified PLAT (tPA) promoter region (−547 to −345) contains a ZEB1 binding site at −419. Values represent relative binding in relation to input (2%), normalised against control (1.0). Data are mean ± s.d. n = 4 samples per group. ***P < 0.001. e PLAT promoter reporter assays in ATIIER:KRASV12 cells with indicated treatments. Values represent relative fold of firefly luciferase in relation to Renilla luciferase, normalised against control (1.0). Data are mean ± s.d. n = 3 samples per group. **P < 0.01. ***P < 0.001. f Protein expression of α-SMA and phospho-Smad2 (p-Smad2) in MRC5 lung fibroblasts with indicated treatments. β-tubulin was used as a loading control
To validate the ZEB1 binding site in the PLAT promoter, we first performed a chromatin immunoprecipitation (ChIP) assay. An anti-ZEB1 antibody was used to precipitate formaldehyde cross-linked ZEB1-DNA complexes in ATIIER:KRASV12 cells treated without or with 4-OHT. The presence of PLAT promoter DNA sequences in the immunoprecipitate was verified by PCR using primers amplifying the region between −547 and −345 upstream of the TSS, and we found RAS activation in ATII cells increased ZEB1 occupancy on the PLAT promoter (Fig. 7d; Supplementary Fig. S6b). We next generated two PLAT promoter constructs (−689 to −1 upstream of the TSS) which were cloned into a pGL3 basic luciferase reporter plasmid and transfected into ATII cells; the pGL3 basic-PLAT (−689 to −1) construct contained the ZEB1 motif whereas this was deleted in the second construct (delta −419 to −414 upstream of the TSS) (pGL3 basic-Δ ZEB1 motif). RAS activation by 4-OHT in ATIIER:KRASV12 cells resulted in a significant increase in pGL3 basic-PLAT (−689 to −1) luciferase activity. Under the same conditions, luciferase activity was not increased using pGL3 basic-Δ ZEB1 motif (Fig. 7e). These data confirm that PLAT (tPA) is a transcriptional target of ZEB1 in response to RAS activation in ATII cells.
Consistent with a previous report [25], tPA synergistically promoted TGFβ-induced α-SMA expression in human lung fibroblasts (Supplementary Fig. S6c). Like ZEB1, PLAT RNAi (Supplementary Fig. S6d) in ATII cells completely abolished the effects of CM from RAS-activated ATII cells on TGFβ-induced α-SMA expression in fibroblasts (Fig. 7f), demonstrating tPA as a key paracrine factor secreted by ATII cells undergoing RAS-induced EMT. These results provide clear evidence that a ZEB1-tPA axis is involved in the paracrine signalling between ATII cells undergoing RAS-induced EMT and fibroblasts to augment their differentiation into myofibroblasts caused by TGFβ.
Finally, in view of the requirement for exogenous TGFβ to demonstrate an effect of the 4-OHT-treated ATII CM on fibroblasts, we investigated whether ATII cells in fibrotic tissue in vivo or those undergoing injury/repair in vitro expressed endogenous TGFβ. Using a publicly available dataset [24], we found that the major TGFB isoform expressed by alveolar epithelial cells in vivo was TGFB2 and that this was expressed at significantly higher levels in IPF compared with control lung tissue (Supplementary Fig. S7a). In contrast with the study in kidney [14], the data also revealed that Snail2 is up-regulated in IPF vs. control lung epithelial cells, but not Snail1 or Twist (Supplementary Fig. S7b). As we have previously shown that scrape-wounding of bronchial epithelial cells stimulates release of TGFβ2 independently of EGFR activation [26], we examined whether damage of ATII cells similarly affected TGFB2 expression. This showed that scrape-wounded ATII cells expressed more TGFB2 and this increased in proportion to the extent of injury (Supplementary Fig. S7c). These data suggest that damaged ATII cells are a potential a source of TGFβ in vivo.
Discussion
Fibrotic diseases are a major cause of morbidity and mortality worldwide and their prevalence is increasing with an ageing population. Abnormal wound healing responses appear to make major contributions to the scarring process, but the underlying pathological mechanisms are unclear, especially the role of EMT. In this study, we have used a variety of approaches to show that activation of EGFR–RAS–ERK signalling in ATII cells induces EMT via the transcriptional regulator ZEB1. Importantly, beyond its effects on the epithelial cell phenotype, we have identified that ZEB1 is a regulator of paracrine signalling between lung epithelial cells and fibroblasts, as ATII cells undergoing RAS-induced EMT secrete tPA to augment TGFβ-induced myofibroblast differentiation (Fig. 8). This may be an important profibrotic event as, relative to epithelial cells, the ability of fibroblasts to synthesise ECM is orders of magnitude greater.
Fig. 8.
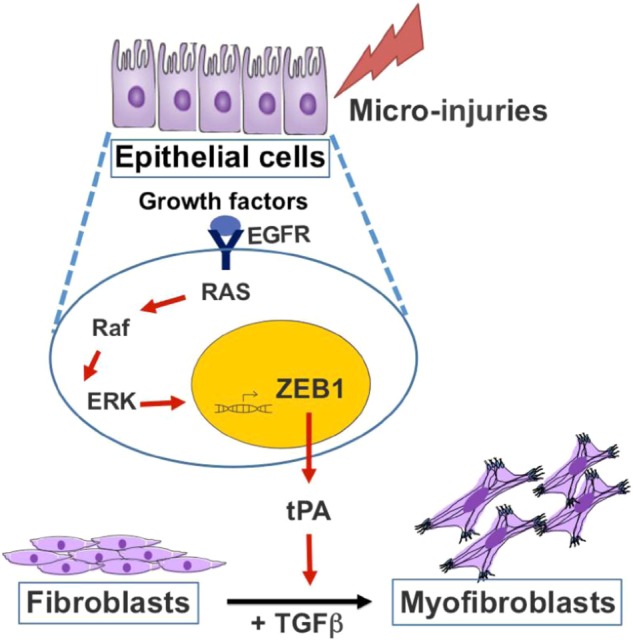
Diagram summarising a critical role of ZEB1-tPA axis regulated by EGFR–RAS–ERK pathway in the development of lung fibrosis (details provided in Discussion)
Consistent with previous findings [27, 28], we found strong expression of ZEB1 in the epithelium in proximity to fibroblastic foci in IPF lung tissue. However, we also found ZEB1 was expressed in epithelial cells of thickened alveolar septae where ECM deposition was evident. This suggests that ZEB1 is induced as an early response to alveolar epithelial injury and that, by regulating the expression of factors involved in paracrine signalling, ZEB1 may promote TGFβ-induced fibroblast activation in IPF. While this may be a normal physiological response to injury, persistent epithelial injury and/or failure to resolve the lesion may sensitise the underlying fibroblasts to drive a pathologic profibrogenic response. In line with this, exposure of human lung cells to nickel (Ni), an environmental and occupational pollutant linked to lung fibrosis [29], caused ZEB1-dependent EMT, which was irreversible even after the termination of Ni exposure [30]. Thus, it is conceivable that repetitive environmental exposures to metals such as Ni could lead to deregulation of ZEB1 to cause persistent EMT and exaggerated profibrogenic crosstalk during the initiation of IPF.
EMT in the ATII cells was strongly induced by EGFR activation. The EGFR is a transmembrane receptor tyrosine kinase activated by members of the EGF family, including EGF and TGFα [31]. EGFR dimerisation activates one or more downstream effectors including the ERK, PI3K/AKT, signal transducer and activator of transcription (STAT) and mammalian target of rapamycin (mTOR) pathways through receptor autophosphorylation and cytoplasmic protein binding [32–34]. These in turn act as critical mediators of airway and alveolar homoeostasis, with aberrant activation within one or more pathway components capable of driving a variety of respiratory pathologies, including lung fibrosis [33, 35]. The EGFR pathway has been implicated in lung fibrosis through studies in which transgenic mice that constitutively express TGFα in epithelial cells develop progressive lung fibrosis [36, 37]. Conversely, mice deficient in TGFα that lack normal EGFR signalling or that are treated with EGFR pathway inhibitors exhibit resistance to bleomycin-induced lung fibrosis [38]. In IPF patients, EGFR mutations [39] or increased expression of the EGFR [40] have been reported. Our evidence that an EGFR–RAS–ERK–ZEB1 axis may contribute to the early stages of lung fibrosis suggests that inhibiting EGFR signalling may be of clinical relevance for regulating human fibrotic lung disease.
A key finding of our study was identification that ZEB1 controls tPA expression and that this affects the sensitivity of fibroblast activation induced by TGFβ. While tPA is a key activator of fibrinolysis, it also has direct cellular effects by virtue of its ability to bind to the low-density lipoprotein (LDL) receptor-related protein-1 (LRP-1), triggering LRP-1 tyrosine phosphorylation and recruitment of β1-integrin signalling involving integrin-linked kinase (ILK) [25]. In this context, tPA acts as a survival factor that protects fibroblasts/myofibroblasts from apoptosis, and it has previously been implicated in kidney fibrosis [25], but no other fibrotic conditions. In keeping with a role of tPA in human lung fibrosis was the identification in a publicly available dataset [24] that there is an increased expression of PLAT (tPA) by IPF epithelial cells.
EMT converts epithelial cells into migratory and/or invasive mesenchymal cells, and is well established during development and carcinogenesis, however, its role in fibrosis has been more controversial [1, 3, 41]. Although human IPF tissue studies have demonstrated co-localisation of epithelial and mesenchymal markers [27, 28, 42], the number of fibroblasts arising from epithelial cells was small in some mouse lineage tracing studies [11, 12], suggesting effects of EMT beyond direct phenotypic conversion into matrix-producing cells. Recent studies in renal fibrosis have reported that tubular epithelial cells undergoing EMT relay signals to the interstitium, which promote myofibroblast differentiation and fibrogenesis, without directly contributing to the myofibroblast population [14, 43–45]. In tubulointerstitial renal fibrosis, TGFβ induces EMT via Snail1, and then Snail1 induces TGFβ expression generating an autocrine loop that sustains the progression of the disease by influencing the differentiation of fibroblasts into myofibroblasts [14]. In contrast with studies in kidney, our analysis of publicly available transcriptomic datasets of IPF lung tissue, identified the EGFR–ERK pathway as the top-ranked pathway with 150 of 458 pathway candidates being significantly overrepresented in the IPF dataset, highlighting the potential importance of this pathway in IPF pathogenesis. Building on these observations, our in vitro studies of EGFR–RAS–ERK-induced EMT, identified ZEB1 as the main transcription factor that controlled EMT as well as paracrine signalling through regulation of tPA expression, which potentiated fibroblast differentiation in the presence of TGFβ. However, as these paracrine effects required exogenous TGFβ, this raised the question of the source of TGFβ in lung fibrosis in vivo. While many cell types produce TGFβ isoforms which can also be stored as latent growth factor bound in the ECM [46–48], we focussed on the epithelium and found increased expression of TGFB2, as well as SNAI2 in IPF epithelial cells using publicly available datasets, with scrape wounding of ATII cells also inducing TGFB2 expression in vitro. The increased epithelial TGFB2 signature highlights the potential for EGF (ZEB1) and TGFβ (Snail2) to synergize in paracrine activation of the underlying fibroblasts.
Together with previous findings in kidney fibrosis [14, 43], our study helps to provide a unified concept for the role of EMT in fibrosis: persistent EMT of epithelial cells may dysregulate paracrine signalling between epithelial and mesenchymal cells, so creating a profibrogenic microenvironment which leads to the development of fibrosis. Based on the relative low levels of ECM biosynthesis by epithelial cells and the relatively small numbers of mesenchymal cells identified in lineage tracing studies [11, 12, 14, 43], this mechanism may be more important than direct conversion of epithelial cells into mesenchymal cells. While the finer details of these paracrine mechanisms may vary according to disease and tissue location, our identification of ZEB1 as a key regulator of EGF/RAS-induced EMT and an enhancer of paracrine signalling mediating the crosstalk between ATII cells and lung fibroblasts may help to find drug targets or biomarkers to intervene or predict the progression of pulmonary fibrosis.
Methods
Lung tissue sampling
All human lung experiments were approved by the Southampton and South West Hampshire and the Mid and South Buckinghamshire Local Research Ethics Committees, and all subjects gave written informed consent. Clinically indicated IPF lung biopsy tissue samples and non-fibrotic control tissue samples (macroscopically normal lung sampled remotely from a cancer site) were deemed surplus to clinical diagnostic requirements. All IPF samples were from patients subsequently receiving a multidisciplinary diagnosis of IPF according to international consensus guidelines [49].
Cell culture, reagents and transfections
Primary parenchymal lung fibroblast cultures were established from IPF or control lung tissue as described previously [50]. Fibroblasts were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 50 units/ml penicillin, 50 μg/ml streptomycin, 2 mM L-glutamine, 1 mM sodium pyruvate and 1× non-essential amino acids (DMEM/FBS) (all from Life Technologies).
Primary human ATII cells were isolated from macroscopically normal regions of surgically resected lung parenchyma as described previously [50, 51]. The alveolar epithelial cells were resuspended in fresh DCCM-1 (Biological Industries Ltd.) supplemented with 10% new-born calf serum (NBCS) (Life Technologies), 1% penicillin, 1% streptomycin and 1% L-glutamine (all from Sigma Aldrich) and plated on collagen 1 (PureCol 5005-b, Advanced BioMatrix Inc.) coated 96 well plates at 60% density; purity of the cultures was determined by staining for alkaline phosphatase.
ATIIER:KRASV12 cells [17, 18] were cultured in DCCM-1 (Biological Industries Ltd.) supplemented with 10% NBCS (Life Technologies), 1% penicillin, 1% streptomycin and 1% L-glutamine (all from Sigma Aldrich). To induce RAS activation in ATIIER:KRASV12 cells, 250 nM 4-OHT (Sigma-Aldrich) was added [17, 18]. MRC5 cells were obtained from the European Collection of Authenticated Cell Cultures (ECACC) and were cultured in DMEM (Thermo Fisher Scientific). Both cell culture media were supplemented with 10% FBS (Thermo Fisher Scientific), 1% penicillin/streptomycin and glutamine (Thermo Fisher Scientific). All cells were kept at 37 °C and 5% CO2. For 3D culture, ATIIER:KRASV12 cells were cultured as previously described [23] in Matrigel (BD Biosciences). TGFα was from Fisher Scientific UK Ltd. TGFβ1 was from PeproTech. EGF and recombinant human tPA protein were from Bio-Techne. AKT VIII and U0126 were from Sigma Aldrich. No mycoplasma contamination was detected in the cell lines used.
Short interfering RNA (siRNA) oligos against ZEB1 (MU-006564-02-0002), SNAI1 (Snail1) (MU-010847-00-0002), SNAI2 (Snail2) (MU-017386-00-0002) and PLAT (tPA) (MU-005999-01-0002) were purchased from Dharmacon. Sequences are available from Dharmacon, or on request. As a negative control, we used siGENOME RISC-Free siRNA (Dharmacon). ATIIER:KRASV12 cells were transfected with the indicated siRNA oligos at a final concentration of 35 nM using DharmaFECT 2 reagent (Dharmacon).
Western blot analysis
Western blot analysis was performed with lysates from cells with urea buffer (8 M urea, 1 M thiourea, 0.5% CHAPS, 50 mM DTT and 24 mM Spermine). Primary antibodies were from Santa Cruz (β-actin, sc-47778; ZEB1, sc-25388; ZEB2, sc-48789; E-cadherin, sc-21791; Snail2, sc-10436), Abcam (β-tubulin, ab6046), Cell Signalling Technology (α-SMA, 14968; phospho-AKT, 9271; phospho-ERK, 9101; Snail1, 3879; Snail2, 9585; TWIST, 46702; Phospho-Smad2, 3104; β-tubulin, 86298), BD Transduction Laboratories (E-cadherin, 610405; Vimentin, 550513) and Millipore (proSP-C, AB3786; tPA, 05-883). Signals were detected using an Odyssey imaging system (LI-COR), and evaluated by ImageJ 1.42q software (National Institutes of Health).
qRT-PCR
Total RNA was isolated using RNeasy mini kit (Qiagen) according to manufacturer’s instructions and quantified using a Nanodrop Spectrophotometer 2000c (Thermo Fisher Scientific). Real-time quantitative RT-PCR was carried out using gene-specific primers (QuantiTect Primer Assays, Qiagen) for CDH1 (E-cadherin) (QT00080143), SNAI1 (Snail1) (QT00010010), SNAI2 (Snail2) (QT00044128), ZEB1 (QT00008555), ZEB2 (QT00008554), TWIST (QT00011956), VIM (QT00095795), COL1A1 (QT00037793), COL3A1 (QT00058233), FN1 (QT00038024), ACTA2 (α-SMA) (QT00088102), PLAT (tPA) (QT00075761), TGFB1 (QT00000728), TGFB2 (QT00025718), GAPDH (QT01192646) or ACTB (β-actin) (QT01680476) with QuantiNova SYBR Green RT-PCR kits (Qiagen). Relative transcript levels of target genes were normalised to GAPDH or ACTB (β-actin).
Immunofluorescence microscopy
Cells were fixed in 4% PBS-paraformaldehyde for 15 min, incubated in 0.1% Triton X-100 for 5 min on ice, then in 0.2% fish skin gelatin in PBS for 1 h and stained for 1 h with an anti-Prosurfactant Protein C (proSP-C) antibody (1:100, Millipore AB3786, rabbit polyclonal) or anti-ZEB1 (1:100, Santa Cruz sc-25388, rabbit polyclonal). Protein expression was detected using Alexa Fluor (1:400, Molecular Probes) for 20 min. DAPI (Invitrogen) was used to stain nuclei (1:1000). Rhodamine-phalloidin was used to visualise filamentous actin (F-actin) (Molecular Probes). For immunofluorescence staining of 3D cultures from ATIIER:KRASV12 cells, spheres were fixed with 4% PBS-paraformaldehyde for 40 min, permeabilised in 0.5% Triton X-100 for 10 min on ice and stained with rhodamine-phalloidin for 1 h at room temperature. Spheres were counterstained with DAPI. Samples were observed using a confocal microscope system (Leica SP8). Acquired images were analysed using Photoshop (Adobe Systems) according to the guidelines of the journal.
Immunohistochemistry, haematoxylin and eosin (H/E) and tinctorial stains
Control or IPF lung tissues (n = 3 donors) were fixed and embedded in paraffin wax; tissue sections (4 µm) were processed and stained as previously described [20]. Briefly, the tissue sections were de-waxed, rehydrated and incubated with 3% hydrogen peroxide in methanol for 10 min to block endogenous peroxidase activity. Sections were then blocked with normal goat serum and incubated at room temperature with a primary antibody against ZEB1 (1:500, Sigma), followed by a biotinylated secondary antibody (1:500, Vector Laboratories Ltd., UK); antibody binding was detected using streptavidin-conjugated horse-radish peroxidase and visualised using DAB (DAKO) before counterstaining with Mayer’s Haematoxylin. For H/E stain, Shandon Varistain 24-4 automatic slide stainer (Thermo Fisher Scientific) was used. For tinctorial stain, Trichrome stain (Abcam ab150686) was used according to the manufacturers’ instructions. Images were acquired using an Olympus Dotslide Scanner VS110.
Chromatin immunoprecipitation (ChIP)
ChIP assays were carried out using SimpleChIP enzymatic chromatin IP kits (Cell Signalling Technology) as per the manufacturer’s instructions. Briefly, ATIIER:KRASV12 cells with indicated treatments were incubated for 10 min with 1% formaldehyde solution at room temperature, followed by incubation with 125 mM glycine. Antibodies used for ChIP were as follows: rabbit anti-ZEB1 (PA5-28221, Invitrogen, rabbit polyclonal, 5 μg per IP sample), normal rabbit IgG (2729, Cell Signalling Technology, 5 μg per IP sample). For the ZEB1 binding site at position −419 of the human PLAT (tPA) promoter, the primers amplifying the region between −547 and −345 were as follows: forward 5′-GGAAAGTCCCCGGAGGCCACCTA-3′ and reverse 5′-TGGAACACTTTGTGTGGTGGC-3′. DNA fragments were quantified by qPCR. PCR products were analysed in a 1.5% agarose gel by ethidium bromide staining.
Luciferase constructs and luciferase reporter assays
The human PLAT (tPA) promoter (sequence −689 to −1 upstream of the TSS) was amplified from human genomic DNA by PCR, and was subsequently cloned into pGL3 basic vector (Promega), termed pGL3 basic-PLAT (−689 to −1). The putative ZEB1 binding site, positioned −419 to −414 on the human PLAT promoter, was removed from pGL3 basic-PLAT (−689 to −1) construct to create the pGL3 basic-Δ ZEB1 motif construct.
For the luciferase reporter assays, ATIIER:KRASV12 cells were transfected using Lipofectamine 3000 (Invitrogen) with 80 ng of phRL-CMV (Promega), which constitutively expresses the Renilla luciferase reporter, plus 600 ng of pGL3 basic-PLAT (−689 to −1) or pGL3 basic-Δ ZEB1 motif per well in the presence or absence of 4-OHT. Finally, the transcriptional assay was carried out using the Dual-Luciferase reporter assay system (Promega) following the manufacturer’s protocol.
Quantitative proteomic analysis of the secretome and the subsequent data analysis
Serum-free CM from ATIIER:KRASV12 cells treated without or with 4-OHT (250 nM, 24 h) were analysed using an enrichment strategy based upon Strataclean resin (Agilent) in combination with the quantitative label-free approach, LC-MSE, to provide in-depth proteome coverage and estimates of protein concentration in absolute amounts [52] (details provided in Supplementary Methods).
Raw data were processed and collated into a single.csv document. Values were then normalised to total fmol of each sample multiplied by 10,000. Pseudo-counts were applied to the normalised values to replace missing ones, to allow for full statistical analysis to be completed [53]. We first sorted the normalised values in each column in order of abundance, in ascending order, then the minimum value of each sample identified. This minimum was used to replace all missing values in the data set. A two-tailed, unpaired Student’s t-test was used to compare two groups for independent samples. P < 0.05 was considered statistically significant.
In order to highlight their implications in IPF, differentially expressed proteins/genes identified in the quantitative secretome analysis were searched in LGEA web portal (https://research.cchmc.org/pbge/lunggens/mainportal.html) for their levels in pulmonary epithelial cells from control and IPF lung tissue.
Bioinformatics
IPF transcriptomic data was downloaded from the NCBI’s Gene Expression Omnibus (GEO). We used data from GSE24206 [15], a microarray study comparing samples from 11 IPF patients undergoing lung transplantation or diagnostic biopsy to six normal lung samples taken from lung transplantation donors. Microarray series matrix files were imported into R, and differential expression analysis comparing normal to IPF samples performed using the R package limma [54]. Data were log-transformed before analysis. To correct for multiple testing, a Benjamini–Hochberg FDR of 5% was applied to the data, and a Q-value cut-off of 0.02 was used to determine significance. Differentially expressed gene lists were input into the human Consensus Pathways Database, which determined pathways with differentially expressed genes overrepresented in this database. A 5% FDR was used as above.
Statistical analysis and repeatability of experiments
Each experiment was repeated at least twice. Unless otherwise noted, data are presented as mean and s.d., and a two-tailed, unpaired Student’s t-test was used to compare two groups for independent samples. P < 0.05 was considered statistically significant.
Electronic supplementary material
Acknowledgements
This project was supported by the Academy of Medical Sciences/the Wellcome Trust Springboard Award [SBF002/1038], Wessex Medical Trust and AAIR Charity. LY was supported by China Scholarship Council. FC was supported by Medical Research Foundation [MRF-091-0003-RG-CONFO]. CH was supported by Gerald Kerkut Charitable Trust and University of Southampton Central VC Scholarship Scheme. MGJ was supported by the Wellcome Trust [100638/Z/12/Z]. DED was supported by British Lung Foundation [BLF-RG14-14]. YW was supported by the Ministry of Science and Technology of China National Key Research and Development Projects [2016YFC0904701] and the National Natural Science Foundation of China [81772827]. Instrumentation in the Centre for Proteomic Research is supported by the BBSRC [BM/M012387/1] and the Wessex Medical Trust. We thank Carine Fixmer, Maria Lane, Benjamin Johnson and the nurses of the Southampton Biomedical Research Unit for their help in the collection of human samples, supported by the Wessex Clinical Research Network and the National Institute of Health Research, UK.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by C. Borner
These authors contributed equally: Liudi Yao, Franco Conforti, Charlotte Hill.
Contributor Information
Donna E. Davies, Email: D.E.Davies@soton.ac.uk
Yihua Wang, Email: yihua.wang@soton.ac.uk.
Electronic supplementary material
The online version of this article (10.1038/s41418-018-0175-7) contains supplementary material, which is available to authorized users.
References
- 1.Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 2.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 3.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–76. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 4.Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46:795–806. doi: 10.1183/09031936.00185114. [DOI] [PubMed] [Google Scholar]
- 5.Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet. 2017;389:1941–52. doi: 10.1016/S0140-6736(17)30866-8. [DOI] [PubMed] [Google Scholar]
- 6.DeMaio L, Buckley ST, Krishnaveni MS, Flodby P, Dubourd M, Banfalvi A, et al. Ligand-independent transforming growth factor-β type I receptor signalling mediates type I collagen-induced epithelial-mesenchymal transition. J Pathol. 2012;226:633–44. doi: 10.1002/path.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim KK, Wei Y, Szekeres C, Kugler MC, Wolters PJ, Hill ML, et al. Epithelial cell α3β1 integrin links β-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J Clin Invest. 2009;119:213–24. doi: 10.1172/JCI36940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006;103:13180–5. doi: 10.1073/pnas.0605669103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180:657–65. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Degryse AL, Tanjore H, Xu XC, Polosukhin VV, Jones BR, Boomershine CS, et al. TGFβ signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol. 2011;300:L887–97. doi: 10.1152/ajplung.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humphreys BD, Lin SL, Kobayashi A, Hudson TE, Nowlin BT, Bonventre JV, et al. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol. 2010;176:85–97. doi: 10.2353/ajpath.2010.090517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, et al. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci USA. 2011;108:E1475–83. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, et al. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–53. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015;21:989–97. doi: 10.1038/nm.3901. [DOI] [PubMed] [Google Scholar]
- 15.Meltzer EB, Barry WT, D’Amico TA, Davis RD, Lin SS, Onaitis MW, et al. Bayesian probit regression model for the diagnosis of pulmonary fibrosis: proof-of-principle. BMC Med Genomics. 2011;4:70. doi: 10.1186/1755-8794-4-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamburov A, Stelzl U, Lehrach H, Herwig R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013;41:D793–800. doi: 10.1093/nar/gks1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molina-Arcas M, Hancock DC, Sheridan C, Kumar MS, Downward J. Coordinate direct input of both KRAS and IGF1 receptor to activation of PI3 kinase in KRAS-mutant lung cancer. Cancer Discov. 2013;3:548–63. doi: 10.1158/2159-8290.CD-12-0446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coelho MA, de Carne Trecesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD-L1 mRNA. Immunity. 2017;47:1083–99. doi: 10.1016/j.immuni.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 20.Wang Y, Bu F, Royer C, Serres S, Larkin JR, Soto MS, et al. ASPP2 controls epithelial plasticity and inhibits metastasis through β-catenin-dependent regulation of ZEB1. Nat Cell Biol. 2014;16:1092–104. doi: 10.1038/ncb3050. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Ngo VN, Marani M, Yang Y, Wright G, Staudt LM, et al. Critical role for transcriptional repressor Snail2 in transformation by oncogenic RAS in colorectal carcinoma cells. Oncogene. 2010;29:4658–70. doi: 10.1038/onc.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dajee M, Tarutani M, Deng H, Cai T, Khavari PA. Epidermal Ras blockade demonstrates spatially localized Ras promotion of proliferation and inhibition of differentiation. Oncogene. 2002;21:1527–38. doi: 10.1038/sj.onc.1205287. [DOI] [PubMed] [Google Scholar]
- 23.Yu W, Fang X, Ewald A, Wong K, Hunt CA, Werb Z, et al. Formation of cysts by alveolar type II cells in three-dimensional culture reveals a novel mechanism for epithelial morphogenesis. Mol Biol Cell. 2007;18:1693–700. doi: 10.1091/mbc.e06-11-1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1:e90558. doi: 10.1172/jci.insight.90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu K, Wu C, Mars WM, Liu Y. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor-related protein 1-mediated integrin signaling. J Clin Invest. 2007;117:3821–32. doi: 10.1172/JCI32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Puddicombe SM, Polosa R, Richter A, Krishna MT, Howarth PH, Holgate ST, et al. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J. 2000;14:1362–74. doi: 10.1096/fasebj.14.10.1362. [DOI] [PubMed] [Google Scholar]
- 27.Chilosi M, Calio A, Rossi A, Gilioli E, Pedica F, Montagna L, et al. Epithelial to mesenchymal transition-related proteins ZEB1, β-catenin, and β-tubulin-III in idiopathic pulmonary fibrosis. Mod Pathol. 2017;30:26–38. doi: 10.1038/modpathol.2016.147. [DOI] [PubMed] [Google Scholar]
- 28.Park JS, Park HJ, Park YS, Lee SM, Yim JJ, Yoo CG, et al. Clinical significance of mTOR, ZEB1, ROCK1 expression in lung tissues of pulmonary fibrosis patients. BMC Pulm Med. 2014;14:168. doi: 10.1186/1471-2466-14-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitamura H, Ichinose S, Hosoya T, Ando T, Ikushima S, Oritsu M, et al. Inhalation of inorganic particles as a risk factor for idiopathic pulmonary fibrosis–elemental microanalysis of pulmonary lymph nodes obtained at autopsy cases. Pathol Res Pract. 2007;203:575–85. doi: 10.1016/j.prp.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 30.Jose Cynthia C., Jagannathan Lakshmanan, Tanwar Vinay S., Zhang Xiaoru, Zang Chongzhi, Cuddapah Suresh. Nickel exposure induces persistent mesenchymal phenotype in human lung epithelial cells through epigenetic activation of ZEB1. Molecular Carcinogenesis. 2018;57(6):794–806. doi: 10.1002/mc.22802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Linggi B, Carpenter G. ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 2006;16:649–56. doi: 10.1016/j.tcb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 32.Jorissen RN, Walker F, Pouliot N, Garrett TP, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exp Cell Res. 2003;284:31–53. doi: 10.1016/S0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 33.Vallath S, Hynds RE, Succony L, Janes SM, Giangreco A. Targeting EGFR signalling in chronic lung disease: therapeutic challenges and opportunities. Eur Respir J. 2014;44:513–22. doi: 10.1183/09031936.00146413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scaltriti M, Baselga J. The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res. 2006;12:5268–72. doi: 10.1158/1078-0432.CCR-05-1554. [DOI] [PubMed] [Google Scholar]
- 35.Burgel PR, Nadel JA. Epidermal growth factor receptor-mediated innate immune responses and their roles in airway diseases. Eur Respir J. 2008;32:1068–81. doi: 10.1183/09031936.00172007. [DOI] [PubMed] [Google Scholar]
- 36.Korfhagen TR, Swantz RJ, Wert SE, McCarty JM, Kerlakian CB, Glasser SW, et al. Respiratory epithelial cell expression of human transforming growth factor-alpha induces lung fibrosis in transgenic mice. J Clin Invest. 1994;93:1691–9. doi: 10.1172/JCI117152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hardie WD, Bruno MD, Huelsman KM, Iwamoto HS, Carrigan PE, Leikauf GD, et al. Postnatal lung function and morphology in transgenic mice expressing transforming growth factor-alpha. Am J Pathol. 1997;151:1075–83. [PMC free article] [PubMed] [Google Scholar]
- 38.Madtes DK, Elston AL, Hackman RC, Dunn AR, Clark JG. Transforming growth factor-alpha deficiency reduces pulmonary fibrosis in transgenic mice. Am J Respir Cell Mol Biol. 1999;20:924–34. doi: 10.1165/ajrcmb.20.5.3526. [DOI] [PubMed] [Google Scholar]
- 39.Stella GM, Inghilleri S, Pignochino Y, Zorzetto M, Oggionni T, Morbini P, et al. Activation of oncogenic pathways in idiopathic pulmonary fibrosis. Transl Oncol. 2014;7:650–5. doi: 10.1016/j.tranon.2014.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tzouvelekis A, Ntolios P, Karameris A, Vilaras G, Boglou P, Koulelidis A, et al. Increased expression of epidermal growth factor receptor (EGF-R) in patients with different forms of lung fibrosis. BioMed Res Int. 2013;2013:654354. doi: 10.1155/2013/654354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kage H, Borok Z. EMT and interstitial lung disease: a mysterious relationship. Curr Opin Pulm Med. 2012;18:517–23. doi: 10.1097/MCP.0b013e3283566721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lomas NJ, Watts KL, Akram KM, Forsyth NR, Spiteri MA. Idiopathic pulmonary fibrosis: immunohistochemical analysis provides fresh insights into lung tissue remodelling with implications for novel prognostic markers. Int J Clin Exp Pathol. 2012;5:58–71. [PMC free article] [PubMed] [Google Scholar]
- 43.Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21:998–1009. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ovadya Y, Krizhanovsky V. A new Twist in kidney fibrosis. Nat Med. 2015;21:975–7. doi: 10.1038/nm.3938. [DOI] [PubMed] [Google Scholar]
- 45.Huang S, Susztak K. Epithelial plasticity versus EMT in kidney fibrosis. Trends Mol Med. 2016;22:4–6. doi: 10.1016/j.molmed.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelley J. Cytokines of the lung. Am Rev Respir Dis. 1990;141:765–88. doi: 10.1164/ajrccm/141.3.765. [DOI] [PubMed] [Google Scholar]
- 47.Moore B, Murphy RF, Agrawal DK. Interaction of TGF-β with immune cells in airway disease. Curr Mol Med. 2008;8:427–36. doi: 10.2174/156652408785160943. [DOI] [PubMed] [Google Scholar]
- 48.Robertson IB, Horiguchi M, Zilberberg L, Dabovic B, Hadjiolova K, Rifkin DB. Latent TGF-β-binding proteins. Matrix Biol. 2015;47:44–53. doi: 10.1016/j.matbio.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Conforti F, Davies ER, Calderwood CJ, Thatcher TH, Jones MG, Smart DE, et al. The histone deacetylase inhibitor, romidepsin, as a potential treatment for pulmonary fibrosis. Oncotarget. 2017;8:48737–54. doi: 10.18632/oncotarget.17114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Witherden IR, Vanden Bon EJ, Goldstraw P, Ratcliffe C, Pastorino U, Tetley TD. Primary human alveolar type II epithelial cell chemokine release: effects of cigarette smoke and neutrophil elastase. Am J Respir Cell Mol Biol. 2004;30:500–9. doi: 10.1165/rcmb.4890. [DOI] [PubMed] [Google Scholar]
- 52.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics. 2006;5:144–56. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 53.Wang J, Li L, Chen T, Ma J, Zhu Y, Zhuang J, et al. In-depth method assessments of differentially expressed protein detection for shotgun proteomics data with missing values. Sci Rep. 2017;7:3367. doi: 10.1038/s41598-017-03650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.