

Abstract
Heteroaryl thioethers, comprised of pyridines and diazines, are an important class of compounds with relevance to medicinal chemistry. Metal-catalyzed cross-couplings and SNAr are traditionally used to form C–S bonds in these systems but are limited by available halogenated precursors. An alternative approach is presented where pyridines and diazines are transformed into heterocyclic phosphonium salts and then C–S bonds are formed by adding thiolate nucleophiles. The process is 4-selective for pyridines, simple to execute and can be used to make derivatives of complex pharmaceuticals.
Keywords: Pyridines, Diazines, C-S Bonds, Heteroaryl thioethers, Phosphonium salts, Late-stage functionalization
Graphical Abstract
1. Introduction
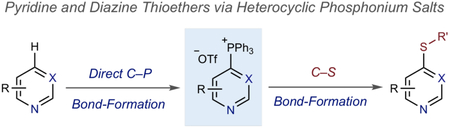
Adding heteroatom substituents to pyridines and diazines is a way to tune the steric and electronic properties of these heterocycles. Forming C–S bonds is an example of this strategy, and the resulting heteroaryl thioethers are commonly found in therapeutic compounds (Figure 1A).la-e Furthermore, the thioether moiety is a platform to synthesize higher oxidation state sulfoxide and sulfone derivatives.1f Methods to form heteroaryl ethers usually rely on metal-catalyzed cross-couplings or SNAr reactions of halogenated precursors.2,3 However, these strategies are often limited by the lack of selective methods to halogenate a broad range of pyridines and diazines. As a result, there are large numbers of potentially valuable heteroaryl thioethers that are inaccessible to medicinal chemists. Our laboratory recently disclosed a general approach to directly transform pyridines and diazines into phosphonium salts and subsequently react them with heteroatom nucleophiles to form C–O, C–S and C–N bonds.4 Herein, we present a detailed account of a two-step protocol to form heteroaryl thioethers (Figure 1B). The reaction has a broad scope, in both the thiol and heterocycle components, and generally forms the C–S bond with exclusive regioselectivity. Simple experimental protocols are employed and the strategy can be applied for late-stage functionalization of complex bioactive compounds.
Fig. 1.

Biologically active heteroaryl ethers and our strategy for C–S bond formation.
2. Results and Discussion
We began our study with phosphonium salt 1a, formed according to reported procedure by sequentially adding Tf2O, PPh3 and DBU to a solution of 2-phenylpyridine in dichloromethane at −78 °C,4a and a range of distinct thiols as coupling partners. The procedure involves deprotonating the thiol at 0 °C with sodium hydride in THF followed by adding the phosphonium salt and stirring at room temperature until the reaction is complete. As shown in Table 1, a range of aliphatic thiols of varying steric and electronic dispositions can be used in the coupling protocol.
Table 1.
C-S Bond-formation: Thiol scope.a
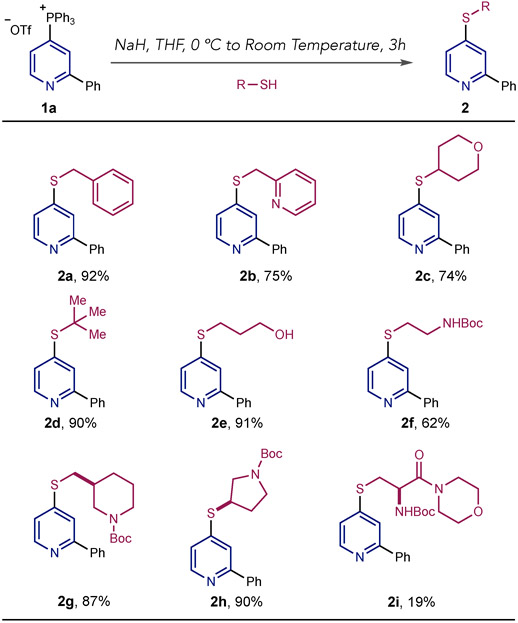 |
Isolated yields are shown. Typical reaction conditions: 1a (0.5 mmol), thiol (0.55 mmol), NaH (0.55 mmol) and THF (2.0 mL).
Primary benzylic and heterobenzylic thiols are effective with the corresponding thioethers formed in good yields (2a & 2b). A secondary pyranthiol and tert-butyl thiol were also accommodated without difficulty demonstrating that the reaction is not overly sensitive to the steric demands of the thiolate nucleophile (2c & 2d). 1,3-Thiopropanol reacted with complete chemoselectivity to form thioether 2e in good yield; a similar example of chemoselectivity was observed in 2f where the thiol reacted in preference to the carbamate group. Saturated amine heterocycles are common constituents of pharmaceutical compounds; a protected piperidine containing a primary thiol is an excellent substrate for this reaction (2g) and a pyrolidine-thiol was also effective (2h). Finally, a cysteine amide derivative could be successfully coupled to the pyridine, albeit in lower yield. Two potential mechanisms are under consideration for C–S bond-formation. First path A, where the thiolate adds to the phosphonium group resulting in a thiophosphorane that undergoes ligand coupling to form the thioether (Fig. 2).5 Related C–O couplings have been postulated via analogous alkoxyphosphoranes intermediates.6 Second, an SNAr pathway, with PPh3 as a leaving group (Fig. 2, path B),7 is also possible, and mechanistic studies into this reaction are ongoing in our laboratory.
Fig. 2.

Potential mechanistic pathways for C–S bond formation.
Next, the phosphonium salt formation-thiol addition sequence was examined with a range of pyridines and diazines (Table 2). In all but one case, the phosphonium salt is installed with exclusive regioselectivity with C–P bond-formation selective for the 4-position of pyridines. Examples 2j-2l show that C–S bond-formation will outcompete SNAr displacement of the 2-halo substituent with only minor amounts of double thiolate addition observed in each case.8 2,2-Bipyridine is an effective substrate in this protocol forming thioether derivative 2m, in moderate yield. Tetrahydroquinoline, containing a 2,3-disubstituion pattern, smoothly undergoes C–S bond formation (2n). A 3-phenyl substituent is no impediment to regioselective salt formation and thiol addition (2o). Furthermore, a 3,4,5-substituted pyridine can be formed with exclusive thiol addition at the 4-position of the pyridine in excellent yield (2p). Diazines are also amenable; a thioether derivative of a pyrimidine was formed with only minor amounts of 2-isomer observed during phosphonium salt formation (2q). Similarly, quinoxaline can be thiolated via the two-step process, forming 2r with complete regiocontrol.
Table 2.
C-S Bond-formation: Azaarene scope.a
 |
Isolated yields of single regioisomers (unless stated otherwise) with yields of phosphonium salts in parentheses. Typical reaction conditions for C-S bond formation: 1a (0.5 mmol), thiol (0.55 mmol), NaH (0.55 mmol) and THF (2.0 mL).
To investigate the viability of this strategy as a method for late-stage functionalization, we selected three distinct bioactive molecules to test the C–S bond-forming protocol.9 Etoricoxib can effectively be converted into a thioether analogue without difficulty (2s). Loratadine, an allergy treatment, is also effective in this two-step protocol to form thioether 2t. Benzyl protected cinchonidine, where the 4-position of the quinoline is blocked, undergoes C–S bond formation at the 2-position of the heteroaromatic forming 2u in reasonable overall yield.
3. Conclusion
We have shown that heteroaryl ethers can be formed from pyridines and diazines by selectively generating phosphonium salts and subsequent reactions with thiolate nucleophiles. A range of thiols can be employed, including chemoselective reactions with other functional groups, amine heterocycles and amino acid derivatives. The heterocyclic component can be varied with a variety of substituted pyridines and diazines applicable. Complex bioactive molecules are also amenable to this strategy representing a means for late-stage thiolation of pharmaceuticals. Due to the broad applicability and simplicity of this approach, we anticipate that it should be particularly relevant to medicinal chemists.
4. Experimental Section
4.1. General Experimental Considerations
Unless stated, all starting materials are either known compounds or were obtained from commercial sources and used without purification. Reactions were carried out under an inert atmosphere of nitrogen unless stated. Reaction progress was monitored by TLC, 1H NMR spectra taken from reaction samples or LCMS analysis. Tetrahydrofuran (THF), toluene, diethyl ether and dichloromethane were degassed and passed through a solvent purification system (alumina columns). 1,2-Dichloroethane (DCE), 1,4-dioxane, chloroform, chlorobenzene and acetone were purchased anhydrous from Sigma Aldrich chemical company. Flash chromatography was performed on Silicycle silica gel (Silaflash P60 (230-400 mesh)) with the indicated solvent system.
1H NMR spectra were recorded at ambient temperature on a Varian 400 MR spectrometer (400 MHz) or an Agilent Inova 400 (400 MHz) spectrometer. Chemical shifts (δ) are reported in ppm and quoted to the nearest 0.01 ppm relative to the residual protons in CDCl3 (7.26 ppm), C6D6 (7.16 ppm), (CD3)2SO (2.50 ppm), CD3OD (3.31 ppm) or CD3CN (1.94 ppm) and coupling constants (J) are quoted in Hertz (Hz). Data are reported as follows: Chemical shift (number of protons, multiplicity, coupling constants). Coupling constants were quoted to the nearest 0.1 Hz and multiplicity reported according to the following convention: s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, sext = sextet, sp = septet, m = multiplet, br = broad. 13C NMR spectra were recorded at ambient temperature on a Varian 400 MR spectrometer (100 MHz) or an Agilent Inova 400 (100 MHz) spectrometer. Chemical shift (δ) was measured in ppm and quoted to the nearest 0.1 ppm relative to the residual solvent peaks
4.2. Experimental Methods and Characterization Data
4.2.1. Formation of phosphonium salts
Phosphonium salts 1a, 1j-1o, 1q, 1t, and 1u have been reported previously.4a,4b
4.2.1.1. (3-cyano-5-(3-fluorophenyl)pyridine-4-yl)triphenylphosphonium trifluormethanesulfonate (1p)
To a solution of 5-(3-fluorophenyl)nicotinonitrile (1.29 g, 6.51 mmol) in CH2Cl2 (65 mL) at −78 °C was added Tf2O (1.09 mL, 6.51 mmol) dropwise over 5 minutes. The reaction was stirred for 30 minutes before PPh3 (1.88 g, 7.16 mmol) was added in one portion. The reaction was subjected to three rapid cycles of vacuum/nitrogen backfill and stirred for a further 30 minutes at − 78 °C. DBU (0.97 mL, 6.51 mmol) was added dropwise, the cooling bath was removed and the reaction was allowed to warm to room temperature while stirring (approximately 15 minutes). The reaction mixture was quenched with H2O (65 mL) and the mixture transferred to a separatory funnel. The mixture was diluted with CH2Cl2 and the resulting organic layer was washed three times with H2O. The organic layer was dried (MgSO4), filtered and concentrated to approximately 10 mL. The concentrated solution was added to an excess of Et2O (− 20 °C) and then placed in a −20 °C refrigerator for around 1 hour. The resulting suspension was filtered on a frit, the solid washed with chilled Et2O ((− 20 °C) and dried in vacuo to provide the title compound 1p (3.00 g, 4.93 mmol, 76% yield) as a white solid. mp 98-112 °C; IR νmax/cm−1 (film): 3065, 1585, 1439, 1260, 1150, 1099, 1029, 997, 719, 684, 636, 549; 1H NMR (400 MHz, CDCl3) δ: 9.10 (1H, dd, J = 4.9, 1.2 Hz), 8.83 (1H, dd, J = 5.5, 1.1 Hz), 7.92-7.44 (15H, m), 7.02-6.92 (1H, m), 6.84-6.73 (2H, m), 6.70 (1H, d, J = 8.9 Hz); 13C NMR (100 MHz, CDCl3) δ: 162.59 (d, J = 247.8 Hz), 152.82, 147.29, 140.07, 137.63 (d, J = 7.9 Hz), 135.63, 130.31 (d, J = 8.4 Hz), 128.72, 127.93, 125.41 (d, J = 3.1 Hz), 116.68 (d, J, 22.5 Hz), 115.98 (d, J = 21.0 Hz), 115.08; 19F NMR (365 MHz, CDCl3), −78.1; 31P NMR (162 MHz, CDCl3) 21.4; m/z HRMS (DART) found [M-OTf]+ 459.1425, C30H21FN2P+ requires 459.1421.
4.2.1.2. Triphenyl(quinoxalin-2-yl)phosphonium trifluoromethanesulfonate (1r)
To a solution of quinoxaline (52 mg, 0.40 mmol) in CH2Cl2 (4 mL) at −78 °C was added Tf2O (67 μL, 0.40 mmol) dropwise over 5 minutes. The reaction was stirred for 30 minutes before PPh3 (113 mg, 0.44 mmol) and NaOAc (50 mg, 0.60 mmol) were added in one portion. The reaction was subjected to three rapid cycles of vacuum/nitrogen backfill and stirred for a further 30 minutes at − 78 °C before being heated at 40 °C for 1 hour. DBU (60 μL, 0.40 mmol) was added dropwise at room temperature, and the reaction was stirred for a further hour at 40 °C. The reaction mixture was quenched with H2O (10 mL) and the mixture transferred to a separatory funnel. The mixture was diluted with CH2Cl2 and the resulting organic layer was washed three times with H2O. The organic layer was dried (MgSO4), filtered and concentrated to approximately 10 mL. The concentrated solution was added to an excess of Et2O (− 20 °C) and then placed in a −20 °C refrigerator for around 12 hours. The resulting suspension was filtered, dissolved in 10 mL CH2Cl2 and the precipitation process was repeated. The resulting solid was filtered on a frit and dried in vacuo to provide the title compound 1r (138 mg, 0.26 mmol, 64% yield) as a white solid. mp 130-132 °C; IR νmax/cm−1 (film): 3063, 2360, 1484, 1438, 1262, 1109, 1030, 634; 1H NMR (400 MHz, CDCl3) δ: 8.99 (1H, s, H1), 8.27-8.22 (2H, m, H3 and H4), 8.09-8.01 (2H, m, H2 and H5), 7.93-7.90 (3H, m, H8), 7.79-7.72 (12H, m, H6 and H7); 13C NMR (100 MHz, CDCl3) δ: 145.86 (d, J = 25.4 Hz), 143.38 (d, J = 2.8 Hz), 142.70 (d, J = 17.3 Hz), 140.83 (d, J = 111.6 Hz), 136.16 (d, J = 3.1 Hz), 134.98, 134.66 (d, J = 10.5 Hz), 133.08, 130.86 (d, J = 13.0 Hz), 130.19 (d, J = 2.0 Hz), 129.85 (d, J = 2.3 Hz), 120.76 (q, J = 319.5 Hz), 116.03 (d, J = 88.1 Hz); 19F NMR (365 MHz, CDCl3) δ: −78.17; 31P NMR (162 MHz, CDCl3) δ: 13.54; m/z HRMS (DART) found [M-OTf]+ 391.1355, C26H20N2P+ requires 391.1359.
4.2.1.3. (5-chloro-6’-methyl-3-(4-(methylsulfonyl)phenyl)-[2,3’-bipyridin]-4’-yl)triphenylphosphonium trifluormethanesulfonate (1s)
To a solution of 5-chloro-6'-methyl-3-(4-(methylsulfonyl)phenyl)-2,3'-bipyridine (384 mg, 1.07 mmol) in CH2Cl2 (11 mL) at −78 °C was added Tf2O (180 μL, 1.07 mmol) dropwise over 5 minutes. The reaction was stirred for 30 minutes before PPh3 (309 mg, 1.18 mmol) and NaOAc (88 mg, 1.07 mmol) were added in one portion. The reaction was subjected to three rapid cycles of vacuum/nitrogen backfill and stirred for a further 30 minutes at −78 °C. DBU (160 μL, 1.07 mmol was added dropwise, the cooling bath was removed and the reaction was allowed to warm to room temperature while stirring (approximately 15 minutes). The reaction mixture was quenched with H2O (30 mL) and the mixture transferred to a separatory funnel. The mixture was diluted with CH2Cl2 and the resulting organic layer was washed three times with H2O. The organic layer was dried (MgSO4), filtered and concentrated to approximately 10 mL. The concentrated solution was added to an excess of Et2O (− 20 °C) and then placed in a −20 °C refrigerator 1 hour. The resulting solid was filtered on a frit and dried in vacuo to provide the title compound 1s (603 mg, 0.78 mmol, 73% yield) as a white solid. mp 157-163 °C; IR νmax/cm−1 (film): 3062, 1709, 1577, 1542, 1485, 1436, 1311, 1261, 1223, 1150, 1101, 1030, 921, 888, 715, 690, 636; 1H NMR (400 MHz, CDCl3) 8: 8.28 (1H, d, J = 7.1 Hz, H1), 8.10 (2H, d, J = 8.2 Hz, H6), 7.86-7.62 (16H, m, H4, H7, H8, and H9), 7.51-7.45 (3H, m, H3 and H5), 7.20 (1H, d, J = 16.5 Hz, H2), 3.14 (3H, s, H10), 2.54 (3H, s, H11); 13C NMR (100 MHz, CDCl3) δ: 160.84 (d, J = 11.2 Hz), 152.42 (d, J = 7.3 Hz), 147.53 (d, J = 2.2 Hz), 146.09, 141.53, 141.02, 138.92, 135.62, 134.84 (d, J = 2.9 Hz), 134.17 (d, J = 10.0 Hz), 133.29 (d, J = 3.6 Hz), 132.10, 130.76 (d, J = 10.2 Hz), 130.03 (d, J = 13.1 Hz), 129.86, 128.55, 128.19 (d, J = 86.2 Hz), 120.77 (q, J = 321.1 Hz), 119.34 (d, J = 91.8 Hz), 43.96, 24.55; 19F NMR (365 MHz, CDCl3) δ: − 78.14; 31P NMR (162 MHz, CDCl3) δ: 25.54; m/z LRMS (ESI + APCI) found [M-OTf]+ 619.2, C36H29N2O2PS+ requires 619.1.
4.2.2. General procedure for the thiol addition reaction to form heteroaryl thioethers 2a-2u.
An oven dried 8 mL vial with a septa cap was charged with sodium hydride (60% dispersion in mineral oil, 1.1 equiv) and placed under a nitrogen atmosphere. THF (0.25 M) was added, the suspension was cooled to 0 °C and the thiol (1.1 equiv) was added dropwise over 5 minutes (if the thiol was a solid or viscous liquid, it was added as a 0.5 M solution in THF to an equivalent volume 0.5 M solution of NaH in THF). The resulting thick slurry was stirred for 30 minutes at 0 °C before the septa cap was briefly removed and the phosphonium salt (1.0 equiv) was added in one portion. The reaction was subjected to three rapid cycles of vacuum/nitrogen backfill, the ice bath removed and the reaction stirred for 3 hours while warming to room temperature. The reaction was quenched with H2O, the aqueous layer was separated and extracted with CH2Cl2 (3x). The combined organic extracts were washed with a saturated aqueous solution of brine, dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography under the stated conditions to provide the heteroaryl thioether product.
4.2.2.1. 4-(benzylthio)-2-phenylpyridine (2a)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), triphenyl(2-phenylpyridin-4-yl)phosphonium trifluoromethanesulfonate (1a, 283 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel: 15% EtOAc in hexanes to 10% EtOAc in hexanes) afforded the title compound (2a) as a white powder (128 mg, 0.46 mmol, 92% yield). mp 48-52 °C; IR νmax/cm−1 (film): 3060, 3040, 3027, 2922, 1560, 1533, 1495, 1455, 1377, 770, 709; 1H NMR (400 MHz, CDCl3) δ: 8.47 (1H, d, J = 5.3 Hz), 7.90 (2H, d, J = 7.7 Hz), 7.54 (1H, m), 7.49-7.27 (8H, m), 7.08 (1H, dd, J = 5.2, 1.7 Hz), 4.27 (2H, s); 13C NMR (100 MHz, CDCl3) δ: 157.27, 149.50, 149.16, 139.07, 135.73, 129.11, 128.82, 128.72, 128.71 (2C), 126.96, 119.25, 117.90, 35.92; m/z HRMS (DART) found [M+H]+ 278.1001, C18H16NS+ requires 278.0998.
4.2.2.2. 2-phenyl-4-((pyridine-3-ylmethyl)thio)pyridine (2b)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), pyridine-3-ylmethane thiol (62 μL, 0.55 mmol), triphenyl(2-phenylpyridin-4- yl)phosphonium trifluoromethanesulfonate (1a, 283 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel: 50% EtOAc in hexanes) afforded the title compound (2b) as a yellow oil (104 mg, 0.38 mmol, 75% yield). IR νmax/cm−1 (film): 3049, 2918, 1569, 1534, 1466, 1435, 1381, 772, 730, 693; 1H NMR (400 MHz, CDCl3) δ: 8.59 (1H, d, J = 4.7 Hz), 8.45 (1H, d, J = 5.4 Hz) 7.95-7.89 (2H, m), 7.70-7.63 (2H, m), 7.51-7.37 (4H, m), 7.20 (1H, dd, J = 7.2, 4.9 Hz), 7.13 (1H, dd, J = 5.3, 0.7 Hz), 4.41 (2H, s); 13C NMR (100 MHz, CDCl3) δ: 157.20, 156.64, 149.40, 149.09, 148.93, 138.98, 137.00, 129.10, 128.70, 126.93, 122.86, 122.48, 119.26, 117.84, 37.48; m/z HRMS (DART) found [M+H]+ 279.0948, C17H15N2S+ requires 279.0950.
4.2.2.3. 2-phenyl-4-((tetrahydro-2H-pyran-4-yl)thio)pyridine (2c)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), tetrahydro-2H-pyran-4-thiol (63 μL, 0.55 mmol), triphenyl(2-phenylpyridin-4-yl)phosphonium trifluoromethanesulfonate (1a, 283 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel, gradient elution: 40% EtOAc in hexanes) afforded the title compound (2c) as a white solid (92 mg, 0.37 mmol, 74% yield). mp 62-64 °C; IR νmax/cm−1 (film): 3071, 2956, 2914, 2863, 2839, 1565, 1533, 1461, 1440, 1379, 1126, 1083, 1007, 981, 818, 772, 698; 1H NMR (400 MHz, CDCl3) δ: 8.50 (1H, d, J = 5.3 Hz), 7.97-7.91 (2H, m), 7.57 (1H, d, J = 1.3 Hz), 7.50-7.38 (3H, m), 7.09 (1H, dd, J = 5.3, 1.7 Hz), 4.00 (2H, dt, J = 11.9, 3.9 Hz), 3.66-3.49 (3H, m), 2.09-1.99 (2H, m), 1.83-1.70 (2H, m); 13C NMR (100 MHz, CDCl3) δ:157.57, 149.39. 147.62, 138.98, 129.19, 128.76, 120.65, 119.50, 67.03, 40.54, 32.69; m/z HRMS (DART) found [M+H]+ 272.1101, C17H15N2S+ requires 272.1104.
4.2.2.4. 4-(tert-butylthio)-2-phenylpyridine (2d)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), tert-butyl thiol (62 μL, 0.55 mmol), triphenyl(2-phenylpyridin-4-yl)phosphonium trifluoromethanesulfonate (1a, 283 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel: 5% EtOAc in hexanes) afforded the title compound (2d) as a white crystalline solid (109 mg, 0.45 mmol, 90% yield). mp 56-62 °C; IR νmax/cm−1 (film): 3042, 2976, 2967, 2955, 2921, 2856, 1568, 1533, 1463, 1443, 1375, 1364, 1161, 847, 775, 686, 618; 1H NMR (400 MHz, CDCl3) δ: 8.61 (1H, d, J = 5.1 Hz), 7.99 (2H, d, J = 7.6 Hz), 7.83 (1H, m), 7.52-7.40 (3H, m), 7.35 (1H, d, J = 5.0, 1.7 Hz), 1.41 (9H, s); 13C NMR (100 MHz, CDCl3) δ: 157.55, 149.35, 144.77, 138.91, 129.17, 128.78, 128.20, 126.97, 126.90, 47.08, 31.24; m/z HRMS (DART) found [M+H]+ 244.1169, C15H18NS+ requires 244.1154.
4.2.2.5. 3-((2-phenylpyridin-4-yl)thio)propan-1-ol (2e)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), 3-mercaptopropan-1-ol (48 μL, 0.55 mmol), triphenyl(2-phenylpyridin-4-yl)phosphonium trifluoromethanesulfonate (1a, 283 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel, dry load: 50% EtOAc in hexanes) afforded the title compound (2e) as a yellow oil (111 mg, 0.45 mmol, 91% yield). IR νmax/cm−1 (film): 3285 (br), 2923, 2850, 1571, 1533, 1465, 1444, 1382, 1056, 907, 773, 730, 694; 1H NMR (400 MHz, CDCl3) δ: 8.48 (1H, d, J = 5.3 Hz), 7.95 (2H, d, J = Hz), 7.57 (1H, m), 7.50-7.38 (3H, m), 7.09 (1H, dd, J = 5.2, 1.3 Hz), 3.83 (2H, t, J = 6.1 Hz). 3.18 (2H, t, J = 7.1 Hz), 2.00 (2H, app t, J = 6.5 Hz); 13C NMR (100 MHz, CDCl3) δ: 157.26, 149.97, 139.03, 129.15, 128.73, 127.02, 119.02, 117.82, 60.74, 31.28, 27.21; m/z HRMS (DART) found [M+H]+ 246.0961, C14H16NOS+ requires 246.0947.
4.2.2.6. tert-butyl (2-((2-phenylpyridin-4-yl)thio)ethyl)carbamate (2f)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), tert-butyl (2-mercaptoethyl)carbamate (93 μL, 0.75 mmol), triphenyl(2-phenylpyridin-4-yl)phosphonium trifluoromethanesulfonate (1a, 283 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel: 30% EtOAc in hexanes) afforded the title compound (2f) as an off-white amorphous solid (102 mg, 0.31 mmol, 62% yield). IR νmax/cm−1 (film): 3343, 2976, 2930, 1699, 1571, 1534, 1506, 1365, 1251, 1164, 908, 773, 730, 694; 1H NMR (400 MHz, CDCl3) δ: 8.47 (1H, d, J = 5.3 Hz), 7.98 (2H, d, J = 7.5 Hz), 7.64 (1H, m), 7.48-7.34 (3H, m), 7.10 (1H, dd, J = 5.3, Hz), 3.43 (2H, q, J = 6.6 Hz). 3.17 (2H, t, J = 6.8 Hz), 2.15 (1H, s). 1.43 (9H, s); 13C NMR (100 MHz, CDCl3) δ: 157.41, 149.22, 148.73, 138.96, 129.13, 128.67, 127.06, 119.13, 117.69, 79.72, 53.42, 39.69, 30.59, 28.34; m/z HRMS (DART) found [M+H]+ 331.1488, C18H23N2O2S+ requires 331.1475.
4.2.2.7. tert-butyl (S)-3-(((2-phenylpyridin-4-yl)thio)methyl)piperidine-1-carboxylate (2g)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), tert-butyl (S)-3-(mercaptomethyl)piperidine-1-carboxylate (127 mg, 0.55 mmol in 1 mL of THF), triphenyl(2-phenylpyridin-4-yl)phosphonium trifluoromethanesulfonate (1a, 283 mg, 0.50 mmol) and THF (1.0 mL). Flash column chromatography (silica gel: 30% EtOAc in hexanes) afforded the title compound (2g) as a yellow oil (167 mg, 0.44 mmol, 87% yield). IR νmax/cm−1 (film): 2975, 2929, 2854, 1681, 1571, 1535, 1424, 1365, 1261, 1241, 1163, 907, 772, 728, 694; 1H NMR (400 MHz, CDCl3) δ: 8.49 (1H, 1H, dd, J = 5.3, 0.5 Hz), 7.97-7.92 (2H, m), 7.53 (1H, dd, J = 1.8, 0.6 Hz), 7.50-7.39 (3H, m), 7.05 (1H, dd, J = 5.3, 1.8 Hz), 4.22-3.73 (2H, m), 3.06-2.64 (4H, m), 2.06-1.94 (1H, m), 1.91-1.79 (1H, m), 1.75-1.63 (1H, m), 1.53-1.40 (10H, m), 1.39-1.27 (1H, m); 13C NMR (100 MHz, CDCl3) δ: 157.29, 154.74, 149.62, 149.19, 139.07, 129.12, 128.71, 126.96, 119.08, 117.87, 79.56, 48.92 (br), 44.30 (br), 35.18, 34.18 (br), 28.40, 24.24 (br); m/z HRMS (DART) found [M+H]+ 385.1913, C22H29N2O2S+ requires 385.1944.
4.2.2.8. tert-butyl (R)-3-((2-phenylpyridin-4-yl)thio)pyrrolidine-1-carboxylate (2h)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), tert-butyl (R)-3-mercaptopyrrolidine-1-carboxylate (112 mg, 0.5 mmol, in 1.0 mL of THF), triphenyl(2-phenylpyridin-4-yl)phosphonium trifluoromethanesulfonate (1a, 283 mg, 0.50 mmol) and THF (1.0 mL). Flash column chromatography (silica gel: 30% EtOAc in hexanes) afforded the title compound (2h) as a clear oil (160 mg, 0.45 mmol, 90% yield). IR νmax/cm−1 (film): 2976, 2877, 2246, 1685, 1570, 1535, 1400, 1365, 1163, 1115, 908, 772, 728, 694; 1H NMR (400 MHz, CDCl3) δ: 8.50 (1H, br s), 7.93 (2H, d, J = 7.3 Hz), 7.53 (1H, s), 7.50-7.36 (3H, m), 7.06 (1H, d, J = 5.1 Hz), 4.04-3.76 (2H, m), 3.68-3.25 (3H, m), 2.43-2.28 (1H, m), 2.07-1.91 (1H,m), 1.45 (9H, s); 13C NMR (100 MHz, CDCl3) δ: 157.67, 154.30, 149.52, 148.39, 139.01, 129.32, 128.85, 127.07, 119.98, 118.76, 79.87, 51.88, 51.62(rot), 47.75, 44.53(rot), 42.42, 41.71(rot), 32.06, 31.41(rot), 28.56; m/z HRMS (DART) found [M+H]+ 357.1670, C20H35N2O2S+ requires 357.1631.
4.2.2.9. tert-butyl (R)-(1-morpholino-1-oxo-3-((2-phenylpyridin-4-yl)thio)propan-2-yl)carbamate (2i)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 11 mg, 0.28 mmol), tert-butyl (R)-(3-mercapto-1-morpholino-1-oxopropan-2-yl)carbamate (80 mg, 0.28 mmol, in 1.0 mL of THF), triphenyl(2-phenylpyridin-4-yl)phosphonium trifluoromethanesulfonate (1a, 142 mg, 0.25 mmol) and THF (1.0 mL). Flash column chromatography (silica gel: 50% EtOAc in hexanes) afforded the title compound (2i) as a clear oil (21 mg, 0.05 mmol, 19% yield). IR νmax/cm−1 (film): 3062, 2984, 2903, 1784, 1677, 1523, 1161, 1138, 1079, 984, 847, 776, 687; 1H NMR (400 MHz, CDCl3) δ: 8.50 (1H, d, J = 5.1 Hz), 8.08 (2H, d, J = 7.4 Hz), 7.76 (1H, m), 7.56-7.37 (3H, m), 7.15 (1H, d, J = 5.1 Hz), 4.90 (1H, m), 3.81-3.16 (10H, m), 1.43 (9H, s); 13C NMR (100 MHz, CDCl3) δ: 169.01, 157.89, 155.04, 149.55, 148.40, 138.91, 129.45, 128.87, 127.28, 119.22, 117.11, 80.71, 66.70, 48.66, 46.69, 33.26, 28.46; m/z HRMS (DART) found [M+H]+ 444.1944, C23H30N3O4S+ requires 444.1952.
4.2.2.10. 4-(benzylthio)-2-chloropyridine (2j)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (68 μL, 0.58 mmol), (2-chloropyridin-4-yl)triphenylphosphonium trifluormethane sulfonate (1j, 262 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel: 5% EtOAc in hexanes) afforded the title compound (2j) as a white solid (106 mg, 0.45 mmol, 90% yield). mp 58-62 °C; IR νmax/cm−1 (film): 3068, 3028, 3007, 2920, 1568, 1522, 1456, 1370, 1150, 1078, 821, 794, 777, 713, 685; 1H NMR (400 MHz, CDCl3) δ: 815 (1H, d, J = 5.3 Hz), 7.42-7.27 (4H, m), 7.15 (1H, d, J = 1.5 Hz), 7.02 (1H, dd, J = 5.4, 1.7 Hz), 4.21 (2H, s); 13C NMR (100 MHz, CDCl3) δ: 152.33, 151.65, 148.79, 134.85, 128.90, 128.70, 127.92, 120.24, 119.43, 35.78; m/z HRMS (DART) found [M+H]+ 236.0307, C12H11ClNS+ requires 236.0295.
4.2.2.11. 4-(benzylthio)-2-bromopyridine (2k)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (68 μL, 0.58 mmol), (2-bromopyridin-4-yl)triphenylphosphonium trifluormethane sulfonate (1k, 284 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel, dry load: 5% EtOAc in hexanes) afforded the title compound (2k) as a white solid (114 mg, 0.41 mmol, 81% yield). mp 59-63 °C; IR νmax/cm−1 (film): 3063, 2918, 1560, 1515, 1453, 1365, 1245, 1069, 981, 821, 765, 712, 678; 1H NMR (400 MHz, CDCl3) δ: 8.12 (1H, dd, J = 5.4, 0.5 Hz), 7.41-7.27 (5H, m), 7.05 (1H, dd, J = 5.4, 1.7 Hz), 4.21 (2H, s); 13C NMR (100 MHz, CDCl3) δ: 152.12, 149.15, 142.29, 134.81, 128.90, 128.72, 127.93, 123.90, 119.79, 35.78; m/z HRMS (DART) found [M+H]+ 278.9718, C12H11BrNS+ requires 279.9790.
4.2.2.12. 4-(benzylthio)-2-fluoropyridine (2l)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), (2-fluoropyridin-4-yl)triphenylphosphonium trifluormethane sulfonate (1l, 284 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel: 5% EtOAc in hexanes) afforded the title compound (2l) as a white amorphous solid (62 mg, 0.28 mmol, 57% yield). IR νmax/cm−1 (film): 3063, 3027, 2918, 1560, 1515, 1453, 1365, 1244, 1069, 821, 765, 712, 677; 1H NMR (400 MHz, CDCl3) δ: 7.99 (1H, d, J = 5.4 Hz), 7.42-7.27 (5H, m), 7.05 (1H, dt, J = 5.5, 1.7 Hz), 4.23 (2H, s); 13C NMR (100 MHz, CDCl3) δ: 163.98 (d, J = 237.49 Hz), 154.65, (d, J = 8.6 Hz), 146.97, (d, J = 16.2 Hz), 135.01, 128.99, 128.77, 127.99, 118.63, (d, J = 3.7 Hz), 105.61 (d, J = 40.4 Hz), 35.910; 19F NMR (365 MHz, CDCl3), −68.26; m/z HRMS (DART) found [M+H]+ 220.0591, C12H11FNS+ requires 220.0591.
4.2.2.13. 4-(benzylthio)-2,2’-bipyridine (2m)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), [2,2'-bipyridin]-4-yltriphenylphosphonium trifluoromethanesulfonate (1m, 283 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel, dry load: 10% EtOAc in hexanes) afforded the title compound (2m) as a white solid (85 mg, 0.31 mmol, 61% yield). mp 99-103 °C; IR νmax/cm−1 (film): 3064, 2918, 1574, 1561, 1534, 1446, 1378, 987, 789, 716, 705, 697; 1H NMR (400 MHz, CDCl3) δ: 8.68 (1H, d, J = 5.3 Hz), 8.45 (1H, d, J = 5.3 Hz), 8.39-8.34 (2H, m), 7.81 (1H, tdd, J = 7.7, 1.6, 1.2 Hz), 7.44 (2H, d, J = 7.6 Hz), 7.37-7.27 (4H, m), 7.13 (1H, dd, J = 5.2, 1.7 Hz), 4.32 (2H, s); 13C NMR (100 MHz, CDCl3) δ: 155.77, 155.71, 150.13, 149.11, 148.64, 136.90, 135.65, 128.89, 128.74, 127.63, 123.86, 121.26, 120.63, 117.92, 35.77; m/z HRMS (DART) found [M+H]+ 279.0958, C15H17N2S+ requires 279.0950.
4.2.2.14. 4-(benzylthio)-5,6,7,8-tetrahydroquinoline (2n)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), triphenyl(5,6,7,8-tetrahydroquinolin-4-yl)phosphonium trifluoromethanesulfonate (1n, 272 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (alumina: 10% EtOAc in hexanes) afforded the title compound (2n) as a white solid (96 mg, 0.38 mmol, 75% yield). mp 126-132 °C; IR νmax/cm−1 (film): 3028, 2934, 2861, 1560, 1435, 711; 1H NMR (400 MHz, CDCl3) δ: 8.19 (1H, d, J = 5.3 Hz), 7.44-7.23 (5H, m), 6.92 (1H, d, J = 5.3 Hz), 4.17 (2H, s), 2.89 (2H, t, J = 6.0 Hz), 2.61 (2H, t, J = 6.0 Hz), 1.90-1.77 (4H, m); 13C NMR (100 MHz, CDCl3) δ: 156.26, 148.38, 146.08, 135.58, 128.84, 128.80, 128.75, 127.63, 116.18, 35.57, 32.91, 25.92, 22.68, 22.54; m/z HRMS (DART) found [M+H]+ 256.1161, C16H18NS+ requires 256.1154.
4.2.2.15. 4-(benzylthio)-3-phenylpyridine (2o)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), triphenyl(3-phenylpyridin-4-yl)phosphonium trifluoromethanesulfonate (1o, 283 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (alumina: 15% EtOAc in hexanes) afforded the title compound (2o) as a white powder (120 mg, 0.44 mmol, 87% yield). mp 93-97 °C; IR νmax/cm−1 (film): 3024, 2920, 2645, 1563, 1453, 1390, 764, 699; 1H NMR (400 MHz, CDCl3) δ: 8.38 (1H, d, J = 5.4 Hz), 8.29 (1H, s), 7.49-7.19 (11H, m), 4.10 (2H, s); 13C NMR (100 MHz, CDCl3) δ: 149.18, 148.29, 147.78, 136.63, 135.42, 135.23, 129.32, 128.85, 128.73, 128.47, 128.29, 127.67, 119.11, 36.21; m/z HRMS (DART) found [M+H]+ 278.1000, C18H16NS+ requires 278.0998.
4.2.2.16. 4-(benzylthio)-5-(3-fluorophenyl)nicotinonitrile (2p)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), (1p, 3-cyano-5-(3-fluorophenyl)pyridin-4-yl)triphenylphosphonium trifluoromethanesulfonate (304 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel: 15% EtOAc in hexanes) afforded the title compound (2p) as a yellow oil (149 mg, 0.47 mmol, 93% yield). IR νmax/cm−1 (film): 3063, 3030, 2230, 1613, 1584, 1545, 1398, 1204, 908, 733, 696; 1H NMR (400 MHz, CDCl3) δ: 8.76 (1H, s), 8.53, (1H, s), 7.47-7.39 (1H, m), 7.24-7.12 (4H, m), 7.06-6.67 (3H, m), 6.92 (1H, d, J = 9.3 Hz), 4.10 (2H, s); 13C NMR (100 MHz, CDCl3) δ: 162.45 (d, J = 246.5 Hz), 152.81, 147.28, 140.05, 137.62 (d, J = 7.9 Hz), 135.62, 130.30 (d, J = 8.3 Hz), 128.94, 128.71, 127.92, 125.40 (d, J = 3.0 Hz), 116.67 (d, J = 22.4 Hz), 115.96 (d, J = 20.9 Hz), 115.06, 39.47; m/z HRMS (DART) found [M+H]+ 321.0870, C19H14FN2S+ requires 321.0856.
4.2.2.17. 4-(benzylthio)-5-(4-methoxyphenyl)pyrimidine (2q)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), (5-(4-methoxyphenyl)pyrimidin-4-yl)triphenylphosphonium trifluoromethanesulfonate (1q, 298 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel: 20% EtOAc in hexanes) afforded the title compound (2q) as a yellow oil (136 mg, 0.44 mmol, 88% yield). IR νmax/cm−1 (film): 3028, 2932, 2835, 1610, 1558, 1522, 1381, 1248, 1118, 1033, 831, 765, 700; 1H NMR (400 MHz, CDCl3) δ: 8.91 (1H, s), 8.24 (1H, s). 7.38-7.30 (4H, m), 7.29-7.18 (3H, m), 6.97-6.92 (2H, m), 4.41 (2H, s), 3.82 (3H, s); 13C NMR (100 MHz, CDCl3) δ: 167.97, 160.10, 156.26, 153.64, 137.00, 132.82, 130.25, 129.25, 128.53, 127.31, 126.54. 114.23. 55.29, 34.30; m/z HRMS (DART) found [M+H]+ 309.1036, C18H17N2OS+ requires 309.1056.
4.2.2.18. 2-(benzylthio)quinoxaline (2r)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), triphenyl(quinoxalin-2-yl)phosphonium trifluoromethanesulfonate (1r, 270 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (silica gel: 5% EtOAc in hexanes) afforded the title compound (2r) as an amorphous tan solid (115 mg, 0.46 mmol, 91% yield). IR νmax/cm−1 (film): 3060, 3028, 1539, 1494, 1247, 1150, 1082, 961, 757, 697; 1H NMR (400 MHz, CDCl3) δ: 8.56 (1H, s), 8.01-7.92 (2H, m), 7.69 (1H, t, J = 7.4 Hz), 7.61 (1H, t, J = 7.3 Hz), 7.46 (2H, d, J = 7.4 Hz), 7.33-7.18 (3H, m), 4.57 (2H, s); 13C NMR (100 MHz, CDCl3) δ: 155.62, 144.53, 142.63, 139.98, 137.33, 130.19, 129.25, 129.18, 128.57, 128.09, 127.80, 127.39, 33.68; m/z HRMS (DART) found [M+H]+ 253.0804, C15H13N2S+ requires 253.0794.
4.2.2.19. 4’-(benzylthio)-5-chloro-6’-methyl-3-(4-(methylsulfonyl)phenyl)-2,3’-bipyridine (2s)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 11 mg, 0.28 mmol), benzyl thiol (33 μL, 0.28 mmol), (5-chloro-6’-methyl-3-(4-(methylsulfonyl)phenyl)-[2,3]bipyridine]-4’-yl) triphenylphosphonium trifluoromethanesulfonate (1s, 192 mg, 0.25 mmol) and THF (1.0 mL). Flash column chromatography (basic alumina: 30% EtOAc in hexanes) afforded the title compound (2s) as a white amorphous solid (78 mg, 0.16 mmol, 65% yield). IR νmax/cm−1 (film): 3061, 1708, 1570, 1544, 1481. 1301, 1252, 1140, 1028, 911, 882, 690, 640; 1H NMR (400 MHz, CDCl3) δ: 8.68 (1H, d, J = 2.2 Hz), 7.94, (1H, s), 7.8107.72 (3H, m), 7.34-7.21 (7H, m), 7.01 (1H, s), 4.08 (2H, s), 3.03 (3H, s), 2.47 (3H, s); 13C NMR (100 MHz, CDCl3) δ: 158.25, 151.80, 149.17, 148.51, 148.24, 143.14, 140.13, 137.60, 136.84, 135.28, 131.89, 130.11, 129.93, 128.95, 128.89, 127.93, 127.62, 119.12, 44.56, 36.24, 24.57; m/z HRMS (DART) found [M+H]+ 481.0792, C25H22ClN2O2S2+ requires 481.0806.
4.2.2.20. ethyl-4-(4-benzylthio)-8-chloro-5, 6-dihydro-11H-benzo[5,6]cyclohepta[1,2-b]pyridine-11-ylidene)piperidine-1-carboxylate (2t)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), (8-chloro-11-(1-(ethoxycarbonyl)piperidin-4-ylidene)-6,11-dihydro-5H-benzo[5,6]cyclohepta[1,2-b]pyridin-4-yl)triphenylphosphonium trifluoromethanesulfonate (1t, 397 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (basic alumina, dry load, gradient elution: 20% EtOAc in hexanes to 30% EtOAc in hexanes) afforded the title compound (2t) as a white solid (129 mg, 0.36 mmol, 51% yield). mp 200-203 °C; IR νmax/cm−1 (film): 2989,2901, 1694, 1548, 1431, 1216, 1108, 1001, 764, 712, 697; 1H NMR (400 MHz, CDCl3) δ: 8.23 (1H, d, J = 5.3 Hz), 7.40-7.24 (5H, m), 7.16-7.05 (3H, m), 6.99 (1H, d, J = 5.3 Hz), 4.18-4.08 (4H, m), 3.88-3.68 (2H, m), 3.42-3.31 (1H, m), 3.18-3.04 (3H, m), 2.92-2.71 (2H, m), 2.52-2.39 (1H, m), 2.38-2.23 (3H, m), 1.24 (3H, t, J = 7.1 Hz); 13C NMR (100 MHz, CDCl3) δ: 156.28, 155.61, 149.07, 146.31, 139.75, 137.95, 137.67. 135.37, 134.25, 133.07, 130.35, 129.87, 128.95, 128.94, 128.88, 127.91, 126.33, 117.96, 61.45, 44.88, 36.28, 30.97, 30.75, 28.79, 14.83; m/z HRMS (DART) found [M+H]+ 505.1705, C29H30ClN2O2S+ requires 505.1711.
4.2.2.21. (1S,2S4S,5R)-2-((R)-benzyloxy)(2-(benzylthio)quinoline-4-yl)methyl)-5-vinylquinuclidine (2u)
Prepared according to general procedure using sodium hydride (60% dispersion in mineral oil, 22 mg, 0.55 mmol), benzyl thiol (65 μL, 0.55 mmol), (4-((R)-(benzyloxy)((1S,2S,4S,5R)-5-vinylquinuclidin-2-yl)methyl)quinolin-2-yl)triphenylphosphonium trifluoromethanesulfonate (1u, 397 mg, 0.50 mmol) and THF (2.0 mL). Flash column chromatography (basic alumina: 30% EtOAc in hexanes) afforded the title compound (2u) as a yellow amorphous solid (157 mg, 0.31 mmol, 62% yield). IR νmax/cm−1 (film): 3063, 3029, 2928, 2863, 1591, 1549, 1452, 1290, 1094, 907, 758, 729, 697; 1H NMR (400 MHz, CDCl3) δ: 8.09-7.95 (2H, m), 7.66 (1H, dd, J = 8.2, 7.1 Hz), 7.55-7.42 (3H, m), 7.39-7.18 (9H, m), 5.80-5.66 (1H, m), 5.19 (1H, br s), 4.99-4.85 (2H, m), 4.61 (2H, s), 4.45 (1H, d, J = 11.4 Hz), 4.37 (1H, d, J = 11.4 Hz), 3.44-3.28 (1H, m), 3.17-3.00 (2H, m), 2.72-2.54 (2H, m), 2.30-2.18 (1H, m), 1.86-1.39 (4H, m); 13C NMR (100 MHz, CDCl3) δ: 158.90, 148.85, 145.88, 142.07, 138.48, 137.89, 129.48, 129.32, 129.13, 128.59, 128.55, 127.88, 127.17, 125.47, 124.57, 123.39 (br), 118.05 (br), 114.31, 81.17 (br), 71.51, 60.67, 57.22, 43.28, 40.18, 34.03, 28.06, 27.85, 22.40 (br); m/z HRMS (DART) found [M+H]+ 507.2484, C33H35N2OS+ requires 507.2465.
Supplementary Material
Acknowledgments
We acknowledge Colorado State University for startup funds and the ACS Petroleum Research Fund (ACS PRF56878-DNI1) for supporting this project.
Footnotes
Supplementary Material
Supplementary data related to this article can be found at https://doi.org/10.1016/j.tet.2017.12.040.
References and notes
- 1.(a) Feng M; Tang B; Liang S; Jiang X Curr. Top. Med. Chem 2016, 16, 1200; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ilardi EA; Vitaku E; Njardarson JT; J. Med. Chem 2014, 57, 2832; [DOI] [PubMed] [Google Scholar]; (c) Tan PK; Liu S; Gunic E; Miner JN Sci. Rep 2017, 7, 665; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu L; Stelmach JE; Swaminathan R; Natarajan R; Chen MH; Singh SB; Schwartz CD; Fitzgerald CE; O’Keefe SJ; Zaller DM; Schmatz DM; Doherty JB Bioorg. Med. Chem. Lett 2003, 13, 3979; [DOI] [PubMed] [Google Scholar]; (e) Berndt A; Miller S; Williams O; Le DD; Houseman BT; Pacold JI; Gorrec F; Hon WC; Ren P; Liu Y; Rommel C; Gaillard P; Ruckle T; Schwarz MK; Shokat KM; Shaw JP; Williams RL Nat. Chem. Biol 2011, 6, 117; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Hopkins AL; Ren J; Milton J; Hazen RJ; Chan JH; Stuart DI; Stammers DK J. Med. Chem 2004, 47, 5912. [DOI] [PubMed] [Google Scholar]
- 2.(a) Beletskaya IP; Ananikov VP Chem. Rev 2011, 111, 1596; [DOI] [PubMed] [Google Scholar]; (b) Lee C-F; Liu Y-C, Badsara S Chem. Asian J 2014, 9, 706; [DOI] [PubMed] [Google Scholar]; (c) Ling R; Yoshida M; Mariano PS J. Org. Chem 1996, 61, 4439–4449; [DOI] [PubMed] [Google Scholar]; (d) Sujatha A; Thomas AM; Thankachan AP; Anilkumar G Arkivoc 2014, 2015, 1; [Google Scholar]; (e) Hartwig JF Acc. Chem. Rev 2008, 41, 1534; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Murata M; Buchwald SL Tetrahedron 2004, 60, 7397; [Google Scholar]; (g) Sayah M; Organ MG Chem. Eur. J 2011, 17, 11719; [DOI] [PubMed] [Google Scholar]; (h) Oderinde MS; Frenette M; Robbins DW; Aquila B; Johannes JW J. Am. Chem. Soc 2016, 138, 1760; [DOI] [PubMed] [Google Scholar]; (i) Jouffroy M; Kelly CB; Molander GA Org. Lett 2016, 18, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Representative examples of SNAr reactions: Zhang X; Lu G-P; Cai C Green Chem 2016, 18, 5580; [Google Scholar]; (b) Beugelmans R; Bois-Choussy M; Boudet B Tetrahedron 1983, 39, 4153; [Google Scholar]; (c) Furukawa N; Ogawa S; Kawai T J. Chem. Soc. Perkin Trans. 1 1984, 1839; [Google Scholar]; (d) Bhagwat A; Campi EM; Potdar MK; Jackson RW; Hearn MTW Green Chem. Lett. Rev 2012, 5, 595; [Google Scholar]; (e) Omar WAE; Heiskanen JP; Hormi OEO J. Heterocyclic Chem 2008, 45, 593; [Google Scholar]; (f) Takashiro E; Nakamura Y; Fujimoto K Tetrahedron Lett 1999, 40, 5565; [Google Scholar]; (g) Cherng Y-J Tetrahedron 2002, 58, 4931. [Google Scholar]
- 4.(a) Hilton MC; Dolewski RD; McNally A J. Am. Chem. Soc 2016, 138, 13806; [DOI] [PubMed] [Google Scholar]; (b) Zhang X; McNally A Angew. Chem. Int. Ed 2017, 56, 9833; [DOI] [PubMed] [Google Scholar]; (c) Anders E; Markus F Tetrahedron Lett 1987, 28, 2675. [Google Scholar]; (d) Anders E; Markus F Chem. Ber 1989, 122, 112; [Google Scholar]; (e) Anders E; Markus F Chem. Ber 1989, 122, 119; [Google Scholar]; Haas M; Goerls H; Anders E Synthesis 1998, 195; [Google Scholar]; (f) Bull JA; Mousseau JJ; Pelletier G; Charette AB Chem. Rev. 2012, 112, 2642. [DOI] [PubMed] [Google Scholar]; (g) Sammes MP; Leung CWF; Mak CK; Katritzky AR J. Chem. Soc. Perkin Trans. 1 1981, 1585. [Google Scholar]
- 5.(a) Finer J-P Ligand Coupling Reactions with Heteroaromatic Compounds. Tetrahedron Organic Chemistry Series; Pergamon Press: Oxford, 2009; Vol. 18, p 95; [Google Scholar]; (b) Byrne PA; Ortin Y; Gilheany DG Chem. Commun 2014, 51, 1147. [DOI] [PubMed] [Google Scholar]
- 6.(a) Razuvaev GA; Osanova NA J. Organomet. Chem 1972, 38, 77; [Google Scholar]; (b) Razuvaev GA; Osanova NA; Brilkina TG; Zinovjeva TI; Sharutin VV J. Organomet. Chem 1975, 99, 93; [Google Scholar]; (c) Eyles CT; Trippett SJ Chem. Soc. C 1966, 67; [Google Scholar]; (d) Grayson M; Keough P-TJ J. Am. Chem. Soc 1960, 82, 3919. [Google Scholar]
- 7.Bedford MS; Yang X; Jolly KM; Binnicker RL; Cramer SB; Keen CE; Mairena CJ; Patel AP; Rivenbank MT; Galabura Y; Luzinov I; Smith RC Polym. Chem 2015, 6, 900. [Google Scholar]
- 8.1H NMR yields of phosphonium ion displacement with double thiolate addition shown in parentheses: 2j – 92% (1%); 2k – 93% (2%), 2l – 84% (8%). 1,3,5-trimethoxybenzene was used as an internal standard.
- 9.Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW Chem. Soc. Rev 2016, 45, 546. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.