

CRISPR guide RNA scaffolds have been adapted to carry multiple binding sites for fluorescent proteins to enhance brightness for live cell imaging of genomic loci. However, many of these modifications result in guide RNA instability and thus lower genome labeling efficiency than anticipated. Here we introduce CRISPR-Sirius, based on octet arrays of aptamers conferring both enhanced guide RNA stability and brightness and provide initial biological applications of this new platform.
How chromosomes are spatially and dynamically organized within the nucleus and how genomic 3D structures govern transcription and other nuclear processes is an area of substantial current interest1–2. Chromosome conformation capture (3C)-based techniques measure the contact frequency between locus pairs in a given cell population1. However, the spatial distance between locus pairs often differs among individual cells when observed in fixed cells via DNA fluorescence in situ hybridization (FISH)3. Live cell imaging can identify these spatial and temporal features of genomic elements at the single-cell level, providing novel information unavailable in static datasets.
The CRISPR-Cas9 system was repurposed for tracking chromosomal loci in living cells4, and several multicolor CRISPR-based imaging systems were subsequently developed5–9. In our “CRISPRainbow” system7, six loci each containing >100 copies of the dCas9 target sites on six distinct chromosomes were visualized simultaneously in living cells. Nevertheless there are very few high-copy chromosome-specific loci in human genome shown in Supplementary Fig. 1a-1b and at our webserver named CRISPRbar (http://genome.ucf.edu/CRISPRbar/). Hence, it becomes essential to generate a more sensitive, multicolor CRISPR-based imaging system.
We previously established that the stability of guide RNAs determines the labeling efficiency10. Using the “Broccoli” system10, 11(Fig. 1a) to visualize RNA in living cells, we found that insertion of RNA aptamers at the 3’-end of the guide RNA scaffold results in much lower guide RNA levels than insertion in the tetraloop (Supplementary Fig. 2a and 2b). As shown in Fig. 1b and 1c, a similar effect on the labeling efficiency was observed when targeting to the pericentromeric region of chromosome 9 (C9–1), which contains thousands of target sites5. Thus, we suspect both the optimal structure of multiplexed RNA aptamers and their insertion site in the guide RNA scaffold are key parameters for the efficient live cell labeling.
Figure 1. Development of CRISPR-Sirius, a bright and multicolor DNA imaging system.
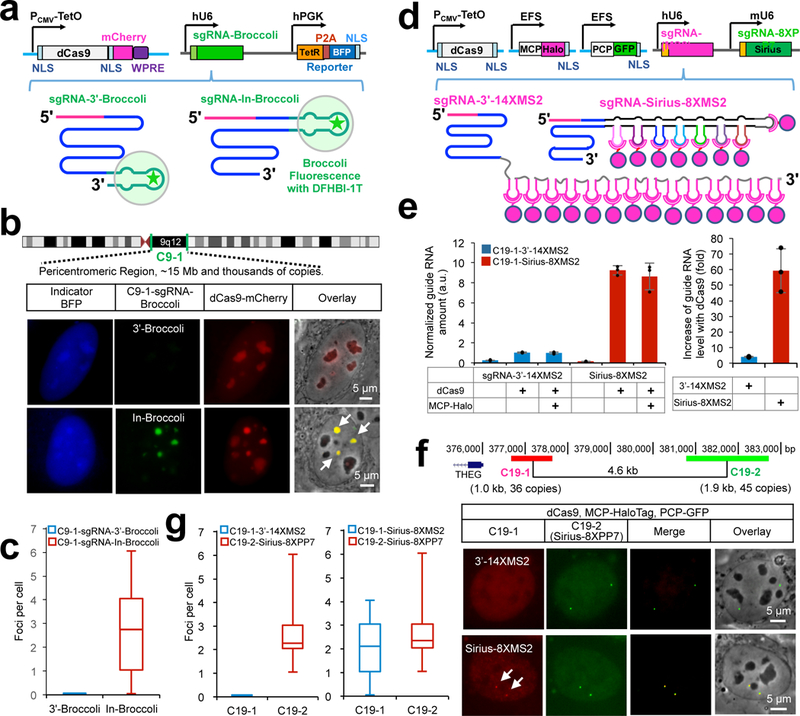
(a) Diagram of the CRISPR sgRNA-Broccoli system and the details of this system are described in the Supplementary Fig. 2a.
(b) Visualization of C9–1 (an pericentromeric region on chromosome 9, upper row). Localization of BFP report (blue), dCas9-mCherry (red), and C9–1-sgRNA–Broccoli (green) expressed in U2OS cells. The C9–1 foci are indicated by the arrows. Scale bar: 5 μm. The images with the same color were scaled to the same minimal and maximal levels.
(c) Box plot showing the number of foci per cell for C9–1 loci using the sgRNAs-3’-Broccoli and sgRNA-In-Broccoli. The line within the boxplot represents the mean; the outer edges of the box are the 10th and 90th percentiles; the whiskers extend to the minimum and maximum values; n=125 cells (left) and 119 cells (right).
(d) Diagram of the strategies for multiplexed RNA aptamers tagging to sgRNA. dCas9 expression was under the control of CMV-TetO promoter, while MCP-HaloTag and PCP-GFP was expressed via elongation factor 1α (EFS) promoter. Dual sgRNAs were cloned into the same plasmid. SgRNA-Sirius-8XPP7 was used an internal control, while sgRNA-3’−14XMS2 and sgRNA-Sirius-8XMS2 were used for direct comparison.
(e) RT-PCR analysis of sgRNA levels. The guide RNA levels of C19–1-sgRNA-3’−14XMS2 and C19–1sgRNA-Sirius-8XMS2 in the presence or absence of dCas9 or MCP-HaloTag were measured by RT-PCR. All data are presented as the mean ± s.d.; n=3 independent experiments; black dots represent individual data points.
(f) C19–1 and C19–2 targets (upper row) were used for dual color CRISPR imaging. Localization of C19–2 (green) as an internal control, C9–1 (red) was tested using C19–1-sgRNA-3’−14XMS2 or C19–1-sgRNA-Sirius-8XMS2 in U2OS cells. The C19–1 foci are indicated by the arrows. Scale bar: 5 μm. The images with the same color were scaled to the same minimal and maximal levels.
(g) Box plot showing the number of foci per cell for C19–1 and C19–2 loci counts when using C19–1-sgRNA-3’−14XMS2 or C19–1-sgRNA-Sirius-8XMS2 along with C19–2-Sirius-8XPP7. n=124 cells (left panels) and 116 cells (right panels).
MS2 and PP7 RNA aptamers have previously been inserted into the CRISPR RNA scaffold for imaging of genomic loci7. Here we tested inserting octets of MS2 aptamers into the tetraloop (sgRNA-In-8XMS2, Supplementary Fig. 3a) for signal amplification and found that this resulted in barely detectable labeling of FBN3 repeats located in intron 10 of human FBN3 gene, which consists of 22 copies of the target sites in a ~800 bp region (Supplementary Fig. 3d and 3e). We therefore rationally designed thermostable octets of MS2 aptamers linked by three-way junctions to create stable RNA secondary structures and generated sgRNA-In-8XMS2T (Supplementary Fig. 3b) and observed the U2OS cells displayed 1–3 labeled foci (Supplementary Fig. 3d and 3e). We then proceeded to introduce mutations into individual MS2 hairpins to minimize both misfolding of the transcripts and minimize recombination during virus production12. We named this design CRISPR sgRNA-Sirius (Supplementary Fig. 3c). This modification resulted in higher percentage of cells displaying labeling of the 1–4 target foci (Supplementary Fig. 3d and 3e). Supplementary Fig. 4 illustrates the flowchart for rational design of the Sirius-8XMS2 scaffold.
It has been reported that 14 copies of the MS2 (14XMS2) introduced at 3’-end of the guide RNA scaffold (sgRNA-3’−14XMS2) can be used for detection of low copy-number target sites13. We directly compared the stability and labeling efficiency of sgRNA-3’−14XMS2 and sgRNA-Sirius-8XMS2 (Fig. 1d). As shown in Supplementary Fig. 5a-5c, C19–1 signals were not detectable when sgRNA-3’−14XMS2 was used. On the contrary, majority of the cells had 2 foci when sgRNA-Sirus-8XMS2 was used. To probe the intracellular stability of these engineered guide RNAs, we performed real time PCR analysis (Fig. 1e). This revealed a low level of sgRNA-3’−14XMS2 or sgRNA-Sirius-8XMS2 in absence of dCas9, which is consistent with our previous finding that guide RNAs are very unstable without dCas910. In the presence of dCas9, sgRNA-Sirius-8XMS2 level strikingly increased ~60 fold, whereas the level of sgRNA-3’−14XMS2 only increased ~4 fold (Fig. 1e). The presence of MCP-HaloTag14 didn’t affect the guide RNA levels in either sgRNA-3’−14XMS2 or sgRNA-Sirius-8XMS2. These results indicated that the instability of sgRNA-3’−14XMS2 is the bottleneck for its labeling efficiency, which is consistent our previous findings that the sgRNA level is the rate limiting of CRISPR-based labeling10. We further compared the labeling efficiency of sgRNA-3’−14XMS2 and sgRNA-Sirius-8XMS2 by introducing sgRNA-Sirius-8XPP7 in the same plasmid for targeting another locus as an internal control for dual-color detection (Fig. 1d). As shown in Fig. 1f and 1g, more than 90% of cells showed the C19–1 signals from sgRNA-Sirius-8XMS2 in these C19–2 positive cells, while none of them showed C19–1 signals from sgRNA-3’−14XMS2. Similar results were obtained when the C19–1-sg16XMS2 was tested in the same way (Supplementary Fig. 6). To confirm the increased sensitivity, we compared the foci brightness of CRISPR-Sirius or CRISPRainbow by labeling the FBN3 intronic repeat (22 copies of target sites, Supplementary Fig. 7a) in U2OS cell. As seen in Supplementary Fig. 7b and 7c, the FBN3 target signals with CRISPR sgRNA-Sirius-8XMS2 or 8XPP7 were considerably brighter than the signals from CRISPRainbow-2XMS2 or 2XPP7 and also observed that the average ratio of signal intensity to nuclear background increased as well (Supplementary Fig. 7d). Additionally, we also generated CRISPR sgRNA-Sirius-4X(MS2-PP7) (Supplementary Fig. 8a) as a tricolor platform to visualize multiple distinct loci on the same chromosome. Using this latter system, two subtelomeric regions (T1 and T2) and one pericentromeric region (PR1) of chromosome 19 could be visualized simultaneously and the T1 to T2 inter-locus distance could be measured (Supplementary Fig. 8b).
To measure the spatial distance and dynamics of pairs of loci ranging from kilobases to megabases apart on chromosome 19, we mined chromosome 19-specific repeats containing ≥5 target site copies. These repeats were classified by their genomic locations and copy numbers of target sites (Supplementary Fig. 9a-9c). Previous CRISPRainbow system allowed us to detect repeats having ≥100 copies7, now we tested all repeats having ≥20 copies by CRISPR sgRNA-Sirius and found 26 out of these 46 loci displayed detectable signals in human U2OS cells (Supplementary Fig. 10). Seven locations (four intergenic DNA regions (IDRs), two intronic regions (TCF3 and FBN3) and one pericentromeric region (PR1)) distributed on the p-arm of chromosome 19 were chosen for further analysis of loci pairs with distinct length scales (Fig. 2a). We created a dual-guide RNA expression vector for one-step generation of each pair of guide RNAs (Supplementary Fig. 11). IDR3 labeled by CRISPR-Sirius-8XPP7 was used as the common reference locus in all cases while CRISPR-Sirius-8XMS2 was used to label the other loci (IDR1, IDR2, TCF3, IDR4, FBN3 and PR1, see Fig. 2a). All six pairs of loci were readily visualized in individual cells. Consistent with the copy number variation (CNV) from whole genome sequences of U2OS cells (Fig. 2a), two foci were detected for IDR1, IDR2, TCF3, IDR3, IDR4 and FBN3 in these presumably G1 cells and only one site was detected for PR1, possibly due to the existence of a deletion mutation in one allele (Supplementary Fig. 12a-12b and Fig. 2b). There was a considerably higher percentage of cells with 3–4 foci for TCF3, IDR4 and FBN3 than IDR1–3 and PR1 (to the latter displaying 2 foci) suggesting that the former loci might have the earlier replication timing15 (Fig. 2b). In further analysis we quantified the average inter-locus distance of each pair (Fig. 2c), which revealed the distinct correlation of spatial distances and genomic distances at the kilobase and megabase scales, suggesting a diversity of chromatin folding states. The observation at the megabase scale is consistent with the results of a DNA FISH analysis in fixed cells16. To further validate the loci detected by the CRISPR-Sirius system, we used two different guide RNAs targeting to the same locus, viz. IDR3. As shown in Supplementary Fig. 12c and Supplementary video 1-3, two different guide RNAs targeting to the same IDR3 perfectly overlapped in their spatial localization and co-movements. We also analyzed the dynamic behaviors of IDR1/IDR3 locus pair and observed a spatiotemporal pattern (Fig. 2d and Supplementary video 4-6) indicative of sister chromatid separation/fusion events17, tracked here on a time resolution of seconds.
Figure 2. Imaging locus pairs from kilobases to megabases by CRISPR-Sirius.
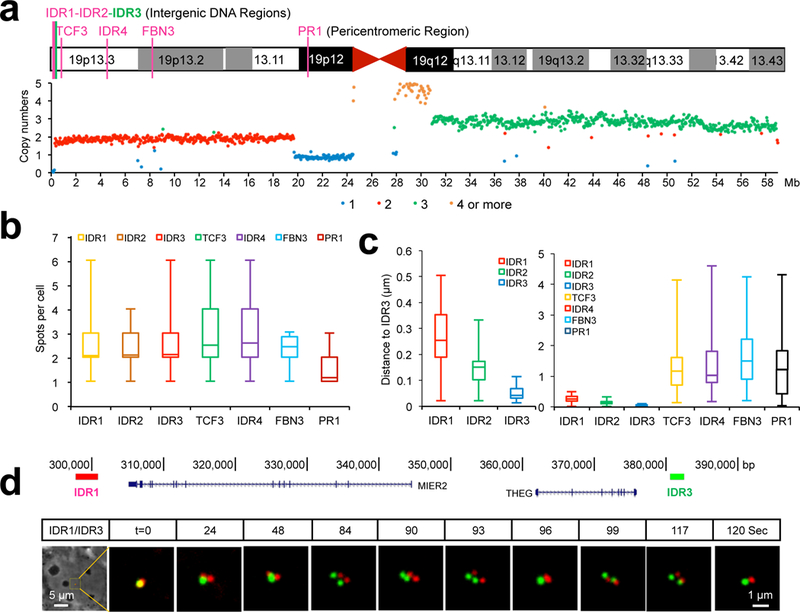
(a) Schematic of seven unique loci on human chromosome 19 used in this study consisting of 4 intergenic DNA regions (IDRs), 2 intronic regions (TCF3 and FBN3) and 1 pericentromeric region (PR1). Shown beneath chromosome map are the copy numbers of each locus.
(b) Box plot showing the number of foci per cell for the seven loci described above. n=178, 173, 227, 271, 291, 183 and 336 cells from left to right respectively.
(c) Box plot showing the variance of the observed inter-locus distance of each locus pair. n=39 loci for IDR1, 63 loci for IDR2, 24 loci for IDR3, 54 loci for TCF3, 54 loci for IDR4, 78 loci for FBN3 and 53 loci for PR1.
(d) Time lapse of the IDR1/IDR3 locus pair over two minutes. The diagram shows the genomic distance between IDR1 and IDR3 (upper panel). Phase contrast images and dual-color time lapse images were shown in the bottom panel.
Microscopy and 3C-based methods have revealed that genomes are spatially organized in a hierarchical manner in the nucleus, with implications for cellular functions, but in a static mode1. Live-cell DNA imaging becomes an essential approach to uncover the dynamic features of genomic regions at different spatial and temporal scales. Here we developed a thermostable CRISPR-Sirius system allowing to efficiently label a series of genomic loci on the same chromosome. This work provides a foundation for study of the dynamics of genes, promoters, enhancers and various genomic elements in space and time during development and disease in live cells. It will now be worthwhile to explore more stable and fluorescent RNA origami18 positioned in the tetraloop of guide RNA scaffold to expand the color range and boost the sensitivity yet further. Moreover, to the extent that the mechanism of sister chromatid resolution at fine time scales has not been probed, our initial findings (Fig. 2d) prompt investigation of whether the observed dynamics of sister chromatid separation and fusion occur in the G2 phase, and are driven by DNA loop extrusion19 or phase separation20.
METHODS
Mining chromosome-specific repeats for the human genome
Human reference genome (assembly GRC h37/hg19) (genome.ucsc.edu) was analyzed to find target regions and design gRNAs. Bioinformatics tool Tandem Repeat Finder21 was used to identify tandem repeats with repeats period length smaller or equal to 2000 bp in the human genome. Bioinformatics tool Jellyfish22 was used to identify tandem repeats with repeat length longer than 2000 bp in human genome. Jellyfish was used to search for the 15-mers in the identified repeat regions. All the tandem repeat regions with more than 5 non-overlapping copies of one 15-mer were selected. The non-overlapping repetitive 15-mers with CRISPR PAM sequences ending with NGG or starting with CCN were examined for their specificity. The 15-mers that had more than 20% of copies within in other 50 kb regions were discarded. The 15-mers containing “TTTT” or ending with “TNGG” were filtered out due to the potential pre-termination on sgRNA expression under the U6 promoter4. The distribution of unique repeats in human genome was shown in Supplementary Fig. 1 and chromosome 19-specific unique repeats identified by the above-mentioned bioinformatics pipeline was shown in Supplementary Fig. 9. The copy numbers shown in the figures were defined as the maximal non-overlapping target sites from a single sgRNA in the region.
Design CRISPR sgRNA-Sirius scaffolds
To design a stable RNA scaffold accommodating multiple RNA aptamers and compatible for insertion into sgRNA, the aptamers were linked by tandem three way junctions23. For CRISPR Sirius-8XMS2, we randomized the linker of three way junctions between each MS2 stem loop and made the synonymous mutations of 8XMS224 in the scaffold. To design the variants, we used the consensus sequences as shown below:

where Y was replaced with C or U, the D was replaced with A, G or U, the S with C or G, R with G or A, and N with any nucleotide. The Sirius-8XMS2 scaffold was designed to avoid a repeating 8-mer in the sequences and to optimize RNA secondary structures. The detailed design of Sirius-8XMS2 is shown in Supplementary Fig. 4. The RNA sequence was iteratively evolved by increasing the thresholds (X) for candidate sub-optimal structures. mFold25 was used to fold the RNA sequence and compute minimum free energy (MFE) and suboptimal free energy (SFE). Initially, all mutable residues were replaced in the base-paring manner shown in the Supplementary Fig. 4 while preserving the A-U or C-G pairs. If the generated sequence contained any repetitive 8-mer, all the mutable residues were mutated again. The sequences were then folded with the initial sub-optimally percentage X=5%. If there was unique structure within the SFE structure, the sequence was fixed and stored. The process was then continued to increase the sub-optimally threshold (X) by 1.0 and the sequence was folded again. Unstable regions were identified if any other structure was predicted within the SFE. Those regions were then marked for further mutation and the process continued until the sub-optimally percentage exceeds 10% or the number of iterations exceeded a given threshold (1000). For CRISPR Sirius-8XPP7, we adapted the three-way junction linkers from CRISPR Sirius-8XMS2 and used the PP7 aptamer mutants that had the least reduction on the PCP binding26, resulting in the CRISPR Sirius-8XPP7. The CRISPR Sirius-4X(MS2-PP7) was generated by alternative MS2 and PP7 in the RNA aptamer octet.
Identification of copy number variation of U2OS cells
The mapped pair-end whole genome sequencing reads for the osteosarcoma cell line (U2OS)27 were downloaded. The bam file was sorted by reference coordinates using samtools28. Control-FREEC29 was used to find the copy number alternations from the sorted bam file. Control-FREEC can detect copy number alternations and allelic imbalance from sequencing data without requiring control data. The window size for Control-FREEC was set to 50,000 bps. Different ploidy numbers were used: 2,3,4, and 5. The ploidy number 3 was able to explain the most observed copy number alternations (0.847636) and it was selected subsequently.
Plasmid construction
The expression vector for dCas9 (nuclease-dead) from S. pyogenes was that originally constructed from pHAGE-TO-DEST5 into which mCherry, GFP or P2A-HSA (Heat Stable Antigen) was inserted at the C-terminus resulting in pHAGE-TO-dCas9-mCherrry, pHAGE-TO-dCas9-GFP and pHAGE-TO-dCas9-P2A-HSA respectively. PCP-GFP7 expressed from pHAGE-EFS-PCP-GFPnls was previously described and HaloTag14 were subcloned to replace the GFP in the pHAGE-EFS-MCP-GFPnls plasmid. The expression vector for guide RNAs was based on the pLKO.1 lentiviral expression system, Hygromycin, TetR-P2A-BFP or PUR-P2A-BFP was inserted right after the PGK promoter to generate pLH-sgRNA, pTetR-P2A-BFPnls-sgRNA or pPUR-P2A-BFPnls-sgRNA respectively. A series of modified sgRNA cassettes under the control of human or mouse U6 promoters used in this study are listed in Supplementary Table 1. The sgRNA-3’−14XMS2 and sg16XMS2 were subcloned from sg14x(MS2) MUC4.113. The one-step generation of paired guide RNAs was performed by simultaneously subcloning into the cassettes hU6-sgRNA-Sirius-8XMS2 and mU6-sgRNA-Sirius-8XPP7 into pPUR-P2A-BFPnls vector, resulting the dual-guide RNA expression vector pPUR-P2A-BFPnls- hU6-sgRNA-Sirius-8XPP7-mU6-sgRNA-Sirius-8XMS2, containing the CcdB gene between two Bbs I sites in each cassette with different cohesive sites. The details of the cloning strategy were shown in Supplementary Fig. 11. The dCas9 and sgRNA-Sirius expression vector reported here will be deposited at Addgene.
Cell culture and transfection
Human osteosarcoma U2OS cells were cultured on 35 mm glass bottom dishes (MatTek) at 37°C in Dulbecco-modified Eagle’s Minimum Essential Medium (DMEM; Life Technologies) containing high glucose and supplemented with 10% (vol/vol) fetal bovine serum. For transfection, typically 20 ng each of PCP-GFP and MCP-HaloTag, 200 ng of dCas9 plasmid DNA and 1 μg of plasmid DNA for desired guide RNAs were co-transfected using Lipofectamine 2000 (Life Technologies) and the cells were incubated for another 24–72 hours before imaging.
Quantitative real-time PCR
Cells were transfected as described in previous sections. Briefly, 200 ng of dCas9 plasmid DNA, 50 ng of MCP-Halo and 1 μg of total guide RNA plasmid DNA were cotransfected using Lipofectamine 3000 (Thermo Fisher Scientific), and the cells were incubated for another 48–72 h before harvest. RNA was extracted with an RNeasy Plus Mini Kit (QIAGEN) and then subjected to RT-PCR using the following primers and probe (Integrated DNA Technologies) for C19–1-Sirus-8XMS2–guide RNA: Forward primer: 5′-GGCAGTAGCAAGTTTAAATAAG−3′; complementary to nt 315–336 of the RNA; Probe: 5′-TTCAAGTTGATAACGGACTAGC−3′; complementary to nt 337–358 of the RNA; Reverse primer: 5′-GACTCGGTGCCACTTT−3′; complementary to 374–359 nt of the RNA. The target sequence is located at nt 40–100 of C19–1-3’−14XMS2-guide RNA. Identical reagents and concentration were used to detect C19–1-3’−14XMS2-guide RNA except five nt of the forward primer at 5’ end was replaced to optimize the annealing temperature. The forward primer for C19–1-3’−14XMS2-guide RNA was 5′-CAGCATAGCAAGTTTAAATAAG−3′; complementary to nt 35–56 of the RNA. For BFP RNA from the same plasmid carrying guide RNA: Forward primer: 5′-CGCCAAGACCACATATAGATCC−3′; complementary to nt 531–552 of the RNA; Probe: 5′-ACCCGCTAAGAACCTCAAGATGCC−3′; complementary to nt 558–581 of the RNA; Reverse primer: 5′-TGGCCTCCTTGATTCTTTCC−3′; complementary to 628–609 nt of the RNA. The DNA Primetime qPCR kit (Hs.PT.39a.22214847, Integrated DNA Technologies) were used for quantification of β-actin mRNA. BFP RNA produced from the same plasmid of guide RNA (Supplementary Table 1) was used as the calibration standard for transfected plasmid DNA. All data were normalized for the cell number using β-actin mRNA as the internal reference.
Lentivirus production and transduction
HEK293T cells were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM; Fisher Scientific) containing high glucose and supplemented with 1% GlutaMAX (Life Technologies), 10% fetal bovine serum (Hycolne FBS, Thermo Scientific) and 1% each penicillin and streptomycin (Life Technologies). 24 hours before transfection, approximately 5×105 cells were seeded in 6-well plates. For each well, 0.5 μg of pCMV-dR8.2 dvpr (Addgene), 0.3 μg of pCMV-VSV-G (Addgene), each constructed to carry HIV LTRs, and 1.5 μg of plasmid containing the gene of interest were co-transfected by using TransIT transfection reagent (Mirus) according to manufacturer’s instructions. After 48 hours, the virus was collected by filtration through a 0.45 μm polyvinylidene fluoride filter (Pall Laboratory). The virus was immediately used or stored at −80 °C. For lentiviral transduction, U2OS cells maintained as described above were transduced by Spinfection in 6-well plates with lentiviral supernatant for 2 days and ~2×105 cells were combined with 1 ml lentiviral supernatant and centrifuged for 30 minutes at 1200 x g.
Flow cytometry and stable cell selection
Cells expressing the desired fluorescent Cas9 and/or guide RNA were selected using a FACSAria cell sorter (BD Bioscience) equipped with 405, 488, 561 and 640 nm excitation lasers, and the emission signals were detected by using filters at 450/50 nm (wavelength/bandwidth) for the Brilliant Violet 421-conjugated anti-mouse CD24 antibody (BioLegend) staining of the HSA, 530/30 nm for PCP-GFP and 582/15 nm for MCP-HaloTag stained with HaloTag-JF549. For the sorting of dCas9 signals, 1 μl of the Brilliant Violet 421-conjugated anti-mouse CD24 antibody was added in a 100 μl cell solution for 30 minutes before FACS. For sorting of MCP-HaloTag, HaloTag-JF549 was added to the cells at 2 nM 18–24 hours before sorting. Single cells were sorted into single wells of 96-well plates containing 1% GlutaMAX, 20 % fetal bovine serum and 1% penicillin and streptomycin in chilled DMEM medium. Positive clones of U2OSdCas9-HSA/PCP-GFP/MCP-HaloTag were selected from 96-well plates 10 days later. To generate stable cell lines in which the IDR2/IDR3 locus pair was labeled, the U2OSdCas9-HSA/PCP-GFP/MCP-HaloTag cell line was transduced for 48 hours by lentivirus for PUR-P2A-BFP-hU6-IDR2-sgRNA-Sirius-8XMS2-mU6-IDR3-sgRNA-Sirius-8XPP7 for 48 hours. Cells were then selected with 1μg/ml puromycin for 3–5 days before sorting for BFP, using filters at 405 nm excitation and 450/50 nm emission. The resulting cell lines was simply named U2OSIDR2/IDR3. The stable cell lines with other locus pairs were generated by the same procedures.
Fluorescence microscopy
A Leica DMIRB microscope was equipped with an EMCCD camera (Andor iXon-897), mounted with a 2x magnification adapter and 100x oil objective lens (NA 1.4), and resulting in a total 200x magnification equal to a pixel size of 80 nm in the images was used. The microscope stage incubation chamber was maintained at 37 °C in HEPES-buffered DMEM with 10% FBS. GFP was excited with an excitation filter at 470/28 nm (Semrock) and its emission was collected using an emission filter at 512/23 nm (Semrock). HaloTag-JF549 was excited at 556/20 nm (Semrock) and its emission was collected in a 630/91 nm channel. Imaging data were acquired by MetaMorph acquisition software (Molecular Devices). Image size was adjusted to show individual nuclei and intensity thresholds were set on the basis of the ratios between nuclear focal signals to background nucleoplasmic fluorescence. To detect loci numbers, maximum intensity projection of Z-series images was performed. To quantify the spatial distance or track the dynamics, only pairs of loci lying in the same foci plane were analyzed.
Imaging processing
The images were analyzed by the Fiji (http://fiji.dc/Fiji) and Mathematica (Wolfram) software. Images from the green and red channels were registered by using 0.1 μm coverglass-absorbed TetraSpeck fluorescent microsphere (Invitrogen) as a standard sample. Intensity quantification in Supplementary Fig. 7d was performed as following
where IR is the intensity ratio between the labeled FBN3 loci (IS) and nucleoplasm (IN). The background fluorescence intensity (IB) from a dark region in the same image were subtracted. In live cell tracking, the specific genomic loci signals were identified and tracked by using the TrackMate plugin30. The 2D Gaussian fittings for precise measurement of spatial distance of locus pairs in Fig. 2c were performed by Mathematica and graphs were generated by OriginPro (OriginLab) or Excel.
Statistical analysis
All box plots and bar graphs were generated using the OriginPro or Excel. The line within the box plot represents the mean, the outer edges of the box are the 10th and 90th percentiles and the whiskers extend to the minimum and maximum values. In the bar graphs, all data are shown as the mean ± s.d. and individual data points were overlaid on the graphs. The exact n values used to calculate statistics are described in the associated figure legends. All the images and videos shown in the figures were repeated at least 3 times independently with similar results.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Rita Strack and Samie Jaffrey from Cornell University for the Broccoli aptamer. HaloTag JF-549 was a gift from the laboratory of Luke Lavis lab at Janelia Research Campus, Howard Hughes Medical Institute. sg14x(MS2) MUC4.1 was a gift from Mazhar Adli (Addgene plasmid # 101153). We are grateful to Jon Goguen of UMass Medical School for microscopy support, and thank Magda Kordon, Ying Feng and Yang Zhao in the Pederson lab for help in some of the imaging and data processing. We also thank Aviva Joseph for providing the Heat Stable Antigen (HSA) labeling assay. This work was supported in part by the Vitold Arnett Professorship Fund to T.P., NIH grant U01 DA-040588 to P. Kaufman, J. Dekker and T.P., NIH grant R01 GM102515 to S.Z., NIH grant U01 EB021238 to D.G. and NIH grant (K99 GM126810) to L-C.T. We are grateful to Job Dekker, Erik Sontheimer and Scot Wolfe at UMass Medical School for critical comments on the manuscript and strong support during the study.
Footnotes
METHODS
Methods, including statements of data availability and any associated accession codes and references, are available in the online version of the paper.
Note: Supplementary Information and Source Data files are available in the online version of the paper.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Data availability
All other data supporting the findings of this study are available within the paper and the associated supplementary files. Source data for Figs. 1, 2, S3, S5, S6 and S7 are available online. The dCas9 and sgRNA-Sirius expression vector information will be available at Addgene.
Reporting summary
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
REFERENCES
- 1.Dekker J et al. Nature 549, 219–226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bickmore WA & van Steensel B Cell 152, 1270–1284 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Finn, E. et al. bioRxiv doi.org/10.1101/171801
- 4.Chen B et al. Cell 155, 1479–1491 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma H et al. Proc. Natl. Acad. Sci. USA. 112, 3002–3007 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen B et al. Nucleic Acids Res. 44, e75 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma H et al. Nat. Biotechnol. 34, 528–530 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu Y et al. Nat. Commun. 7, 11707 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shao S et al. Nucleic Acids Res. 44, e86 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma H et al. J. Cell Biol. 214, 529–537 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grigory S et al. J. Am. Chem. Soc. 136, 16299–16308 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu B et al. Genes Dev. 29, 876–886 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qin P et al. Nat. Commun. 8, 14725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grimm JB et al. Nat. Methods 12, 244–250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benjamin D et al. Nature 515, 402–405 (2014).25409831 [Google Scholar]
- 16.Wang S et al. Science 353, 598–602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stanyte R et al. J. Cell Biol. DOI: 10.1083/jcb.201801157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geary C et al. Science 345, 799–804 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Goloborodko A et al. eLife 5, e14864 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hnisz D et al. Cell 169, 13–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benson G Nucleic Acids Res. 27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marçais G & Kingsford C Bioinformatics 27, 764–770 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filonov GS, Kam CW, Song W & Jaffrey SR Chem. Biol. 22, 649–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schneider D, Tuerk C & Gold LJ Mol. Biol. 228, 862–869 (1992). [DOI] [PubMed] [Google Scholar]
- 25.Zuker M Nucleic Acids Res. 31, 3406–3415 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lim F & Peabody DS Nucleic Acids Res. 30, 4138–4144 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akan P et al. Genome Med. 4, 86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H et al. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boeva V et al. Bioinformatics 28, 423–425 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tinevez JY et al. TrackMate: Methods 115, 80–90 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.