

Abstract
Pancreatic cancer (PC) is a highly aggressive tumor, often difficult to diagnose and treat. Aspartate β-hydroxylase (ASPH) is a type II transmembrane protein and the member of α-ketoglutarate-dependent dioxygenase family, found to be overexpressed in different cancer types, including PC. ASPH appears to be involved in the regulation of proliferation, invasion and metastasis of PC cells through multiple signaling pathways, suggesting its role as a tumor biomarker and therapeutic target. In this review, we briefly summarize the possible mechanisms of action of ASPH in PC and recent progress in the therapeutic approaches targeting ASPH.
Keywords: Aspartate β-hydroxylase, pancreatic cancer, Notch signaling pathway, Mitochondrial DNA, NK cell
INTRODUCTION
Pancreatic cancer (PC) is an aggressive malignancy with a high mortality rate [1-4]. In the United States (US), a 5-year relative survival rate was estimated to be only 8% [4]. Despite improvements in the diagnosis and management of PC over the last few decades [1,2], PC was reported to be the seventh cause of cancer-related death in China [3]. According to the most recent American Cancer Society (ACS) report, in the US, the number of new PC cases and deaths was 55,440 and 44,330, respectively in 2018, and PC was the fourth leading cause of cancer death in 2015 [4]. Also, by 2030, PC is projected to become the second leading cause of cancer-related death in the US [5,6]. Radical surgery combined with neoadjuvant chemotherapy is considered to be the most effective treatment for PC. However, due to the absence of early symptoms, 80–85% of PC patients are diagnosed at the stage of locally advanced or distant metastatic, unresectable disease. Moreover, clinical and preclinical data indicate that PC metastases develop during the early stages of pathogenesis, before the primary tumor can even be detected [5]. Thus, to improve the diagnosis, treatment and outcomes of PC patients it is necessary to better understand the molecular mechanisms of PC onset, progression and metastasis and to identify targetable pathways.
Aspartate β-hydroxylase (ASPH) is a highly conserved dioxygenase enzyme found to be overexpressed in multiple malignancies, including PC. ASPH appears to be involved in the regulation of proliferation, invasion and metastasis of PC cells through multiple signaling pathways, suggesting its role as a tumor biomarker and therapeutic target. In this review, we briefly summarize the possible mechanisms of action of ASPH in PC and recent progress in the therapeutic approaches targeting ASPH.
THE STRUCTURE AND FUNCTION OF ASPH
ASPH was first described in 1989, it is a type II transmembrane protein of ~86 kDa in size and the member of α-ketoglutarate-dependent dioxygenase family [7-13]. ASPH has a very low expression in normal adult tissue and is predominately expressed during embryogenesis, to promote cell migration for organ development [7,8]. The ASPH gene is 214,085 bases long and has 33 exons. By alternative splicing, it encodes four protein isoforms: ASPH, junctin (structural protein of sarcoplasmic reticulum), humbug (ASPH-type junctate that lacks the catalytic domain), and junctin-type junctate [14,15]. The ASPH protein consists of four domains: amino or N-terminal cytoplasmic domain, transmembrane domain, a highly charged region that projects into the lumen of endoplasmic reticulum (ER), and COOH-terminal catalytic domain [14]. Different studies showed that the Wnt/β-catenin, insulin (IN)/insulin-like growth factor 1 (IGF-1)/insulin receptor substrate 1 (IRS1)/phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt), and IN/IGF-1/IRS1/mitogen-activated protein kinase (MAPK)/extracellular-signal-regulated kinase (ERK) signaling pathways play an important role in the transcriptional regulation of ASPH (Figure 1) [16,17]. When Wnt signaling is aberrantly activated, Wnt ligand binds to Frizzled (FZD) cell-surface receptors and low density lipoprotein (LDL)-receptor-related proteins 5 and 6 (LRP5 and LRP6) which leads to the degradation of the β-catenin destruction complex (contains adenomatous polyposis coli [APC] and AXIN) and inhibition of glycogen synthase kinase 3β (GSK3β), and consequent accumulation of β-catenin in the cytoplasm. Subsequently, β-catenin moves into the nucleus where it interacts with T-cell factor/lymphoid enhancer-binding factor (TCF/LEF) proteins to form a transcriptional regulatory complex and activate the transcription of target genes [16]. Among the proposed target genes is IRS1, where the TCF/LEF/β-catenin complex upregulates its expression possibly by binding to TCF consensus binding elements (enhancers) located in the first intron of the IRS1 gene and downstream of its transcriptional start site [18,19]. The overexpressed IRS1 can relay signals from activated IN/IGF-1 receptors to downstream effector cascades such as the ERK/MAPK and PI3K/Akt signaling, and thus upregulate the expression of ASPH as a downstream target of these pathways [19]. Namely, binding of IN and IGF-1 to insulin receptor (IR) and IGF-1 receptor (IGF1R), respectively leads to the autophosphorylation of the receptor on tyrosine residues and activation of the intrinsic tyrosine kinase. The kinase catalyzes the phosphorylation of tyrosine (Y-P) on intracellular IRS1 and activates PI3K, which then phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP2) on the 3C position and generates phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 interacts with protein kinases such as phosphoinositide-dependent kinase 1 (PDK1) which initiates a cascade of phosphorylation events, finally leading to the activation of Akt and/or atypical protein kinase C (PKC) [18,20]. In addition to the PI3K cascade, tyrosine phosphorylation of IRS1 can result in the activation of the downstream MAPK pathway, i.e., PY-IRS1 interacts with growth factor receptor-bound protein 2 (Grb2) and synaptophysin (Syp) proteins leading to the sequential activation of p21ras, mitogen-activated protein kinase kinase (MAPKK), and MAPK [21,22].
FIGURE 1.
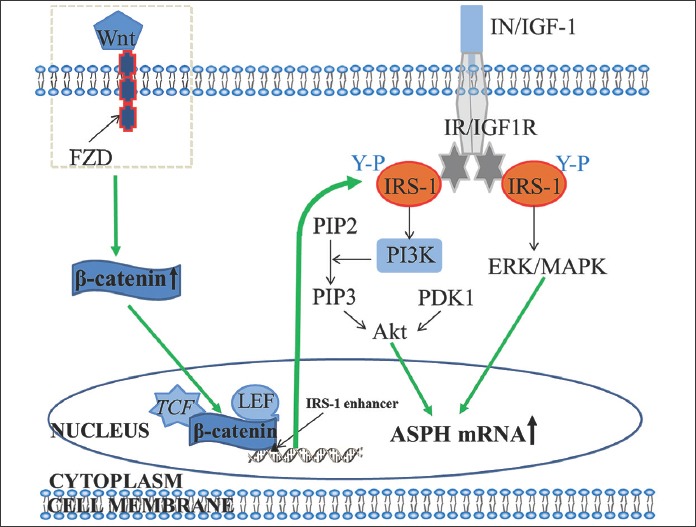
The role of the Wnt/β-catenin, insulin (IN)/insulin-like growth factor 1 (IGF-1)/insulin receptor substrate 1 (IRS1)/phosphatidylinositol-3-kinase (PI3K)/protein kinase B (Akt), and IN/IGF-1/IRS1/mitogen-activated protein kinase (MAPK)/extracellular-signal-regulated kinase (ERK) signaling pathways in the transcriptional regulation of aspartate β-hydroxylase (ASPH). The binding of Wnt ligand to Frizzled (FZD) cell-surface receptors and low density lipoprotein (LDL)-receptor-related proteins 5 and 6 (LRP5 and LRP6) leads to the accumulation of β-catenin in the cytoplasm. β-catenin then moves into the nucleus where it interacts with T-cell factor/lymphoid enhancer-binding factor (TCF/LEF) proteins to form a transcriptional regulatory complex and activate the transcription of target genes such as IRS1. The overexpressed IRS1 relays signals from activated IN/IGF-1 receptors to downstream effector cascades such as the ERK/MAPK and PI3K/Akt signaling, and thus upregulate the expression of ASPH as a downstream target of these pathways. Namely, binding of IN and IGF-1 to insulin receptor (IR) and IGF-1 receptor (IGF1R), respectively leads to the activation of the intrinsic tyrosine kinase. The kinase catalyzes the phosphorylation of tyrosine (Y-P) on intracellular IRS1 and activates PI3K, which then phosphorylates phosphatidylinositol (4,5)-bisphosphate (PIP2) and generates phosphatidylinositol (3,4,5)-trisphosphate (PIP3). PIP3 interacts with phosphoinositide-dependent kinase 1 (PDK1), initiating a cascade of phosphorylation events and leading to the activation of Akt. The tyrosine phosphorylation of IRS1 can also result in the activation of the downstream MAPK pathway, i.e., PY-IRS1 interacts with growth factor receptor-bound protein 2 (Grb2) and synaptophysin (Syp) proteins leading to the sequential activation of p21ras, mitogen-activated protein kinase kinase (MAPKK), and MAPK [16,19,21,22].
One of the downstream targets of IRS1-mediated signaling pathways is ASPH. For example, de la Monte et al. [22] found that the stimulation of insulin and IGF-1 increased ASPH mRNA and protein expression, and consequently the motility of human hepatocellular carcinoma (HCC) cell lines, which was mediated by the ERK/MAPK and PI3K/Akt pathways [22].
ASPH is rarely expressed in normal adult tissue, except placental trophoblastic cells [23-25]; however, its overexpression has been observed in a number of malignancies, including cholangiocarcinoma, HCC, lung, breast and colon cancer, as well as in the neoplasms of the nervous system [22,26]. Moreover, in HCC patients, Wang et al. [25] showed a significant association between ASPH overexpression and higher recurrence and lower survival rate following surgery. Also, ASPH overexpression could predict worse surgical outcome in the early-stage HCC patients [25]. The overexpression of ASPH was also observed in PC, and its important role in the promotion of proliferation, migration, invasion, and malignant transformation of PC cells, through multiple signaling pathways, was suggested [27].
THE MECHANISM OF ASPH IN PC
ASPH activates the Notch signaling pathway
The mechanisms how ASPH affects cell proliferation and tumor invasion/metastasis in PC are not completely clear. Dong et al. [27] indicated that ASPH activates the Notch signaling pathway as the mechanism of malignant transformation in PC cells. For example, they showed that activated Notch1 and hairy and enhancer of split-1 (HES1), which is a Notch responsive gene, were overexpressed in the cytoplasm and nuclei of pancreatic ductal adenocarcinoma (PDAC) cells compared to adjacent normal tissues [27].
The Notch signaling cascade is a highly conserved pathway with a critical role in cell-cell signaling and the control of cell fate determination during embryogenesis. Due to the diverse functions of Notch pathway, including the maintenance of stem cell populations and the regulation of cell proliferation, survival, apoptosis and differentiation, it plays an important role in the development and progression of human cancers. In mammals, four different Notch receptors exist (NOTCH1–4), which respond to five different ligands. Four of these ligands Jagged (JAG) 1 and 2 and Delta-like (DLL) 1 and 4 may act in cis to inhibit Notch receptor or in trans to interact with neighboring cells, whereas the fifth ligand DLL3 has the cis-inhibitory function. While the expression and activation of Notch receptors and ligands appear to be downregulated in normal adult pancreas tissues, as in many other cancer types, they are activated during pancreatic tumorigenesis and may act as oncogenes or tumor suppressors [28]. Numerous studies have shown that Notch signaling is associated with the occurrence and progression of PC [29-33]. For example, the activation of Notch signaling pathway can promote the epithelial-to-mesenchymal transition (EMT) and facilitate the invasion and metastasis of cancer cells in PC tissues [34].
ASPH catalyzes the hydroxylation of aspartyl and asparaginyl residues present in epidermal growth factor (EGF)-like domains of various proteins, including Notch receptors and ligands (Figure 2) [27,35,36]. The C-terminal catalytic domain of ASPH contains the amino acid (AA) sequence M670HPGTH675. The AA H675 is involved in Fe2+ coordination and is critical for the enzymatic activity of ASPH [14,27]. After ASPH overexpression is induced, the enzyme interacts with the EGF-like repeats in Notch receptor extracellular domain (ECD), promoting the interaction between Notch receptor and its ligand. The receptor-ligand interaction induces a conformational change of the Notch receptor, leading to S2 cleavage by tumor necrosis factor-α-converting enzyme (TACE/ADAM17) and then S3 cleavage by the presenilin/γ-secretase complex. The S3 cleavage releases the active Notch intracellular domain (NICD) from the plasma membrane, which then enters the nucleus and mediates the conversion of the CSL [CBF1–Su(H)–LAG1] repressor complex into a transcriptional activation complex and the recruitment of mastermind-like 1 (MAML1) coactivator protein, leading to the transcriptional activation of a number of downstream target genes, including those from HES and hairy-related transcription factor (HRT or HEY) families, cyclin D1 (CCND1), c-myc, cyclooxygenase-2 or prostaglandin-endoperoxide synthase 2 (PTGS2), matrix metalloproteinase-9 (MMP9) and vascular endothelial growth factor (VEGFA). The overall effect of these processes in PC is the promotion of cell proliferation, migration, invasion, tumor growth, and metastasis [34,37].
FIGURE 2.
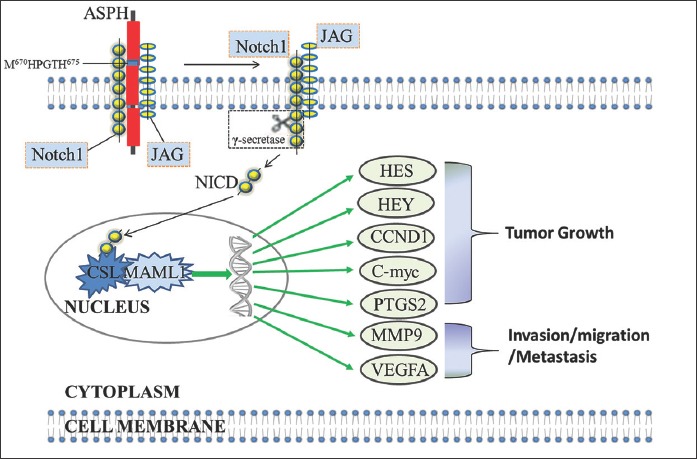
The proposed mechanism how aspartate β-hydroxylase (ASPH) may affect the progression of pancreatic cancer (PC) through the activation of Notch signaling pathway [27]. ASPH catalyzes the hydroxylation of aspartyl and asparaginyl residues present in epidermal growth factor (EGF)-like domains of Notch receptors and ligands. The C-terminal catalytic domain of ASPH contains the amino acid (AA) sequence M670HPGTHM675. After ASPH overexpression is induced, the enzyme interacts with the EGF-like repeats in Notch receptor extracellular domain (ECD), promoting the interaction between Notch receptor and its ligand (e.g., Jagged [JAG]). The receptor-ligand interaction induces a conformational change of the Notch receptor, leading to S2 and S3 cleavage. The S3 cleavage releases the active Notch intracellular domain (NICD) from the plasma membrane, which then enters the nucleus and mediates the conversion of the CSL [CBF1–Su(H)–LAG1] repressor complex into a transcriptional activation complex and the recruitment of mastermind-like 1 (MAML1) coactivator protein, leading to the transcriptional activation of a number of downstream target genes, including those from hairy and enhancer of split (HES) and hairy-related transcription factor (HRT or HEY) families, cyclin D1 (CCND1), c-myc, cyclooxygenase-2 or prostaglandin-endoperoxide synthase 2 (PTGS2), matrix metalloproteinase-9 (MMP9) and vascular endothelial growth factor (VEGFA).
ASPH promotes mitochondrial DNA D-loop mutations by inhibiting H2A histone family, member X (H2AX)-mitochondrial transcription factor A (mtTFA) signal
Somatic mitochondrial DNA (mtDNA) mutations have been detected in various tumor types, including PC [38-41]. In HCC tissues and cell lines, the overexpression of ASPH was significantly correlated with decreased copy numbers of displacement loop (D-loop) and nicotinamide adenine dinucleotide (NADH) dehydrogenase subunit 1, and increased somatic mutations in the D-loop [38]. The D-loop is a noncoding region of mtDNA which contains the origin of replication for heavy (H) mtDNA strand and the promoters for the transcription of H and light (L) strands [41].
In HCC cell lines, Tang et al. [38] demonstrated that the overexpressed ASPH disrupts the mtDNA integrity and affects mitochondrial function through H2AX-mtTFA signal (Figure 3) [38]. mtTFA is a key regulator of mtDNA transcription and is also important in the maintenance and repair of mtDNA. H2AX has been suggested to function as a shuttle protein transporter with a critical role in mitochondrial protein transport [42-44]. mtTFA was identified as protein that is transported by H2AX [44] from the cytoplasm to the mitochondria, to participate in the replication, transcription and repair of mtDNA (Figure 3A) [42,43]. However, it appears that overexpressed ASPH competes with mtTFA for binding to H2AX, thus blocking the translocation of mtTFA into the mitochondria and resulting in reduced binding of mtTFA to the D-loop [38,44]. This ultimately disrupts the function of mtTFA in mtDNA replication, transcription and repair (Figure 3B), leading to increased mutations in the D-loop and other mtDNA regions, reduced mtDNA copy number and decreased expression of mitochondrial respiratory chain enzymes. These alterations affect the mitochondrial function, resulting in aberrant mitochondrial membrane potential, decreased adenosine triphosphate (ATP) production and increased levels of reactive oxygen species (ROS) [38,45]. In primitive neuroectodermal tumor 2 (PNET2) human neuronal cells exposed to H2O2, Lawton et al. [46] showed that ASPH, hypoxia-inducible factor 1-alpha (HIF-1α) and neuronal migration were stimulated by the mild oxidative stress and suggested that the cross-talk between these molecules within a hydroxylation-regulated signaling pathway, which ultimately affects cell motility and migration, is transiently driven by fluctuations in oxidative stress and chronically regulated by the insulin/IGF signaling [46]. Changes in cellular microenvironment can lead to increased oxidative stress which is characterized by the overproduction of ROS in mitochondria, leading to increased mutations in mtDNA and disruption of its stability and function [45], possibly resulting in tumor development. However, due to low occurrence of somatic mtDNA D-loop mutations in a series of PC, Navaglia et al. [41] suggested that those molecular changes are epiphenomena, probably related to the damaging effects of ROS, rather than a direct cause of PC [41,47].
FIGURE 3.
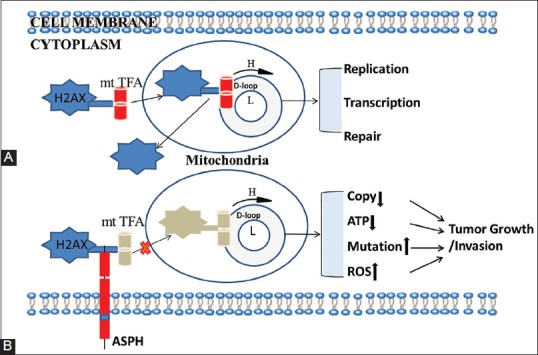
Aspartate β-hydroxylase (ASPH) disrupts the mitochondrial DNA (mtDNA) integrity and affects mitochondrial function through H2A histone family, member X (H2AX)-mitochondrial transcription factor A (mtTFA) signal in hepatocellular carcinoma (HCC) cells [38]. (A) H2AX transfers mtTFA from the cytoplasm to the mitochondria to participate in the replication, transcription, and repair of mtDNA. (B) ASPH competes with mtTFA for binding to H2AX, consequently blocking the binding between mtTFA and mtDNA displacement (D)-loop. This ultimately disrupts the function of mtTFA in mtDNA replication, transcription and repair, leading to increased mutations in the D-loop and other mtDNA regions, reduced mtDNA copy number and decreased expression of mitochondrial respiratory chain enzymes. These alterations affect the mitochondrial function, resulting in aberrant mitochondrial membrane potential, decreased adenosine triphosphate (ATP) production and increased levels of reactive oxygen species (ROS), and promoting tumor growth. H: Heavy mtDNA strand; L: Light mtDNA strand.
ASPH suppresses the natural killer (NK) cell-surveillance activity
NK cells play a pivotal role in immune surveillance of tumors and exert their function by producing cytokines such as interferon-γ (IFN-γ) and cytolytic proteins (perforin and granzymes), and by expressing NK cell-activating receptors (e.g., NKG2D, NKp30, and NKp44) [48]. IFN-γ is a critical molecule for innate and adaptive immunity and has antiviral, immunostimulatory, immunoregulatory, and antitumor properties [49]. Perforins do not only generate poly-perforin tubular channels (pores) on tumor cell membrane, increasing membrane permeability, but also mediate delivery of granzymes which induce apoptosis of target cells [48]. NKG2D triggers cytotoxicity by recognizing ligands (induced-self proteins) overexpressed by transformed and infected cells. Together with NKp30 and NKp44, it is the most important molecule in NK cell-mediated tumor cell lysis [50]. In MP2 PC cells, the cytotoxic activity of NK cells was enhanced by the combined effect of curcuminoids, omega-3 fatty acids, and antioxidants [51].
The activation and function of NK cells is controlled by two different categories of receptors, activating and inhibitory receptors, which are expressed on the surface of NK cells [52,53]. In normal conditions, the inhibitory receptors on NK cells interact with their ligands (mostly major histocompatibility complex [MHC] class I molecules), suppressing the activation of NK cells [54]. In cancer cells, these types of ligands can be downregulated and the ligands of activating receptors upregulated, leading to the engagement of activating receptors and consequent activation of NK cells. Finally, activated NK cells can rapidly kill tumor cells by releasing molecules such as IFN-γ, perforins and granzymes (Figure 4) [55].
FIGURE 4.
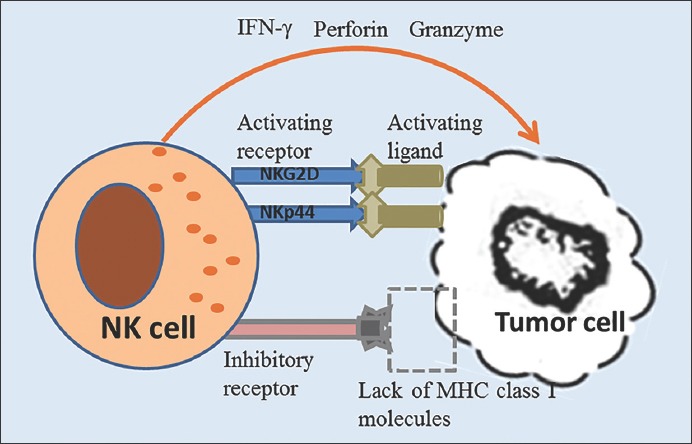
Natural killer (NK) cell-surveillance activity in tumor. The activation and function of NK cells is controlled by two different categories of receptors, activating and inhibitory receptors. In normal conditions, the inhibitory receptors on NK cells interact with their ligands (mostly major histocompatibility complex [MHC] class I molecules), suppressing the activation of NK cells. In cancer cells, these types of ligands can be downregulated and the ligands of activating receptors upregulated, leading to the engagement of activating receptors (e.g., NKG2D and NKp44) and consequent activation of NK cells. Activated NK cells can rapidly kill tumor cells by releasing molecules such as interferon-γ (IFN-γ), perforins and granzymes [52-55].
A recent study showed that a recombinant ASPH (rASPH) had a negative effect on the activity and function of primary human NK cells. Namely, the rASPH reduced the viability and cytotoxicity of NK cells in a time and dose-dependent manner, inhibited NK cell aggregation, promoted apoptosis and necrosis, reduced the mRNA expression of INF-γ, mRNA and protein expression of activating receptors NKG2D and NKp44, and mRNA expression of inhibitory receptor NKG2A [56]. Overall, it appears that one of the mechanisms of ASPH in promoting tumor formation and viability is by inhibiting NK cell-surveillance activity [56]. Other studies also demonstrated that the inhibition of NK cell activating receptor expression and function, e.g., by methylprednisolone or histone deacetylase inhibitors, significantly reduces NK cell cytotoxicity [57,58], allowing tumor cells to escape immune surveillance.
THERAPEUTIC APPROACHES TARGETING ASPH
ASPH has been proposed as an important biological target to control tumor cell migration and invasion, as its overexpression has been observed in 70–90% of human tumors, including PC [22,27]. After ASPH overexpression is induced in tumor cells, the protein transfers from the ER to the plasma membrane, where it is exposed to extracellular environment and, thus, could be used as a tumor associated antigen (TAA) in immunotherapy [59].
The activation of both cluster of differentiation (CD)4+ T cells and CD8+ cytotoxic T cells (CTLs) is required for a sustained anti-tumor response [59-61]. In an experimental murine model, ASPH-loaded dendritic cells (DCs) had a substantial anti-tumor effect on HCC, and both CD4+ and CD8+ cells contributed to these effects [59]. Furthermore, in peripheral blood mononuclear cells (PBMCs) derived from HCC patients, ASPH protein-loaded DCs could also activate CD4+ T cell and CD8+ CTLs, via ASPH-derived human leukocyte antigen (HLA) class I- and class II-restricted peptides [61]. These findings indicate the usefulness of ASPH as a molecular target in immunotherapy, especially in HCC. For example, ASPH-DCs immunotherapy could potentially delay the recurrence of HCC following surgical resection [59]. Similarly, Noda et al. [62] showed that immunization with ASPH-loaded DCs had a cytotoxic effect on cholangiocarcinoma cells in vitro, suppressed intrahepatic tumor growth and metastasis in rats, and was associated with an increased infiltration of CD3+ lymphocytes into the tumor [62]. However, the effect of ASPH-loaded DCs on immune response in PC remains to be investigated.
Recently, molecular targeted therapy against ASPH has gained considerable attention. Dong et al. [27] reported that MO-I-1100, a small molecule inhibitor (SMI) of β-hydroxylase, reduced ASPH activity by 80%, inhibited ASPH-induced proliferation, migration, invasion and colony formation, and suppressed Notch signaling in PC [27]. Sturla et al. [63] showed that SMI MO-I-1100 and MO-I-1151 significantly reduced viability and directional motility of glioblastoma multiforme (GBM) cells, and similar effects were observed in GBM cells using lentivirus-sh-ASPH construct, confirming the role of ASPH in these processes [63]. Revskaya et al. indicated that radiolabeled human monoclonal antibody (mAb) PAN-622 targeting ASPH on the surface of cancer cells is a promising approach in imaging and, possibly, treatment of metastatic breast cancer [64]. In addition, antisense oligodeoxynucleotide inhibition of ASPH expression significantly reduced the motility of cholangiocarcinoma cells [65] and small interfering RNAs (siRNAs) targeting the exon 2 of ASPH gene inhibited the expression of ASPH and reduced directional motility in HCC cells [22]. In another study, mAb against the ASPH C-terminal (ASPH-C) increased antibody-dependent cellular cytotoxicity of NK cells on HeLa, MCF-7 and HepG2 cells, suggesting the use of this mAb in cancer immunotherapy [66]. Considering the above-described findings, ASPH may play an important role in the development of therapeutic agents for PC.
CONCLUSION
ASPH is overexpressed in many cancer types, including PC. It plays an important role in tumor development and progression by activating the Notch signaling pathway, promoting mtDNA D-loop mutations/disrupting mitochondrial function, and inhibiting the NK cell activity. Different studies demonstrated the potential of ASPH as a biomarker and therapeutic target in cancer. Recently, our study has found that the Notch signaling pathway is pivotal for exosome secretion and biological activity in MIA-Paca2 cell lines. Hence, we hypothesize that ASPH stimulates PC cells to secrete/release specific exosomes and acquire invasive properties by activating the Notch signaling. Overall, it appears that the ASPH-mediated regulation of PC development, progression and metastasis may be more complex than originally thought.
ACKNOWLEDGMENTS
This work was supported by grant from National Natural Science Foundation of China (NSFC number 81470887).
DECLARATION OF INTERESTS
The authors declare no conflict of interests.
REFERENCES
- [1].Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. https://doi.org/10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- [2].Vincent A, Herman J, Schulick R, Hruban RH, Goggins M. Pancreatic cancer. Lancet. 2011;378(9791):607–20. doi: 10.1016/S0140-6736(10)62307-0. https://doi.org/10.1016/S0140-6736(10)62307-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lin QJ, Yang F, Jin C, Fu DL. Current status and progress of pancreatic cancer in China. World J Gastroenterol. 2015;21(26):7988–8003. doi: 10.3748/wjg.v21.i26.7988. https://doi.org/10.3748/wjg.v21.i26.7988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. https://doi.org/10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- [5].Tsai S, Evans DB. Therapeutic advances in localized pancreatic cancer. JAMA Surg. 2016;151(9):862–8. doi: 10.1001/jamasurg.2016.1113. https://doi.org/10.1001/jamasurg.2016.1113. [DOI] [PubMed] [Google Scholar]
- [6].Rahib L, Smith BD, Izeberg R, Osenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21. doi: 10.1158/0008-5472.CAN-14-0155. https://doi.org/10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- [7].Ronke RS, VanDusen WJ, Garsky VM, Jacobs JW, Sardana MK, Stern AM, et al. Aspartyl beta-hydroxylase: In vitro hydroxylation of a synthetic peptide based on the structure of the first growth factor-like domain of human factor IX. Proc Natl Acad Sci U S A. 1989;86(10):3609–13. doi: 10.1073/pnas.86.10.3609. https://doi.org/10.1073/pnas.86.10.3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Stenflo J, Holme E, Lindstedt S, Chandramouli N, Huang LH, Tam JP, et al. Hydroxylation of aspartic acid in domains homologous to the epidermal growth factor precursor is catalyzed by a 2-oxoglutarate-dependent dioxygenase. Proc Natl Acad Sci U S A. 1989;86(2):444–7. doi: 10.1073/pnas.86.2.444. https://doi.org/10.1073/pnas.86.2.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shimizu K, Chiba S, Kumano K, Hosoya N, Takahashi T, Kanda Y, et al. Mouse jagged1 physically interacts with notch2 and other notch receptors. Assessment by quantitative methods. J Biol Chem. 1999;274(46):32961–9. doi: 10.1074/jbc.274.46.32961. https://doi.org/10.1074/jbc.274.46.32961. [DOI] [PubMed] [Google Scholar]
- [10].Zimrin AB, Pepper MS, McMahon GA, Nguyen F, Montesano R, Maciag T. An antisense oligonucleotide to the notch ligand jagged enhances fibroblast growth factor-induced angiogenesis in vitro. J Biol Chem. 1996;271(51):32499–502. doi: 10.1074/jbc.271.51.32499. https://doi.org/10.1074/jbc.271.51.32499. [DOI] [PubMed] [Google Scholar]
- [11].McGinnis K, Ku GM, VanDusen WJ, Fu J, Garsky V, Stern AM, et al. Site-directed mutagenesis of residues in a conserved region of bovine aspartyl (asparaginyl) beta-hydroxylase: Evidence that histidine 675 has a role in binding Fe2+ Biochemistry. 1996;35(13):3957–62. doi: 10.1021/bi951520n. https://doi.org/10.1021/bi951520n. [DOI] [PubMed] [Google Scholar]
- [12].Wands JR, Lavaissiere L, Moradpour D, de la Monte S, Mohr L, Nicolau C, et al. Immunological approach to hepatocellular carcinoma. J Viral Hepat. 1997;4(Suppl 2):60–74. doi: 10.1111/j.1365-2893.1997.tb00181.x. https://doi.org/10.1111/j.1365-2893.1997.tb00181.x. [DOI] [PubMed] [Google Scholar]
- [13].Ho SP, Scully MS, Krauthauser CM, Wexler EJ, Stow MD, Dinchuk JE, et al. Antisense oligonucleotides selectively regulate aspartyl beta-hydroxylase and its truncated protein isoform in vitro but distribute poorly into A549 tumors in vivo. J Pharmacol Exp Ther. 2002;302(2):795–803. doi: 10.1124/jpet.302.2.795. https://doi.org/10.1124/jpet.302.2.795. [DOI] [PubMed] [Google Scholar]
- [14].Dinchuk JE, Henderson NL, Burn TC, Huber R, Ho SP, Link J, et al. Aspartyl beta-hydroxylase (Asph) and an evolutionarily conserved isoform of Asph missing the catalytic domain share exons with junctin. J Biol Chem. 2000;275(50):39543–54. doi: 10.1074/jbc.M006753200. https://doi.org/10.1074/jbc.M006753200. [DOI] [PubMed] [Google Scholar]
- [15].Treves S, Feriotto G, Moccagatta L, Gambari R, Zorzato F. Molecular cloning, expression, functional characterization, chromosomal localization, and gene structure of junctate, a novel integral calcium binding protein of sarco(endo)plasmic reticulum membrane. J Biol Chem. 2000;275(50):39555–68. doi: 10.1074/jbc.M005473200. https://doi.org/10.1074/jbc.M005473200. [DOI] [PubMed] [Google Scholar]
- [16].Tomimaru Y, Koga H, Yano H, de la Monte SM, Wands JR, Kim M. Up-regulation of TCF-4 isoform responsive target genes in hepatocellular carcinoma. Liver Int. 2013;33(7):1100–12. doi: 10.1111/liv.12188. https://doi.org/10.1111/liv.12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Cantarini MC, de la Monte SM, Pang M, Tong M, D’Errico A, Trevisani F, et al. Aspartyl-asparagyl beta hydroxylase over-expression in human hepatoma is linked to activation of insulin-like growth factor and notch signaling mechanisms. Hepatology. 2006;44(2):446–57. doi: 10.1002/hep.21272. https://doi.org/10.1002/hep.21272. [DOI] [PubMed] [Google Scholar]
- [18].Chung W, Kim M, de la Monte S, Longato L, Carlson R, Slagle BL, et al. Activation of signal transduction pathways during hepatic oncogenesis. Cancer Lett. 2016;370(1):1–9. doi: 10.1016/j.canlet.2015.09.016. https://doi.org/10.1016/j.canlet.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bommer GT, Feng Y, Iura A, Giordano TJ, Kuick R, Kadikoy H, et al. IRS1 regulation by Wnt/beta-catenin signaling and varied contribution of IRS1 to the neoplastic phenotype. J Biol Chem. 2010;285(3):1928–38. doi: 10.1074/jbc.M109.060319. https://doi.org/10.1074/jbc.M109.060319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol. 2002;12(2):65–71. doi: 10.1016/s0962-8924(01)02207-3. https://doi.org/10.1016/S0962-8924(01)02207-3. [DOI] [PubMed] [Google Scholar]
- [21].Mohr L, Tanaka S, Wands JR. Ethanol inhibits hepatocyte proliferation in insulin receptor substrate 1 transgenic mice. Gastroenterology. 1998;115(6):1558–65. doi: 10.1016/s0016-5085(98)70036-8. https://doi.org/10.1016/S0016-5085(98)70036-8. [DOI] [PubMed] [Google Scholar]
- [22].de la Monte S, Tamaki S, Cantarini MC, Ince N, Wiedmann M, Cater J, et al. Aspartyl-(asparaginyl)-b-bydroxylase regulates hepatocellular carcinoma invasiveness. J Hepatol. 2006;44(5):971–83. doi: 10.1016/j.jhep.2006.01.038. https://doi.org/10.1016/j.jhep.2006.01.038. [DOI] [PubMed] [Google Scholar]
- [23].Patel N, Khan AO, Mansour A, Mohamed JY, Al-Assiri A, Haddad R, et al. Mutations in ASPH cause facial dysmorphism, lens dislocation, anterior-segment abnormalities, and spontaneous filtering blebs, or Traboulsi syndrome. Am J Hum Genet. 2014;94(5):755–9. doi: 10.1016/j.ajhg.2014.04.002. https://doi.org/10.1016/j.ajhg.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang Z, Li Y, Kong D, Sarkar FH. The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr Drug Targets. 2010;11(6):745–51. doi: 10.2174/138945010791170860. https://doi.org/10.2174/138945010791170860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wang K, Liu J, Yan ZL, Li J, Shi LH, Cong WM, et al. Overexpression of aspartyl-(asparaginyl)-beta-hydroxylase in hepatocellular carcinoma is associated with worse surgical outcome. Hepatology. 2010;52(1):164–73. doi: 10.1002/hep.23650. https://doi.org/10.1002/hep.23650. [DOI] [PubMed] [Google Scholar]
- [26].Ince N, de la Monte SM, Wands JR. Overexpression of human aspartyl (asparaginyl) beta-hydroxylase is associated with malignant transformation. Cancer Res. 2000;60(5):1261–6. [PubMed] [Google Scholar]
- [27].Dong X, Lin Q, Aihara A, Li Y, Huang CK, Chung W, et al. Aspartate beta-hydroxylase expression promotes a malignant pancreatic cellular phenotype. Oncotarget. 2015;6(2):1231–48. doi: 10.18632/oncotarget.2840. https://doi.org/10.18632/oncotarget.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Avila JL, Kissil JL. Notch signaling in pancreatic cancer: Oncogene or tumor suppressor? Trends Mol Med. 2013;19(5):320–7. doi: 10.1016/j.molmed.2013.03.003. https://doi.org/10.1016/j.molmed.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yen WC, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, et al. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res. 2015;21(9):2084–95. doi: 10.1158/1078-0432.CCR-14-2808. https://doi.org/10.1158/1078-0432.CCR-14-2808. [DOI] [PubMed] [Google Scholar]
- [30].Hu H, Zhou L, Awadallah A, Xin W. Significance of Notch1-signaling pathway in human pancreatic development and carcinogenesis. Appl Immunohistochem Mol Morphol. 2013;21(3):242–7. doi: 10.1097/PAI.0b013e3182655ab7. https://doi.org/10.1097/PAI.0b013e3182655ab7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Vo K, Amarasinghe B, Washington K, Gonzalez A, Berlin J, Dang TP. Targeting notch pathway enhances rapamycin antitumor activity in pancreas cancers through PTEN phosphorylation. Mol Cancer. 2011;10:138. doi: 10.1186/1476-4598-10-138. https://doi.org/10.1186/1476-4598-10-138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Chiorean EG, Coveler AL. Pancreatic cancer: Optimizing treatment options, new, and emerging targeted therapies. Drug Des Devel Ther. 2015;9:3529–45. doi: 10.2147/DDDT.S60328. https://doi.org/10.2147/DDDT.S60328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gao J, Lang B, Wang ZW. Role of Notch signaling pathway in pancreatic cancer. Am J Cancer Res. 2017;7(2):173–86. [PMC free article] [PubMed] [Google Scholar]
- [34].Espinoza I, Pochampally R, Xing F, Watabe K, Miele L. Notch signaling: Targeting cancer stem cells and epithelial-to-mesenchymal transition. Onco Targets Ther. 2013;6:1249–59. doi: 10.2147/OTT.S36162. https://doi.org/10.2147/OTT.S36162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013;341(1):41–5. doi: 10.1016/j.canlet.2013.08.027. https://doi.org/10.1016/j.canlet.2013.08.027. [DOI] [PubMed] [Google Scholar]
- [36].Wang J, Han F, Wu J, Lee SW, Chan CH, Wu CY, et al. The role of Skp2 in hematopoietic stem cell quiescence, pool size, and self-renewal. Blood. 2011;118(20):5429–38. doi: 10.1182/blood-2010-10-312785. https://doi.org/10.1182/blood-2010-10-312785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat Rev Cancer. 2011;11(5):338–51. doi: 10.1038/nrc3035. https://doi.org/10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- [38].Tang C, Hou Y, Wang H, Wang K, Xiang H, Wan X, et al. Aspartate β-hydroxylase disrupts mitochondrial DNA stability and function in hepatocellular carcinoma. Oncogenesis. 2017;6(7):e362. doi: 10.1038/oncsis.2017.64. https://doi.org/10.1038/oncsis.2017.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kassauei K, Habbe N, Mullendore ME, Karikari CA, Maitra A, Feldmann G. Mitochondrial DNA mutations in pancreatic cancer. Int J Gastrointest Cancer. 2006;37(2-3):57–64. doi: 10.1007/s12029-007-0008-2. https://doi.org/10.1007/s12029-007-0008-2. [DOI] [PubMed] [Google Scholar]
- [40].Jones JB, Song JJ, Hempen PM, Parmigiani G, Hruban RH, Kern SE. Detection of mitochondrial DNA mutations in pancreatic cancer offers a “mass”-ive advantage over detection of nuclear DNA mutations. Cancer Res. 2001;61(4):1299–304. [PubMed] [Google Scholar]
- [41].Navaglia F, Basso D, Fogar P, Sperti C, Greco E, Zambon CF, et al. Mitochondrial DNA D-loop in pancreatic cancer: Somatic mutations are epiphenomena while the germline 16519 T variant worsens metabolism and outcome. Am J Clin Pathol. 2006;126(4):593–601. doi: 10.1309/GQFCCJMH5KHNVX73. https://doi.org/10.1309/GQFCCJMH5KHNVX73. [DOI] [PubMed] [Google Scholar]
- [42].Kang D, Kim SH, Hamasaki N. Mitochondrial transcription factor A (TFAM): Roles in maintenance of mtDNA and cellular functions. Mitochondrion. 2007;7(1-2):39–44. doi: 10.1016/j.mito.2006.11.017. https://doi.org/10.1016/j.mito.2006.11.017. [DOI] [PubMed] [Google Scholar]
- [43].Campbell CT, Kolesar JE, Kaufman BA. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim Biophys Acta. 2012;1819(9-10):921–9. doi: 10.1016/j.bbagrm.2012.03.002. https://doi.org/10.1016/j.bbagrm.2012.03.002. [DOI] [PubMed] [Google Scholar]
- [44].Choi YS, Hoon Jeong J, Min HK, Jung HJ, Hwang D, Lee SW, et al. Shot-gun proteomic analysis of mitochondrial D-loop DNA binding proteins: Identification of mitochondrial histones. Mol Biosyst. 2011;7(5):1523–36. doi: 10.1039/c0mb00277a. https://doi.org/10.1039/c0mb00277a. [DOI] [PubMed] [Google Scholar]
- [45].Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–98. doi: 10.1038/nrc3365. https://doi.org/10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lawton M, Tong M, Gundogan F, Wands JR, de la Monte SM. Aspartyl-(asparaginyl) beta-hydroxylase, hypoxia-inducible factor-alpha and Notch cross-talk in regulating neuronal motility. Oxid Med Cell Longev. 2010;3(5):347–56. doi: 10.4161/oxim.3.5.13296. https://doi.org/10.4161/oxim.3.5.13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Garcea G, Dennison AR, Steward WP, Berry DP. Role of inflammation in pancreatic carcinogenesis and the implications for future therapy. Pancreatology. 2005;5(6):514–29. doi: 10.1159/000087493. https://doi.org/10.1159/000087493. [DOI] [PubMed] [Google Scholar]
- [48].Smyth MJ, Cretney E, Kelly JM, Westwood JA, Street SE, Yagita H, et al. Activation of NK cell cytotoxicity. Mol Immunol. 2005;42(4):501–10. doi: 10.1016/j.molimm.2004.07.034. https://doi.org/10.1016/j.molimm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- [49].Lieberman LA, Hunter CA. Regulatory pathways involved in the infection-induced production of IFN-gamma by NK cells. Microbes Infect. 2002;4(15):1531–8. doi: 10.1016/s1286-4579(02)00036-9. https://doi.org/10.1016/S1286-4579(02)00036-9. [DOI] [PubMed] [Google Scholar]
- [50].Zwirner NW, Fuertes MB, Girart MV, Domaica CI, Rossi LE. Cytokine-driven regulation of NK cell functions in tumor immunity: Role of the MICA-NKG2D system. Cytokine Growth Factor Rev. 2007;18(1-2):159–70. doi: 10.1016/j.cytogfr.2007.01.013. https://doi.org/10.1016/j.cytogfr.2007.01.013. [DOI] [PubMed] [Google Scholar]
- [51].Fiala M. Curcumin and omega-3 fatty acids enhance NK cell-induced apoptosis of pancreatic cancer cells but curcumin inhibits interferon-γ production: Benefits of omega-3 with curcumin against cancer. Molecules. 2015;20(2):3020–6. doi: 10.3390/molecules20023020. https://doi.org/10.3390/molecules20023020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. 2015;15(4):243–54. doi: 10.1038/nri3799. https://doi.org/10.1038/nri3799. [DOI] [PubMed] [Google Scholar]
- [53].Sun C, Sun H, Zhang C, Tian Z. NK cell receptor imbalance and NK cell dysfunction in HBV infection and hepatocellular carcinoma. Cell Mol Immunol. 2015;12(3):292–302. doi: 10.1038/cmi.2014.91. https://doi.org/10.1038/cmi.2014.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Fang F, Xiao W, Tian Z. NK cell-based immunotherapy for cancer. Semin Immunol. 2017;31:37–54. doi: 10.1016/j.smim.2017.07.009. https://doi.org/10.1016/j.smim.2017.07.009. [DOI] [PubMed] [Google Scholar]
- [55].Paul S, Lal G. The molecular mechanism of natural killer cells function and its importance in cancer immunotherapy. Front Immunol. 2017;8:1124. doi: 10.3389/fimmu.2017.01124. https://doi.org/10.3389/fimmu.2017.01124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Huyan T, Li Q, Ye LJ, Yang H, Xue XP, Zhang MJ, et al. Inhibition of human natural killer cell functional activity by human aspartyl β-hydroxylase. Int Immunopharmacol. 2014;23(2):452–9. doi: 10.1016/j.intimp.2014.09.018. https://doi.org/10.1016/j.intimp.2014.09.018. [DOI] [PubMed] [Google Scholar]
- [57].Chiossone L, Vitale C, Cottalasso F, Moretti S, Azzarone B, Moretta L, et al. Molecular analysis of the methylprednisolone-mediated inhibition of NK-cell function: Evidence for different susceptibility of IL-2- versus IL-15-activated NK cells. Blood. 2007;109(9):3767–75. doi: 10.1182/blood-2006-07-037846. https://doi.org/10.1182/blood-2006-07-037846. [DOI] [PubMed] [Google Scholar]
- [58].Ogbomo H, Michaelis M, Kreuter J, Doerr HW, Cinatl Jr J. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett. 2007;581(7):1317–22. doi: 10.1016/j.febslet.2007.02.045. https://doi.org/10.1016/j.febslet.2007.02.045. [DOI] [PubMed] [Google Scholar]
- [59].Shimoda M, Tomimaru Y, Charpentier K.P, Safran H, Carlson RI, Wands J. Tumor progression-related transmembrane protein aspartate-β-hydroxylase is a target for immunotherapy of hepatocellular carcinoma. J Hepatol. 2012;56(5):1129–35. doi: 10.1016/j.jhep.2011.12.016. https://doi.org/10.1016/j.jhep.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Solar P, Sytkowski AJ. Differentially expressed genes associated with cisplatin resistance in human ovarian adenocarcinoma cell line A2780. Cancer Lett. 2011;309(1):11–8. doi: 10.1016/j.canlet.2011.05.008. https://doi.org/10.1016/j.canlet.2011.05.008. [DOI] [PubMed] [Google Scholar]
- [61].Tomimaru Y, Mishra S, Safran H, Charpentier KP, Martin W, De Groot AS, et al. Aspartate-β-hydroxylase induces epitope-specific T cell responses in hepatocellular carcinoma. Vaccine. 2015;33(10):1256–66. doi: 10.1016/j.vaccine.2015.01.037. https://doi.org/10.1016/j.vaccine.2015.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Noda T, Shimoda M, Ortiz V, Sirica AE, Wands JR. Immunization with aspartate-β-hydroxylase-loaded dendritic cells produces antitumor effects in a rat model of intrahepatic cholangiocarcinoma. Hepatology. 2012;55(1):86–97. doi: 10.1002/hep.24629. https://doi.org/10.1002/hep.24629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sturla LM, Tong M, Hebda N, Gao J, Thomas JM, Olsen M, et al. Aspartate-β-hydroxylase (ASPH): A potential therapeutic target in human malignant gliomas. Heliyon. 2016;2(12):2–31. doi: 10.1016/j.heliyon.2016.e00203. https://doi.org/10.1016/j.heliyon.2016.e00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Revskaya E, Jiang Z, Morgenstern A, Bruchertseifer F, Sesay M, Walker S, et al. A radiolabeled fully human antibody to human aspartyl (asparaginyl) β-hydroxylase is a promising agent for imaging and therapy of metastatic breast cancer. Cancer Biother Radiopharm. 2017;32(2):57–65. doi: 10.1089/cbr.2016.2141. https://doi.org/10.1089/cbr.2016.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Maeda T, Sepe P, Lahousse S, Tamaki S, Enjoji M, Wands JR, et al. Antisense oligodeoxynucleotides directed against aspartyl (asparaginyl) beta-hydroxylase suppress migration of cholangiocarcinoma cells. J Hepatol. 2003;38(5):615–22. doi: 10.1016/s0168-8278(03)00052-7. https://doi.org/10.1016/S0168-8278(03)00052-7. [DOI] [PubMed] [Google Scholar]
- [66].Huyan T, Li Q, Dong DD, Yang H, Xue XP, Huang QS. Development of a novel anti-human aspartyl-(asparaginyl) β-hydroxylase monoclonal antibody with diagnostic and therapeutic potential. Oncol Lett. 2017;13(3):1539–46. doi: 10.3892/ol.2017.5642. https://doi.org/10.3892/ol.2017.5642. [DOI] [PMC free article] [PubMed] [Google Scholar]