

Abstract
While the role of the ascending dopaminergic system in brain function and dysfunction has been a subject of extensive research, the role of the descending dopaminergic system in spinal cord function and dysfunction is just beginning to be understood. Adenosine plays a key role in the inhibitory control of the ascending dopaminergic system, largely dependent on functional complexes of specific subtypes of adenosine and dopamine receptors. Combining a selective destabilizing-peptide strategy with a proximity ligation assay and patch-clamp electrophysiology in slices from male mouse lumbar spinal cord, the present study demonstrates the existence of adenosine A1-dopamine D1 receptor heteromers in the spinal motoneuron by which adenosine tonically inhibits D1 receptor-mediated signaling. A1-D1 receptor heteromers play a significant control of the motoneuron excitability, represent main targets for the excitatory effects of caffeine in the spinal cord and can constitute new targets for the pharmacological therapy after spinal cord injury, motor aging-associated disorders and Restless Legs Syndrome.
Keywords: Adenosine A1 receptor, dopamine D1 receptor, receptor heteromers, spinal cord, motoneuron
Introduction
Adenosine is a ubiquitous neuromodulator in the central nervous system (CNS), which is involved in numerous functions. More general functions include the regulation of arousal and its role in neuroprotection. Thus, adenosine is one of the main endogenous homeostatic sleepiness-promoting substances [1, 2] and it is intensively released following a cellular insult, acting as an endogenous distress signal that modulates tissue damage and repair [3]. Still general but circuit specific, adenosine plays a very significant role in the modulation of dopaminergic transmission, with implications for psychomotor activity and reinforcement [4]. Adenosine functions depend on its ability to activate specific G protein-coupled receptors (GPCRs), which include the Gs protein-coupled adenosine A2A and A2B receptors (A2AR and A2BR) and the Gi protein-coupled adenosine A1 and A3 receptors (A1R and A3R) [5].
The modulatory role of adenosine on dopaminergic transmission depends largely on the existence of antagonistic interactions mediated by specific subtypes of adenosine and dopamine receptors, the so-called A2AR-dopamine D2 receptor (D2R) and A1R-dopamine D1 receptor (D1R) interactions [6]. These interactions are respectively segregated in the two main populations of striatal neurons, the GABAergic striatopallidal and the GABA striatonigral efferent neurons [6]. Apart from the endogenous neurotransmitters, these specific adenosine-dopamine receptor interactions are involved in the central effects of caffeine, a non-selective A1R-A2AR competitive antagonist and the most consumed psychoactive drug in the world [4, 7]. The psychostimulant effects of caffeine depend therefore on its ability to counteract the tonic inhibitory effect of endogenous adenosine on central dopaminergic neurotransmission. It is now well established that A2AR-D2R interactions depend on their ability to form functional heteromers with a heterotetrameric structure composed of A2AR and D2R homodimers functionally coupled to their respective cognate Gs and Gi proteins, and that these heteromers constitute a mayor population of A2AR and D2R in the striatum [8, 9]. On the other hand, heteromerization of the Gi-coupled A1R with the Gs-coupled D1R has not been unequivocally demonstrated, although behavioral, functional and biochemical antagonistic interactions between A1R and D1R ligands have been extensively reported in several artificial cell systems and in the experimental animal [10-29]. Nevertheless, A1R-D1R heteromers cannot yet be included in the so far short list of GPCR heteromers that fulfill the criteria for their identification in native tissues [30-31].
We have recently found a significant antagonistic interaction between A1R and D1R ligands in the mouse spinal cord that mediates the ability of caffeine to enhance locomotor-related activity by acting on spinal circuits [32], although the molecular mechanisms and cellular localization remained to be determined. In the present study, first, A1R-D1R heteromerization is clearly demonstrated in mammalian transfected cells using new biophysical techniques and the proximity ligation assay. Then, using synthetic peptides with the amino acid sequence of specific transmembrane domains (TMs) of the D1R we demonstrate that the antagonistic interaction between A1R and D1R ligands depends on A1R-D1R heteromerization. Finally, we specifically identify the presence of A1R-D1R heteromers in spinal motoneurons, where they mediate the modulatory control by adenosine and dopamine and the strong spinal pharmacological effects of caffeine and selective A1R antagonists.
Materials and Methods
Expression vectors and fusion proteins
Sequences encoding amino acid residues 1-155 and 156-238 of YFP Venus protein were subcloned in pcDNA3.1 vector to obtain YFP Venus hemi-truncated proteins. The cDNAs for A1R, D1R, CB1R or serotonin 5HT2AR and 5HT2BR, cloned into pcDNA3.1, were amplified without their stop codons using sense and antisense primers harboring: EcoRI and BamHI sites to clone 5HT2BR and D1R, EcoRI and XhoI to clone 5HT2AR or EcoRI and KpnI to clone A1R and CB1R. The amplified fragments were subcloned to be in-frame with restriction sites of pRLuc-N1 (PerkinElmer, Wellesley, MA) or pEYFP-N1 (enhanced yellow variant of GFP; Clontech, Heidelberg, Germany) vectors to provide plasmids that express proteins fused to Rluc or YFP on the C-terminal end (A1R-Rluc, D1R-YFP or 5HT2BR-YFP). For BiFC experiments, the cDNA for A1R, D1R, CB1R and 5HT2AR were also subcloned into pcDNA3.1-nVenus or pcDNA3.1-cVenus to provide a plasmid that expresses the receptor fused to the hemitruncated nYFP Venus or hemitruncated cYFP Venus on the C-terminal end of the receptor (A1R-nVenus, D1R-cVenus, CB1R-nVenus, 5HT2AR-cVenus).
HIV TAT-fused TM peptides
Peptides, with the amino acid sequence of TMs of the D1R were used as heteromer destabilizing agents [9, 33-35]. To allow intracellular delivery, a peptide can be fused to the cell-penetrating HIV transactivator of transcription (TAT) peptide (YGRKKRRQRRR) [36]. HIV TAT fused to a TM GPCR peptide can be inserted effectively into the plasma membrane as a result of both the penetration capacity of the TAT peptide and the hydrophobic property of the TM peptide [33]. To obtain the right orientation of the membrane-inserted peptide, HIV TAT peptide was fused to the C-terminus of peptides with the amino acid sequence of TM 5 and TM 7 of D1R (TM5 and TM7 peptides, respectively). All peptides were synthesized by Genemed Synthesis, Inc. Their sequences were: TM5, TYAISSSLISFYIPVAIMIVTYTSIYYGRKKRRQRRR; TM7, FDVFVWFGWANSSLNPIIYAFNADFYGRKKRRQRRR.
Cell cultures and transient transfection
Human embryonic kidney 293T (HEK-293T) cells obtained from ATCC were grown in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 2 mM L-glutamine, 100 μg/ml sodium pyruvate, 100 U/ml penicillin/streptomycin, MEM Non-Essential Amino Acids Solution (1/100) and 5% (v/v) heat inactivated Fetal Bovine Serum (FBS) (all supplements were from Invitrogen, Paisley, Scotland, UK). Mouse fibroblast Ltk- cell lines stably transfected with human D1R cDNA (D1R cells) or with both human D1R and human A1R cDNAs (A1R-D1R cells) were previously obtained and characterized [12]. A1R-D1R cells were cultured in Dulbecco's minimal essential medium with 4.5 mg/ml glucose and 0.11 mg/ml sodium pyruvate supplemented with 10% fetal calf serum, 2 mM glutamine, 100 units/ml penicillin, 100 units/ml streptomycin, 200 mg/ml G418, and 300 mg/ml hygromycin. D1R cells were cultured as described for A1R-D1R cells, but without hygromycin. All cells were maintained at 37°C in an atmosphere of 5% CO2. For transient transfection, HEK-293T cells growing in 6-well dishes were transfected with the corresponding fusion protein cDNA by the PEI (PolyEthylenImine, Sigma, Steinheim, Germany) method. Cells were incubated (4 h) with the corresponding cDNA together with PEI (5.47 mM in nitrogen residues) and 150 mM NaCl in a serum-starved medium. After 4 h, the medium was changed to a fresh complete culture medium. Forty-eight h after transfection, cells were washed twice in quick succession in HBSS (containing, in mM: 137 NaCl, 5 KCl, 0.34 Na2HPO4 ×12 H2O, 0.44 KH2PO4, 1.26 CaCl2×2H2O, 0.4 MgSO4 ×7H2O, 0.5 MgCl2, 10 HEPES, pH 7.4), supplemented with 0.1% glucose (w/v), detached, and re-suspended in the same buffer. To control the cell number, sample protein concentration was determined using a Bradford assay kit (Bio-Rad, Munich, Germany) using bovine serum albumin dilutions as standards.
Bimolecular Fluorescence complementation assay (BiFC)
HEK-293T cells expressing the receptor fused to the YFP Venus N-terminal fragment (n-YFP) and the receptor fused to the YFP Venus C-terminal fragment (c-YFP) were treated with vehicle or the indicated TAT-fused TM peptides (4 μM) for 4 h at 37°C. To quantify protein-reconstituted YFP Venus expression, cells (20 μg protein) were distributed in 96-well microplates (black plates with a transparent bottom; Porvair, King's Lynn, UK), and emission fluorescence at 530 nm was read in a Fluo Star Optima Fluorimeter (BMG Labtechnologies, Offenburg, Germany) equipped with a high-energy xenon flash lamp, using a 10 nm-bandwidth excitation filter at 400 nm reading. Protein fluorescence expression was determined as fluorescence of the sample minus fluorescence of non-transfected cells. Cells expressing A1R-nVenus and nVenus or D1R-cVenus and cVenus showed similar fluorescence levels to non-transfected cells.
Bioluminescence Resonance Energy Transfer (BRET) assay
HEK-293T cells growing in six-well plates were transiently co-transfected with a constant amount of cDNA encoding the receptor fused to Rluc protein and with increasingly amounts of cDNA corresponding to the receptor fused to YFP protein. To quantify receptor-YFP expression, cells (20 μg protein) were distributed in 96-well microplates (black plates with a transparent bottom) and fluorescence at 530 nm was read as described above. Receptor-fluorescence expression was determined as fluorescence of the sample minus the fluorescence of cells expressing only the BRET donor. For BRET measurements, cells (20 μg protein) were distributed in 96-well microplates (Corning 3600, White plates; Sigma) and BRET signal was collected 1 minute after addition of 5 μM coelenterazine H (Molecular Probes, Eugene, OR) using a Mithras LB 940 that allows the integration of the signals detected in the short-wavelength filter at 485 nm (440–500 nm) and the long-wavelength filter at 530 nm (510–590 nm). To quantify receptor-Rluc expression luminescence readings were also performed after 10 minutes of adding 5 μM of coelenterazine H. Both fluorescence and luminescence of each sample were measured before every experiment to confirm similar donor expressions (about 150,000 luminescent units) while monitoring the increase acceptor expression (10,000–70,000 fluorescent units). The net BRET is defined as [(long-wavelength emission)/(short-wavelength emission)]-Cf where Cf corresponds to [(long-wavelength emission)/(short-wavelength emission)] for the Rluc construct expressed alone in the same experiment. BRET is expressed as milli BRET units (mBU; net BRET × 1,000). In BRET curves BRET was expressed as a function of the ratio between fluorescence and luminescence × 100 (YFP/Rluc). To calculate maximum BRET (BRETmax) and BRET50 from saturation curves, data was fitted using a nonlinear regression equation and assuming a single phase with GraphPad Prism software (San Diego, CA, USA).
cAMP accumulation
Homogeneous time-resolved fluorescence energy transfer (HTRF) assays were performed using the Lance Ultra cAMP kit (PerkinElmer, Waltham, Massachusetts, US), based on competitive displacement of an europium chelate-labelled cAMP tracer bound to a specific antibody conjugated to acceptor beads. We first established the optimal cell density for an appropriate fluorescent signal. This was done by measuring the TR-FRET signal determined as a function of forskolin concentration using different cell densities. Forskolin dose-response curves were related to the cAMP standard curve in order to establish which cell density provides a response that covers most of the dynamic range of the cAMP standard curve. Cells were not treated or treated with vehicle or 4 μm of the indicated TAT-fused TM peptides for 4 h at 37°C in an atmosphere of 5% CO2. Cells were then grown (1,000 cells/well in white ProxiPlate 384-well microplates, PerkinElmer) in medium containing 50 μM zardaverine, pretreated at 25°C for 15 min with vehicle or the indicated receptor antagonist, stimulated with agonists for 10 min before adding 20 μM forskolin or vehicle and incubated for an additional 15-min period. Fluorescence at 665 nm was analyzed on a PHERAstar Flagship microplate reader equipped with an HTRF optical module (BMG Lab technologies, Offenburg, Germany).
Spinal cord dissection
Experiments were performed using spinal cords of 0- to 11-day old (P0-P11) CD1 mice (Charles River laboratories). The animal protocol was approved by the Animal Use and Care Committee at the University of Puerto Rico and was in accordance with National Institutes of Health guidelines. Animals were killed by rapid decapitation, and their spinal cords were isolated by ventral laminectomy.
Proximity Ligation Assay (PLA) in cells and in spinal cord slices
For PLA assay in cells, HEK-293 cells expressing A1R and D1R or LTK- cells growing on glass coverslips were treated with vehicle or medium containing 4 μM of the indicated TAT-fused TM peptides for 4 h at 37°C in an atmosphere of 5% CO2. Cells were fixed in 4% paraformaldehyde for 15 min, washed with PBS containing 20 mM glycine, permeabilized with the same buffer containing 0.05% Triton X-100, and successively washed with PBS. For PLA assay in spinal cord slices, mouse spinal cord was dissected, treated for 4 h with ice-cold oxygenated (O2/CO2: 95/5%) Krebs-HCO3- buffer (124 mM NaCl, 4 mM KCl, 1.25 mM KH2PO4, 1.5 mM MgCl2, 1.5 mM CaCl2, 10 mM glucose, and 26 mM NaHCO3, pH 7.4), supplemented with vehicle or 4 μM of the indicated TAT-TM peptides for 4 h. Then, a lumbar section of spinal cord was fixed by immersion with 4% paraformaldehyde solution for 1 h, at 4°C. Samples were then washed in 50 mM Tris-HCl, 0.9% NaCl pH 7.8 buffer (TBS), cryopreserved in a 30% sucrose solution for 48 h at 4°C and stored at -20°C until sectioning. 20-μm-thick slices were cut coronally on a freezing cryostat (Leica Jung CM-3000) and mounted on slide glass. Samples were thawed at 4°C, washed in TBS, permeabilized with TBS containing 0.01% Triton X-100 for 10 min and successively washed with TBS. In all cases putative heteromers were detected using the Duolink II in situ PLA detection Kit (OLink; Bioscience, Uppsala, Sweden) following the instructions of the supplier. To detect A1R-D1R heteromers, a mixture of equal amounts of rabbit anti-A1R antibody (1:200 AB3460, Abcam, Billerica, MA, USA,) and guinea pig anti-D1R antibody (1:200 D1R-GP-Af500, Frontier Institute) were used and incubated with a PLA probe anti-rabbit plus and a PLA probe anti-guinea pig minus. Cells and slices were processed for ligation and amplification with a Detection Reagent Red and were mounted using a DAPI-containing mounting medium. The samples were observed in a Leica SP2 confocal microscope (Leica Microsystems, Mannheim, Germany) equipped with an apochromatic 63× oil-immersion objective (N.A. 1.4), and a 405 nm- and a 561 nm-laser line. For each field, a stack of two channels (one per staining) and 9 to 15 Z stacks with a step size of 1 μm were acquired. Images were opened and processed with Image J confocal. Quantification of the ratio between the number of red spots and the number of cells containing spots (r) and the percentage of cells containing one or more red spots versus total cells (blue nucleus) was performed in both cell preparations and spinal cord slices. A total of 200–350 cells from 5–8 different fields were counted for cell preparations and a total of 1,500–3,000 cells from 5–10 different fields from three different animals were counted for the spinal cord slices. The ImageJ confocal program using the Fiji package (http://pacific.mpi-cbg.de/) was used. Nuclei and red spots were counted on the maximum projections of each image stack. After getting the projection, each channel was processed individually. The blue nuclei and red dots were segmented by filtering with a median filter, subtracting the background, enhancing the contrast with the contrast limited adaptive histogram equalization (CLAHE) plug-in, and finally applying a threshold to obtain the binary image and the regions of interest (ROIs) around each nucleus.
Immunohistochemistry
Spinal cords were dissected, fixed for 1 h with 4% paraformaldehyde (PFA) and cryopreserved in a 30% sucrose solution for 48 h at 4°C. The lumbar sections of the spinal cord were embedded in low-melting agarose gel and 300 μm-thick of floating sections were made with a vibrating microtome (Leica Microsystems). After washing with 1 × PBS, sections were blocked with 20% normal donkey serum (NDS) and 1% bovine serum albumin (BSA) in 1 × PBS for 1 h and then incubated with the following primary antibodies overnight at 4°C: rabbit-anti-A1R (1:300; Abcam), goat-anti-D1R (1:100; Frontier Institute, Japan). After washing with 1 × PBS, the sections were incubated with corresponding secondary antibodies: Alexa Fluor 488-conjugated donkey anti-rabbit (1:500; Invitrogen), rhodamine-conjugated rabbit anti-goat (1:1000; Invitrogen) for 2 h at room temperature. For nuclear labeling, slices were incubated with Hoechst (1:10,000; Invitrogen) for 20 min. Images were acquired using a confocal microscope (Nikon A1R+).
Slice preparation for path-clamp electrophysiology
Spinal cords were isolated under ice-cold (4°C) oxygenated (95% O2 and 5% CO2) glycerol-based modified artificial cerebrospinal fluid (∼300 mOsmol/kg H2O), which contained, in mM: 222 glycerol, 3.08 KCl, 1.18 KH2PO4, 1.25 MgSO4, 2.52 CaCl2, 25 NaHCO3, and 11 D-glucose; all obtained from Sigma. The isolated spinal cords were incubated for 10 minutes in an oxygenated artificial cerebrospinal fluid (ACSF; ∼280 mosmol/kg H2O) which contained, in mM: 111 NaCl, 3.08 KCl, 1.18 KH2PO4, 1.25 MgSO4, 2.52 CaCl2, 25 NaHCO3, and 11 D-glucose. Transverse lumbar spinal cord slices (300-μm thick) were made with a vibrating microtome (Leica Microsystems) in an ice cold HEPES based solution which contained, in mM: 101 NaCl, 3.8 KCl, 1.3 MgCl2(6H2O), 1.2 MgSO4, 1.2 KH2PO4, 1.0 CaCl2, 10 Hepes and 25 D-glucose. Slices were then transferred to ACSF at 32°C for 30 min and at room temperature for an extra 30 min before recording at room temperature (20-23°C) with constant perfusion (3-2 ml/min) of ACSF. Patch-clamp electrode resistance and contents were different for whole cell recording (WCR) and perforated patch recording (PPR). The spinal neurons were visualized using a 60X water immersion objective (Nikon) with differential interference contrast (DIC) optics (Nikon). Drugs were added at a 2 to 3 ml/min perfusion rate.
Patch-clamp electrophysiology
The intracellular solution for WCR contained, in mM: 138 K-gluconate, 10 Hepes, 5 ATP-Mg, 0.3 GTP-Li and 0.0001 CaCl2. The intracellular solution for PPRs contained, in mM: 138 K-gluconate, 10 Hepes, 10 KCl and 2 MgCl2. Both intracellular solutions were adjusted with KOH at pH 7.4. To block most synaptic input, neurons were isolated from rapid synaptic inputs with a combination of DL-2-amino-5-phosphonopentanoic acid (AP-5; 10 μM; Tocris) and CNQX disodium salt hydrate (10 μM; Tocris) to block glutamatergic synapses; picrotoxin (10 μM; Tocris) to block GABAergic synapses, and strychnine (10 μM; Sigma) to block glycinergic synapses. Spontaneous excitatory postsynaptic potentials (EPSPs) and/or inhibitory postsynaptic potentials (IPSPs) disappeared in 2 to 3-min after addition of the blocking solution. WCR and PPR were made with thick-walled, unfilamented borosilicate glass (1.5-mm outer diameter, 1.0-mm inner diameter; PG52151-4, WPI) on a vertical puller (PC-10; Narishige); with a resistance of 6-7 MΩ for WCR and 3-5 MΩ for PPR. For PPR, the tip of the pipette was first filled with PPR intracellular solution by placing the pipette tip side down into a 1.5-ml Eppendorf cap filled with ∼1-ml intracellular solution and applying 5 to 7 ml of negative pressure with a 10-ml syringe for 1 s. The pipette was then backfilled with a combination of PPR intracellular solution, amphotericin B (Sigma) and Pluronic F-127 (Sigma). To prepare the solution, 1.2-mg amphotericin B was dissolved in 20 μl DMSO (Sigma) and added to 1 mg Pluronic F-127 dissolved in 40 μl DMSO. The 60-μl amphotericin B-Pluronic F-127-DMSO mix was added and vortexed in 1 ml intracellular solution. The ionophore mix was stored at room temperature and replaced every hour as needed.
For WCR, cells were approached with application of positive air pressure (80-100 mm Hg), whereas for PPR the cells were approached with a low application of positive air pressure (10-20 mm Hg) to the patch pipette to reach cells up to several layers below the surface but to avoid forcing the ionophore mix to the tip, which would impede seal formation. Close to the clearly visible membrane, the positive pressure was converted to a slight suction to obtain a gigaOhm seal (1-1.5 GΩ). The intact membrane patch in the pipette was observed and the patch properties where compare during the experiment to confirm the stability of the perforated patch configuration. All current-clamp recordings were made with a MultiClamp 700B amplifier (Molecular Devices) controlled by Clampex (pCLAMP 9.0; Molecular Devices). Data were sampled at 10 kHz and low-pass filtered at 2 kHz. The voltage threshold for action potential (AP) generation was measured as the peak of the second derivative of voltage with time during the rising phase of the AP. To measure the membrane rheobase, all neurons were held below threshold at -60 to -70 mV with a bias current. The minimal amount of current necessary for spike generation was defined as the rheobase. APs with peak amplitude above 0 mV were included in the analysis. To determine the F-I plot, 1-s current injections of increasing amplitude were delivered, and the average spike frequency during a step was determined by counting the number of spikes during the each 1-s step and plotted against the injected current amplitude. The first 1-s step was always determined by the amount of current that would produce three APs and the second 1-s step was determined by the amount of current that would produce at least one additional AP. The increment between the first and second steps would then be applied in the subsequent steps. The voltage threshold for AP generation was measured as the peak of the second derivative of voltage with time at the beginning of the action potential. Spike after hyperpolarization (AHP) amplitude was measured from the AP threshold to the minimal voltage after the action potential. Data analysis was performed with Spike2 (Cambridge Electronic Design, Cambridge, UK), CorelDraw (Softonic), Sigma Plot (Systat Software) and Excel (Microsoft, Seattle, WA). Data are given as mean ± S.E.M. The firing frequency was determined by a custom-made program in Spike (courtesy of Dr. Thomas Cleland, Cornell University) defined as the time (s) between each AP at each depolarizing current step, which was them converted to frequency (1/s = Hz) and plotted against the injected current.
The following compounds were used: dopamine (Sigma), caffeine (Sigma), the A1R antagonist 1,3-dipropyl-8- cyclopentylxanthine (DPCPX; Tocris), the A1R agonist 2-chloro-N6-cyclopentyladenosine (CPA; Tocris) and the D1R agonist SKF81297 hydrobromide (Tocris). TAT-fused TM peptides TM5 and TM7 (4 μM) were applied to the perfusion (2-3 ml/min rate) after the patch was made and before starting drug perfusion. Caffeine was prepared everyday (store at 4°C) in de-ionized ultrapure water and later dissolved in normal-ACSF whereas the adenosine and dopamine receptor agonists and antagonists were prepared as stock solutions (stored at -20°C) in dimethyl sulphoxide (DMSO) or de-ionized ultrapure water (depending on solubility) and later diluted in the working normal-ACSF solution.
Statistical analyses
Quantitative data were expressed as the mean ± SEM. No statistical methods were used to predetermine sample sizes, but sample sizes are comparable to those reported in previous publications [8, 9, 37, 38]. Statistical analyses were performed using Sigma Plot and Past3 and GraphPad Prism software. Differences among more than two groups of results were analyzed by one-way or repeated-measures ANOVA (followed by Dunnett's or Bonferroni's comparisons). Differences between experimental group pairs were analyzed with paired or non-paired t test.
Results
Identification of A1R-D1R heteromers in transiently transfected HEK-293T cells
Bioluminescent Resonance Energy Transfer (BRET) and Bimolecular Fluorescence Complementation (BiFC) techniques were first used to demonstrate A1R-D1R heteromerization in vitro. In both techniques, two biosensors that can only interact when they are in very close proximity are separately fused to the two putatively interacting receptors. In BRET (Figure 1A), a bioluminescence donor (such as Renilla luciferase; Rluc) transfers energy to a fluorescence acceptor molecule (such as Yellow Fluorescence Protein; YFP). Saturable BRET curves were obtained in several preparations of HEK-293T cells transfected with a constant amount of A1R-Rluc cDNA and increasing amounts of D1R-YFP cDNA (Figure 1A; BRETmax = 37 ± 5 mBU and BRET50 = 2 ± 1 of YFP/Rluc ratio), suggestive of specific intermolecular interactions (Marullo and Bouvier, 2007). In contrast, linear plots were obtained in cells transfected with a constant amount of A1R-Rluc and increasing amounts of the serotonin 5-HT2B receptor fused to YFP (5-HT2BR; Figure 1A), indicative of random-collisions typical of non-specific interactions [39]. Further support for the specificity of A1R-D1R heteromerization was obtained by using BiFC, where two complementary halves of YFP (Venus variant; the c-terminal moiety of YFP, cYFP, and the n-terminal moiety of YFP, nYFP) are separately fused to the two putative interacting receptors. Fluorescence is obtained after reconstitution of functional YFP (Figure 1B) [9, 34, 35]. Clear fluorescent values could be detected in HEK-293T cells co-transfected with A1R-nYFP and D1R-cYFP cDNAs, which were used as comparative control. Those values were significantly lower when A1R-nYFP was co-transfected with serotonin 5-HT2A receptor (5-HT2AR)-cYFP, when D1R-cYFP was co-transfected with cannabinoid CB1 receptor (CB1R)-nYFP or by adding a synthetic peptide with the amino acid sequence of transmembrane domain (TM) 5, but not TM 7, of the D1R (TM5 and TM7 peptides, respectively); oneway ANOVA followed by Dunnett's comparisons (F(4, 24) = 35.89, p < 0.0001, Figure 1B).
Figure 1. A1R-D1R heteromer expression in transiently transfected HEK-293T cells.
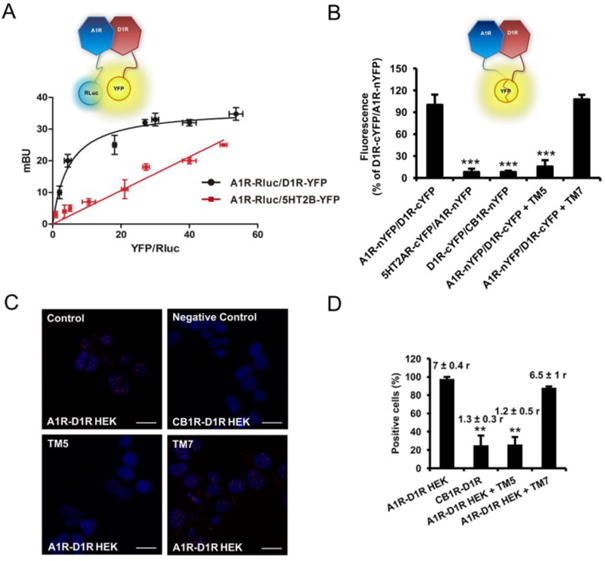
In (A) BRET saturation experiments were performed in HEK-293T cells transfected with 0.5 μg of A1R-Rluc cDNA and increasing amounts of D1R-YFP cDNA (1 μg to 5 μg, black curve) or, as negative control, with 0.5 μg of A1R-Rluc cDNA and increasing amounts of 5HT2BR-YFP cDNA (0.5 μg to 5 μg, red line). The relative amount of BRET is given as a function of 100 × the ratio between the fluorescence of the acceptor (YFP) and the luciferase activity of the donor (Rluc). BRET is expressed as milli BRET units (mBU) and is given as the mean ± SD of 5–6 experiments grouped as a function of the amount of BRET acceptor. At the top a schematic representation of BRET is given. In (B), BiFC experiments were performed in HEK-293T cells transfected with cDNA (4 μg each one) of A1R-nYFP and D1R-cYFP or, as negative controls, 5HT2A-nYFP and D1R-cYFP or D1R-cYFP and CB1R-nYFP. Cells were treated for 4 h with vehicle or 4 μM of D1R TM5 or TM7 peptides and fluorescence at 530 nm was read. Values are mean ± S.E.M. of 5-6 experiments (100% represents 30000 fluorescence units); ***: p < 0.001, as compared with A1R-nYFP and D1R-cYFP expressing cells not treated with peptides (one-way ANOVA followed by Dunnett's comparisons). In (C and D), HEK-293T cells transfected with 0.3 μg of A1R and 0.5 μg of D1R cDNA (A1R-D1R HEK) or with 0.4 μg of CB1R and 0.5 μg of D1R cDNA as negative control (CB1R-D1R HEK) were treated for 4 h with vehicle or with 4 μM of D1R TM5 or TM7 peptides before performing proximity ligation assays. In (C), confocal microscopy images (superimposed sections) are shown in which A1R-D1R heteromers appear as red spots in vehicle and TM7 treated cells and not in cells treated with TM5 peptide or in the negative control. In all cases, cell nuclei were stained with DAPI (blue). Scale bars = 20 μM. In (D), the percentage of cells showing red spots related to the total cell number determined as stained blue nuclei is given in each case as well as the ratio (r) between the number of red spots and cells showing spots (top columns). Values are mean ± S.E.M. of n = 5-6; **: p < 0.01 as compared with cells not treated with peptides (one-way ANOVA followed by Dunnett's comparisons).
Additional evidence of heteromer formation involving TM 5 of the D1R was provided by using the proximity ligation assay (PLA). This technique permits the direct detection of molecular interactions between two endogenous proteins or transfected proteins without the need of fusion proteins, allowing its use in native tissues [8, 9, 35]. A1R-D1R complexes were observed as red punctate staining in HEK-293T cells expressing A1R and D1R, but not in cells expressing CB1R and D1R, as negative control (Figure 1C, top panels). Pretreatment of cells with TM5 of D1R, but not with TM7, significantly decreased PLA staining (Figure 1C, bottom panels), decreasing both the percentage of stained cells (showing one or more red spots) and the mean number (r) of red spots per stained cell; one-way ANOVA followed by Dunnett's comparisons (F(3, 14) = 21.21, p < 0.0001, Figure 1D). These results demonstrate that, in fact, the A1R-D1R complexes correspond to A1R-D1R heteromers, where TM5 of D1R forms part of the heteromer interface.
Identification of functional A1R-D1R heteromers in stably transfected Ltk- cells
An important characteristic of a GPCR heteromer is that their functional properties are demonstrably different from those of its individual components [40]. A common consequence of GPCR heteromerization is a specific integrated signaling upon co-activation of the molecularly different protomers as compared to their separate activation [30, 31, 40]. In order to control receptor expression and to study wild-type receptors, not fused to biosensors, the biochemical properties of A1R-D1R heteromers were analyzed in a fibroblast LtK- cells stably transfected with A1R and D1R (A1R-D1R Ltk-cells; [12]). The heteromer expression was first evaluated by PLA. A1R-D1R complexes, observed as red punctate staining, were very significantly increased in A1R-D1R Ltk- cells as compared with the D1R Ltk- cell line that did not express A1R, used as negative control (Figure 2A, top panels). The same increase could be observed in the presence of TM7 of D1R, but not in the presence of TM5 (Figure 2A, bottom panels); one-way ANOVA followed by Dunnett's comparisons (F (3, 43) = 32.00, p < 0.0001, Figure 2B). These results demonstrate the presence of A1R-D1R heteromers also in A1R-D1R Ltk- cells, with TM5 of D1R forming part of the heteromer interface.
Figure 2. A1R-D1R heteromer expression and adenylyl cyclase signaling in fibroblast Ltk- cells.
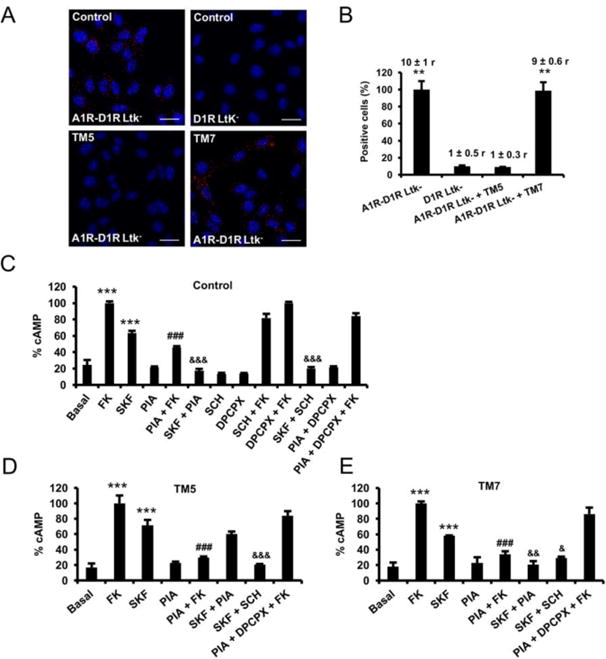
In (A and B), Ltk- cells expressing A1R and D1R (A1R-D1R Ltk) or only D1R as negative control (D1R Ltk-) were treated for 4 h with vehicle or with 4 μM of D1R TM5or TM7 peptides before performing proximity ligation assays. In (A), confocal microscopy images (superimposed sections) are shown in which heteromers appear as red spots in vehicle and TM7 treated cells and not in cells treated with TM5 peptide or in the negative control. In all cases, cell nuclei were stained with DAPI (blue). Scale bars = 20 μM. In (B) the percentage of cells showing spots related to the total cells determined as stained blue nuclei is given in each case as well as the ratio (r) between the number of red spots and cells showing spots (top columns). Values are mean ± S.E.M. of n = 8-16; **: p < 0.01 as compared with D1R Ltk-cells (one-way ANOVA followed by Dunnett's comparisons). In (C-E), A1R-D1R Ltk- were pre-treated for 4 h with vehicle (C) or with 4 μM of D1R TM 5 (D) or TM 7 (E) peptides. Cells were then treated for 10 min with vehicle or with A1R antagonist DPCPX (1 μM) or the D1R antagonist SCH23390 (1 μM) prior being stimulated with medium, the A1R agonist R-PIA (100 nM) or the D1R agonist SKF38393 (200 nM) in the absence or in the presence of 20 μM forskolin (FK). Values are mean ± S.E.M. of n = 4–5 and are expressed as percentage of FK treated cells in each condition (100% represents 80-100 pmols cAMP/106 cells) ***: p < 0.001 versus basal; ###: p < 0.001 versus FK; &&&, && and &: p < 0.001, p < 0.01 and p < 0.05 versus SKF38393, respectively (one-way ANOVA followed by Bonferroni's comparisons).
To study the functional characteristics of A1R-D1R heteromer we determined cAMP production in A1R-D1R Ltk- cells. As expected, the D1R agonist (SKF38393; 200 nM) produced a significant cAMP accumulation, according to the Gs coupling to D1R, while the A1R agonist (R-PIA; 100 nM) significantly decreased forskolin-induced cAMP, according to the Gi coupling to A1R (Figure 2C); the effects of SKF38393 and R-PIA were selectively counteracted by the respective selective antagonists SCH23390 (1 μM) and DPCPX (1 μM) (Figure 2C), which did not significantly modify basal cAMP or forskolin-induced cAMP on their own; and upon co-administration with both agonists, R-PIA was able to counteract SKF38393-induced adenylyl cyclase activation; one-way ANOVA followed by Bonferroni's comparisons (F(12, 26) = 144.1, p < 0.0001, Figure 2C). The effect of forskolin, and the A1R and D1R agonists, alone or in combination, were performed in the presence of either TM5 (Figure 2C) or TM7 (Figure 2D) of D1R. Importantly, the only significantly difference as compared with the experiments without peptides was the inability of R-PIA to counteract SKF38393 effect in the presence of TM5, as analyzed by one-way ANOVA followed by Bonferroni's comparisons (F(7, 16) = 35.63, p < 0.0001, Figure 2D; F(7, 16) = 40.25, p < 0.0001, Figure 2E). These results therefore demonstrate that the ability of A1R activation to counteract D1R agonist-mediated adenylyl cyclase activation depends on A1R-D1R heteromerization, as recently demonstrated for the A2AR-D2R heteromer [35].
Identification of A1R-D1R heteromers in mouse spinal motoneurons
We then investigated the possible existence of A1R-D1R heteromers in the mouse spinal cord, which could mediate our recently described A1R-D1R interactions involved in the ability of caffeine to enhance spinally-generated locomotor activity [32]. In order to identify the cellular location of these potential receptor heteromers in the spinal cord, we performed immunohistochemical experiments using antibodies directed toward A1R and D1R in postnatal 0-5 (P0-P5) mouse lumbar spinal cord slices, localization of the network controlling hind-limb locomotion [41-46]. A rhodamine-labeled secondary antibody directed toward D1R antibody revealed positive staining (red fluorescence) throughout the central, medial and lateral ventral region of the lumbar spinal cord (Figs. 3A and 3B, middle panels). The same slices were also processed for A1R immunohistochemistry with an Alexa Fluor 488-labeled secondary antibody. Positive A1R staining (green fluorescence) was principally located within the ventral-lateral region (Rexed lamina IX) of the lumbar spinal cord within the motoneurons, which were identified by cell size and location (Figs. 3A and 3B, left panels). Overlay of the A1R and D1R immunostaining confirmed that co-labeling of both receptors was mainly found within lumbar motoneurons [47, 48], which were visually identified by their location, size and clustering (Figs. 3A and 3B, right panels). These results suggested that A1R-D1R heteromers could be localized in lumbar motoneurons.
Figure 3. Immunohistochemical co-localization of A1R and D1R in the ventral lumbar spinal cord.
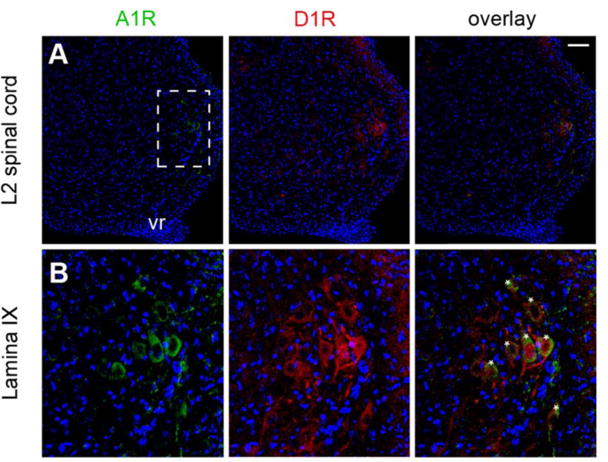
In (A), low-magnification confocal images of a ventrolateral L2 section (P3 mouse), showing the distributions of A1R (green fluorescence from Alexa Fluor 488-conjugated secondary antibodies), D1R (red fluorescence from rhodamine-conjugated secondary antibodies) and the overlay of both fluorescent labels. Hoechst staining allows identification of cell nuclei (blue channel). In (B), higher-magnification of the framed area in (A), showing the co-localization of A1R and D1R in lamina IX of the ventral spinal cord, where motoneurons can be identified by size and position (asterisks); vr: ventral root; scale bar: 50 μM.
The expression of A1R-D1R heteromers in lumbar spinal cord was then demonstrated by PLA in slices from mouse spinal cord (lumbar region) from P4, P6 and P11. A1R-D1R complexes were detected as red spots and were quantified as percentage of cells showing one or more red spots and as the mean number of red spots per stained cell in lamina IX (motoneurons) and laminae VIII and X (interneurons) (Figs. 4A and 4B). Irrespective of the postnatal period, A1R-D1R complexes were significantly increased in cells from lamina IX, corresponding to motoneurons, as compared to cells from laminae VIII and X (Figs. 4A and 4B); one-way ANOVA followed by Dunnett's comparisons (F (9, 82) = 64.84, p < 0.0001, Figure 4B). Importantly, pretreatment of the spinal cord slices with the TM5 peptide of D1R involved in the A1R-D1R heteromer interface (see above), but not with TM7, significantly decreased PLA staining (Figure 4C), decreasing both the percentage of stained cells and the mean number of red spots per stained cell; one-way ANOVA followed by Dunnett's comparisons (F (2, 24) = 16.50, p < 0.0001, Figure 4D). These results demonstrated the existence of A1R-D1R heteromers in the mouse spinal motoneurons.
Figure 4. A1R-D1R heteromer expression in mouse spinal cord motoneurons.
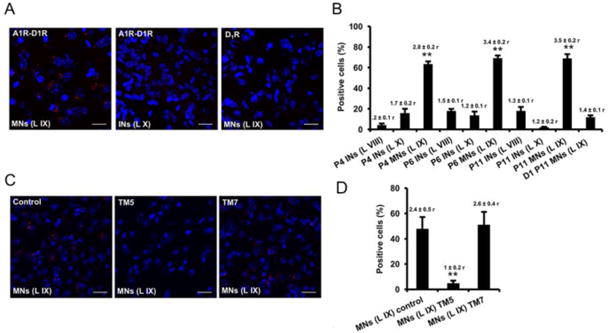
In situ proximity ligation assays were performed using slices from mouse lumbar spinal cord from P4, P6 and P11 animals. Slices from lamina IX (motoneurons) and laminae VIII and X (interneurons) were analyzed using specific primary antibodies directed against A1R and D1R (A1R-D1R) or only against D1R as negative control (D1). Slices were treated for 4 h with vehicle (A, B and control in C and D) or with 4 μM of D1R TM 5 or TM7 peptides (C, D). In (A and C), confocal microscopy images (superimposed sections) from (A) laminae IX and X from P11 animals in which heteromers appear as red spots in lamina IX, but not in lamina X or in negative controls, and from (C) lamina IX from P5 animals in which heteromers appear as red spots in non peptide treated slices (control) and treated with D1R TM 7 but not in slices treated with TM5. In all cases, cell nuclei were stained with DAPI (blue). Scale bars = 20 μM. In (B and D) the percentage of cells showing red spots related to the total cell number determined as stained blue nuclei is given in each case as well as the ratio (r) between the number of red spots and cells showing spots (top columns). Values are mean ± S.E.M. of n =7-15. **: p < 0.01 as compared with the negative control D1 in (B); ** p < 0.01 as compared with the control non-treated with peptides in (D) (one-way ANOVA followed by Dunnett's comparisons).
Functional properties of A1R-D1R heteromers in mouse spinal motoneurons
After establishing the anatomical localization of A1R-D1R heteromers in the spinal motoneuron we investigated their functional and pharmacological significance by intracellular electrophysiological recordings using WCR and PPR. Caffeine (50 μM) did not show significant excitatory effects when applied alone (data not shown), but it significantly increased excitability of the spinal motoneuron when co-applied with dopamine (3 μM); repeated-measures ANOVA followed by Dunnett's comparisons (F (2, 18) = 3.64, p = 0.0468, Figure 5A3). Figure 5A1 shows a representative response of a motoneuron to increasing pA depolarizing steps under control, dopamine, and dopamine + caffeine conditions. We also generated F–I plots of the average spike frequency over a range of current step amplitudes; a representative example is given in Figure 5A2, where dopamine + caffeine causes a parallel upward shift in the frequency response to the current step. The quantified averaged response from 10 motoneurons is shown in Figure 5A3. The result of dopamine + caffeine was mimicked by dopamine (3 μM) + the A1R antagonist DPCPX (1 μM) (Figure 5B). DPCPX significantly increased excitability of the spinal motoneuron when co-applied with dopamine (3 μM); repeated-measures ANOVA followed by Dunnett's comparisons (F (2, 6) = 21.85, p = 0.0018, n = 4 motoneurons; Figure 5B3). The effect of DPCPX was apparently more effective than that of caffeine, which could be related to the non-selective antagonism of caffeine, which could also block A2AR and functionally act opposite to A1R. In fact, A2AR have also been identified in spinal motoneurons, although with a low expression unless under ischemic conditions [49]. The same qualitative effects of caffeine or DPCPX on dopamine could be explained by the ability of caffeine or the A1R antagonist DPCPX to potentiate the depolarizing effects of dopamine within the A1R-D1R heteromers of the spinal motoneuron. This was demonstrated in experiments using the destabilizing peptide TM5 of D1R and TM7 as a negative control, according to our results with peptides in cultured cells and in PLA ex vivo (see above). In these experiments, we tested the effect of caffeine (50 μM) in the presence of the D1R receptor agonist SKF81297 (1 μM). The selective D1R agonist was used instead of dopamine to address in parallel the question of the selective involvement of the D1R in the effects of dopamine plus caffeine. As expected, in the presence of the non-destabilizing peptide TM7 (4 μM), SKF81297 + caffeine, but not SKF81297 alone, significantly increased neuronal excitability. The evoked spike frequency was unchanged in the presence of SKF81297 + TM7 but it was significantly increased when caffeine was applied in the presence of SKF81297 + TM7 (Figure 5C1). The representative F–I plot shows that SKF81297 + caffeine + TM7 caused a parallel upward shift in the frequency response to the current steps (Figure 5C2). The quantified averaged response from 6 motoneurons is shown in Figure 5C3 (repeated-measures ANOVA followed by Dunnett's comparisons (F (2, 10) = 8.46, p < 0.0078). The effect of SKF81297 (in the presence of the inactive peptide TM7) was apparently more effective than that of dopamine, which could be related to the non-selective agonism of dopamine, which could also activate D2-like receptors (D2Ror D3R) and act functionally opposite to D1R. In fact, D2-like receptors have also been identified in spinal motoneurons [50]. On the other hand, there was a trend for the opposite effect, a decrease in the neuronal excitability, when caffeine was applied in the presence of SKF81297 + TM5 (Figure 5D). The quantified averaged response from 4 motoneurons is shown in Figure 5D3. A repeated-measures ANOVA followed by Dunnett's comparisons did not show significant differences between control, SKF81297 and the SKF81297 + caffeine in the presence of TM5 of D1R (F (2, 6) = 4.54, p = 0.0628, Figure 5D3).
Figure 5. A1R-D1R heteromer-dependent excitatory effects of caffeine in the spinal motoneuron.
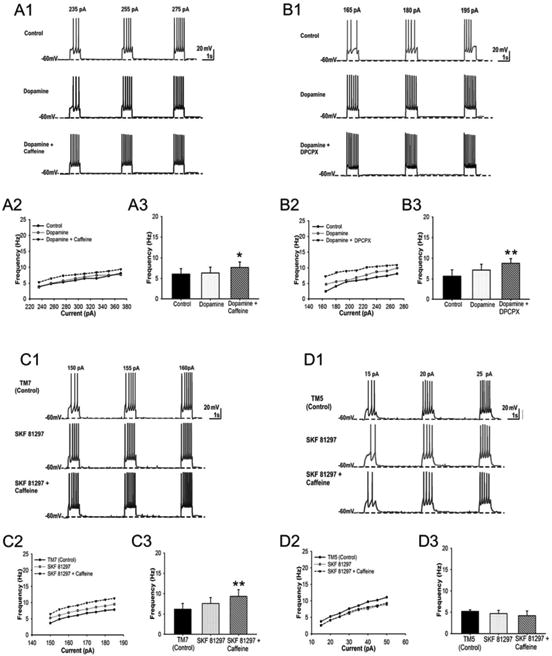
WCR and PPR recordings of spinal motoneurons with application of dopamine (3 μM) or the D1R agonist SKF81297 (1 μM) and caffeine (50 μM) or the selective A1R antagonist DPCPX (1 μM). In (A), the effect of caffeine + dopamine is compared with the effect of dopamine alone and with the non-drug control conditions. In (B), the effect of DPCPX plus dopamine is compared with the non-drug control conditions. In (C and D), the effect of caffeine + SKF81297 is compared with the effect of SKF81297 alone and with non-drug conditions and with the application of synthetic peptides with the sequence of TM5 (C) and TM7 (D) of the D1R. (A1, B1, C1 and D1) show representative responses of a motoneuron to increasing pA depolarizing steps; (A2, B2, C2 and D2) show F–I plots of the average spike frequency over a range of current step amplitudes; (A3, B3, C3 and D3) show the quantified averaged response from several motoneurons in mean ± S.E.M. of n = 5–10. Repeated-measures ANOVA followed by Dunnett's comparisons or paired t test showed significant increases in spike frequency of the motoneurons treated with caffeine + dopamine (*: p < 0.05), DPCPX + dopamine (***: p < 0.001), and caffeine + SKF81297 in the presence of TM7 of D1R (**: p < 0.01), but not TM5, as compared with control. Specific parameters (threshold, rheobase and AP amplitude; see Materials and Methods) for each group of experiments are shown as Supplementary Information.
The cellular specificity of these interactions was also demonstrated by parallel experiments in spinal interneurons (tested from lamina VII, VIII or X) (Figure S1). SKF81297 (1 μM) + Caffeine (50 μM) co-application did not produced an increase, but a non-significant decrease in excitability as compared to the control condition. Figure S1A1 shows a representative response of an interneuron to increasing pA depolarizing steps under control, SKF81297 (1 μM) or SKF81297 + caffeine (50 μM) conditions. The representative F–I plot shows a trend for a parallel downward shift in the frequency response to the current steps (Figure S1A2), but the quantified averaged response from 15 interneurons did not show significant differences between the three different conditions (repeated-measures ANOVA followed by Dunnett's comparisons (F(2,10) = 1.32, p = 0.3086, Figure S1A3).
Discussion
The present study demonstrates that A1R-D1R heteromers play a significant role in the control of the excitability of spinal motoneurons. More specifically, we demonstrate: (1) that A1R-D1R heteromers are specifically localized in the spinal motoneurons (not in interneurons); (2) that A1R-D1R heteromers mediate the ability of adenosine to exert a tonic inhibitory modulation of dopamine-mediated increase in neuronal excitability; and (3), that the psychostimulant effects of caffeine in spinal motoneurons depends on its ability to counteract the tonic inhibitory effect of endogenous adenosine via A1R-D1R heteromers, which extends to the spinal cord the previously established supraspinal mechanism of these psychostimulant effects. More specifically, A2AR-D2R and A1R-D1R heteromers segregated in two different striatal populations have been invoked as main targets for the central effects of caffeine. However, only striatal A2AR-D2R heteromers have been undoubtedly identified [9, 51, 52], while A1R-D1R heteromerization remained still to be unequivocally demonstrated. The use of specific destabilizing peptides, as demonstrated by BiFC, PLA and signaling experiments in transfected cells, allowed us to demonstrate both the ability of A1R and D1R to heteromerize (with TM5 of D1R forming part of the heteromeric interface) and the dependence of A1R-D1R heteromerization for the ability of A1R activation to inhibit D1R agonist-induced adenylyl cyclase activation. We also recently demonstrated a dependence of A2AR-D2R heteromerization for the ability of the Gi-coupled D2R to inhibit Gs-coupled A2AR-mediated adenylyl cyclase activation [35]. The present results therefore confirm that the canonical antagonistic interaction at adenylyl cyclase level, by which a Gi-coupled receptor counteracts adenylyl cyclase activation mediated by a Gs-coupled receptor, is a functional property of receptor heteromers [35]. In addition, with the use of specific destabilizing peptides we have identified A1R-D1R heteromers in the spinal motoneuron (PLA experiments) and, more importantly, we have established their pharmacological relevance to modulate neuronal excitability (patch-clamp experiments). The significant pharmacological interactions between A1R and D1R ligands that depend on A1R-D1R heteromerization observed in the present study demonstrate that a significant population of the motoneuron D1R forms heteromers with A1R.
We can now provide the neuronal correlate of our recent study on the effects of caffeine on the NMDA/serotonin/dopamine-induced fictive locomotor behavior in neonatal mouse lumbar spinal cord [32]. In that study, caffeine or the A1R antagonist DPCPX were found to accelerate the ongoing locomotor rhythm (measured by extracellular ventral root recording), unless in the absence of dopamine or in the presence of a selective D1R antagonist or a blocker of cAMP-dependent protein kinase (PKA), demonstrating a dependence on A1R-D1R antagonistic interactions [32]. However, we did not resolve the neuronal localization of these pharmacological interactions nor we established their dependence on A1R-D1R heteromerization (see Introduction). Using patch-clamp electrophysiology, applying dopamine or the D1R agonist SKF81297, did not show a significant increase in the intrinsic excitability of motoneurons. Based on our previous study [32], we assumed this could be related to a tonic inhibition exerted by adenosine present in the slice preparation mediated by the A1R-D1R heteromer. In fact, caffeine, which did not produce a significant effect on its own, significantly increased the excitability of motoneurons when co-applied with dopamine or SKF81297. Also in complete correlation with the results obtained by extracellular ventral root recording on fictive locomotor behavior in neonatal mouse lumbar spinal cord [32], the effects of caffeine were reproduced by the A1R antagonist DPCPX. More importantly, the effect of caffeine plus SKF81297 was specifically counteracted by a TM peptide that selectively destabilized A1R-D1R heteromers both in transfected mammalian cells and in the mouse spinal motoneuron. These results therefore indicate that the ability of caffeine to stimulate locomotor activity in the mammalian spinal cord is mediated by A1R-D1R heteromers localized in the spinal motoneuron.
The monoamines serotonin, norepinephrine and dopamine are potent modulators of neuronal networks within the spinal cord, including central pattern generators responsible for locomotion [53-55]. Currently, studies into pharmacological therapy after spinal cord injury (SCI) focuses on the combination of monoaminergic drugs with electrical and physical training, such as the use of quizapine, a broad-spectrum serotonin agonist, with step training and epidural stimulation in animal models and patients [56-58]. Spinal dopaminergic neurotransmission and more specifically D1R are also being considered as potential new targets to promote recovery of locomotor function following SCI (reviewed in [55]). It has already been established that D1-like receptors (D1R and D5R) and D2-like receptors (D2R, D3R and D4R) are mostly segregated in the spinal cord and that they play opposite modulatory effects on spinal central pattern generator of locomotion [54, 55]. Although there are differences among species, in the mouse, D2-like receptors are highly expressed in laminae I-II of the dorsal horn, where they mediate inhibitory effects, although they are also expressed, with less density, in the ventral horn [50]. On the other hand, D1-like receptors, are highly expressed in the ventral horn, in lamina IX, including the motoneurons, where they mediate excitatory effects [48, 54, 55]. In addition, adenosine has been shown to also modulate the mouse spinal locomotor network and also suppress neuronal cell death induced by ischemia in rat spinal motoneurons [32, 59-62]. The present results provide a conceptual new approach, targeting the A1R-D1R heteromer. Thus, it has already been inferred that pharmacological targeting of receptor heteromers could become an important area for developing more selective drugs with reduced side effects [30]. The present results can also have implications for Parkinson's disease (PD) and other motor aging-associated disorders. Normal aging is associated with a decrease in motor function [63] and a concomitant increase in muscle stiffness and tone [64], which we have recently suggested to depend on an aging-dependent D1R upregulation in the spinal cord with a lack of concomitant increased expression of inhibitory receptors [65]. Finally, recent preclinical evidence indicates that alterations in the adenosinergic system, and particularly downregulation of A1R, play a key pathogenetic role in Restless Legs Syndrome (RLS) [66, 67]. Depending on the spinal cord pathology, either selective A1R antagonists (SCI and aging disorders) or drugs that would produce an increase in the endogenous tone of adenosine (RLS), by acting on the A1R-D1R heteromer in the spinal cord, could indirectly modulate D1R-mediated excitability of the spinal motoneuron.
Supplementary Material
Acknowledgments
We would like to thank Dr. Thomas Cleland (Cornell University) for providing the scripts for electrophysiological data analysis, Joselyne Álvarez-González, Garrett Seale, Amelia Merced and Andrea Husch for technical assistance. This work is supported by COBRE Center for Neuroplasticity (NIH NIGMS 1P20GM103642), intramural funds of the National Institute of Drug Abuse, NSF (DBI-1337284), RCMI (NIMHD 8G12-MD007600), RISE Program (R25GM061151-13), and grants from the Spanish “Ministerio de Economía y Competitividad” and European Regional Development Funds of the European Union (SAF2014-54840-R and SAF2017-87629-R), the “Fundació La Marató de TV3” (20140610), and Government of Catalonia (2017-SGR-1497).
References
- 1.McCarley RW. Neurobiology of REM and NREM sleep. Sleep Med. 2007;8(4):302–330. doi: 10.1016/j.sleep.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Ferré S. Role of the central ascending neurotransmitter systems in the psychostimulant effects of caffeine. J Alzheimers Dis. 2010;20(Suppl 1):S35–S49. doi: 10.3233/JAD-2010-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007;14(7):1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- 4.Ferré S. Mechanisms of the psychostimulant effects of caffeine: implications for substance use disorders. Psychopharmacology. 2016;233(10):1963–1979. doi: 10.1007/s00213-016-4212-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fredholm BB, IJzerman AP, Jacobson KA, Linden J, Müller CE. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors--an update. Pharmacol Rev. 2011;63(1):1–34. doi: 10.1124/pr.110.003285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferré S, Fredholm BB, Morelli M, Popoli P, Fuxe K. Adenosine-dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997;20(10):482–487. doi: 10.1016/s0166-2236(97)01096-5. [DOI] [PubMed] [Google Scholar]
- 7.Ferré S. An update on the mechanisms of the psychostimulant effects of caffeine. J Neurochem. 2008;105(4):1067–1079. doi: 10.1111/j.1471-4159.2007.05196.x. [DOI] [PubMed] [Google Scholar]
- 8.Navarro G, Aguinaga D, Moreno E, Hradsky J, Reddy PP, Cortés A, Mallol J, Casadó V, Mikhaylova M, Kreutz MR, Lluís C, Canela EI, McCormick PJ, Ferré S. Intracellular calcium levels determine differential modulation of allosteric interactions within G protein-coupled receptor heteromers. Chem Biol. 2014;21(11):1546–1556. doi: 10.1016/j.chembiol.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonaventura J, Navarro G, Casadó-Anguera V, Azdad K, Rea W, Moreno E, Brugarolas M, Mallol J, Canela EI, Lluís C, Cortés A, Volkow ND, Schiffmann SN, Ferré S, Casadó V. Allosteric interactions between agonists and antagonists within the adenosine A2A receptor-dopamine D2 receptor heterotetramer. Proc Natl Acad Sci USA. 2015;112(27):E3609–E3618. doi: 10.1073/pnas.1507704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferré S, Popoli P, Giménez-Llort L, Finnman UB, Martínez E, Scotti de Carolis A, Fuxe K. Postsynaptic antagonistic interaction between adenosine A1 and dopamine D1 receptors. Neuroreport. 1994;6(1):73–76. doi: 10.1097/00001756-199412300-00020. [DOI] [PubMed] [Google Scholar]
- 11.Ferré S, O'Connor WT, Svenningsson P, Bjorklund L, Lindberg J, Tinner B, Stromberg I, Goldstein M, Ogren SO, Ungerstedt U, Fuxe K. Dopamine D1 receptor-mediated facilitation of GABAergic neurotransmission in the rat strioentopenduncular pathway and its modulation by adenosine A1 receptor-mediated mechanisms. Eur J Neurosci. 1996;8(7):1545–1553. doi: 10.1111/j.1460-9568.1996.tb01617.x. [DOI] [PubMed] [Google Scholar]
- 12.Ferré S, Torvinen M, Antoniou K, Irenius E, Civelli O, Arenas E, Fredholm BB, Fuxe K. Adenosine A1 receptor-mediated modulation of dopamine D1 receptors in stably cotransfected fibroblast cells. J Biol Chem. 1998;273(8):4718–4724. doi: 10.1074/jbc.273.8.4718. [DOI] [PubMed] [Google Scholar]
- 13.Ferré S, Rimondini R, Popoli P, Reggio R, Pèzzola A, Hansson AC, Andersson A, Fuxe K. Stimulation of adenosine A1 receptors attenuates dopamine D1 receptor-mediated increase of NGFI-A, c-fos and jun-B mRNA levels in the dopamine-denervated striatum and dopamine D1 receptor-mediated turning behaviour. Eur J Neurosci. 1999;11(11):3884–3892. doi: 10.1046/j.1460-9568.1999.00810.x. [DOI] [PubMed] [Google Scholar]
- 14.Popoli P, Giménez-Llort L, Pezzola A, Reggio R, Martínez E, Fuxe K, Ferré S. Adenosine A1 receptor blockade selectively potentiates the motor effects induced by dopamine D1 receptor stimulation in rodents. Neurosci Lett. 1996;218(3):209–213. doi: 10.1016/s0304-3940(96)13143-8. [DOI] [PubMed] [Google Scholar]
- 15.Rimondini R, Ferré S, Giménez-Llort L, Ogren SO, Fuxe K. Differential effects of selective adenosine A1 and A2A receptor agonists on dopamine receptor agonist-induced behavioural responses in rats. Eur J Pharmacol. 1998;347(2-3):153–158. doi: 10.1016/s0014-2999(98)00107-1. [DOI] [PubMed] [Google Scholar]
- 16.Mayfield RD, Jones BA, Miller HA, Simosky JK, Larson GA, Zahniser NR. Modulation of endogenous GABA release by an antagonistic adenosine A1/dopamineD1 receptor interaction in rat brain limbic regions but not basal ganglia. Synapse. 1999;33(4):274–281. doi: 10.1002/(SICI)1098-2396(19990915)33:4<274::AID-SYN4>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 17.Ginés S, Hillion J, Torvinen M, Le Crom S, Casadó V, Canela EI, Rondin S, Lew JY, Watson S, Zoli M, Agnati LF, Verniera P, Lluis C, Ferré S, Fuxe K, Franco R. Dopamine D1 and adenosine A1 receptors form functionally interacting heteromeric complexes. Proc Natl Acad Sci USA. 2000;97(15):8606–8611. doi: 10.1073/pnas.150241097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Florán B, Barajas C, Florán L, Erlij D, Aceves J. Adenosine A1 receptors control dopamine D1-dependent [(3)H]GABA release in slices of substantia nigra pars reticulata and motor behavior in the rat. Neuroscience. 2002;115(3):743–751. doi: 10.1016/s0306-4522(02)00479-7. [DOI] [PubMed] [Google Scholar]
- 19.Le Crom S, Prou D, Vernier P. Autocrine activation of adenosine A1 receptors blocks D1A but not D1B dopamine receptor desensitization. J Neurochem. 2002;82(6):1549–1552. doi: 10.1046/j.1471-4159.2002.01115.x. [DOI] [PubMed] [Google Scholar]
- 20.Schwienbacher I, Fendt M, Hauber W, Koch M. Dopamine D1 receptors and adenosine A1 receptors in the rat nucleus accumbens regulate motor activity but not prepulse inhibition. Eur J Pharmacol. 2002;444(3):161–169. doi: 10.1016/s0014-2999(02)01622-9. [DOI] [PubMed] [Google Scholar]
- 21.Toda S, Alguacil LF, Kalivas PW. Repeated cocaine administration changes the function and subcellular distribution of adenosine A1 receptor in the rat nucleus accumbens. J Neurochem. 2003;87(6):1478–1484. doi: 10.1046/j.1471-4159.2003.02121.x. [DOI] [PubMed] [Google Scholar]
- 22.Batista LC, Prediger RD, Morato GS, Takahashi RN. Blockade of adenosine and dopamine receptors inhibits the development of rapid tolerance to ethanol in mice. Psychopharmacology. 2005;181(4):714–721. doi: 10.1007/s00213-005-0014-7. [DOI] [PubMed] [Google Scholar]
- 23.Cao Y, Xie KQ, Zhu XZ. The enhancement of dopamine D1 receptor desensitization by adenosine A1 receptor activation. Eur J Pharmacol. 2007;562(1-2):34–38. doi: 10.1016/j.ejphar.2007.01.090. [DOI] [PubMed] [Google Scholar]
- 24.Uustare A, Reinart R, Rinken A. Modulation of dopamine D1 receptor signaling by adenosine A1 receptors in Sf9 cells requires expression of Gi proteins. Neurosci Lett. 2006;406(3):169–173. doi: 10.1016/j.neulet.2006.07.051. [DOI] [PubMed] [Google Scholar]
- 25.Yabuuchi K, Kuroiwa M, Shuto T, Sotogaku N, Snyder GL, Higashi H, Tanaka M, Greengard P, Nishi A. Role of adenosine A1 receptors in the modulation of dopamine D1 and adenosine A2A receptor signaling in the neostriatum. Neuroscience. 2006;141(1):19–25. doi: 10.1016/j.neuroscience.2006.04.047. [DOI] [PubMed] [Google Scholar]
- 26.Sakiyama Y, Hatano K, Kato T, Tajima T, Kawasumi Y, Ito K. Stimulation of adenosine A1 receptors decreases in vivo dopamine D1 receptor binding of [11C]SCH23390 in the cat striatum revealed by positron emission tomography. Ann Nucl Med. 2007;21(8):447–453. doi: 10.1007/s12149-007-0048-5. [DOI] [PubMed] [Google Scholar]
- 27.Hobson BD, O'Neill CE, Levis SC, Monteggia LM, Neve RL, Self DW, Bachtell RK. Adenosine A1 and dopamine D1 receptor regulation of AMPA receptor phosphorylation and cocaine-seeking behavior. Neuropsychopharmacology. 2013;38(10):1974–1983. doi: 10.1038/npp.2013.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen J, Zhang L, Song WL, Meng T, Wang X, Chen L, Feng LY, Xu YC, Shen JK. Design, synthesis and biological evaluation of bivalent ligands against A(1)-D(1) receptor heteromers. Acta Pharmacol Sin. 2013;34(3):441–452. doi: 10.1038/aps.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mango D, Bonito-Oliva A, Ledonne A, Cappellacci L, Petrelli R, Nisticò R, Berretta N, Fisone G, Mercuri NB. Adenosine A1 receptor stimulation reduces D1 receptor-mediated GABAergic transmission from striato-nigral terminals and attenuates l-DOPA-induced dyskinesia in dopamine-denervated mice. Exp Neurol. 2014;261:733–743. doi: 10.1016/j.expneurol.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 30.Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ, Milligan G, Pin JP, Guitart X. G protein-coupled receptor oligomerization revisited: functional and pharmacological perspectives. Pharmacol Rev. 2014;66(2):413–434. doi: 10.1124/pr.113.008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomes I, Ayoub MA, Fujita W, Jaeger WC, Pfleger KD, Devi LA. G Protein-Coupled Receptor Heteromers. Annu Rev Pharmacol Toxicol. 2016;56:403–425. doi: 10.1146/annurev-pharmtox-011613-135952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Acevedo J, Santana-Almansa A, Matos-Vergara N, Marrero-Cordero LR, Cabezas-Bou E, Díaz-Ríos M. Caffeine stimulates locomotor activity in the mammalian spinal cord via adenosine A1 receptor-dopamine D1 receptor interaction and PKA-dependent mechanisms. Neuropharmacology. 2016;101:490–505. doi: 10.1016/j.neuropharm.2015.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He SQ, Zhang ZN, Guan JS, Liu HR, Zhao B, Wang HB, Li Q, Yang H, Luo J, Li ZY, Wang Q, Lu YJ, Bao L, Zhang X. Facilitation of mu-opioid receptor activity by preventing delta-opioid receptor-mediated codegradation. Neuron. 2011;69:120–13. doi: 10.1016/j.neuron.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 34.Guitart X, Navarro G, Moreno E, Yano H, Cai NS, Sánchez-Soto M, Kumar-Barodia S, Naidu YT, Mallol J, Cortés A, Lluís C, Canela EI, Casadó V, McCormick PJ, Ferré S. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: the dopamine D1-D3 receptor heterotetramer. Mol Pharmacol. 2014;86:417–429. doi: 10.1124/mol.114.093096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Navarro G, Cordomí A, Casadó-Anguera V, Moreno E, Cai NS, Cortés A, Canela EI, Dessauer CW, Casadó V, Pardo L, Lluis C, Ferré S. Evidence for functional pre-coupled complexes of receptor heteromers and adenylyl cyclase. Nat Commun. 2018;9:1242. doi: 10.1038/s41467-018-03522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 37.Zhong G, Díaz-Ríos M, Harris-Warrick RM. Serotonin modulates the properties of ascending commissural interneurons in the neonatal mouse spinal cord. J Neurophysiol. 2006;95:1545–1555. doi: 10.1152/jn.01103.2005. [DOI] [PubMed] [Google Scholar]
- 38.Zhong G, Díaz-Ríos M, Harris-Warrick RM. Intrinsic and functional differences among commissural interneurons during fictive locomotion and serotonergic modulation in the neonatal mouse. J Neurosci. 2006;26:6509–6517. doi: 10.1523/JNEUROSCI.1410-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marullo S, Bouvier M. Resonance energy transfer approaches in molecular pharmacology and beyond. Trends Pharmacol Sci. 2007;28:362–365. doi: 10.1016/j.tips.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 40.Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, Fuxe K, George SR, Javitch JA, Lohse MJ, Mackie K, Milligan G, Pfleger KD, Pin JP, Volkow ND, Waldhoer M, Woods AS, Franco R. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5:131–134. doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kjaerulff O, Barajon I, Kiehn O. Sulphorhodamine-labelled cells in the neonatal rat spinal cord following chemically induced locomotor activity in vitro. J Physiol. 1994;478:265–273. doi: 10.1113/jphysiol.1994.sp020248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kjaerulff O, Kiehn O. Distribution of networks generating and coordinating locomotor activity in the neonatal rat spinal cord in vitro: a lesion study. J Neurosci. 1996;16:5777–5794. doi: 10.1523/JNEUROSCI.16-18-05777.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kiehn O, Johnson BR, Raastad M. Plateau properties in mammalian spinal interneurons during transmitter-induced locomotor activity. Neuroscience. 1996;75:263–273. doi: 10.1016/0306-4522(96)00250-3. [DOI] [PubMed] [Google Scholar]
- 44.Tresch MC, Kiehn O. Coding of locomotor phase in populations of neurons in rostral and caudal segments of the neonatal rat lumbar spinal cord. J Neurophysiol. 1999;82:3563–3574. doi: 10.1152/jn.1999.82.6.3563. [DOI] [PubMed] [Google Scholar]
- 45.Butt SJ, Harris-Warrick RM, Kiehn O. Firing properties of identified interneuron populations in the mammalian hindlimb central pattern generator. J Neurosci. 2002;22:9961–9971. doi: 10.1523/JNEUROSCI.22-22-09961.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Butt SJ, Kiehn O. Functional identification of interneurons responsible for left-right coordination of hindlimbs in mammals. Neuron. 2003;38:953–963. doi: 10.1016/s0896-6273(03)00353-2. [DOI] [PubMed] [Google Scholar]
- 47.Deuchars SA, Brooke RE, Deuchars J. Adenosine A1 receptors reduce release from excitatory but not inhibitory synaptic inputs onto lateral horn neurons. J Neurosci. 2001;21:6308–6320. doi: 10.1523/JNEUROSCI.21-16-06308.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu H, Clemens S, Sawchuk M, Hochman S. Expression and distribution of all dopamine receptor subtypes (D(1)-D(5)) in the mouse lumbar spinal cord: a real-time polymerase chain reaction and non-autoradiographic in situ hybridization study. Neuroscience. 2007;149:885–897. doi: 10.1016/j.neuroscience.2007.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cassada DC, Tribble CG, Long SM, Kaza AK, Linden J, Rieger JM, Rosin D, Kron IL, Kern JA. Adenosine A2A agonist reduces paralysis after spinal cord ischemia: correlation with A2A receptor expression on motor neurons. Ann Thorac Surg. 2002;74:846–849. doi: 10.1016/s0003-4975(02)03793-1. [DOI] [PubMed] [Google Scholar]
- 50.Yokoyama C, Okamura H, Nakajima T, Taguchi J, Ibata Y. Autoradiographic distribution of [3H]YM-09151-2, a high-affinity and selective antagonist ligand for the dopamine D2 receptor group, in the rat brain and spinal cord. J Comp Neurol. 1994;344:121–136. doi: 10.1002/cne.903440109. [DOI] [PubMed] [Google Scholar]
- 51.Azdad K, Gall D, Woods AS, Ledent C, Ferré S, Schiffmann SN. Dopamine D2 and adenosine A2A receptors regulate NMDA-mediated excitation in accumbens neurons through A2A-D2 receptor heteromerization. Neuropsychopharmacology. 2009;34(4):972–986. doi: 10.1038/npp.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernández-Dueñas V, Taura JJ, Cottet M, Gómez-Soler M, López-Cano M, Ledent C, Watanabe M, Trinquet E, Pin JP, Luján R, Durroux T, Ciruela F. Untangling dopamine-adenosine receptor–receptor assembly in experimental parkinsonism in rats. Dis Model Mech. 2015;8:57–63. doi: 10.1242/dmm.018143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jordan LM, Liu J, Hedlund PB, Akay T, Pearson KG. Descending command systems for the initiation of locomotion in mammals. Brain Res Rev. 2008;57:183–191. doi: 10.1016/j.brainresrev.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 54.Clemens S, Belin-Rauscent A, Simmers J, Combes D. Opposing modulatory effects of D1-and D2-like receptor activation on a spinal central pattern generator. J Neurophysiol. 2012;107:2250–2259. doi: 10.1152/jn.00366.2011. [DOI] [PubMed] [Google Scholar]
- 55.Sharples SA, Koblinger K, Humphreys JM, Whelan PJ. Dopamine: a parallel pathway for the modulation of spinal locomotor networks. Front Neural Circuits. 2014;8:55. doi: 10.3389/fncir.2014.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fong AJ, Cai LL, Otoshi CK, Reinkensmeyer DJ, Burdick JW, Roy RR, Edgerton VR. Spinal cord transected mice learn to step in response to quipazine treatment and robotic training. J Neurosci. 2005;25:11738–11747. doi: 10.1523/JNEUROSCI.1523-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roy RR, Harkema SJ, Edgerton VR. Basic concepts of activity-based interventions for improved recovery of motor function after spinal cord injury. Arch Phys Med Rehabil. 2012;93:1487–1497. doi: 10.1016/j.apmr.2012.04.034. [DOI] [PubMed] [Google Scholar]
- 58.Krucoff MO, Rahimpour S, Slutzky MW, Edgerton VR, Turner DA. Enhancing Nervous System Recovery through Neurobiologics, Neural Interface Training, and Neurorehabilitation. Front Neurosci. 2016;10:584. doi: 10.3389/fnins.2016.00584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miyazaki N, Nakatsuka T, Takeda D, Nohda K, Inoue K, Yoshida M. Adenosine modulates excitatory synaptic transmission and suppresses neuronal death induced by ischaemia in rat spinal motoneurones. Pflugers Arch. 2008;457:441–451. doi: 10.1007/s00424-008-0542-1. [DOI] [PubMed] [Google Scholar]
- 60.Witts EC, Panetta KM, Miles GB. Glial-derived adenosine modulates spinal motor networks in mice. J Neurophysiol. 2012;107:1925–1934. doi: 10.1152/jn.00513.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Witts EC, Nascimento F, Miles GB. Adenosine-mediated modulation of ventral horn interneurons and spinal motoneurons in neonatal mice. J Neurophysiol. 2015;114:2305–2315. doi: 10.1152/jn.00574.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Acton D, Miles GB. Stimulation of glia reveals modulation of mammalian spinal motor networks by adenosine. PLoS One. 2015;10:e0134488. doi: 10.1371/journal.pone.0134488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allen EN, Cavanaugh JE. Loss of motor coordination in an aging mouse model. Behav Brain Res. 2014;26:119–125. doi: 10.1016/j.bbr.2014.03.032. [DOI] [PubMed] [Google Scholar]
- 64.Cham R, Perera S, Studenski SA, Bohnen NI. Striatal dopamine denervation and sensory integration for balance in middle-aged and older adults. Gait Posture. 2007;26:516–525. doi: 10.1016/j.gaitpost.2006.11.204. [DOI] [PubMed] [Google Scholar]
- 65.Keeler BE, Lallemand P, Patel MM, de Castro Brás LE, Clemens S. Opposing aging-related shift of excitatory dopamine D1 and inhibitory D3 receptor protein expression in striatum and spinal cord. J Neurophysiol. 2016;115:363–369. doi: 10.1152/jn.00390.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Quiroz C, Gulyani S, Ruiqian W, Bonaventura J, Cutler R, Pearson V, Allen RP, Earley CJ, Mattson MP, Ferré S. Adenosine receptors as markers of brain iron deficiency: Implications for Restless Legs Syndrome. Neuropharmacology. 2016;111:160–168. doi: 10.1016/j.neuropharm.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferré S, Quiroz C, Guitart X, Rea W, Seyedian A, Moreno E, Casadó-Anguera V, Díaz-Ríos M, Casadó V, Clemens S, Allen RP, Earley CJ, García-Borreguero D. Pivotal Role of Adenosine Neurotransmission in Restless Legs Syndrome. Front Neurosci. 2018;11:722. doi: 10.3389/fnins.2017.00722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.