

Abstract
The unabated rise in bacterial resistance to conventional antibiotics, coupled with collateral damage to normal flora incurred by overuse of broad-spectrum antibiotics, necessitates the development of new antimicrobials targeted against pathogenic organisms. Here, we explore the antibacterial outcomes and mode of action of a prochelator that exploits the production of β-lactamase enzymes by drug-resistant bacteria to convert a non-toxic compound into a metal-binding antimicrobial agent directly within the microenvironment of pathogenic organisms. Compound PcephPT contains a cephalosporin core linked to 2-mercaptopyridine N-oxide (pyrithione) via one of its metal-chelating atoms, which minimizes its pre-activation interaction with metal ions and its cytotoxicity. Spectroscopic and chromatographic assays indicate that PcephPT releases pyrithione in the presence of β-lactamase-producing bacteria. The prochelator shows enhanced antibacterial activity against strains expressing β-lactamases, with bactericidal efficacy improved by the presence of lowmicromolar copper in the growth medium. Metal analysis shows that cell-associated copper accumulation by the prochelator is significantly lower than that induced by pyrithione itself, suggesting that the location of pyrithione release influences biological outcomes. Lowmicromolar (4–8 μg/mL) MIC values of PcephPT in ceftriaxone-resistant bacteria compared with LD50 values greater than 250 μM in mammalian cells suggests favorable selectivity. Further investigation into the mechanisms of prochelators will provide insight for the design of new antibacterial agents that manipulate cellular metallobiology as a strategy against infection.
Keywords: prochelator, resistance, β-lactamase, antibacterial, chelation, copper
Graphical Abstract
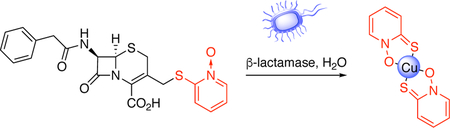
Bacterial resistance to conventional antibiotics has risen dramatically over the past decade, depleting treatment options and fundamentally altering the approach to infection prevention and treatment.1 In particular, the rise of methicillin-resistant Staphylococcus aureus (MRSA) and extended-spectrum β-lactamase- (ESBL) and carbapenemase-producing Gram-negative bacteria such as Escherichia coli, Klebsiella pneumoniae, Enterobacter cloacae, Pseudomonas aeruginosa, and Acinetobacter baumannii-calcoaceticus complex have challenged clinicians in the prevention and treatment of a range of infections, including those of the skin and soft tissues, bloodstream, lungs, and urinary tract. The development of novel anti-infective chemotypes with new mechanisms of action is critical to addressing the antibiotic resistance problem. Here, we investigate the antibacterial outcomes and mode of action of a prochelator that leverages two unique aspects of drug-resistant bacteria: their own resistance mechanisms coupled with the distinctive metallobiology at the host-pathogen interface.
Metal ions are fundamental to life, and thus, the acquisition and utilization of metal ions are battlegrounds in the fight between host defense and microbial virulence.2 Selective interference in microbial acquisition and utilization of metals is consequently an opportunity for anti-infectives to decrease the viability of invading pathogens.3–5 Given the essentiality of iron to microbes, iron chelators have been explored as potential antimicrobials but with limited success.6–9 Iron chelators can also inadvertently promote infection by becoming appropriated as siderophore mimics that supply iron to bacteria, and non-siderophore-like chelators do not sufficiently deplete iron to prevent microbial growth.9–11
Unlike iron restriction, an emerging picture shows that host immune cells increase levels of copper (Cu) during infection.12–17 Under normal circumstances, Cu concentrations in biological systems are tightly regulated; however, localized elevation of Cu levels plays an important role in both host antimicrobial defense18 and bacterial antimicrobial resistance.5, 19, 20 By overburdening the invading microorganisms’ tolerance for Cu, mammalian hosts use Cu’s toxic reactivity as a weapon to intensify cell killing of the pathogen, likely via mismetallation of bacterial enzymes and promotion of reactive oxygen species.21 Compounds that synergize with Cu along the host-pathogen interface present new opportunities to develop antimicrobials.22–26 The prochelator approach takes advantage of this conditional change in host Cu during the response to infection as one prong of a two-pronged approach that targets potent antimicrobial agents specifically against pathogenic, drug-resistant bacteria.
The second prong in this prochelator approach co-opts β-lactamase enzymes to selectively direct Cu toxicity against drug-resistant organisms. Bacterial production of β-lactamases is a major mechanism of resistance to the large family of β-lactam drugs, which includes the penicillins, cephalosporins, and carbapenems. β-lactamases are widely distributed among bacteria, including key organisms that represent emerging threats. To explore the mode of action of prochelators that specifically target antimicrobial-resistant microbes, we synthesized prochelator PcephPT (phenylacetamido-cephem-pyrithione), which contains a metal-chelating antimicrobial agent linked to a cephalosporin core via one of the metal-binding atoms. We show here that, linked in the prochelator state, the small molecule has minimal metal chelation ability and mammalian cytotoxicity. However, the linkage is broken in the presence of β-lactamases, releasing the toxic agent directly in the microenvironment of drug-resistant organisms (Scheme 1).
Scheme 1. A double-pronged approach to selectively target drug-resistant bacteria.

Prochelator PcephPT releases a metal-binding antimicrobial agent, pyrithione, in the presence of drug-resistant bacteria that produce β-lactamase enzymes. The metal-binding agent synergizes with bioavailable copper to exacerbate microbial killing.
Results and Discussion
Prochelator design and synthesis
We define a prochelator as a compound with limited metal affinity that, in response to a specific trigger, removes a chemical masking group to yield a metal-chelating agent. Prochelators have been designed to respond to a variety of stimuli, including reactive oxygen species, specific enzymes, and light.27–29 The prochelator used in this study contains a masking group that responds to β-lactamase activity.
The β-lactamase enzymes produced by antibiotic-resistant bacteria hydrolyze β-lactam rings to render such drugs ineffective at inhibiting their intended targets: penicillin-binding proteins, which are central to bacterial cell wall biosynthesis.30, 31 In the case of cephalosporins, scission of the β-lactam ring triggers elimination of the 3’-substituent.32, 33 A variety of cephalosporin analogs have been designed to take advantage of this reactivity to release bioactive molecules, including quinolones, halogenated dipeptides, thiols, umbelliferone, 5-fluorouracil, and others.34–43 In particular, O’Callaghan et al.44 explored the antibacterial activity of a cephalosporin that released pyrithione (PT), a metal-chelating agent with excellent broad-spectrum antibacterial and antifungal activity that synergizes with Cu to induce toxicity.45–49 We utilized a similar PT-releasing molecule, PcephPT, for our studies on the metal-dependent antibacterial mode of action of β-lactamase-activated prochelators. We hypothesized that attaching PT to a cephalosporin at one of its metal-chelating atoms would greatly reduce its metal affinity prior to enzymatic activation. In this way, manipulation of metals would preferentially occur at the site of drug-resistant infections with minimal interference in the metallobiology of either the host or non-β-lactamase-producing commensal bacteria. A final design consideration for prochelator PcephPT was the identity of the substituent at the 7-position of the cephalosporin core, as this position affects cellular uptake, enzyme selectivity, associated binding, and hydrolysis kinetics for different classes of β-lactamases.40, 50 We therefore chose to study a prochelator with a phenylacetamido group at this position based on its reported ability to serve as a substrate for multiple classes of β-lactamase.50, 51 Compared to similar β-lactam conjugates,39, 44 PcephPT is unique in that it is intentionally designed as an efficient substrate for β-lactamase in order to study the metal-binding consequences of the released chelator.
PcephPT was synthesized by reacting cephalosporin starting material 1 with 2-mercaptopyridine N-oxide in the presence of potassium trimethyl silanolate to functionalize the C-3 position (Scheme S1). This base was chosen to prevent Δ3 → Δ2 isomerization, a problem frequently encountered in cephalosporin synthesis.52, 53 Deprotection of the carboxylate using trifluoroacetic acid in phenol yielded the final product.54 Because 2-mercaptopyridine N-oxide is capable of nucleophilic attack by either its oxygen or sulfur atom, formation of both O- and S-linked isomers is possible; however, the presence of heat and oxygen during the final step promoted rearrangement to a single isomer, consistent with reports by Hay et. al in which alkylated pyrithiones rearranged to a single isomer.55 The13C NMR chemical shifts at C-3’ of the cephalosporin and C-2 of the PT moiety confirmed the identity of the product as the S-linked isomer only.
Stability of prochelator PcephPT and conversion to chelator PT in the presence of β-lactamase
Chromatographic and spectroscopic assays were conducted to test the stability of PcephPT under various conditions. As shown in supplementary Figures S3 and S4, PcephPT is stable for hours in phosphate-buffered saline but rapidly releases PT in the presence of commercially available β-lactamase P99 (an Ambler class C β-lactamase) from E. cloacae. We also tested the stability of PcephPT in the presence of high concentrations of Cu, since hydrolysis of β-lactams is known to be catalyzed by metal ions.56–58 While concentrations below 10 μM CuCl2 did not catalyze noticeable degradation of 100 μM PcephPT in water, 100 μM CuCl2 indeed induced significant PcephPT hydrolysis, consistent with known metal-dependent β-lactam hydrolysis (Figure S5). However, solutions of PcephPT prepared in LB medium were found to be stable, with no discernable signs of degradation even when challenged with 100 μM CuCl2 and incubated at 37 °C for over 20 h. We attribute this improved stability to the more neutral pH of LB medium compared to water and the presence of other metal-coordinating species in the LB medium that make Cu less available to catalyze hydrolysis of PcephPT. These results confirm that PcephPT is inherently stable under the cellular assay conditions and concentrations used in these studies.
A calcein fluorescence assay was utilized in order to evaluate the relative metal-binding ability of PcephPT compared to PT. The fluorescence intensity of the probe calcein is quenched by more than 90% upon binding Cu(II). As shown in Figure 1, addition of PT to Cu-loaded calcein restores calcein fluorescence, whereas PcephPT does not compete with calcein for Cu binding. However, when incubated with 10 equivalents of PcephPT and 0.1 U/mL of β-lactamase from E. cloacae, there is an 85% return in calcein fluorescence, which correlates well with the 90% return observed for PT itself at the same concentration. This experiment confirms that the minimal affinity of the prochelator for Cu can be dramatically increased upon exposure to β-lactamase.
Figure 1. Relative Cu binding assay with PcephPT and PT against calcein.

Solutions of 2 μM calcein and 2.4 μM CuCl2 in pH 7.4 PBS were equilibrated at room temperature for 2 h prior to addition of 20 μM PT, 20 μM PcephPT, and/or 0.1 U/mL β-lactamase. Fluorescence emission at 535 nm (λex = 485 nm) was recorded after 1 h.
Prochelator PcephPT is selectively turned over by bacteria that produce β-lactamase.
In order to test the activity and probe the mode of action of PcephPT across a range of β-lactamase classes, we transformed plasmids containing genes encoding each of the β-lactamases listed in Table 1 (or a plasmid lacking β-lactamase for the null strains) into E. coli K-12 strain MG1655 and E. coli K1 strain UTI89.
Table 1.
Substrate scope and expected hydrolysis of PcephPT by the β-lactamases expressed in MG1655 and UTI89 E. coli background strains.
| Name | Ambler Class | Active Site | Substrates | Expected to cleave PcephPT? |
|---|---|---|---|---|
| null | – | – | No | |
| OXA-1 | D | Ser | Penicillins | No |
| TEM-1 | A | Ser | Penicillins, some cephalosporins | Yes |
| CTX-M-1 | A | Ser | Penicillins, cephalosporins | Yes |
| CMY-2 | C | Ser | Penicillins, cephalosporins | Yes |
| NDM-1 | B | Zn | Penicillins, cephalosporins, carbapenems | Yes |
The turnover of prochelator PcephPT was measured in bacterial cultures of E. coli MG1655 to demonstrate that it is cleaved by β-lactamases expressed in cells. To quantify the amount of the prochelator present in the medium initially (at less than 1 h) and at 20 h, samples for LC-MS analysis were extracted from spent media of bacterial cultures that had been incubated with PcephPT for the designated time. As shown in Figure 2, the concentration of recovered prochelator did not change after 20 h incubation in the β-lactamase-null strain. The persistence of intact PcephPT under these conditions further confirms the inherent stability of the compound and indicates its resistance to degradation by non-specific hydrolytic processes present in E. coli MG1655. Incubation with the strain expressing OXA-1 did not diminish PcephPT concentration within the first hour, although levels were reduced with some variability by the 20 h timepoint, consistent with its classification as a penicillinase with low efficiency for cephalosporin substrates. All strains expressing cephalosporinase enzymes, on the other hand, showed a dramatic decrease in recoverable prochelator concentration over time. CMY-2 and NDM-1 showed nearly complete prochelator disappearance in the time it took to collect and extract the sample for the initial timepoint. Consistent with the disappearance of PcephPT in these strains, the appearance of PT was observed in the absorbance spectrum of these bacterial suspensions (Figure S7). The disappearance of the prochelator and the appearance of PT in these experiments are attributed to specific enzymatic cleavage and are consistent with the expected substrate scope of each enzyme.
Figure 2. Quantification of PcephPT in cultures of E. coli MG1655 expressing β-lactamases.

The concentration decreases most in strains expressing enzymes capable of cleaving cephalosporins. [PcephPT]initial = 50 μM. Concentration determined by integration of UV chromatogram at 280 nm relative to internal standard. An example UV chromatogram is shown in Figure S6.
Prochelator PcephPT inhibits growth of β-lactamase-expressing E. coli.
The antibacterial activities of PcephPT, PT, cephalothin, and ceftriaxone were evaluated in the twelve E. coli strains by broth microdilution. In brief, E. coli were treated with serial dilutions of compounds and incubated in LB medium for 20 h at 37 °C while shaking at 200 rpm. The concentration of compound that completely inhibited visible growth as measured by OD600 at 20 h was recorded as the minimum inhibitory concentration (MIC) (Table 2). Clinically used β-lactam antibiotics cephalothin and ceftriaxone served as controls for comparison with PcephPT and PT and assessment of β-lactam drug resistance. As expected, the first-generation drug, cephalothin, inhibited the β-lactamase-null strains as well as those expressing the penicillinase OXA-1, but was ineffective (MICs ≥ 140 μM) at inhibiting growth of the four strains expected to have cephalosporinase activity (Table 1). Consistent with its expected narrower substrate scope for cephalosporins, the TEM-1 strain was resistant to cephalothin but susceptible to ceftriaxone, the third-generation drug. The differential response of the TEM-1 strain to two cephalosporin drugs highlights the variability of substrate specificity of β-lactamases for β-lactam compounds. The CTX-M-1, CMY-2, and NDM-1 strains, on the other hand, were resistant to both cephalothin and ceftriaxone (MIC ≥ 70 μM).
Table 2. Minimum inhibitory concentrations (MICs) of PcephPT, PT, cephalothin, and ceftriaxone against E. coli MG1655 and E. coli UTI89 expressing different β-lactamases.
Bacteria were incubated in LB medium at 37 °C while shaking at 200 rpm for 20 h. Growth was evaluated by OD600. Concentrations tested: 140 μM, 70 μM, 35 μM, 17.5 μM, 8.8 μM, 4.4 μM, 2.2 μM, 1.1 μM. For PcephPT, 2.2 μM = 1 μg/mL (see Table S1 for MIC values in μg/mL). MICs within one dilution factor were not considered significantly different. At least two biological replicates were performed with a minimum of at least four total technical replicates.
| Strain | β-lactamase | PcephPT(μM) | Pyrithione (μM) | Cephalothin (μm) | Ceftriaxone (μm) | ||||
|---|---|---|---|---|---|---|---|---|---|
| compound only | + 10 μM CuCl2 | + 2 mM BCS | compound Only | + 10 μM CuCl2 | + 2 mM BCS | compound only | compound only | ||
| MG1655 | null | 70 | 70 | 35 | 17.5 | 17.5 | 140 | 35 | <1 |
| OXA-1 | 70 | 70 | 70 | 35 | 17.5 | >140 | 70 | <1 | |
| TEM-1 | 35 | 17.5 | 140 | <1 | |||||
| CTX-M-1 | 17.5 | 8.8 | >140 | >140 | |||||
| CMY-2 | 17.5 | 8.8 | >140 | >140 | |||||
| NDM-1 | 17.5 | 17.5 | >140 | 17.5 | 17.5 | >140 | >140 | 70 | |
| UTI89 | null | 35 | 35 | 35 | 35 | 17.5 | >140 | 35 | <1 |
| OXA-1 | 35 | 70 | 70 | 35 | 35 | >140 | 35 | <1 | |
| TEM-1 | 70 | 140 | >140 | 35 | 35 | >140 | >140 | <1 | |
| CTX-M-1 | 17.5 | 17.5 | >140 | 35 | 35 | >140 | >140 | >140 | |
| CMY-2 | 17.5 | 17.5 | >140 | 17.5 | 17.5 | >140 | >140 | >140 | |
| NDM-1 | 17.5 | 17.5 | >140 | 35 | 35 | >140 | >140 | 140 | |
In contrast to conventional cephalosporins, both PcephPT and PT inhibited growth of all strains tested with MIC values of 70 μM or better. PcephPT showed particularly promising antibacterial activity against strains resistant to both cephalothin and ceftriaxone. As shown in Table 2, the MIC of PcephPT in strains expressing CTX-M-1, CMY-2, and NDM-1 β-lactamases was 17.5 μM, or 8 μg/mL. On a concentration basis, these values are similar to those obtained with PT, a result that is consistent with the hypothesis that the antibacterial mode of action of PcephPT is related to its release of PT when hydrolyzed by a β-lactamase. However, PcephPT was also modestly effective in strains that either did not express a β-lactamase, or that showed low substrate turnover of PcephPT, indicating that PT release is not its sole mechanism of action. Its modest efficacy (MICs of 35–70 μM, or 16–32 μg/mL) against the null, OXA-1 and TEM-1 strains suggests that PcephPT retains some activity as an inhibitor of penicillin-binding proteins due to its cephalosporin core.
Cu improves bactericidal outcome of PT treatment.
The biological activity of PT has been associated with its ability to bind a variety of metal ions, including those of important biometals Fe, Cu, and Zn.48, 49 To test the influence of these metals on PT efficacy in our system, additional MIC testing was performed in representative examples from our collection of strains. Co-treating MG1655 null or CTX-M-1-expressing strains with PT and 10 μM supplemental CuCl2, FeCl3, or ZnCl2 added to LB medium did not shift the MICs by more than one dilution factor of the unsupplemented PT treatment condition (data not shown). To measure bactericidal activity, bacterial cultures treated for 20 h with growth-inhibiting concentrations of each compound were spotted on fresh LB agar plates. Colony-forming units (CFU) were enumerated after 24 h of incubation. Absence of CFUs indicates bactericidal activity, whereas visible colonies indicate that some cells were not killed by treatment. Interestingly, colony regrowth was observed for all PT treatment conditions except PT with Cu, most notably in the MG1655 strains (Figure S8). This observation suggests that the presence of Cu improves the bactericidal effect of PT treatment.
To further probe the Cu dependence on PT bactericidal activity, Cu-deficient conditions were created by adding the extracellular Cu-sequestering agent bathocuproinedisulfonic acid (BCS) to the growth medium. As shown in Figure 3 for null and CTX-M-1 strains, bacterial growth that is normally inhibited at the MIC of PT recovered in a manner that depended on BCS concentration, suggesting that PT loses efficacy when it cannot access Cu. Addition of 2 mM BCS showed nearly complete abrogation of PT activity across these tested strains and was therefore selected for additional MIC determinations across the full panel of strains. In the presence of 2 mM BCS, PT was ineffective at inhibiting bacterial growth in all cases, with MIC values greater than or equal to the highest PT dose tested (140 μM) (Table 2). As expected, the results for PT did not depend on whether β-lactamase was expressed. Given the positive reduction potential and high affinity of BCS for Cu(I), these conditions keep free Cu levels buffered below 10−19 M and therefore effectively inaccessible to micromolar levels of the weaker binding PT.49, 59 This analysis combined with the results from Figure 3 is consistent with the notion that PT binds and mobilizes Cu under regular growth conditions to use as part of its mode of action.48, 49
Figure 3. BCS rescues cell growth during treatment with PT.

As the concentration of BCS in the medium increases, bacterial growth is rescued from PT treatment. Treatment conditions: 17.5 μM PT for MG1655 strains, 35 μM PT for UTI89 strains; 37 °C, 200 rpm for 20 h.
Ability of PcephPT to kill cephalosporin-resistant bacteria depends on both β-lactamase and Cu.
The exploratory experiments described in the previous section on potential metal-modulated activity of PT against E. coli revealed a clear dependence on Cu availability for maximal bactericidal outcomes. The MIC of PcephPT was therefore re-evaluated in the presence of supplemental Cu and BCS (Table 2). Neither Cu nor BCS at these concentrations affected the efficacy of cephalothin or ceftriaxone, which was expected because these β-lactams do not release a metal chelating moiety (data not shown). These conditions also failed to substantively shift the MIC of PcephPT against the null and OXA-1 strains of MG1655 and UTI89 (MICs remained within one dilution factor). Similar to PT, the MIC of PcephPT was not significantly affected by supplemental Cu, even in strains that cleave it. On the other hand, the Cu-deficient conditions created with 2 mM BCS resulted in MIC values higher than the 140 μM highest dose tested for strains capable of hydrolyzing PcephPT (TEM-1, CTX-M-1, CMY-2, and NDM-1). Taken together, Cu deprivation by BCS showed a consistent trend of rescued growth for both PT and, in strains capable of cleaving it, PcephPT. This observation indicates that availability of Cu may play a role in the antibacterial mechanisms of PT and PcephPT.
We next measured the bactericidal effects of PcephPT and PT treatment conditions. Consistent with the known bactericidal activity of β-lactam drugs, no colonies were detected for PcephPT in the MG1655 null strain (Figure 4). Variable numbers of colonies, however, were apparent for all the β-lactamase-producing strains treated with either PcephPT or PT, indicating that the minimum bactericidal concentration (MBC) is greater than the MIC for these conditions. Similar results were observed with UTI89 (Figure S9). The outcomes in the β-lactamase-producing strains changed distinctively for both compounds when co-treated with Cu, however, with a few sporadic colony appearances for the UTI89 strains and no colonies detected for MG1655 strains. Combined, these results support the conclusion that PcephPT acts as a bactericidal β-lactam drug when no β-lactamase is present to cleave it, but relies on release of PT to inhibit bacterial growth when hydrolyzed. The mode of action of PT itself seems to depend on availability of Cu, the presence of which improves bactericidal outcomes. PcephPT recapitulates the Cu-dependent mode of action of PT in the strains expressing a β-lactamase.
Figure 4. Ability of PcephPT to kill MG1655 bacteria depends on both the presence of β-lactamase and availability of Cu.

Bacteria treated with minimal growth-inhibiting concentrations of PcephPT or PT with and without supplemental CuCl2 (10 μM) in liquid medium for 20 h were plated on fresh agar and incubated at 37 °C for 24 h prior to enumeration of colonies. Each point on the graph is an individual replicate. 0 h CFU/mL values: null = 5900, TEM-1 = 38000, CTX-M-1 = 35000, CMY-2 = 19000, NDM-1 = 47000. CFUs above 12500 were not enumerated and are grouped collectively on the plot as >12500.
CopA knockout strains are hypersensitive to PcephPT and PT with Cu.
The dependence on Cu availability for PT and PcephPT bactericidal activity suggests that E. coli mutants deficient in Cu resistance mechanisms should be hypersensitive to PT + Cu and PcephPT + Cu exposure. To test this hypothesis, we measured the susceptibility of a Cu-sensitive MG1655 copA knockout mutant, LEM8, to PT. CopA is a Cu(I)-effluxing P1B-type ATPase that translocates Cu(I) from the cytoplasm to the periplasm as an important component of bacterial resistance to Cu stress.60 The MG1655 copA knockout and parent MG1655 strains responded similarly to treatment with PT alone; however, co-treatment of PT with supplemental Cu resulted in a decrease of the MICs to 4.4 μM (Table 3). As expected, co-treatment of PT and 2 mM BCS rescued growth (MIC > 140 μM). Similarly, in MG1655 copA knockout strains expressing a β-lactamase, co-treatment of PcephPT with Cu resulted in a significant decrease in the MIC compared to treatment with PcephPT alone, while BCS rescued growth. The increased sensitivity of the copA knockout strains to PcephPT and PT in the presence of Cu further supports a model in which Cu contributes to the released PT’s antibacterial mode of action.
Table 3. Minimum inhibitory concentrations (MICs) of PcephPT, PT, cephalothin, and ceftriaxone against E. coli MG1655 copA knockout, LEM8, expressing different β-lactamases.
Bacteria were incubated in LB medium at 37 °C while shaking at 200 rpm for 20 h. Growth was evaluated by OD600. Concentrations tested: 140 μM, 70 μM, 35 μM, 17.5 μM, 8.8 μM, 4.4 μM, 2.2 μM, 1.1 μM. For PcephPT, 2.2 μM = 1 μg/mL (see Table S2 for MIC values in μg/mL). MICs within one dilution factor were not considered significantly different. At least two biological replicates were performed with a minimum of at least four total technical replicates.
| Strain | β-lactamase | PcephPT(μM) | Pyrithione (μM) | Cephalothin (μm) | Ceftriaxone (μm) | ||||
|---|---|---|---|---|---|---|---|---|---|
| compound only | + 10 μM CuCl2 | + 2 mM BCS | compound only | + 10 μM CuCl2 | + 2 mM BCS | compound only | compound only | ||
| MG1655 LEM8 copAΔ | null | 35 | 35 | 70 | 17.5 | 4.4 | >140 | 70 | <1 |
| TEM-1 | 35 | 8.8 | >140 | 17.5 | 4.4 | >140 | >140 | <1 | |
| CTX-M-1 | 8.8 | 2.2 | >140 | 17.5 | 4.4 | >140 | >140 | >140 | |
| CMY-2 | 17.5 | 4.4 | >140 | 17.5 | 4.4 | >140 | >140 | >140 | |
| NDM-1 | 17.5 | 4.4 | >140 | 17.5 | 4.4 | >140 | >140 | 70 | |
PT causes more cellular Cu accumulation than PcephPT.
PT is a recognized metal ionophore known to induce cellular hyperaccumulation of metals to which it binds.49 To evaluate how incubation with supplemental Cu influences cell-associated Cu levels as a function of PT and PcephPT treatment, samples from two representative strains (the MG1655 null strain and CMY-2, a rapidly cleaving strain) were analyzed by inductively coupled plasma mass spectrometry (ICP-MS). Bacteria were grown to an OD600 of 0.1, then treated or not with sub-MIC levels of PcephPT or PT, each with and without supplemental Cu. Cells were harvested after 15 min (Figure 5) or 2 h of treatment (Figure S10), washed with chelex-treated water and ethylenediaminetetraacetic acid (EDTA) to remove adventitious metals, and digested in nitric acid prior to ICP-MS analysis.
Figure 5. ICP-MS data comparing cell-associated Cu content of MG1655 null and CMY-2 strains treated for 15 min with PT or PcephPT, with and without Cu.

Cu concentration was normalized to phosphorous content for each sample. Treatment conditions: 4 μM PcephPT, 4 μM PT, 10 μM CuCl2. * indicates p < 0.05 using one-tailed Student’s t-test.
As expected, co-treatment with PT and CuCl2 (red bars) resulted in a 9- to 14-fold increase in cell-associated Cu compared to the Cu-only treatment (dark blue bars). Co-treatment with PcephPT and CuCl2 (dark green bars), on the other hand, caused only a 2.5-fold increase in cell-associated Cu in the CMY-2-expressing strain compared to Cu alone. Even after 2 h, relative concentrations of Cu remain comparable to the 15 min concentrations (Figure S10). As discussed previously, the CMY-2 strain was shown to cleave PcephPT in less than 1 h, so we expect PT to be present at near stoichiometric levels after 2 h of treatment with this strain. It is interesting, therefore, that less Cu accumulated in this strain for PcephPT compared to PT, even though both compounds exhibit similar antibacterial MICs.
The difference in Cu accumulation indicates that PcephPT may behave differently than free PT. One hypothesis is that the location of free PT affects its Cu-importing behavior. Since β-lactamases are located in the periplasm, PcephPT is expected to release PT in the periplasm. PT released in the periplasm may operate through a different mode of action or access a different supply of Cu than extracellular PT. For example, basal levels or a small increase of cell-associated Cu may be sufficient to provide an antibacterial effect. Surprisingly, E. coli co-treated with PcephPT and Cu accumulated the same amount of Cu regardless of whether they contained the highly active CMY-2 enzyme or lacked β-lactamase. Weakening of the cell wall due to cephalosporin activity may account for this observation, although this has not been validated. Further investigation into the role of Cu in the mode of action of PcephPT will reveal more information about how this prochelator exerts its antibacterial function.
Host vs. pathogen toxicity
As the basis of the prochelator strategy is to prevent systemic exposure to potentially harmful metal chelating agents, the toxicities of both the prochelator PcephPT and the chelator PT were evaluated in CCD-19Lu human lung fibroblast cells and HepG2 human liver epithelial cells (Figure 6). Using the CellTox Green cytotoxicity assay, the viability of mammalian cells was monitored as indicated by their membrane integrity. While the cytotoxicity of PT increased steadily with concentration, it did not induce more than 50% cell death at concentrations below 500 μM in either cell line after 24 h of incubation. PcephPT showed even less cytotoxicity and appeared nontoxic below 500 μM. Suspecting that the cytotoxicity apparent at high concentrations of PcephPT in the mammalian cells may be due to PT released by non-specific hydrolysis of PcephPT, we analyzed the spent cell culture medium used for the cytotoxicity assay by LC-MS and found 50–60% loss of detectable, intact prochelator compared to the non-incubated controls after 4 h (Figure S11). While these observations suggest that these mammalian cells are capable of hydrolyzing PcephPT, this activity is less efficient than in β-lactamase-producing bacteria. The large differential between the MIC values of PcephPT in pathogenic bacteria in the low-micromolar range (Table 2) and the LD50 of PcephPT exceeding 500 μM in mammalian cells (Figure 6) indicates a ratio of LD50 to MIC favorable for further development of prochelators as antibacterial agents.
Figure 6. Cytotoxicity of PcephPT in CCD-19Lu and HepG2 cells.

Cytotoxicity of human lung fibroblast cells CCD-19Lu and human liver epithelial cells HepG2 was assessed with CellTox Green after 24 h incubation.
Conclusions
The metal chelator PT has found many antimicrobial applications throughout the years; however, concerns over its inherent toxicity have limited its treatment applications. Our prochelator, PcephPT, has been shown to have low toxicity in mammalian cells, but demonstrates antibacterial activity even in drug-resistant strains expressing clinically relevant β-lactamases. The antibacterial activity of PcephPT correlates with the availability of Cu, suggesting a metal-dependent mode of action. However, PcephPT does not elevate cell-associated Cu as much as free PT, suggesting that hyperaccumulation of Cu within bacteria is not required for conditionally released PT to induce bacterial cell death. These observations could imply that the released PT has metal-independent modes of action; alternatively, in situ release of PT as a metal-interacting agent may be sufficient to manipulate Cu in ways that contribute to antibacterial activity. Many bacteria are sensitive to Cu stress and have evolved redundant mechanisms to mitigate Cu toxicity, including expression of Cu export pumps and periplasmic proteins involved in Cu tolerance.61 Metal chelators like PT may interfere with these systems, especially if the agent is directly released in their proximity. Further investigation into these prochelators’ mechanisms of metal manipulation will lead to advances in the field of targeted, narrow-spectrum antibiotics.
Methods
Materials
Reagent grade 7-amino-3-chloromethyl-3-cephem-4-carboxylic acid p-methoxybenzyl ester (1) was purchased from Ark Pharm Inc.; other reagents were purchased from Sigma Aldrich unless otherwise stated. All solvents were reagent grade and all aqueous solutions were prepared from nanopure water.1H NMR and13C NMR spectra were collected on a Varian 500 MHz spectrometer. Mass spectra were collected on an Agilent 1100 Series spectrometer utilizing an electrospray ionization source, an LC/MSD trap, and a Daly conversion dynode detector; high-resolution mass spectra were acquired with an Agilent G6224 LCMS-TOF using an electrospray ionization source and a Series 1200 HPLC. ICP-MS data were acquired by Kim Hutchison of the North Carolina State University Department of Crop and Soil Sciences using a Perkin Elmer Elan DRCII spectrometer. UV/Vis spectra were recorded on a Cary 50 UV/Vis spectrophotometer.
PcephPT, 2-mercaptopyridine N-oxide (pyrithione, PT) (Chem Impex), cephalothin (Ark Pharm), ceftriaxone (Acros), and ampicillin (Sigma) stock solutions were prepared in DMSO for use in cell assays. Copper (II) chloride (Merck), bathocuproinedisulfonic acid (Sigma), kanamycin (Affymetrix) and tazobactam sodium (Chem Impex) stock solutions were prepared in deionized water, frozen for storage and sterile filtered prior to use in cell assays. NMR analysis showed no tazobactam degradation during storage as a frozen aqueous solution. Aqueous stock solutions of CuCl2 were standardized by EDTA titration in an ammonium buffer to a murexide endpoint and stored at −20 °C. All cell work was performed in a laminar-flow hood using sterile techniques.
Synthesis
2-((((6R,7R)-2-(((4-methoxybenzyl)oxy)carbonyl)-8-oxo-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl)methyl)thio)pyridine 1-oxide (2)
To a mixture of 1 (973.6 mg, 1.999 mmol) and 2-mercaptopyridine N-oxide (306 mg, 2.41 mmol) in MeCN (approx. 60 mL), stirring under N2, was added potassium trimethylsilanolate 90% (305 mg, 2.38 mmol) dissolved in MeCN (5 mL). After stirring for 3 h at r.t., the resulting dark orange slurry was filtered and the flow-through evaporated under reduced pressure to yield an orange oil. The oil was dissolved in CH2Cl2 (30 mL) and washed with saturated NaHCO3 (3 × 15 mL). The organic layer was dried over anhydrous Na2SO4, filtered, and solvent was evaporated under reduced pressure to yield a brown oil, which was washed with hexanes and used in the next step without further purification (914 mg, 1.58 mmol, 79% yield).1H NMR (500 MHz, CDCl3) δ 8.16–8.25 (m, 2H), 7.34–7.39 (m, 5H), 7.29–7.34 (m, 10H), 7.04–7.19 (m, 4H), 7.00–7.04 (m, 1H), 6.94–7.00 (m, 1H), 6.87 (q, J = 8.4, 8.0 Hz, 6H), 6.25 (s, 1H), 6.18 (d, J = 9.1 Hz, 1H), 6.10 (d, J = 8.9 Hz, 1H), 5.80 (dd, J = 9.2, 4.9 Hz, 1H), 5.60 (dd, J = 8.8, 4.0 Hz, 1H), 5.20 (d, J = 2.4 Hz, 2H), 5.17 (d, J = 4.3 Hz, 2H), 5.11 (s, 1H), 5.06 (s, 1H), 4.90 (d, J = 4.9 Hz, 1H), 4.06 (s, 2H), 3.86 (d, J = 14.7 Hz, 1H), 3.80 (s, 5H), 3.79 (s, 4H), 3.58–3.69 (m, 6H), 3.56 (d, J = 7.9 Hz, 2H).13C NMR (DMSO-d6, 125 MHz) δ (ppm): 171.39, 165.35, 161.89, 159.75, 150.24, 138.74, 136.22, 130.79, 130.71, 129.53, 129.45, 128.69, 127.35, 126.96, 126.20, 125.91, 125.50, 122.82, 122.06, 114.33, 114.19, 67.69, 59.58, 58.39, 55.55, 42.06, 42.00, 32.81, 27.89. HR-MS (ESI) (m/z): [M+Na]+ calcd for ([C29H27N3O8S2] + Na)+ , 600.1233, found 600.1233.
2-((((6R,7R)-2-carboxy-8-oxo-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl)methyl)thio)pyridine 1-oxide (PcephPT)
Portions of 2 (914 mg, 1.58 mmol) and phenol (3 g, 30 mmol) were added to a flask and heated to 45 °C. After the phenol melted, trifluoroacetic acid (0.2 mL, 2 mmol) was added and the mixture was allowed to stir for 2.5 h at a temperature maintained between 45–50 °C. MeCN was then added until a brown precipitate formed, which was collected by filtration and discarded. A light-colored precipitate, which formed upon cooling the solution to r.t., was collected, and washed with EtOAc to yield a fine off-white powder (191 mg, 0.417 mmol, 26% yield).1H NMR (500 MHz, DMSO-d6) δ 13.75 (s, 1H), 9.13 (d, J = 8.4 Hz, 1H), 8.31 (d, J = 6.4 Hz, 1H), 7.46 (d, J = 8.6 Hz, 1H), 7.42–7.12 (m, 8H), 5.67 (dd, J = 4.76, 8.37 Hz), 5.12 (d, J = 4.8 Hz, 1H), 4.10, 4.06 (ABq, JAB = 12.5 Hz, 2H), 3.74 (d, J = 17.9 Hz, 1H), 3.56 (d, J =14.3 Hz, 2H), 3.49(d, J = 13.9 Hz, 1H).13C NMR (125 MHz, DMSO-d6) δ 170.9, 164.7, 163.0, 150.1, 138.3, 135.8, 129.0, 128.2, 126.5, 126.2, 125.5, 125.0, 122.2, 121.5, 59.1, 57.8, 41.6, 32.3, 27.3. HRMS (ESI) (m/z): [M+H]+ calcd. for ([C21H18N3O5S2] + H)+ 458.0839, found 458.0828.
Transformation of E. coli clones
Gene blocks for TEM-1 (Ambler A; Accession: AY2141643), CTX-M-1 (Ambler A; Accession: X92506.1), NDM-1 (Ambler B; Accession: AP012208.1), CMY-2 (Ambler C; Accession: FJ621586), and OXA-1 (Ambler D; Accession: AY458016) were obtained from Integrated DNA Technologies (IDT). Using the Zero Blunt Topo PCR Cloning Kit (Thermo Fischer Scientific), the gene blocks were inserted into the pCR-Blunt II-TOPO vector (Kanamycin resistant; KanR) and then transformed into One Shot competent cells. The resulting products were incubated overnight on LB agar containing 100 μg/mL ampicillin and 50 μg/mL kanamycin. Single colonies were screened for the expected inserts, plasmids purified from probable clones, and the inserts sequenced using the Sanger method (Genewiz). The products for each transformation were found to be >99% identical to the accession sequences. Purified plasmid was transformed by electroporation into E. coli K1 strain UTI89, K-12 strain MG1655, and K-12 strain MG1655 LEM8 and transformants were selected on LB agar containing kanamycin and ampicillin. Individual colonies were then grown and stored at −80 °C in 16% glycerol for future use.
The K-12 MG1655 copA knockout, LEM8, was generously provided by Dr. James Imlay.
LC-MS Assays
Degradation studies with Cu:
Triplicate samples were prepared in either unbuffered water or LB medium (1 mL) with 100 μM PcephPT and 0, 1, 10, or 100 μM CuCl2 (added as a dilution from a 100-mM aqueous stock solution) and incubated in closed tubes either at r.t. or in a 37 °C water bath. 200-μL aliquots were taken at the indicated timepoints and immediately diluted to a final volume of 1.00 mL of water containing 5 mM ethylenediaminetetraacetic acid (EDTA) and 35 μM Rofecoxib (as internal standard). LC-MS was run and peaks corresponding to PcephPT (11 min) and internal standard (14 min) were integrated in the UV chromatogram at 280 nm. This wavelength was chosen because PcephPT has a high absorbance at 280 nm. The quantity of PcephPT was reported as a percentage relative to the average amount present in the initial timepoint samples for each condition.
Enzyme assay:
Samples were prepared in 100 μL phosphate-buffered saline, pH 7.4 (Lonza) with 100 μM PcephPT, 10 μM Rofecoxib (as internal standard) and either 0.2 U/mL β-lactamase from E. cloacae (Type III, Sigma Aldrich) or no enzyme and incubated in closed tubes at r.t. Aliquots (20 μL) were taken at the indicated timepoints and immediately added to new tubes containing β-lactamase inhibitor tazobactam (2.5 mM in 5 μL water). These tubes were immediately mixed, sonicated, and frozen until shortly before LC-MS injection. Samples were diluted with 50 μL H2O, LC-MS was run, and peaks corresponding to PcephPT (11 min) and internal standard (14 min) were integrated in the UV chromatogram at 280 nm. The quantity of PcephPT was reported as a percentage relative to the average amount present in the initial timepoint samples for each condition. The ion masses for the peak at 6 min were determined using an unquenched sample, due to co-elution of tazobactam with this peak.
Analysis of compound in spent bacterial medium:
Before each experiment, an LB agar plate was streaked with bacteria from the appropriate frozen stock and incubated overnight at 37 °C. A single colony was used to inoculate LB medium which was then incubated at 37 °C, 200 rpm, for 16–18 h. This overnight culture was diluted 1:1000 in fresh LB medium containing 50 μM PcephPT and used as the working culture. Aliquots (1 mL) were removed and centrifuged for 5 min at 10,000 rpm. Supernatants were transferred to new tubes and acetone (5 mL) was added to each sample. Samples were mixed, stored at −20 °C for 1 h, and centrifuged again for 5 min at 5,000 rpm. Solvent was evaporated under reduced pressure from each supernatant to yield a light-colored residue that was dissolved in 1:1 MeCN:H2O containing 50 μM Rofecoxib as an internal standard. LC-MS was run and peaks corresponding to PcephPT and internal standard were integrated in the UV chromatogram at 280 nm. Concentration was determined by normalizing to the internal standard and to triplicate calibration samples containing 50 μM PcephPT extracted from LB with no bacteria added. A standard curve generated in a separate experiment confirmed the linearity of the relationship between the PcephPT concentration and the ratio of the PcephPT peak area to the internal standard peak area in samples extracted from LB medium.
All LC-MS analyses were performed by using an 18-min gradient starting with 2% and ending with 65% MeCN in H2O. Eluent mixtures contained 3% MeOH and 0.1% formic acid.
Microdilution screening
Prior to all experiments, E. coli carrying the appropriate plasmids were streaked onto LB agar containing 100 μg/mL ampicillin and 50 μg/mL kanamycin. A single colony was used to inoculate 2 mL LB medium containing 100 μg/mL ampicillin and 50 μg/mL kanamycin, which was then incubated at 37 °C, 200 rpm, for 16–18 h. This overnight culture was diluted 1:500 in fresh LB medium and used as the working culture. Compounds to be tested were serially diluted two-fold in LB medium to final concentrations ranging from 0 to 140 μM and plated in a clear, flat-bottomed 96-well plate. The working culture, spiked with nothing, CuCl2, or BCS, as appropriate, was then added to the 96-well plate, giving a final inoculum dilution of 1:1000 (5 × 105 to 1 × 106 CFU/mL) and final volume of 100 μL per well. Wells containing media only were included to verify sterility. Plates were incubated for 20 h at 37 °C, 200 rpm. Plates were covered with AeraSeal film (Sigma-Aldrich) during incubation to minimize evaporation.
Bacterial growth was evaluated by visual inspection corroborated by measuring the optical density at 600 nm (OD600) using a Perkin Elmer Victor3 V multilabel plate reader at 0 and 20 h. The 0-h timepoint data were subtracted from the 20-h data to remove background medium signal. Then, data were normalized to the average of the untreated positive control wells for that plate. The minimum inhibitory concentration (MIC) was defined as the concentration at which no detectable growth occurred after 20 h of incubation (background subtracted, normalized OD600 < 0.010). At least two biological replicates were performed with a minimum of at least four total technical replicates for PcephPT and PT MIC data.
Bactericidal testing
Antibacterial microdilution assays were prepared as described above. After 20 h of incubation at 37 °C, 200 rpm, 4 μL of treated culture that showed no growth (background subtracted, normalized OD600 < 0.010) were spotted on a fresh agar plate containing kanamycin (and ampicillin for β-lactamase-expressing strains) or no antibiotic and allowed to dry. Plates were then incubated for 24 h at 37 °C, after which colony forming units (CFUs) were counted. No difference in colony growth was observed regardless of whether the agar plate contained antibiotic.
ICP-MS
Overnight cultures were prepared as described above and grown for 16–18 h. The overnight culture was then diluted 1:1000 and grown to an OD600 of 0.1. The resulting culture (10 mL) was aliquoted into sterile centrifuge tubes and treated with nothing, PcephPT (4 μM), pyrithione (4 μM), CuCl2 (10 μM), PcephPT and CuCl2, or pyrithione and CuCl2. After 15 min or 2 h of treatment, 5 mL of cell solution was placed in an acid-washed centrifuge tube. Cells were pelleted, washed twice with 1 mL chelex-treated water, then once with 1 mL of 1 mM EDTA. The supernatant was discarded after each wash. Pellets were dried at 80–90 °C for 16–20 h. Samples were digested in 50 μL neat trace metal-grade nitric acid at 80–90 °C for 30 min. Samples were allowed to cool, then 50 μL of chelex-treated water was added to obtain a final nitric acid concentration of 50%. Samples were stored at −20 °C until submission for ICP-MS analysis. Data were normalized to the concentration of phosphorus for each sample.
Mammalian culture
Cell culture reagents including minimal essential medium (MEM), fetal bovine serum (FBS), penicillin streptomycin (pen-strep), Triton X-100, and 0.25% trypsin-EDTA were purchased from Gibco. CellTox Green LDH Cytotoxicity Detection Kit was purchased from Promega. Cell lines were purchased from the ATCC. Human lung fibroblast cells (CCD-19Lu, ATCC CCL-210) and human liver epithelial cells (HepG2, ATCC HB-8065) were allowed to adhere overnight to a clear, tissue-culture treated, flat-bottom, 96-well plate in MEM with glutamine and pyruvate supplemented with 10% (v/v) FBS and 1% non-essential amino acids. After splitting and counting, black wall, clear bottom 96-well plates were seeded at a density of 15,000 cells/well and incubated overnight at 37 °C in a fully humidified atmosphere containing 5% CO2. The growth medium was replaced with serum-free medium containing CellTox Green dye and serial dilutions of the active compounds in the presence and absence of 10 μM CuCl2. After 24 h of incubation, the fluorescence emission at 520 nm was recorded (excitation at 485 nm). Untreated cells served as the negative control while wells containing untreated cells incubated with 1% Triton X-100 served as the positive control.
Supplementary Material
Acknowledgements
This project was supported with funds from the U.S. National Science Foundation (CHE-1152054), National Institutes of Health (GM084176, DK110492, AI121742, GM108494), and Department of Defense (W81XWH-13–0450). J.M.Z.B. acknowledges fellowship support from the Duke Pharmacological Sciences Training Program (T32 GM007105). A.C.J. acknowledges an NSF Graduate Research Fellowship (DGE 1644868). We thank Prof. James Imlay at University of Illinois at Urbana-Champaign for the generous gift of the MG1655 LEM8 copA knockout strain and Dr. George Dubay at Duke University for assistance with mass spectrometry.
Footnotes
The supporting information, including additional Scheme S1, Tables S1–S2, and Figures S1–S11, is available free of charge on the ACS Publications website at: pubs.acs.org.
The authors declare no competing financial interest.
References:
- 1.Centers for Disease Control. (2013) Antibiotic resistance threats in the United States.
- 2.Becker KW, and Skaar EP (2014) Metal limitation and toxicity at the interface between host and pathogen, FEMS Microbiol. Rev 38, 1235–1249, DOI: 10.1111/1574-6976.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng TF, and Nolan EM (2014) Enterobactin-mediated delivery of β-lactam antibiotics enhances antibacterial activity against pathogenic Escherichia coli, J. Am. Chem. Soc 136, 9677–9691, DOI: 10.1021/ja503911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller M, Zhu H, Xu YP, Wu CR, Walz A, Vergne A, Roosenberg J, Moraski G, Minnick A, McKee-Dolence J, Hu JD, Fennell K, Kurt Dolence E, Dong L, Franzblau S, Malouin F, and Mollmann U (2009) Utilization of microbial iron assimilation processes for the development of new antibiotics and inspiration for the design of new anticancer agents, Biometals 22, 61–75, DOI: 10.1007/s10534-008-9185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Djoko KY, Ong C.-l. Y., Walker MJ., and McEwan AG (2015) The role of copper and zinc toxicity in innate immune defense against bacterial pathogens, J. Biol. Chem 290, 18954–18961, DOI: 10.1074/jbc.R115.647099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu D-H, Huang Z-L, Zhou T, Shen C, and Hider RC (2011) In vitro inhibition of bacterial growth by iron chelators, FEMS Microbiol. Lett 314, 107–111, DOI: 10.1111/j.1574-6968.2010.02153.x. [DOI] [PubMed] [Google Scholar]
- 7.Fernandes SS, Nunes A, Gomes AR, de Castro B, Hider RC, Rangel M, Appelberg R, and Gomes MS (2010) Identification of a new hexadentate iron chelator capable of restricting the intramacrophagic growth of Mycobacterium avium, Microb. Infect 12, 287–294, DOI: 10.1016/j.micinf.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 8.Falconer Shannon B., Wang W, Gehrke Sebastian S., Cuneo Jessica D., Britten James F., Wright Gerard D., and Brown Eric D. (2014) Metal-induced isomerization yields an intracellular chelator that disrupts bacterial iron homeostasis, Chem. Biol 21, 136–145, DOI: 10.1016/j.chembiol.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 9.Nazik H, Penner JC, Ferreira JA, Haagensen JA, Cohen K, Spormann AM, Martinez M, Chen V, Hsu JL, Clemons KV, and Stevens DA (2015) Effects of iron chelators on the formation and development of Aspergillus fumigatus biofilm, Antimicrob. Agents Chemother. 59, 6514–6520, DOI: 10.1128/aac.01684-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins HL, Kaufmann SHE, and Schaible UE (2002) Iron chelation via deferoxamine exacerbates experimental salmonellosis via inhibition of the nicotinamide adenine dinucleotide phosphate oxidase-dependent respiratory burst, J. Immunol 168, 3458–3463, DOI: 10.4049/jimmunol.168.7.3458. [DOI] [PubMed] [Google Scholar]
- 11.Chan GC-F, Chan S, Ho P-L, and Ha S-Y (2009) Effects of chelators (deferoxamine, deferiprone and deferasirox) on the growth of Klebsiella pneumoniae and Aeromonas hydrophila isolated from transfusion-dependent thalassemia patients, Hemoglobin 33, 352–360, DOI: 10.3109/03630260903211888. [DOI] [PubMed] [Google Scholar]
- 12.Hodgkinson V, and Petris MJ (2012) Copper homeostasis at the host-pathogen interface, J. Biol. Chem 287, 13549–13555, DOI: 10.1074/jbc.R111.316406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ladomersky E, and Petris MJ (2015) Copper tolerance and virulence in bacteria, Metallomics 7, 957–964, DOI: 10.1039/C4MT00327F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White C, Lee J, Kambe T, Fritsche K, and Petris MJ (2009) A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity, J Biol Chem 284, 33949–33956, DOI: 10.1074/jbc.M109.070201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Achard MES, Stafford SL, Bokil NJ, Chartres J, Bernhardt PV, Schembri MA, Sweet MJ, and McEwan AG (2012) Copper redistribution in murine macrophages in response to Salmonella infection, Biochem. J 444, 51–57, DOI: 10.1042/bj20112180. [DOI] [PubMed] [Google Scholar]
- 16.Johnson MDL, Kehl-Fie TE, and Rosch JW (2015) Copper intoxication inhibits aerobic nucleotide synthesis in Streptococcus pneumoniae, Metallomics, DOI: 10.1039/C5MT00011D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, and Niederweis M (2011) Copper resistance is essential for virulence of Mycobacterium tuberculosis, Proc. Natl. Acad. Sci. USA 108, 1621–1626, DOI: 10.1073/pnas.1009261108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.German N, Doyscher D, and Rensing C (2013) Bacterial killing in macrophages and amoeba: do they all use a brass dagger?, Future Microbiol. 8, 1257–1264, DOI: 10.2217/fmb.13.100. [DOI] [PubMed] [Google Scholar]
- 19.Hao Z, Lou H, Zhu R, Zhu J, Zhang D, Zhao BS, Zeng S, Chen X, Chan J, He C, and Chen PR (2014) The multiple antibiotic resistance regulator MarR is a copper sensor in Escherichia coli, Nat. Chem. Biol 10, 21–28, DOI: 10.1038/nchembio.1380. [DOI] [PubMed] [Google Scholar]
- 20.Cavet JS (2013) Metals in bacterial pathogenicity and immunity, Encyclopedia of Inorganic and Bioinorganic Chemistry, 1–12. [Google Scholar]
- 21.Macomber L, and Imlay JA (2009) The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity, Proc. Natl. Acad. Sci. USA 106, 8344–8349, DOI: 10.1073/pnas.0812808106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borkow G, and Gabbay J (2009) Copper, an ancient remedy returning to fight microbial, fungal and viral infections, Curr. Chem. Biol 3, 272–278, DOI: 10.2174/187231309789054887. [Google Scholar]
- 23.Djoko KY, Goytia MM, Donnelly PS, Schembri MA, Shafer WM, and McEwan AG (2015) Copper(II)-bis(thiosemicarbazonato) complexes as antibacterial agents: insights into their mode of action and potential as therapeutics, Antimicrob. Agents Chemother. 59, 6444–6453, DOI: 10.1128/aac.01289-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Festa RA, Helsel ME, Franz KJ, and Thiele DJ (2014) Exploiting innate immune cell activation of a copper-dependent antimicrobial agent during infection, Chem. Biol 21, 977–987, DOI: 10.1016/j.chembiol.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Speer A, Shrestha TB, Bossmann SH, Basaraba RJ, Harber GJ, Michalek SM, Niederweis M, Kutsch O, and Wolschendorf F (2013) Copper-boosting compounds: a novel concept for antimycobacterial drug discovery, Antimicrob. Agents Chemother. 57, 1089–1091, DOI: 10.1128/aac.01781-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaturvedi KS, Hung CS, Crowley JR, Stapleton AE, and Henderson JP (2012) The siderophore yersiniabactin binds copper to protect pathogens during infection, Nat Chem Biol 8, 731–736, DOI: 10.1038/nchembio.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charkoudian LK, Dentchev T, Lukinova N, Wolkow N, Dunaief JL, and Franz KJ (2008) Iron prochelator BSIH protects retinal pigment epithelial cells against cell death induced by hydrogen peroxide, J. Inorg. Biochem 102, 2130–2135, DOI: 10.1016/j.jinorgbio.2008.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Folk DS, and Franz KJ (2010) A prochelator activated by β-secretase inhibits Aβ aggregation and suppresses copper-induced reactive oxygen species formation, J. Am. Chem. Soc 132, 4994–4995, DOI: 10.1021/ja100943r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franks AT, Wang Q, and Franz KJ (2015) A multifunctional, light-activated prochelator inhibits UVA-induced oxidative stress, Bioorg. Med. Chem. Lett 25, 4843–4847, DOI: 10.1016/j.bmcl.2015.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blair JMA, Webber MA, Baylay AJ, Ogbolu DO, and Piddock LJV (2015) Molecular mechanisms of antibiotic resistance, Nat Rev Micro 13, 42–51, DOI: 10.1038/nrmicro3380. [DOI] [PubMed] [Google Scholar]
- 31.Palzkill T (2013) Metallo-β-lactamase structure and function, Ann. N.Y. Acad. Sci 1277, 91–104, DOI: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smyth TP, O’Donnell ME, O’Connor MJ, and St Ledger JO (2000) β-lactamase-dependent prodrugs—recent developments, Tetrahedron 56, 5699–5707, DOI: 10.1016/S0040-4020(00)00419-1. [Google Scholar]
- 33.Ruddle CC, and Smyth TP (2007) Exploring the chemistry of penicillin as a β-lactamase-dependent prodrug, Org. Biomol. Chem 5, 160–168, DOI: 10.1039/B614758E. [DOI] [PubMed] [Google Scholar]
- 34.Zhao G, Miller MJ, Franzblau S, Wan B, and Möllmann U (2006) Syntheses and studies of quinolone-cephalosporins as potential anti-tuberculosis agents, Bioorg. Med. Chem. Lett 16, 5534–5537, DOI: 10.1016/j.bmcl.2006.08.045. [DOI] [PubMed] [Google Scholar]
- 35.Albrecht HA, Beskid G, Chan KK, Christenson JG, Cleeland R, Deitcher KH, Georgopapadakou NH, Keith DD, and Pruess DL (1990) Cephalosporin 3’-quinolone esters with a dual mode of action, J. Med. Chem 33, 77–86, DOI: 10.1021/jm00163a013. [DOI] [PubMed] [Google Scholar]
- 36.Albrecht HA, Beskid G, Christenson JG, Deitcher KH, Georgopapadakou NH, Keith DD, Konzelmann FM, Pruess DL, and Chen Wei C (1994) Dual-action cephalosporins incorporating a 3’-tertiary-amine-linked quinolone, J. Med. Chem 37, 400–407, DOI: 10.1021/jm00029a012. [DOI] [PubMed] [Google Scholar]
- 37.Albrecht HA, Beskid G, Christenson JG, Durkin JW, Fallat V, Georgopapadakou NH, Keith DD, Konzelmann FM, and Lipschitz ER (1991) Dual-action cephalosporins: cephalosporin 3’-quaternary ammonium quinolones, J. Med. Chem 34, 669–675, DOI: 10.1021/jm00106a031. [DOI] [PubMed] [Google Scholar]
- 38.Mobashery S, and Johnston M (1987) Inactivation of alanine racemase by beta-chloro-L-alanine released enzymically from amino acid and peptide C10-esters of deacetylcephalothin, Biochemistry 26, 5878–5884, DOI: 10.1021/bi00392a045. [DOI] [PubMed] [Google Scholar]
- 39.Greenwood D, and O’Grady F (1976) Dual-action cephalosporin utilizing a novel therapeutic principle, Antimicrob. Agents Chemother. 10, 249–252, DOI: 10.1128/aac.10.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xie HX, Mire J, Kong Y, Chang MH, Hassounah HA, Thornton CN, Sacchettini JC, Cirillo JD, and Rao JH (2012) Rapid point-of-care detection of the tuberculosis pathogen using a BlaC-specific fluorogenic probe, Nat. Chem 4, 802–809, DOI: 10.1038/Nchem.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phelan RM, Ostermeier M, and Townsend CA (2009) Design and synthesis of a β-lactamase activated 5-fluorouracil prodrug, Bioorg. Med. Chem. Lett 19, 1261–1263, DOI: 10.1016/j.bmcl.2008.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hamilton-Miller JMT (1994) Dual-action antibiotic hybrids, J. Antimicrob. Chemother 33, 197–200, DOI: 10.1093/jac/33.2.197. [DOI] [PubMed] [Google Scholar]
- 43.Stone GW, Zhang Q, Castillo R, Doppalapudi VR, Bueno AR, Lee JY, Li Q, Sergeeva M, Khambatta G, and Georgopapadakou NH (2004) Mechanism of action of NB2001 and NB2030, novel antibacterial agents activated by β-lactamases, Antimicrob. Agents Chemother. 48, 477–483, DOI: 10.1128/aac.48.2.477-483.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Callaghan CH, Sykes RB, and Staniforth SE (1976) A new cephalosporin with a dual mode of action, Antimicrob. Agents Chemother. 10, 245–248, DOI: 10.1128/AAC.10.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chandler CJ, and Segel IH (1978) Mechanism of the antimicrobial action of pyrithione: effects on membrane transport, ATP levels, and protein synthesis, Antimicrob. Agents Chemother. 14, 60–68, DOI: 10.1128/aac.14.1.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yasokawa D, Murata S, Iwahashi Y, Kitagawa E, Kishi K, Okumura Y, and Iwahashi H (2010) DNA microarray analysis suggests that zinc pyrithione causes iron starvation to the yeast Saccharomyces cerevisiae, J. Biosci. Bioeng 109, 479–486, DOI: 10.1016/j.jbiosc.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 47.Reeder NL, Xu J, Youngquist RS, Schwartz JR, Rust RC, and Saunders CW (2011) The antifungal mechanism of action of zinc pyrithione, Br. J. Dermatol 165 Suppl 2, 9–12, DOI: 10.1111/j.1365-2133.2011.10571.x. [DOI] [PubMed] [Google Scholar]
- 48.Reeder NL, Kaplan J, Xu J, Youngquist RS, Wallace J, Hu P, Juhlin KD, Schwartz JR, Grant RA, Fieno A, Nemeth S, Reichling T, Tiesman JP, Mills T, Steinke M, Wang SL, and Saunders CW (2011) Zinc pyrithione inhibits yeast growth through copper influx and inactivation of iron-sulfur proteins, Antimicrob. Agents Chemother. 55, 5753–5760, DOI: 10.1128/aac.00724-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Helsel ME, White EJ, Razvi SZ, Alies B, and Franz KJ (2017) Chemical and functional properties of metal chelators that mobilize copper to elicit fungal killing of Cryptococcus neoformans, Metallomics 9, 69–81, DOI: 10.1039/c6mt00172f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng Y, Xie H, Sule P, Hassounah H, Graviss EA, Kong Y, Cirillo JD, and Rao J (2014) Fluorogenic probes with substitutions at the 2 and 7 positions of cephalosporin are highly BlaC-specific for rapid mycobacterium tuberculosis detection, Angew. Chem. Int. Ed 53, 9360–9364, DOI: 10.1002/anie.201405243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Berkel SS, Brem J, Rydzik AM, Salimraj R, Cain R, Verma A, Owens RJ, Fishwick CWG, Spencer J, and Schofield CJ (2013) Assay platform for clinically relevant metallo-β-lactamases, J. Med. Chem 56, 6945–6953, DOI: 10.1021/jm400769b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee M, Hesek D, and Mobashery S (2005) A practical synthesis of nitrocefin, J. Org. Chem 70, 367–369, DOI: 10.1021/jo0487395. [DOI] [PubMed] [Google Scholar]
- 53.Mobashery S, and Johnston M (1986) Preparation of ceph-3-em esters unaccompanied by Δ3 ➝ Δ2 isomerization of the cephalosporin, J. Org. Chem 51, 4723–4726, DOI: 10.1021/jo00374a045. [Google Scholar]
- 54.Torii S, Tanaka H, Taniguchi M, Kameyama Y, Sasaoka M, Shiroi T, Kikuchi R, Kawahara I, Shimabayashi A, and Nagao S (1991) Deprotection of carboxylic esters of β-lactam homologs. Cleavage of p-methoxybenzyl, diphenylmethyl, and tert-butyl esters effected by a phenolic matrix, J. Org. Chem 56, 3633–3637, DOI: 10.1021/jo00011a034. [Google Scholar]
- 55.Hay BP, and Beckwith ALJ (1989) Synthesis of N-(alkyloxy)pyridine-2(1H)-thiones: alkylations of the ambident nucleophile pyridine-2(1H)-thione N-oxide and attempted isomerizations of 2-(alkylthio)pyridine N-oxide, J. Org. Chem 54, 4330–4334, DOI: 10.1021/jo00279a020. [Google Scholar]
- 56.Alekseev VG (2012) Metal complexes of penicillins and cephalosporins (Review), Pharm. Chem. J 45, 679–697, DOI: 10.1007/s11094-012-0703-6. [Google Scholar]
- 57.Chen J, Sun P, Zhou X, Zhang Y, and Huang C-H (2015) Cu(II)–catalyzed transformation of benzylpenicillin revisited: The Overlooked Oxidation, Environ. Sci. Technol 49, 4218–4225, DOI: 10.1021/es505114u. [DOI] [PubMed] [Google Scholar]
- 58.Chen J, Sun P, Zhang Y, and Huang C-H (2016) Multiple roles of Cu(II) in catalyzing hydrolysis and oxidation of β-lactam antibiotics, Environ. Sci. Technol 50, 12156–12165, DOI: 10.1021/acs.est.6b02702. [DOI] [PubMed] [Google Scholar]
- 59.Xiao Z, and Wedd AG (2010) The challenges of determining metal-protein affinities, Nat. Prod. Rep 27, 768–789, DOI: 10.1039/B906690J. [DOI] [PubMed] [Google Scholar]
- 60.Rensing C, Fan B, Sharma R, Mitra B, and Rosen BP (2000) CopA: an Escherichia coli Cu(I)-translocating P-type ATPase, Proc. Natl. Acad. Sci. USA 97, 652–656, DOI: 10.1073/pnas.97.2.652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hernández-Montes G, Argüello JM, and Valderrama B (2012) Evolution and diversity of periplasmic proteins involved in copper homeostasis in gamma proteobacteria, BMC Microbiol. 12, 1–14, DOI: 10.1186/1471-2180-12-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.