

Abstract
PD-L1, frequently expressed in human cancers, engages with PD-1 on immune cells and contributes to cancer immune evasion. As such, antibodies blocking the PD-1/PD-L1 interaction reactivate cytotoxic T cells to eradicate cancer cells. However, a majority of cancer patients fail to respond to PD-1/PD-L1 blockade with unclear underlying mechanism(s). Recent studies revealed that PD-L1 expression levels on tumor cells might affect the clinical response to anti-PD-1/PD-L1 therapies. Hence, understanding molecular mechanisms for controlling PD-L1 expression will be important to improve the clinical response rate and efficacy of PD-1/PD-L1 blockade. In this review, we primarily focus on summarizing PD-L1 regulation and its potential roles in regulating anti-tumor immune response, with purpose to optimize anti-PD-1/PD-L1 therapies, benefiting a wider cancer patient population.
Keywords: Cancer immunotherapy, Checkpoint blockade, PD-1/PD-L1, Tumor-infiltrating lymphocyte, Combination therapies
1. Research advances in cancer immunotherapy
Although conventional therapies including chemotherapy and radiotherapy are used as first-line treatments for most human cancer patients, recent emerging advances in cancer immunotherapies have transformed the landscape of cancer treatment from complementary elements for conventional therapies to central and standard cancer treatment regimens [1, 2]. Over the past several decades, immunotherapies or biotherapeutics such as passive immunization with donor T cells, utilizing immune adjuvants or cytokines with immune-modulating properties, cancer vaccines, chimeric antigen receptors (CARs)-modified T cells, and immune checkpoint blocking antibodies are effective treatments for various forms of human cancer [2, 3]. However, among these cancer immunotherapies, immune checkpoints blockade such as antibodies blocking cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed death 1/programmed death-ligand 1 (PD-1/PD-L1) have stood out and are revolutionizing the field of cancer therapy. Thus, it is believed that the immune checkpoint blockade strategy will profoundly change the direction of basic and clinical cancer research [4, 5]. Distinct from conventional therapies for directly targeting cancer cells, anti-CTLA4 or anti-PD-1/PD-L1 antibodies reactivate the immune system of patients to eradicate tumors, which induce durable and long-lasting anti-tumor immunity in patients with a variety of cancers [6, 7].
CTLA-4 is a coinhibitory receptor expressed on activated T cells, which inhibits T cell proliferation and activation in part through competing with its homolog of T cell costimulatory receptor CD28 binding with CD80 (also named B7-1) and CD86 (also named B7-2) [8]. The group led by Dr. James P. Allison pioneered the research field of CTLA-4 and was the first to theorize that developing CTLA-4 blocking antibodies might activate T cell proliferation and enhance its physiological functions [8]. Subsequently, seminal preclinical studies from the Allison group revealed that checkpoint blockade using a CTLA-4 blocking antibody could lead to durable regression and long-lasting immune response in mouse tumor models, which resulted in the clinical development and evaluation of anti-CTLA-4 blocking antibodies for human cancer therapy [9]. The success of clinical trials with anti-CTLA-4 blocking antibody ipilimumab, which was approved by the US Food and Drug Administration (FDA) in 2011, opened a new era for the field of cancer immunotherapies [10]. Clinical studies demonstrated that CTLA-4 immune checkpoint blockade by ipilimumab led to a durable tumor regression and significantly enhancement of the overall survival of some patients with advanced metastatic melanoma [10]. However, to resolve a relatively low response rate and frequent toxicities including dermatitis, hepatitis, and enterocolitis related to anti-CTLA-4 therapy [11], further studies will be warranted to identify biomarkers for distinguishing which patient population is best suited to receive anti-CTLA-4 immunotherapy.
PD-1 is another coinhibitory receptor expressed on immune cells including T cells, B cells, dendritic cells, natural killer (NK) cells, and tumor-infiltrating lymphocytes (TILs) [12, 13]. It has been reported that there are two ligands for PD-1, PD-L1 (also known as CD274 or B7-H1) and PD-L2 (CD273 or B7-DC) [14, 15]. Given that PD-L1 is more widely expressed than PD-L2 in normal and tumor cells, more studies focused on exploring physiological and pathological functions for PD-1/PD-L1 engagement as well as how to disrupt their interaction for cancer therapy [12]. High expression of PD-L1 is frequently observed on tumor cells, where elevated PD-L1 engagement with PD-1 on T cells subsequently leads to T cell dysfunction and exhaustion, which prevents cytotoxic T cell from effectively targeting tumor cells (Figure 1). Hence, designing and developing PD-1/PD-L1 blocking antibodies to disrupt the PD-1/PD-L1 interaction reactivate T cell function to eradicate cancer cells (Figure 1) [5]. Moreover, clinical studies demonstrated that PD-L1 expression on tumor cells or in the tumor microenvironment has a positive correlation with the response rate for anti-PD-1/PD-L1 therapy, which might serve as a potential, but not perfect, selective marker for patient stratification [16, 17]. Thus, it is necessary to fully understand the upstream pathways that regulate PD-L1 expression on tumor cells, which might help to design new strategies to manipulate the PD-L1 expression to enhance the clinical response rate and efficacy to immunotherapies.
Figure 1: A schematic illustration of the molecular mechanism for reactivating T cells using PD-1/PD-L1 blocking antibodies.
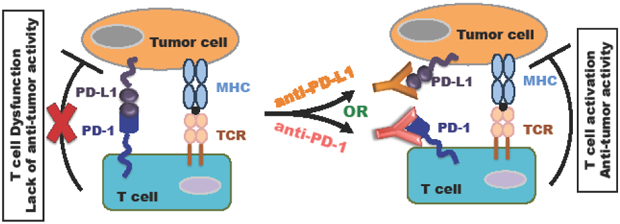
When PD-L1+ tumor cells engage with PD-1+ T cells, this interaction leads to T cell dysfunction and lack of anti-tumor activity. However, blocking PD-1 interaction with PD-L1 using the anti-PD-1 or PD-L1 antibody can reactivate T cells, releasing its anti-tumor activity. The inhibition symbol (⊥) between the T cells and tumor cells represents activated T cells possessing the anti-tumor activity. The red X symbol in the left panel represents T cell dysfunction and lack of anti-tumor activity.
In this review, we summarize recent biochemical research progression on PD-L1 regulation from various aspects, and their potential therapeutic roles in regulating cancer treatment. We also discuss how to further improve the clinical response rate and efficacy of cancer immunotherapy by manipulating PD-L1 expression.
2. Regulation of PD-L1 by genetic alterations and epigenetic modifiers to control cancer immunotherapy
Alterations of genetics and epigenetics play a key role in regulating cancer progression, immune surveillance, and tumor cell evasion [18, 19]. Inhibitors targeting epigenetic modulators such as DNA methyl transferase inhibitors (DNMTi), histone deacetylase inhibitors (HDACi), bromodomain inhibitors, and enhancer of zeste homolog 2 inhibitors (EZH2i) or lysine-specific histone demethylase 1 (LSD1) inhibition enhance immunotherapies through enhancing tumor immunogenicity, restoring cytotoxic T cell functions and reversing the immune suppressive effects of the tumor microenvironment, which synergize with PD-1/PD-L1 or CTLA4 blockade for cancer treatment (Figure 2) [19-24]. Mechanistically, these inhibitors can activate endogenous retroviral elements (ERVs), interferon signaling pathway, or induce neoantigen presentation to stimulate anti-tumor immunity (Figure 2) [19-24]. Moreover, recent studies identified that the PBRM1 and AR1D2 genes, encoding components of the PBAF switch-sucrose nonfermentable (SWI/SNF) chromatin remodeling complex play critical roles in response to immune checkpoint therapies through altering global tumor-cell expression profiles [25, 26]. However, recent studies also showed that genomic alterations and epigenetic pathways could directly control PD-L1 expression to regulate the efficacy of cancer immunotherapies (Figure 3, upper panel).
Figure 2: Inhibition of epigenetic modifiers lead to activate interferon signaling pathways largely through increasing endogenous retroviral elements (ERVs).
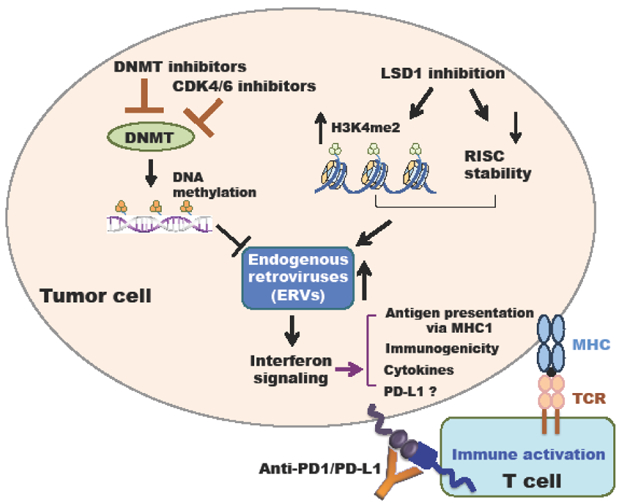
Decreasing DNMT-mediated DNA methylation by DNMT inhibitors or elevating histone H3K4me2 by LSD1 inhibition upregulate the ERVs to activate interferon signaling pathways, leading to promoting antigen presentation via MHC1, immunogenicity, or cytokines production, which synergize with immune-checkpoint blockade such as anti-PD-1/PD-L1.
Figure 3: Genetic, epigenetic and microRNAs regulation of PD-L1.
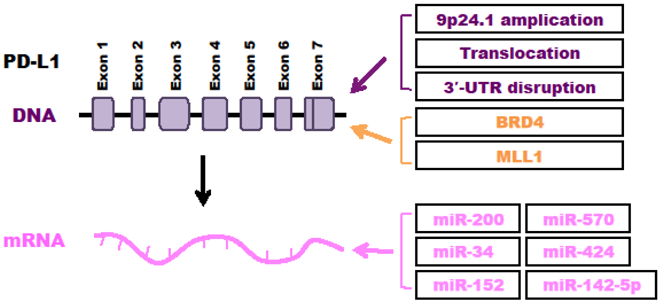
Regulatory mechanisms for PD-L1 by genetic alterations (purple), epigenetic modifiers (orange), and microRNAs (pink) were summarized.
2.1. Genomic alterations regulate PD-L1 and affect the efficacy of PD-1/PD-L1 blockade
Several studies reported that genomic rearrangements including gene amplification and translocation in chromosome 9p24.1 lead to upregulation of PD-L1 and PD-L2 in classic Hodgkin lymphoma (cHL) [27, 28] and primary mediastinal large B-cell lymphoma (PMBCL) [29, 30]. Importantly, these genomic alterations, leading to activate the JAK2-STAT signaling pathway to elevate the PD-L1 transcription and protein abundance, correlate with the relatively high clinical response rate of Hodgkin lymphoma patients to PD-1 blockade [27, 28, 31]. Moreover, amplification of PD-L1, PD-L2 and JAK2 were also identified in gastric cancer [32]. Additionally, Kataoka et al. found a unique genomic structural alteration that can upregulate PD-L1 expression to evade cancer immune surveillance through disrupting the 3’-untranslated region (UTR) of PD-L1 in multiple human cancer types [33]. More importantly, they could recapitulate their findings in mouse tumor models by demonstrating that PD-L1 upregulation by 3’-UTR loss promotes tumor growth and evade immune surveillance mediated by cytotoxic T lymphocytes, which can be effectively suppressed by PD-L1 antibody treatment to restore CD8+ cytotoxic T lymphocytes [33]. These findings together suggest that genomic alterations that upregulate PD-L1 could potentially serve as a genetic marker to distinguish which cancer patients may have greater response to PD-1/PD-L1 immune checkpoint blockade therapy.
2.2. Epigenetic regulation of PD-L1 in anti-tumor immunity
Inhibitors targeting bromodomains and extra-terminal (BET) proteins, such as JQ1 and I-BET762, have an effect on elimination of hematological malignancies such as acute myeloid leukemia (AML), multiple myeloma (MM) and lymphoma [34, 35]. Through their bromodomains, the BET family of proteins bind to acetylated-Lysine motifs present in histones resulting in the recruitment of transcription factors and other chromatin regulators to facilitate gene transcription [35]. Moreover, an implication of BET proteins in solid tumors was also reported [36]. Recently, two independent groups identified that PD-L1 is a direct transcription target of BRD4. Zhu et al. found that BET inhibitors (BETi) significantly suppress PD-L1 expression through screening a panel of inhibitors targeting epigenetic regulators [37]. They further showed that the BET inhibitor JQ1 suppresses PD-L1 expression on tumor cells, dendritic cells and macrophages, leading to retarded tumor progression through enhancing anti-tumor cytotoxic T cell activity [37]. Another group led by Johnstone independently found that the anti-tumor response of BET proteins inhibitors require host immune system [38]. By using genome-wide analysis of the BETi-induced transcriptional response, PD-L1 downregulation was identified as the main mechanism accounting for BETi mediated antitumor effect in a Myc-driven B cell lymphoma model [38]. Mechanistically, they further found that BETi reduces BRD4 occupancy at the PD-L1 locus, leading to PD-L1 transcriptional suppression independent of the transcriptional factor c-Myc. Importantly, they demonstrated that BETi JQ1 synergizes with anti-PD-1 antibodies to suppress the progression of Myc-driven lymphoma in vivo, suggesting that pharmacological BET inhibitors treated in combination with immune modulatory agents might be a novel therapeutic strategy for human cancer treatment [38]. However, although the authors also showed that sustained ectopic expression of PD-L1 reduces the efficacy of JQ1 treatment, it warrants further study to deplete PD-L1 and validate whether these effects of JQ1 treatment is largely dependent on PD-L1 and its downstream targets, Furthermore, recent studies reveal that changes in epigenetic regulation can lead to profound changes in the expression of endogenous retrovirus elements and immune response gene pathways to impact antigen presenting and immunotherapy efficacy (Figure 2). It is possible that JQ1 can potentially function through these pathways in addition to its role in modulating PD-L1 expression.
Additionally, it has been reported that class I HDAC inhibitors upregulated the expression of PD-L1 in melanomas largely through enhancing histone acetylation of the PD-L1 gene, leading to relaxed chromatin state at the PD-L1 gene promoter [39]. Lu et al. demonstrated that histone methyltransferase (HMTase) MLL1 directly binds and catalyzes the tri-methylation of histone H3 on lysine 4 (H3K4me3) on the promoter of PD-L1 to activate the PD-L1 transcription in pancreatic cancer cells [40]. Notably, they showed that pharmacological inhibition or silencing of MLL1 decreases PD-L1 expression, which enhances the efficacy of anti-PD-L1/PD-1 antibody treatment in mouse models bearing pancreatic tumor growth [40]. However, like the JQ1 treatment, suppression of MLL1 also affects many other downstream targets, which warrants further investigations to clarify whether targeting MLL1 is an effective approach to enhance the efficacy of checkpoint immunotherapy against pancreatic cancer in part via regulation of PD-L1 expression.
3. Regulation of PD-L1 at the transcriptional level to modulate anti-tumor immunity
Increasing evidence reveals that various upstream signaling pathways regulate PD-L1 expression at the transcriptional level [41, 42]. These signaling pathways participate in controlling PD-L1 expression largely through activating several key transcriptional factors, which directly bind on the promoter region of PD-L1 to promote its expression [41, 42]. Although previous studies revealed that the IFNγ signaling pathways play the most important role for translational regulation of PD-L1 expression and response to the PD-1/PD-L1 blockade, other signaling pathways can be also activated to upregulate PD-L1 expression for evasion of anti-tumor surveillance under different cellular or tissue circumstances. Here, we document major signaling pathways and transcriptional factors that play critical roles in governing PD-L1 expression to regulate immune surveillance and cancer immunotherapies by targeting PD-1/PD-L1 (Figure 4).
Figure 4: Upstream signaling pathways regulating PD-L1 at transcriptional level.
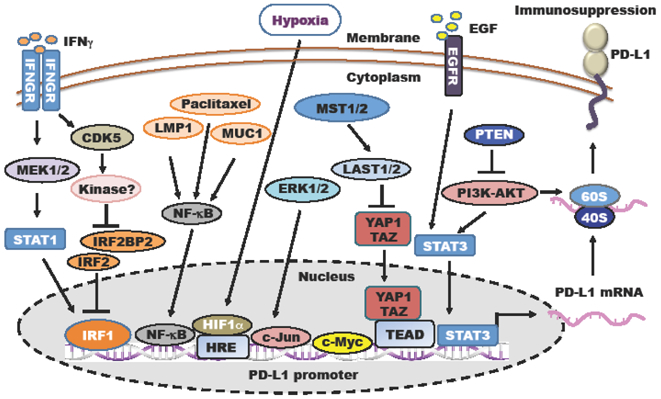
Tumor cells evade anti-tumor effects of T cells in part by elevating PD-L1 mRNA expression at transcriptional level via activation of different upstream signaling pathways.
3.1. The IFNγ-JAK-STAT signaling pathway-mediated regulation of PD-L1
Several studies have shown that PD-L1 expression can be induced by interferon gamma (IFNγ) to allow tumor cells to evade immune surveillance [43-45]. Janus kinase (JAK) and signal transducer and activators of transcription (STAT) are essential for interferon induction through activation of interferon-stimulated response elements (ISREs), gamma interferon activation sites (GASs) and interferon regulatory factor 1 (IRF1) [46]. Upon IFNγ stimulation, the JAK-STAT signaling can activate IRF1, which directly binds to the PD-L1 promoter to induce the transcription of PD-L1, resulting in suppression of anti-tumor immunity [43-45]. A recent study reported that cyclin-dependent kinase 5 (CDK5) helps medulloblastoma (MB) to evade immune surveillance and suppresses anti-tumor immunity [47]. Dorand et al. found that IFN-γ-induced PD-L1 up-regulation in MB requires CDK5 and depletion of CDK5 results in downregulation of PD-L1 expression in tumor cells [47]. Furthermore, CDK5 disruption suppresses the IFNGR signaling in part by recruiting the IRF1 competitive repressors IRF2 and IRF2BP2 to sustain PD-L1 transcriptional repression [47].
3.2. NF-κB pathway regulation of PD-L1
NF-κB is an ubiquitously expressed transcription factor, which is considered a major contributor of tumor development by promoting cell survival, proliferation, angiogenesis, and metastasis [48]. Homo-or hetero-dimerization of NF-κB subunits RelA/p65 and p50 leads to activation of NF-κB, which is required for tumor cells to evade adaptive immunity [49]. Recent studies indicated that activation of NF-κB signaling upregulates PD-L1 to suppress anti-tumor immunity in different cancer settings. In the T-cell lymphoma, PD-L1 upregulation mediated by activation of NF-κB via EBV-driven LMP1 signaling, which correlates with poor prognosis in natural killer/T-cell lymphoma [50]. In ovarian cancer, chemotherapy treatment was demonstrated to induce PD-L1 expression in a NF-κB activation dependent manner, which provides an immunosuppressive tumor microenvironment to evade immune surveillance [51]. Moreover, in non-small cell lung cancer (NSCLC), MUC1-C increased PD-L1 expression through enhancing NF-κB occupancy on the PD-L1 promoter to induce PD-L1 transcription [52].
3.3. Hypoxia-induced expression of PD-L1
Hypoxia is a common feature of most solid tumors, the severity of which contributes to the acquisition of malignant properties in cancer cells such as drug resistance, metastatic ability and immune evasion [53, 54]. The transcription factor hypoxia-inducible factor 1 (HIF-1) mediates adaptive responses to hypoxia, which is carried out through two subunits for HIF-1, HIF-1α and HIF-1β [55]. HIF-1α is the functional subunit, whose stability is regulated by oxygen levels in a pVHL-dependent manner [55]. As such, HIF-1α is upregulated under hypoxic conditions, and plays important roles in tumor cell survival and malignancy. Several studies indicate that hypoxia induces PD-L1 expression in tumor cells, myeloid-derived suppressor cells (MDSCs), dendritic cells, and macrophages, which are largely mediated by HIF-1α [56-58]. Mechanistically, HIF-1α binds to a hypoxia-response element (HRE) in the PD-L1 proximal promoter and induces the transcription of PD-L1 [59]. Hence, hypoxic conditions increase the resistance of tumor cells to cytotoxic T lymphocytes (CTL)-mediated lysis [56]. Selective blocking HIF-1α accumulation in hypoxic cells leads to decreased expression of PD-L1 in tumor cells and results in potent anti-tumor response and attenuation of tumor growth in mice [56].
3.4. c-Myc regulation of PD-L1
The transcription factor c-Myc induces the expression of many genes involved in different functions, including cell proliferation, growth, differentiation, and apoptosis [60]. The c-Myc gene is frequently amplified in many cancer types and upregulation of c-Myc is thought to be one of the driving forces to facilitate development of cancer [60]. Inactivation of c-Myc and other oncogenes in mouse tumor models leads to complete tumor clearance accompanied by the recruitment of CD4+ T cells [61]. Notably, recent studies demonstrate that PD-L1 is a direct transcriptional target of c-Myc [62, 63]. Inactivation of c-Myc in mouse tumor cells decreases expression levels of PD-L1 to improve the anti-tumor immune response. Moreover, re-expression of PD-L1 in c-Myc inactivated tumors suppresses the anti-tumor immunity. Recent results also showed that c-Myc expression significantly correlated with PD-L1 expression in NSCLC patients with poor clinical outcomes [64].
3.5. The effects of the MAPK pathway on PD-L1 expression regulation
Mitogen-activated protein kinase (MAPK) signaling pathways are frequently mutated and over-activated in multiple human cancer types, which have shown promise as therapeutic targets for cancer treatment [65]. MAPK pathways are largely mediated by extracellular-signal-regulated kinase (ERK), c-Jun amino-terminal kinases (JNKs) and p38 [65]. Increasing evidence showed that these MAPK oncogenic signaling pathways promote tumor development and immunotherapy resistance [66]. Pharmacological inhibition of the MAPK pathway components improve the anti-tumor response [66]. Moreover, MAPK signaling pathways are directly involved in regulating PD-L1 expression to evade cancer immune surveillance. The MAPK/ERK pathway increases PD-L1 expression by activating the transcription factor c-Jun [67]. In keeping with this notion, inhibition of MAPK signaling pathways lead to a decrease in PD-L1 expression levels in tumor cells [67, 68].
3.6. The involvement of the PI3K pathway in regulating PD-L1 expression
Gene alterations leading to hyperactivation of the phosphatidylinositol 3-kinase (PI3K) pathway are frequently found in many types of human cancers [69]. These alterations include, but are not limited to, the loss of the phosphatase and tensin homolog (PTEN), mutation and/or upregulation of PI3K catalytic and regulatory subunits, and the upstream activator K-RAS as well as downstream effectors AKT and PDK1 [69]. In addition to inducing cell death, PI3K inhibitors also suppress immune escape largely through stimulation of the T-cell response or modulation of the myeloid cell compartment [70]. Loss of PTEN results in activation of PI3K and increases PD-L1 expression in part by elevating PD-L1 protein translation rate in glioma and lung squamous cell carcinoma, which leads to immune resistance and escape [71, 72]. Additionally, PI3K activation can also induce PD-L1 expression through the transcription factor STAT3 in human melanoma [67].
3.7. EGFR pathway regulation of PD-L1
The epidermal growth factor receptor (EGFR) serves as one of epidermal growth factor family of receptor tyrosine kinases (ErbBs) and is activated following binding with peptide growth factors of the EGF-family of proteins [73]. Inappropriate activation of the EGFR was frequently observed in various cancer types. Activation of the EGFR pathway in tumor cells results in an immunosuppressive lung microenvironment and less responsive to the anti-PD-1 immunotherapy through induction of PD-L1 expression [74]. Further studies showed that the EGFR pathway elevates PD-L1 expression primarily through the IL-6/JAK/STAT3 signaling axis, which can be reduced by treatment with EGFR inhibitors [74, 75].
3.8. Hippo signaling in the regulation of PD-L1 expression
The Hippo tumor suppressive pathway is frequently inactivated in various types of cancers [76]. YAP1 and TAZ function as oncogenes in cancer and form a complex with TEAD, which plays an important role in cell proliferation, apoptotic inhibition, and the epithelial-mesenchymal transition (EMT) [76, 77]. A recent study showed that the Hippo pathway suppresses anti-tumor immunity largely through the LAST1/2 kinases [58]. Additionally, two groups independently reported that YAP1 could bind to the promoter region of PD-L1 to induce PD-L1 expression in lung adenocarcinoma and NSCLC to control immune evasion [78, 79]. Moreover, it has also been reported that TAZ could induce PD-L1 upregulation at the transcriptional level in human cancer cells, which inhibits the anti-tumor activity of T cells in the tumor microenvironment [80].
4. Regulation of PD-L1 expression by microRNAs
MicroRNAs (miRNAs) consisting of 20-22 nucleotides regulate a large number of genes including tumor suppressors and oncogenes through targeting their 3’-UTR to promote cleavage of mRNA transcripts [81]. Recent studies have demonstrated that abnormal expression of miRNAs plays an important role in regulating cancer progression and immune checkpoint blockade mediated cancer therapies [81, 82]. Moreover, increasing evidence demonstrated that miRNAs directly target 3’-UTR of the PD-L1 mRNA to control PD-L1 expression and anti-tumor immunity (Figure 3, lower panel).
Chen et al. showed that microRNA-200 (miR-200), a suppressor of the epithelial-to-mesenchymal transition (EMT) and metastasis, directly targets the 3’-UTR of PD-L1 mRNA and suppresses PD-L1 expression [83]. They further found that the zinc-finger E-box-binding homeobox 1 (ZEB1) factor, an EMT activator and transcriptional repressor of miR-200, elevates PD-L1 expression and metastasis in part through suppressing CD8+ T cell activity in non-small cell lung cancer (NSCLC) [83]. These results suggest that evaluation of the ZEB1/miR-200 axis-mediated regulation of PD-L1 might be a useful marker of lung cancer to guide treatment selection with anti-PD-1/PD-L1 blockade therapies. MiR-34, a downstream target of p53, directly binds to the PD-L1 3’-UTR and inhibits PD-L1 expression [84]. Notably, the authors delivered miR-34a-loaded liposomes (MRX34) in a syngeneic mouse model of lung cancer and found that MRX34, alone or in combination with radiotherapy, dramatically reduces PD-L1 expression on tumor cells and enhances cytotoxic immune T cell populations in the tumor microenvironment [84]. In addition to their functions in lung cancer, miR-200c and miR-34a also target PD-L1 mRNA to decrease PD-L1 expression in acute myeloid leukemia [85, 86]. In gastric cancer, miR-152 and miR-570 were reported to directly bind the 3’-UTR of PD-L1 and suppress PD-L1 expression [87, 88]. In pancreatic cancer, miR-142-5p suppresses PD-L1 expression by binding to its 3’-UTR, which enhances anti-tumor immunity through increasing cytotoxic CD4+ and CD8+ T lymphocytes [89]. In ovarian cancer, miR-424 overcomes chemo-resistance through T-cell immune response activation by directly binding to PD-L1 3’-UTR and decreasing PD-L1 expression [90]. Thus, it appears that various miRNAs regulate PD-L1 mRNA in different tissues to offer functional diversities.
5. Regulation of PD-L1 by post-translational modifications to affect the efficacy of PD-1/PD-L1 blockade
Protein post-translational modifications (PTMs) including ubiquitination, phosphorylation, glycosylation, methylation, and acetylation play critical roles in regulating protein activity, stability, localization, trafficking, and interactions with binding partners including protein, DNA, RNA or lipids [91]. These above mentioned PTMs are essential for controlling various cellular processes including tumor formation, metastasis and anti-tumor immunity [92, 93]. Increasing evidence demonstrated that PD-L1 also undergoes different post-translational modifications to affect its stability, internalization, localization as well as its physiological and pathological functions (Figure 5).
Figure 5: PD-L1 regulation by post-translational modifications (PTMs).
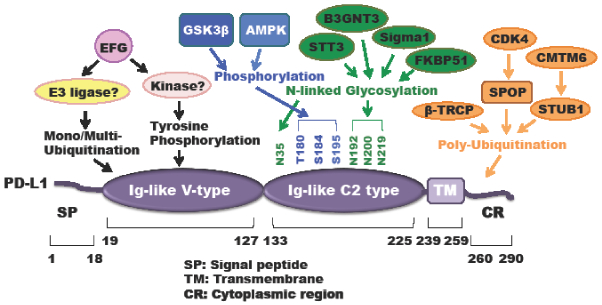
A schematic illustration of various types of PD-L1 PTMs including ubiquitination, phosphorylation, and N-linked glycosylation.
5.1. Poly-ubiquitination regulation of PD-L1 stability to govern anti-PD-1 therapeutic efficacy
The ubiquitin-proteasome system (UPS) promotes protein poly-ubiquitination and subsequent degradation to control various cellular processes and diseases including immune surveillance and tumorigenesis [94, 95]. Degradation by the UPS is catalyzed by three distinct sequential actions of the activating (E1), conjugating (E2), and ligase (E3) enzymes that lead to the covalent attachment of poly-ubiquitin chain to lysine residues on the target protein, which can be recognized by the 26S proteasome complex to promote its degradation [94]. Li et al. showed that the Cullin 1 based ubiquitin E3 ligase adaptor protein β-TRCP promoted PD-L1 poly-ubiquitination and degradation through direct binding with PD-L1 in a 175LSGxxTxxxS184 motif-dependent manner, which requires GSK3β-mediated phosphorylation of PD-L1 at T180 and S184 residues [96]. In keeping with these findings, the dominant-negative form of β-TRCP lacking the F-box or mutation of PD-L1 in the GSK3β phosphorylation motif abolished β-TRCP-mediated PD-L1 poly-ubiquitination and degradation. The Hung group further identified the epidermal growth factor (EGF) signaling as the upstream regulator for the GSK3β/β-TRCP axis to regulate PD-L1 stability [96]. Notably, they demonstrated that the EGFR inhibitor gefitinib could dramatically reduce PD-L1 expression in breast cancer cells through releasing EGF-mediated suppression of GSK3β activation, which increases the activation of tumor-infiltrated CD8+ T-cells and enhances the efficacy of PD-1 antibody therapy in several syngeneic mouse tumor models [96]. Moreover, the authors also mentioned that gefitinib might provide several lines of benefits to this combination therapy such as reducing PD-L1 expression to limit its binding to T-cell receptors, as well as limiting PD-L1 oncogenic potential and reducing EGFR-overexpressing cell survivals [96].
Recently, our group showed that PD-L1 protein abundance fluctuates during cell cycle, and further identified the cell cycle kinase cyclin D-CDK4 activity play a crucial role in destabilizing PD-L1 [97]. Mechanistically, we identified Cullin 3SPOP as the physiological ubiquitin E3 ligase for promoting PD-L1 poly-ubiquitination and degradation. We further demonstrated that the cyclin D-CDK4 stabilizes SPOP in a phosphorylation-dependent manner and treatment with the CDK4/6 inhibitor palbociclib destabilizes SPOP to increase PD-L1 protein abundance to possibly induce immune evasion [97]. Pathologically, cancer patients derived loss-of-function mutations in SPOP fail to promote PD-L1 poly-ubiquitination-mediated degradation, which leads to stabilization of PD-L1 accompanied with reduced numbers of tumor-infiltrating lymphocytes (TILs) in tumors [97]. Notably, the CDK4/6 kinase inhibitor palbociclib synergized with anti-PD-1 therapy to elicit an enhanced therapeutic efficacy as evidenced by dramatically enhancing tumor regression and improving overall survival rates in syngeneic mouse tumor models [97].
5.2. Mono-ubiquitination regulation of PD-L1 upon EGF stimulation
In addition to poly-ubiquitination, proteins can also be modified with mono- or multi-ubiquitination through conjugating a single ubiquitin molecule to one (mono-ubiquitination) or several lysines (multi-ubiquitination), which plays critical roles in regulating various cellular processes, including control of protein stability, localization, endocytosis and trafficking independent of proteasome mediated degradation [98]. EGF treatment induces the mono- and multi-ubiquitination on PD-L1, leading to stabilization of PD-L1 [99]. Moreover, using chemical inhibitors targeting EGFR or ubiquitin E1 activating enzyme dramatically reduced the mono- and multi-ubiquitination of PD-L1 accompanied with decreasing the total PD-L1 level [99]. However, the specific E3 ligase for promoting the mono-ubiquitination of PD-L1 remains unknown and further in-depth studies are needed to address this critical question. Interestingly, Li et al. found that the EGF signaling pathway can also positively regulate PD-L1 stability largely through inactivating GSK3β and suppressing GSK3β-mediated phosphorylation of PD-L1, which prevents the E3 ligase adaptor protein β-TRCP-mediated PD-L1 poly-ubiquitination and subsequent degradation [96]. Hence, further investigation to explore whether there is crosstalk between EGF-induced mono-ubiquitination and GSK3β/β-TRCP-mediated poly-ubiquitination and how to use these findings to design novel combination therapies for human cancer treatment are warranted.
5.3. De-ubiquitination regulation of PD-L1 by CSN5 to modulate cancer immune therapy
Ubiquitination is a reversible process where deubiquitinating enzymes (DUBs) catalytically remove the single ubiquitin or poly-ubiquitin chains from the targeted proteins [100]. Increasing evidence demonstrate that mutation or dysregulated expression of DUBs lead to various human diseases including immune diseases and cancer [101, 102]. Hence, developing selective inhibitors of DUBs has been proposed as new therapeutic strategy for human immune diseases and cancer therapy. Recently, the COP9 signalosome 5 (CSN5) was identified as a DUB for PD-L1 deubiquitination, which results in stabilization of PD-L1 to control T cell suppression [103]. Lim et al. further demonstrated that tumor necrosis factor alpha (TNF-α) stimulation enhances CSN5 expression through inducing the transcriptional factor NF-κB p65 subunit to bind CSN5 promoter and promote its transcription [103]. Upon TNF-α-induced CSN5 binding with PD-L1, CSN5 removes the poly-ubiquitin chain on PD-L1 thereby stabilizing PD-L1, which subsequently promotes tumor immune escape [103]. In keeping with these findings, inhibition of CSN5 by the natural compound curcumin reduced TNF-α/ NF-κB signaling induced PD-L1 stability, which synergized with anti-CTLA-4 therapy to enhance anti-tumor immunity and retard tumor growth [103]
5.4. Phosphorylation of PD-L1 by GSK3β and AMPK to affect PD-L1 protein stability
Dysregulation of kinase-dependent phosphorylation signaling events are frequently observed in many human diseases including neurodegenerative diseases, inflammatory disorders, and cancer [104, 105]. Recently, key kinase signaling pathways such as JAK, mTOR-Akt, and LATS1/2 kinases dependent pathways play critical roles in regulating cancer immune therapy efficacy [58, 106, 107]. As described above, it has been reported that GSK3β directly binds with the C-terminal domain of PD-L1 and phosphorylates T180 and S184 on PD-L1, which is subsequently recognized by β-TRCP to promote PD-L1 poly-ubiquitination and degradation [96]. The authors further demonstrated that activation of GSK3β through inhibition of its upstream negative regulator EGF signaling could dramatically reduce PD-L1 levels and enhance PD-1 blockade therapy in mouse tumor models [96].
AMP-activated protein kinase (AMPK) is an energy sensor that regulates cellular metabolism and energy homeostasis [108]. Moreover, emerging evidence also showed that AMPK activation regulates T cell metabolism and function [109]. However, how the AMPK signaling pathway regulates cancer immunotherapy remains elusive. Recently, Cha et al. found that metformin-activated AMPK directly phosphorylates PD-L1 at the S195 residue to induce abnormal glycosylation, which results in PD-L1 degradation through an ERAD pathway [110]. Notably, this study also showed that metformin combination with the CTLA4 blockade significantly suppresses tumor growth in syngeneic mouse models [110]. Additionally, PD-L1 is also reported to be phosphorylated following EGF treatment [99]. However, the physiological function of EGF-induced phosphorylation of PD-L1 warrants further in-depth investigation.
5.5. Targeting glycosylated PD-L1 for eradication of triple-negative breast cancer (TNBC)
Growing evidence has revealed that protein glycosylation plays fundamental roles in regulating various human diseases including Alzheimer's disease, cardiovascular disease and cancer [111]. N- and O-linked protein glycosylation, which are covalently conjugated with glycans via the nitrogen atom of asparagine or oxygen atom of serine, threonine, hydroxylysine or hydroxyproline residues, respectively, have been widely studied [112]. Recent studies demonstrated that dysregulation of glycosylation might serve as a new cancer hallmark and potential therapeutic target because it has been shown to be related with tumor initiation, angiogenesis, invasion, metastasis and immune surveillance [112, 113]. Hung’s group showed that PD-L1 could be modified by N-linked glycosylation, which stabilizes PD-L1 largely through preventing GSK3β/β-TRCP-mediated PD-L1 poly-ubiquitination and degradation [96]. They also found that the upstream EGF signaling induces PD-L1 N-linked glycosylation and stabilizes PD-L1, which leads to tumor cell immunosuppression [96]. Hence, using the small molecule gefitinib to inhibit EGF signaling could destabilize PD-L1 and promote anti-tumor immunity [96].
Furthermore, EGF signaling upregulates the expression of β-1, 3-N-acetylglucosaminyltransferase (B3GNT3) to promote the N-glycosylation of N192 and N200 sites on PD-L1 in TNBC cells, which is required for binding with PD-1 on cytotoxic T cells and leading to T cell exhaustion [114]. Notably, Hung’s group developed a specific antibody targeting the glycosylated PD-L1 (gPD-L1) and gPD-L1 antibody-drug conjugates (g-PD-L1-ADCs), which blocks PD-L1 interaction with PD-1 and promotes PD-L1 internalization for lysosome-mediated degradation [114]. Moreover, their results showed that g-PD-L1-ADCs could reactivate cytotoxic T cells and exert a bystander effect to kill adjacent cancer cells without PD-L1 expression, suggesting that targeting PD-L1 glycosylation might be a potential strategy to improve cancer immune therapy [114]. Additionally, the N-glycosyltransferase STT3 could stabilize and upregulate PD-L1 in cancer stem cells to evade cancer immune surveillance, which will provide another therapeutic target for suppressing the STT3 enzymatic activity to destabilize PD-L1 to benefit PD-1/PD-L1-mediated checkpoint blockade [115].
5.6. Regulation of PD-L1 protein stability by PD-L1 binding partners
In addition to post-translational modifications mentioned above, two independent groups identified through large-scale genetic screens that CKLF-like MARVEL transmembrane domain containing protein 6 (CMTM6) as the critical positive regulator for PD-L1 [116, 117]. Both groups reported that depletion of CMTM6 dramatically upregulated PD-L1 in multiple human cancer cell lines [116, 117]. They further found that CMTM6 stabilized PD-L1 at the post-translation level via different mechanisms. Burr et al. showed that in addition to at the plasma membrane, CMTM6 also co-localizes with PD-L1 in recycling endosomes, which helps endocytosed PD-L1 recycle thereby preventing lysosome-mediated degradation (Figure 5) [117]. Mezzadra and colleagues demonstrated that CMTM6 interacts with PD-L1 at the cell surface to inhibit PD-L1 poly-ubiquitination, which might be dependent on the ubiquitin E3 ligase STUB1 [116]. Moreover, they also identified that the CMTM4, but not other CMTM family members, plays a complementary function when CMTM6 is deficient in cells [116]. Moreover, the integral membrane scaffolding protein Sigma1 stabilizes and elevates PD-L1 in cancer cells largely through preventing autophagy-mediated degradation of PD-L1 [118], and the co-chaperone FKBP51 stabilizes PD-L1 in glioma (Figure 5) [119]. Interestingly, both Sigma1 and FKBP51s stabilization of PD-L1 is through PD-L1 glycosylation in the endoplasmic reticulum (ER) [118, 119]. Together, these studies suggest that targeting these upstream regulators including CMTM4/6, Sigma1 and FKBP51s to reduce PD-L1 expression might be alternative strategies to enhance PD-1/PD-L1 immune checkpoint blockade, which will likely provide advantage as combination strategies with other immune therapies such as anti-CTLA4 treatment.
6. Regulation of PD-L1 by the DNA damage pathway
6.1. Genomic instability determines sensitivity to immune checkpoint blockade
Genomic instability, driven by exogenous DNA-damaging agents or endogenous DNA repair deficiency, confers cancer cells with an increased propensity of genome alteration, and ranks as one of the most common hallmarks of tumors [120]. Upon double strand breaks (DSB) in normal cells, the primary DNA damage response (DDR) transducer, nuclear kinase ataxia-telangiectasia mutated (ATM) is activated [121], and subsequently transduce this signal to its downstream effector such as the checkpoint kinases ATR, Chk1, Chk2, and the p53 tumor suppressor [122-128], which in turn arrest cell cycle progression to allow DNA damage repair to commence, or promote damage-induced cell apoptosis in instances where DNA damage is too extensive [129-132]. Cancer cells are capable of escaping from immune surveillance by expressing inhibitory proteins to suppress T cell function, such as the well-defined PD-L1 immune checkpoint molecule [133, 134]. Of note, recent studies have suggested a correlation between genomic instability and response to immunotherapy with checkpoint inhibitors largely through generating neoantigens, which puts the interaction between DNA damage and immune system into attention [135-137]. Given the fact that several reviews have discussed this topic [138-142], we will specifically discuss the role of DNA damage in the regulation of PD-L1 expression (Figure 6).
Figure 6: DNA damage signaling pathways regulating PD-L1 at transcriptional level.
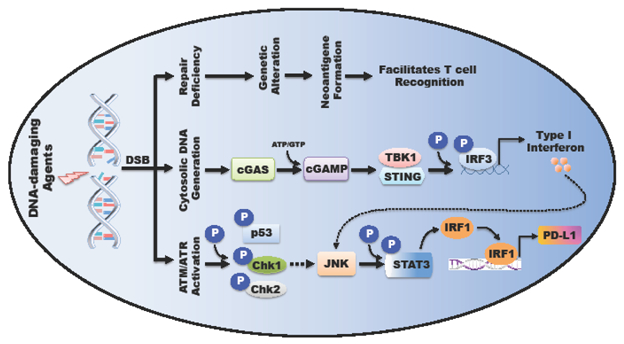
DNA-damaging events-induced double strand break (DSB) triggers (1) ATM/ATR kinase activation; (2) cytosolic DNA generation, both of which finally activate JNK-STAT signaling cascade and up-regulate PD-L1 expression; (3) Insufficient or error-prone repair results in genetic alteration and neoantigen formation, which facilitates T cell recognition.
6.2. Mechanisms of DNA damage regulation of PD-L1 expression
As mentioned above, the JAK-STAT-IRF1 pathway has been shown to regulate PD-L1 expression [45, 143]. DNA damage plays a critical role in modulating the JAK-STAT signaling cascade as well (Figure 6). For example, STAT1 is activated in a p53-dependent manner [144], while JAK1 and STAT3 exhibit enhanced phosphorylation and transcriptional activity upon treatment with DNA damaging agents [145]. These results indicate a potential link between DNA damage and PD-L1 expression. In support of this notion, a recent study suggests DSBs regulates PD-L1 expression in a JAK-STAT-IRF1-dependent manner [146]. Interestingly, STAT3 is able to modulate the DNA damage response pathway as well [147-149], establishing a feedback loop among DNA damage response and JAK-STAT signaling integrity.
In addition, tumor cells harbor cytosolic DNA produced by DSB [150], which will further prime cytosolic DNA sensors, such as the well-defined cyclic GMP-AMP synthase (cGAS) and DNA-dependent protein kinase (DNA-PK) [151, 152]. Recognition of cytosolic DNA by the cGAS-STING pathway and DNA-PK subsequently activates interferon regulatory factor 3 (IRF3) [153, 154], accounting for the causal link between DNA damage and IRF3 activation [155]. Consequently, activation of IRF3 promotes type I interferon production [156, 157], activates JAK-STAT pathway, and elevates PD-L1 expression (Figure 4) [45, 143]. Recent studies suggest that the S-phase DNA damage-induced PD-L1 expression is largely STING-dependent [158].
6.3. Targeting DNA damage and repair for enhancing anti-PD-1/PD-L1 efficacy
Given the observation that mutational burden and neoantigen formation are highly correlated [159], in addition to the increased PD-L1 expression levels in the context of genome instability, we speculate that tumors with higher mutational burden harbor more neoantigens, which facilitates tumor infiltrating T cells recognition; on the other hand, elevated PD-L1 protein levels makes PD-L1 blockade more critical for reactivating cytotoxic T cells to target tumor cells. Indeed, several clinical studies have observed a positive correlation between neoantigen load and PD-L1 expression levels [160, 161], and tumors with higher mutation burden were more susceptible to PD1/PD-L1 blockade [136].
Based on these studied, combination therapy using DNA damage agents and PD1/PD-L1 checkpoint showed encouraging results. For example, ionizing irradiation (IR) upregulates PD-L1 in the tumor microenvironment and administration of anti-PD-L1 enhanced the antitumor immunity in mice [162]; PARP inhibitors upregulate PD-L1 expression and the combination treatment with PARP inhibitors and anti-PD-L1 significantly increased the therapeutic efficacy in vivo [163]; the combination of cGAMP and anti-PD-L1 in combination exerted stronger anti-tumor effects than did either treatment alone [164]. Currently, the study of interaction between DNA damage checkpoint and PD1/PD-L1 immune checkpoint is still in its infancy, and more work needs to be done to establish their relationship and provide theoretical insights for future clinical trials of DNA damaging agents in combination with immunotherapy to combat cancers.
7. Concluding remarks and future perspectives
The success of immunotherapies, especially targeting the PD-1/PD-L1 pathway, has given hope and confidence to better treat and possibly cure cancer [4, 5]. Next, we should explore how to enhance the efficacy and response rate of PD-1/PD-L1 blockade to benefit a greater range of cancer patients. To this end, in-depth investigation and elucidating the molecular mechanisms that regulate the PD-1/PD-L1 pathway will be one of the key steps for designing novel therapeutic strategies to overcome anti-PD-1/PD-L1 resistance and improve the anti-tumor immunity for cancer therapy.
Based on results from clinical trials of anti-PD-1/PD-L1 immunotherapy, it is still debatable whether PD-L1 expression level is a predictive biomarker for efficacy or response rate of PD-1/PD-L1 blockade in at the individual patient level. Although increasing evidence showed that patients with PD-L1-positive (PD-L1+) tumors have a better response rate and overall survival than PD-L1-negative (PD-L1−) patients in different cancer types, it was also reported that there are patients with PD-L1+ tumors exhibiting resistance to anti-PD-1/PD-L1 treatment and minor population patients with PD-L1− tumors that also benefit from these treatments [165-168]. As we discussed above, various cell signaling pathways can regulate PD-L1 expression at the genomic, transcriptional, post-transcriptional, translational, and post-translational levels to affect anti-PD-1/PD-L1 treatment. However, there is still a lack of standard methods to evaluate the quantity of PD-L1 expression on tumor cells necessary to allow a response to PD-1/PD-L1 blockade. Moreover, there is the correlation of PD-L1 expression on tumor cells with response to PD-1/PD-L1 blockade under the adaptive immune resistance that PD-L1 expression is induced by the secreted IFNγ of infiltrated T cells, which were well discussed in recent several reviews [169-171]. Additionally, PD-L1 expression on tumor infiltrating immune cells in tumor microenvironment, particularly macrophages, dendritic cells and stromal cells are also critical for PD-1/PD-L1 blockade [165, 167, 172, 173]. However, in most of current clinical trials there are no distinctions of PD-L1 expression on the cell type such as tumor cells or infiltrating immune cells when tumor biopsies are evaluated for PD-L1 expression. Additionally, it is still warranted to assess whether other important signaling pathways or modifications including ubiquitin-like modification, methylation, or acetylation can regulate PD-L1 expression levels to control anti-tumor immunity (See Outstanding Questions).
Outstanding Questions.
Besides reported ubiquitination, N-linked glycosylation and phosphorylation, whether additional post-translational modifications such as ubiquitin-like modification, methylation, acetylation and succination also can regulate PD-L1 expression to control immunosuppression in cancer patients?
Noncoding RNAs (ncRNAs) including microRNAs (miRNAs), long noncoding RNAs (lncRNAs) and circular RNAs (circRNAs) have been shown to play important roles in regulating gene expression to control tumorigenesis and cancer immunity. However, exception of miRNAs reported in regulating PD-L1 expression and immune surveillance, it is still unclear whether lncRNAs and circRNAs also participate in governing PD-L1 expression to control cancer immune therapy.
Although PD-1 expression was well studied at the transcriptional levels, the upstream signaling pathways governing PD-1 post-translational modifications are less studied. Hence, exploring the upstream signaling pathways controlling PD-1 post-translational modifications such as ubiquitination, glycosylation, acetylation and methylation will be required to understand and design better strategies for improving the response rate and efficacy for PD-1/PD-L1 blockade in future.
Immunomodulatory targets in addition to PD-L1/PD-1 such as LAG3, TIM3, TIGIT, IDO and TGF have been shown to their abilities to regulating anti-tumor immunity in single agent or synergizing with anti-PD-1/PD-L1. However, it warrants in-depth studies to explore how these targets could be regulated in cells and whether manipulating their expression levels can control anti-tumor immune response.
As PD-1 is the important part for targeting PD-1/PD-L1 immuno-checkpoint blockade, manipulating PD-1 expression on T cells might be also critical for enhancing anti-PD-1/PD-L1 immunotherapy. Hence, exploring the upstream signaling pathways controlling PD-1 post-translational modifications such as ubiquitination, glycosylation, acetylation and methylation will be required to understand and design better strategies for improving the response rate and efficacy for PD-1/PD-L1 blockade in future. Moreover, additional immune-modulatory targets such as lymphocyte activation gene-3 (LAG3), T cell-immunoglobulin-mucin domain 3 (TIM3), T Cell Immunoglobulin and ITIM Domain (TIGIT) as well as indoleamine 2, 3-dioxygenase (IDO) or transforming growth factor-β (TGFβ) in tumor microenvironment have been shown their abilities to suppress anti-tumor immunity [11]. Further studies are needed to explore how these targets in tumor microenvironment are regulated at various levels and whether manipulating these targets expression can affect anti-tumor immune response.
Acknowledgments
We sincerely apologize to all those colleagues whose important work was not cited in this paper due to space limitations. We thank Drs. Brian North, Jianping Guo, Wenjian Gan, Xiangpeng Dai and other members of the Wei laboratory for critical reading and discussion of the manuscript. W.W. is an ACS research scholar and a LLS research scholar. This work was supported in part by the NIH grants (W.W., GM094777 and CA177910; J.Z., 1K99CA212292-01).
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mellman I et al. (2011) Cancer immunotherapy comes of age. Nature 480 (7378), 480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sharma P and Allison JP (2015) The future of immune checkpoint therapy. Science 348 (6230), 56–61. [DOI] [PubMed] [Google Scholar]
- 3.Couzin-Frankel J (2013) Breakthrough of the year 2013. Cancer immunotherapy. Science 342 (6165), 1432–3. [DOI] [PubMed] [Google Scholar]
- 4.Mahoney KM et al. (2015) Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov 14 (8), 561–84. [DOI] [PubMed] [Google Scholar]
- 5.Zou W et al. (2016) PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8 (328), 328rv4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melero I et al. (2015) Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat Rev Cancer 15 (8), 457–72. [DOI] [PubMed] [Google Scholar]
- 7.Sharpe AH and Pauken KE (2018) The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol 18 (3), 153–167. [DOI] [PubMed] [Google Scholar]
- 8.Allison JP (2015) Immune Checkpoint Blockade in Cancer Therapy: The 2015 Lasker-DeBakey Clinical Medical Research Award. JAMA 314 (11), 1113–4. [DOI] [PubMed] [Google Scholar]
- 9.Leach DR et al. (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271 (5256), 1734–6. [DOI] [PubMed] [Google Scholar]
- 10.Hodi FS et al. (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363 (8), 711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baumeister SH et al. (2016) Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol 34, 539–73. [DOI] [PubMed] [Google Scholar]
- 12.Keir ME et al. (2008) PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26, 677–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L and Han X (2015) Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 125 (9), 3384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Freeman GJ et al. (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192 (7), 1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Latchman Y et al. (2001) PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2 (3), 261–8. [DOI] [PubMed] [Google Scholar]
- 16.Patel SP and Kurzrock R (2015) PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol Cancer Ther 14 (4), 847–56. [DOI] [PubMed] [Google Scholar]
- 17.Grigg C and Rizvi NA (2016) PD-L1 biomarker testing for non-small cell lung cancer: truth or fiction? J Immunother Cancer 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakravarthi BV et al. (2016) Genomic and Epigenomic Alterations in Cancer. Am J Pathol 186 (7), 1724–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aspeslagh S et al. (2018) Epigenetic modifiers as new immunomodulatory therapies in solid tumours. Ann Oncol 29 (4), 812–824. [DOI] [PubMed] [Google Scholar]
- 20.Dunn J and Rao S (2017) Epigenetics and immunotherapy: The current state of play. Mol Immunol 87, 227–239. [DOI] [PubMed] [Google Scholar]
- 21.Sheng W et al. (2018) LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiappinelli KB et al. (2015) Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 162 (5), 974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Topper MJ et al. (2017) Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 171 (6), 1284–1300 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goel S et al. (2017) CDK4/6 inhibition triggers anti-tumour immunity. Nature 548 (7668), 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan D et al. (2018) A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 359 (6377), 770–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miao D et al. (2018) Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 359 (6377), 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ansell SM et al. (2015) PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 372 (4), 311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roemer MG et al. (2016) PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J Clin Oncol 34 (23), 2690–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Twa DD et al. (2014) Genomic rearrangements involving programmed death ligands are recurrent in primary mediastinal large B-cell lymphoma. Blood 123 (13), 2062–5. [DOI] [PubMed] [Google Scholar]
- 30.Green MR et al. (2010) Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 116 (17), 3268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu WR and Shipp MA (2017) Signaling pathways and immune evasion mechanisms in classical Hodgkin lymphoma. Blood 130 (21), 2265–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Research, N. (2014) Comprehensive molecular characterization of gastric adenocarcinoma. Nature 513 (7517), 202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kataoka K et al. (2016) Aberrant PD-L1 expression through 3'-UTR disruption in multiple cancers. Nature 534 (7607), 402–6. [DOI] [PubMed] [Google Scholar]
- 34.Filippakopoulos P et al. (2010) Selective inhibition of BET bromodomains. Nature 468 (7327), 1067–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferri E et al. (2016) Bromodomains: Structure, function and pharmacology of inhibition. Biochem Pharmacol 106, 1–18. [DOI] [PubMed] [Google Scholar]
- 36.Sahai V et al. (2016) Targeting BET bromodomain proteins in solid tumors. Oncotarget 7 (33), 53997–54009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhu H et al. (2016) BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep 16 (11), 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hogg SJ et al. (2017) BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep 18 (9), 2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woods DM et al. (2015) HDAC Inhibition Upregulates PD-1 Ligands in Melanoma and Augments Immunotherapy with PD-1 Blockade. Cancer Immunol Res 3 (12), 1375–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu C et al. (2017) The MLL1-H3K4me3 Axis-Mediated PD-L1 Expression and Pancreatic Cancer Immune Evasion. J Natl Cancer Inst 109 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun C et al. (2018) Regulation and Function of the PD-L1 Checkpoint. Immunity 48 (3), 434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J et al. (2016) Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol 27 (3), 409–16. [DOI] [PubMed] [Google Scholar]
- 43.Mandai M et al. (2016) Dual Faces of IFNgamma in Cancer Progression: A Role of PD-L1 Induction in the Determination of Pro- and Antitumor Immunity. Clin Cancer Res 22 (10), 2329–34. [DOI] [PubMed] [Google Scholar]
- 44.Abiko K et al. (2015) IFN-gamma from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer 112 (9), 1501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia-Diaz A et al. (2017) Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep 19 (6), 1189–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murray PJ (2007) The JAK-STAT signaling pathway: input and output integration. J Immunol 178 (5), 2623–9. [DOI] [PubMed] [Google Scholar]
- 47.Dorand RD et al. (2016) Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 353 (6297), 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karin M (2009) NF-kappaB as a critical link between inflammation and cancer. Cold Spring Harb Perspect Biol 1 (5), a000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taniguchi K and Karin M (2018) NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 18 (5), 309–324. [DOI] [PubMed] [Google Scholar]
- 50.Bi XW et al. (2016) PD-L1 is upregulated by EBV-driven LMP1 through NF-kappaB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J Hematol Oncol 9 (1), 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peng J et al. (2015) Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-kappaB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res 75 (23), 5034–45. [DOI] [PubMed] [Google Scholar]
- 52.Bouillez A et al. (2017) MUC1-C integrates PD-L1 induction with repression of immune effectors in non-small-cell lung cancer. Oncogene 36 (28), 4037–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown JM and Wilson WR (2004) Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer 4 (6), 437–47. [DOI] [PubMed] [Google Scholar]
- 54.Chouaib S et al. (2017) Hypoxic stress: obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 36 (4), 439–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Semenza GL (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148 (3), 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barsoum IB et al. (2014) A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res 74 (3), 665–74. [DOI] [PubMed] [Google Scholar]
- 57.Samanta D et al. (2018) Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proc Natl Acad Sci U S A 115 (6), E1239–E1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moroishi T et al. (2016) The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 167 (6), 1525–1539 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Noman MZ et al. (2014) PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 211 (5), 781–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McKeown MR and Bradner JE (2014) Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med 4 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rakhra K et al. (2010) CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 18 (5), 485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Casey SC et al. (2016) MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352 (6282), 227–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Atsaves V et al. (2017) PD-L1 is commonly expressed and transcriptionally regulated by STAT3 and MYC in ALK-negative anaplastic large-cell lymphoma. Leukemia 31 (7), 1633–1637. [DOI] [PubMed] [Google Scholar]
- 64.Kim EY et al. (2017) MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer 110, 63–67. [DOI] [PubMed] [Google Scholar]
- 65.Sebolt-Leopold JS and Herrera R (2004) Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer 4 (12), 937–47. [DOI] [PubMed] [Google Scholar]
- 66.Pancione M et al. (2017) Emerging Insight into MAPK Inhibitors and Immunotherapy in Colorectal Cancer. Curr Med Chem 24 (14), 1383–1402. [DOI] [PubMed] [Google Scholar]
- 67.Jiang X et al. (2013) The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin Cancer Res 19 (3), 598–609. [DOI] [PubMed] [Google Scholar]
- 68.Atefi M et al. (2014) Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res 20 (13), 3446–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cantley LC (2002) The phosphoinositide 3-kinase pathway. Science 296 (5573), 1655–7. [DOI] [PubMed] [Google Scholar]
- 70.Okkenhaug K et al. (2016) Targeting PI3K in Cancer: Impact on Tumor Cells, Their Protective Stroma, Angiogenesis, and Immunotherapy. Cancer Discov 6 (10), 1090–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Parsa AT et al. (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 13 (1), 84–8. [DOI] [PubMed] [Google Scholar]
- 72.Xu C et al. (2014) Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 25 (5), 590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sigismund S et al. (2018) Emerging functions of the EGFR in cancer. Mol Oncol 12 (1), 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Akbay EA et al. (2013) Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov 3 (12), 1355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang N et al. (2016) The EGFR pathway is involved in the regulation of PD-L1 expression via the IL-6/JAK/STAT3 signaling pathway in EGFR-mutated non-small cell lung cancer. Int J Oncol 49 (4), 1360–8. [DOI] [PubMed] [Google Scholar]
- 76.Harvey KF et al. (2013) The Hippo pathway and human cancer. Nat Rev Cancer 13 (4), 246–57. [DOI] [PubMed] [Google Scholar]
- 77.Yu FX et al. (2015) Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163 (4), 811–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee BS et al. (2017) Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem Biophys Res Commun 491 (2), 493–499. [DOI] [PubMed] [Google Scholar]
- 79.Kim MH et al. (2018) YAP-Induced PD-L1 Expression Drives Immune Evasion in BRAFi-Resistant Melanoma. Cancer Immunol Res. [DOI] [PubMed] [Google Scholar]
- 80.Janse van Rensburg HJ et al. (2018) The Hippo Pathway Component TAZ Promotes Immune Evasion in Human Cancer through PD-L1. Cancer Res 78 (6), 1457–1470. [DOI] [PubMed] [Google Scholar]
- 81.Nicoloso MS et al. (2009) MicroRNAs--the micro steering wheel of tumour metastases. Nat Rev Cancer 9 (4), 293–302. [DOI] [PubMed] [Google Scholar]
- 82.Wang Q et al. (2017) The Roles of microRNAs in Regulating the Expression of PD-1/PD-L1 Immune Checkpoint. Int J Mol Sci 18 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen L et al. (2014) Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun 5, 5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cortez MA et al. (2016) PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst 108 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pyzer AR et al. (2017) MUC1 inhibition leads to decrease in PD-L1 levels via upregulation of miRNAs. Leukemia 31 (12), 2780–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang X et al. (2015) Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal 27 (3), 443–52. [DOI] [PubMed] [Google Scholar]
- 87.Wang W et al. (2013) A miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Hum Genet 132 (6), 641–8. [DOI] [PubMed] [Google Scholar]
- 88.Wang Y et al. (2017) MicroRNA-152 regulates immune response via targeting B7-H1 in gastric carcinoma. Oncotarget 8 (17), 28125–28134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jia L et al. (2017) miR-142–5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem Biophys Res Commun 488 (2), 425–431. [DOI] [PubMed] [Google Scholar]
- 90.Xu S et al. (2016) miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat Commun 7, 11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walsh G and Jefferis R (2006) Post-translational modifications in the context of therapeutic proteins. Nat Biotechnol 24 (10), 1241–52. [DOI] [PubMed] [Google Scholar]
- 92.Hattori T and Koide S (2018) Next-generation antibodies for post-translational modifications. Curr Opin Struct Biol 51, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martinez-Forero I et al. (2009) Lysine 63 polyubiquitination in immunotherapy and in cancer-promoting inflammation. Clin Cancer Res 15 (22), 6751–7. [DOI] [PubMed] [Google Scholar]
- 94.Zhang J et al. (2014) Functional characterization of Anaphase Promoting Complex/Cyclosome (APC/C) E3 ubiquitin ligases in tumorigenesis. Biochim Biophys Acta 1845 (2), 277–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu J et al. (2016) Post-Translational Modification Control of Innate Immunity. Immunity 45 (1), 15–30. [DOI] [PubMed] [Google Scholar]
- 96.Li CW et al. (2016) Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun 7, 12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang J et al. (2018) Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553 (7686), 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hicke L (2001) Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol 2 (3), 195–201. [DOI] [PubMed] [Google Scholar]
- 99.Horita H et al. (2017) Identifying Regulatory Posttranslational Modifications of PD-L1: A Focus on Monoubiquitinaton. Neoplasia 19 (4), 346–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mevissen TET and Komander D (2017) Mechanisms of Deubiquitinase Specificity and Regulation. Annu Rev Biochem 86, 159–192. [DOI] [PubMed] [Google Scholar]
- 101.Lopez-Castejon G and Edelmann MJ (2016) Deubiquitinases: Novel Therapeutic Targets in Immune Surveillance? Mediators Inflamm 2016, 3481371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.He M et al. (2017) Emerging role of DUBs in tumor metastasis and apoptosis: Therapeutic implication. Pharmacol Ther 177, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lim SO et al. (2016) Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 30 (6), 925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cohen P (2002) Protein kinases--the major drug targets of the twenty-first century? Nat Rev Drug Discov 1 (4), 309–15. [DOI] [PubMed] [Google Scholar]
- 105.Giamas G et al. (2007) Protein kinases as targets for cancer treatment. Pharmacogenomics 8 (8), 1005–16. [DOI] [PubMed] [Google Scholar]
- 106.Waldmann TA and Chen J (2017) Disorders of the JAK/STAT Pathway in T Cell Lymphoma Pathogenesis: Implications for Immunotherapy. Annu Rev Immunol 35, 533–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zeng H (2017) mTOR signaling in immune cells and its implications for cancer immunotherapy. Cancer Lett 408, 182–189. [DOI] [PubMed] [Google Scholar]
- 108.Garcia D and Shaw RJ (2017) AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol Cell 66 (6), 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Blagih J et al. (2015) The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 42 (1), 41–54. [DOI] [PubMed] [Google Scholar]
- 110.Cha JH et al. (2018) Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol Cell 71 (4), 606–620 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pinho SS and Reis CA (2015) Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 15 (9), 540–55. [DOI] [PubMed] [Google Scholar]
- 112.Vajaria BN and Patel PS (2017) Glycosylation: a hallmark of cancer? Glycoconj J 34 (2), 147–156. [DOI] [PubMed] [Google Scholar]
- 113.Salatino M et al. (2018) Glycans Pave the Way for Immunotherapy in Triple-Negative Breast Cancer. Cancer Cell 33 (2), 155–157. [DOI] [PubMed] [Google Scholar]
- 114.Li CW et al. (2018) Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 33 (2), 187–201 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hsu JM et al. (2018) STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun 9 (1), 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mezzadra R et al. (2017) Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549 (7670), 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Burr ML et al. (2017) CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549 (7670), 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Maher CM et al. (2018) Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol Cancer Res 16 (2), 243–255. [DOI] [PubMed] [Google Scholar]
- 119.D'Arrigo P et al. (2017) A regulatory role for the co-chaperone FKBP51s in PD-L1 expression in glioma. Oncotarget 8 (40), 68291–68304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5), 646–74. [DOI] [PubMed] [Google Scholar]
- 121.Paull J.-H.L.a.T.T. (2005) ATM Activation by DNA Double-Strand Breaks Through the Mre11-Rad50-Nbs1 Complex. Science 308 (5721), 551–4. [DOI] [PubMed] [Google Scholar]
- 122.Canman Christine E., D.-S.L., Cimprich Karlene A., Taya Yoichi, Tamai Katsuyuki, Sakaguchi Kazuyasu, Appella Ettore, Kastan Michael B., Siliciano Janet D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281 (5383), 1677–9. [DOI] [PubMed] [Google Scholar]
- 123.Khanna Kum Kum, K.E.K., Kozlov Sergei, Scott Shaun, Gatei Magtouf, Hobson Karen, Taya Yoichi, Gabrielli Brian, Chan Doug, Lees-Miller Susan P. & Lavin Martin F., (1998) ATM associates with and phosphorylates p53- mapping the region of interaction. Nat Genet 20 (4), 398–400. [DOI] [PubMed] [Google Scholar]
- 124.Banin S, L.M., Shieh S-Y, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, and Shiloh Y, Y.Z. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281 (5383), 1674–7. [DOI] [PubMed] [Google Scholar]
- 125.Tibbetts Randal S., K.M.B., Williams Josie M., Sarkaria Jann N., Cliby William A., Shieh Sheau-Yann, Taya Yoichi, Prives Carol, and Abraham Robert T. (1999) A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 13 (2), 152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhao H and Piwnica-Worms H (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21 (13), 4129–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Gatei M et al. (2003) Ataxia-telangiectasia-mutated (ATM) and NBS1-dependent phosphorylation of Chk1 on Ser-317 in response to ionizing radiation. J Biol Chem 278 (17), 14806–11. [DOI] [PubMed] [Google Scholar]
- 128.Jazayeri A et al. (2006) ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 8 (1), 37–45. [DOI] [PubMed] [Google Scholar]
- 129.Di Leonardo Aldo, S.P.L., Clarkin Kris and Wahl Geoffrey M. (1994) DNA damage triggers a prolonged p53-dependent G1 arrest and long-term induction of Cip1 in normal human fibroblasts. Genes Dev 8 (21), 2540–51. [DOI] [PubMed] [Google Scholar]
- 130.Khanna Kum Kum, S.P.J. (2001) DNA double-strand breaks- signaling, repair and the cancer connection. Nat Genet 27 (3), 247–54. [DOI] [PubMed] [Google Scholar]
- 131.Roos WP and Kaina B (2006) DNA damage-induced cell death by apoptosis. Trends Mol Med 12 (9), 440–50. [DOI] [PubMed] [Google Scholar]
- 132.Jackson SP and Bartek J (2009) The DNA-damage response in human biology and disease. Nature 461 (7267), 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Freeman GJ et al. (2000) Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. The Journal of Experimental Medicine 192 (7), 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dong H et al. (2002) Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8 (8), 793–800. [DOI] [PubMed] [Google Scholar]
- 135.Le DT et al. (2015) PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med 372 (26), 2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rizvi Naiyer A., M.D.H., Snyder Alexandra, Kvistborg Pia, Makarov Vladimir, Havel Jonathan J., Lee William, Yuan Jianda, Wong Phillip, Ho Teresa S., Miller Martin L., Rekhtman Natasha, Moreira Andre L., and Ibrahim Fawzia, C.B., Gasmi Billel, Zappasodi Roberta, Maeda Yuka, Sander Chris, Garon Edward B., Merghoub Taha, Wolchok Jedd D., Schumacher Ton N., Chan Timothy A. (2015) Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cance. Science 348 (6230), 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yaghmour G, P.M., Ireland C, Patel K, Nunnery S, Powell D, Baum S, Wiedower E, Schwartzberg LS, Martin MG (2016) Role of genomic instability in immunotherapy with checkpoint inhibitors. Anticancer Research 36 (8), 4033–8. [PubMed] [Google Scholar]
- 138.O'Connor MJ (2015) Targeting the DNA Damage Response in Cancer. Mol Cell 60 (4), 547–60. [DOI] [PubMed] [Google Scholar]
- 139.Minn AJ and Wherry EJ (2016) Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 165 (2), 272–5. [DOI] [PubMed] [Google Scholar]
- 140.Mouw KW et al. (2017) DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov 7 (7), 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Brown JS et al. (2018) Combining DNA damaging therapeutics with immunotherapy: more haste, less speed. Br J Cancer 118 (3), 312–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mouw KW and Konstantinopoulos PA (2018) From checkpoint to checkpoint: DNA damage ATR/Chk1 checkpoint signalling elicits PD-L1 immune checkpoint activation. Br J Cancer 118 (7), 933–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mimura K et al. (2018) PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci 109 (1), 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Youlyouz-Marfak I et al. (2008) Identification of a novel p53-dependent activation pathway of STAT1 by antitumour genotoxic agents. Cell Death Differ 15 (2), 376–85. [DOI] [PubMed] [Google Scholar]
- 145.Yun UJ et al. (2012) DNA damage induces the IL-6/STAT3 signaling pathway, which has anti-senescence and growth-promoting functions in human tumors. Cancer Lett 323 (2), 155–60. [DOI] [PubMed] [Google Scholar]
- 146.Sato H et al. (2017) DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat Commun 8 (1), 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Barry SP et al. (2010) STAT3 modulates the DNA damage response pathway. Int J Exp Pathol 91 (6), 506–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Koganti S et al. (2014) STAT3 interrupts ATR-Chk1 signaling to allow oncovirus-mediated cell proliferation. Proc Natl Acad Sci U S A 111 (13), 4946–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liao XH et al. (2015) STAT3 regulated ATR via microRNA-383 to control DNA damage to affect apoptosis in A431 cells. Cell Signal 27 (11), 2285–95. [DOI] [PubMed] [Google Scholar]
- 150.Shen YJ et al. (2015) Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep 11 (3), 460–73. [DOI] [PubMed] [Google Scholar]
- 151.Ferguson BJ et al. (2012) DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1, e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sun Lijun, J.W., Du Fenghe, Chen Xiang, Chen Zhijian J. (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339 (6121), 786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Karpova Alla Y., M.T., Murray John M., Cantley Lewis C. and Howley Peter M. (2002) Interferon regulatory factor-3 is an in vivo target of DNA-PK. PNAS 99 (5), 2818–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu S et al. (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347 (6227), aaa2630. [DOI] [PubMed] [Google Scholar]
- 155.Kim Taeil, T.Y.K., Song Young-Hwa, Min Irene M., Yim Jeongbin, and Kim Tae Kook (1999) Activation of Interferon Regulatory Factor 3 in Response to DNA-damaging Agents. J Biol Chem 274 (43), 30686–9. [DOI] [PubMed] [Google Scholar]
- 156.Andersen J et al. (2008) IRF-3-dependent and augmented target genes during viral infection. Genes Immun 9 (2), 168–75. [DOI] [PubMed] [Google Scholar]
- 157.Li Peng, J.J.-Y.W., Sum Calvin, Sin Wei-Xiang, Ng Kok-Quan, Koh Mickey B. C., and Chin Keh-Chuang (2011) IRF8 and IRF3 cooperatively regulate rapid interferon-β induction in human blood monocytes. Blood 117, 2847–2854. [DOI] [PubMed] [Google Scholar]
- 158.Parkes EE et al. (2017) Activation of STING-Dependent Innate Immune Signaling By S-Phase-Specific DNA Damage in Breast Cancer. J Natl Cancer Inst 109 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Schumacher TN and Hacohen N (2016) Neoantigens encoded in the cancer genome. Curr Opin Immunol 41, 98–103. [DOI] [PubMed] [Google Scholar]
- 160.Howitt BE et al. (2015) Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol 1 (9), 1319–23. [DOI] [PubMed] [Google Scholar]
- 161.Strickland Kyle C., B.E.H., Shukla Sachet A., Rodig Scott, Ritterhouse Lauren L., Liu Joyce F., Garber Judy E., Chowdhury Dipanjan, Wu Catherine J., D’Andrea Alan D., Matulonis Ursula A., Konstantinopoulos Panagiotis A. (2016) Association and prognostic significance of BRCA1:2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-LPD-L1 in high grade serous ovarian cancer. 7 (12), 13587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Deng L et al. (2014) Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest 124 (2), 687–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jiao S et al. (2017) PARP Inhibitor Upregulates PD-L1 Expression and Enhances Cancer-Associated Immunosuppression. Clin Cancer Res 23 (14), 3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang H et al. (2017) cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A 114 (7), 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Herbst RS et al. (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515 (7528), 563–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Garon EB et al. (2015) Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 372 (21), 2018–28. [DOI] [PubMed] [Google Scholar]
- 167.Taube JM et al. (2014) Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 20 (19), 5064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Brahmer J et al. (2015) Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med 373 (2), 123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ribas A and Hu-Lieskovan S (2016) What does PD-L1 positive or negative mean? J Exp Med 213 (13), 2835–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sharma P et al. (2017) Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168 (4), 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kim TK et al. (2018) Defining and Understanding Adaptive Resistance in Cancer Immunotherapy. Trends Immunol 39 (8), 624–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Juneja VR et al. (2017) PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med 214 (4), 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lau J et al. (2017) Tumour and host cell PD-L1 is required to mediate suppression of anti-tumour immunity in mice. Nat Commun 8, 14572. [DOI] [PMC free article] [PubMed] [Google Scholar]