

Abstract
Recent advances in Ca2+ imaging have given neuroscientists a tool to follow the activity of large numbers of individual neurons simultaneously in vivo in the brains of animals as they are presented with sensory stimulation, respond to environmental challenges, and engage in behaviors. The Ca2+ sensors used to transduce changes in cellular Ca2+ into changes in fluorescence must bind Ca2+ to produce a signal. By binding Ca2+, these sensors can act as buffers, often reducing the magnitude of a Ca2+ change several fold and producing a proportional slowing of the rates of change. Ca2+ probes can thus distort the patterns of activity they are intended to study and modify ongoing Ca2+ signaling functions. Recognizing these factors will enhance the use of in vivo Ca2+ imaging in the investigation of neural circuit function.
Keywords: Calcium imaging, Calcium sensors, In vivo imaging, Two-photon microscopy
The impact of Ca2+ probes on Ca2+ signals
Any molecule that binds Ca2+ can influence its dynamics. All fluorescent Ca2+ sensors currently in use, including both synthetic and genetically-encoded probes, report the presence of Ca2+ through a binding induced change in fluorescence. If the sensor binds to an appreciable fraction of cellular Ca2+ then it will act as a buffer to limit the magnitude of changes in Ca2+ concentration and reduce their speed. This action will depend critically on the affinity and concentration of the sensor. If a sensor’s buffering is strong, it will provide a distorted picture of neuronal activity and perturb a neuron’s Ca2+ signaling. Although the buffering actions of Ca2+ probes are well recognized [1–3], they are often overlooked and underestimated. In fact, the buffering action of a probe can dominate observed Ca2+ signals. In this Opinion we revisit some early evidence for Ca2+ buffering by probes, and review the well-established framework for the quantitative analysis of buffering strength. We then discuss the magnitudes of these effects and their impact on experiments, concluding with suggestions for estimating the impact of Ca2+ probes on Ca2+ imaging experiments.
The buffering action of a Ca2+ probe is illustrated in Fig. 1 taken from an early fluorometric Ca2+ imaging experiment [4]. The Ca2+-sensitive fluorescent dye fura-2 was loaded into a chromaffin cell through a patch pipette (Fig. 1A). The loading was monitored by observing the fluorescence elicited by 360 nm light, a wavelength that excites both Ca2+-bound and free fura-2 roughly equally, so this signal does not depend on Ca2+. This trace shows that the fura-2 from the patch pipette fills the cell in about 5 minutes. At the plateau in fluorescence, the concentration of fura-2 in the cell reached that in the patch pipette, 400µM.As the loading progressed, voltage pulses of a set amplitude and duration were applied repeatedly to open Ca2+ channels and raise intracellular free Ca2+ ([Ca2+]i).These Ca2+ rises were signaled by transient reductions in fluorescence elicited by 390 nm light, a wavelength that preferentially excites the free species of fura-2. The [Ca2+]i computed from the ratio of the two spectral channels illustrates two important consequences of buffering by the Ca2+ sensor: the voltage-induced increase in [Ca2+]i declines from nearly 0.5 µM early in the loading process to about 50nM at the plateau; and the decay time increases from a few seconds to nearly half a minute. Note that the final voltage pulse in the sequence was 500msec instead of 50msec to demonstrate that a large [Ca2+]i transient could still be evoked but with a slow decay. This experiment demonstrates convincingly that the sensor strongly buffers intracellular Ca2+. This study also reversed the buffering effect by establishing a new patch clamp recording with a lower fura-2 concentration (Fig. 1B). The [Ca2+]i transients then reverted to their larger and aster character seen early in the loading process. Similar probe loading sequences were reported in septal neurons [5] and proximal dendrites of neocortical pyramidal cells [6].
Figure 1. Ca2+ buffering by the Ca2+ sensor fura-2 in chromaffin cells.
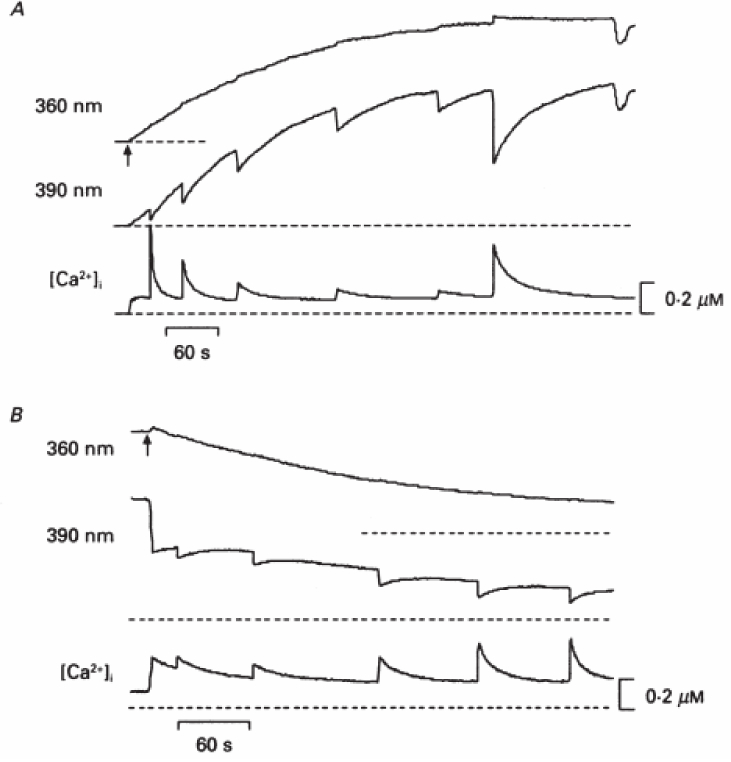
A. In a patch clamped chromaffin cell, the fluorescence of the dye is excited with two different wavelengths nearly simultaneously by rapid switching between excitation filters. The fluorescence at the Ca2+-independent wavelength (360 nm, top trace) gives the time course of fura-2 loading into the cell. Voltage pulses of 50msec from −70 mV to 10mV were applied at irregular intervals during the loading, and dips in the Ca -sensitive wavelength (390 nm, middle trace) reported Ca rises. The final fluorescence change was elicited by a longer voltage pulse of 500 msec. The intracellular free Ca2+ concentration, [Ca2+]i, computed from the ratio of fluorescence elicited by the two wavelengths (bottom trace), illustrates the decrease in amplitude of the [Ca2+]i rise and increase in decay time as [fura-2] in the cell rises to a final concentration of 400 µM. B. After removing the patch electrode containing 400 µM fura-2, a second recording was made from the same cell using a patch pipette containing 50 µM fura-2. The decline in fluorescence at 360 nm tracked the reduction in fura-2 concentration. As the dye concentration fell, the Ca2+ rises evoked by 20msec pulses became larger and the decays became more rapid. Extracellular Ca2+ was 5mM. Reproduced with permission from Ne her and Augustine, 1992 [4].
Quantifying buffering actions
The buffering actions of fluorescent Ca2+ sensors came to light in studies of the role of Ca2+ in exocytosis [4] and excitation-contraction coupling [7]. This work played an essential conceptual role in elucidating how Ca2+ serves as a signal in these processes, and led to the development of quantitative tools for understanding the impact of buffers on Ca2+ fluorescent imaging. The amount of buffering by a Ca2+-binding molecule, B, under a particular set of experimental conditions can be expressed as the buffering capacity, k[1, 4, 7].
| (1) |
K is the ratio of the change in bound Ca2+ to the change in free Ca2+, and Kd is the dissociation constant of B. The binding sites of endogenous Ca2+ buffers saturate as Ca2+ rises [8–11], so k decreases as [Ca2+] rises. But κ provides a useful index of the impact of Ca2+ increments at a given ambient concentration. The experiment illustrated in Fig. 1A used a high concentration of fura-2 (400 µM) in order to demonstrate clearly the buffering action of the dye. For this concentration fura-2 contributes a κ of 889 (with Kd = 200nM and a nominal resting [Ca2+]i of 100nM). Thus, considering only fura-2, the sensor binds 99.9% of entering Ca2+ at rest, leaving only about 0.1% as free ion in solution.
Many proteins and metabolites also bind Ca2+, and these endogenous buffers work together with a sensor in an additive fashion to shape Ca2+ transients. Taking the endogenous and sensor buffering capacities as e and Ks, respectively, it can be shown that when a Ca2+ addition produces a change in total Ca2+ of Δ[Ca2+]Total, free Ca2+ changes by [4]
| (2) |
The two buffers prolong the observed decay time, τobs, by the same factor
| (3) |
where τ0 is the decay time in the absence of any buffer. The effects of the endogenous buffers on a Ca2+ signal, through ke, are biological, while the effects of the sensor, through Ks, are artifacts. Given this additive action of buffers, we can anticipate that a Ca2+ sensor will dominate Ca2+ dynamics as Ks surpasses Ke.
Implications for Ca2+ imaging experiments
The above reasoning indicates that one can assess the impact of the Ca2+ buffering action of a probe by comparing ks and ke. ke values have a range of roughly 50–500 [1]. A value of 100 is fairly typical, although recent work in one particular cell type gave a value of only 20 [12]. According to Eq. 1, with [Ca2+]i = 100 nM fura-2 will have a ks = 20 at a concentration of only 450 nM. Most imaging systems could not detect the fluorescence of a dye at such a low concentration. Even a more typical k value of 100 corresponds to [fura-2] = 2.25 µM, which is on a Ca2+ signal, through ke, are biological, while the effects of the sensor, through ks, are artifacts. Given this additive action of buffers, we can anticipate that a Ca2+ sensor will dominate Ca2+ dynamics as κs surpasses ke The above reasoning indicates that one can assess the impact of the Ca2+ buffering action of a probe by comparing ks and ke. ke values have a range of roughly 50–500 [1]. value of 100 is fairly typical, although recent work in one particular cell type gave a value of only 20 [12]. According to Eq. 1, with [Ca2+]i = 100nM fura-2 will have a ks = 20 at a concentration of only 450nM. Most imaging systems could not detect the fluorescence of a dye at such a low till usually too low to use. Thus, if there is enough sensor to image Ca2+, then there is almost certainly enough to produce major distortions of Ca2+ signals. In hippocampal axons filled with known concentrations by patch loading, extrapolating to zero sensor yielded action potential-induced Ca2+ rises that were 20-fold higher and decays that were 20-fold faster than those seen in fura-2-AM loaded axons [10]. In the absence of a sensor, Ca2+ transients decay in tens of milliseconds in spines, thin dendritic shafts [13], and nerve terminals [10]; in less than 100msec in thick dendrites adjacent to the soma [6]; and perhaps hundreds of milliseconds in cell bodies [5, 14]. Thus, the decay times of seconds often seen in Ca2+ imaging experiments in vivo suggest that probe buffering is a dominant force. The strong buffering action of the probe in typical experiments can have serious consequences. For instance, by dominating Ca2+ dynamics a probe could alter the timing of responses in sequential activations of different cells in a circuit Slowing down Ca2+ changes could mask timing differences and create the appearance of synchrony.
Ca2+ probes can also disrupt Ca2+-dependent cellular signaling and thus alter cellular function. Injecting chelators into the squid giant synaptic terminal reduced release [15], so one should expect Ca2+ probes to have a similar action. GCaMP6m expression in the calyx of Held synaptic terminal decreased synaptic vesicle release and slowed short-term depression [16]. Transgenic mice expressing GCaMP6 had a higher incidence of epileptiform events [17], probably because lower rises in intracellular Ca2+ reduced the activation of Ca2+-activated K+ channels.
There has been a surge of studies using Ca2+ imaging to monitor electrical activity in vivo during behavior [e.g. 18–21]. In many of these studies, there seems to be little indication that the buffering action of the Ca2+ sensor is fully recognized. The Ca2+ sensor is introduced either by bolus injection of the AM-ester form of the dye [22], or by expression of a protein that serves as a genetically-encoded Ca2+ sensor [23, 24]. With both methods the cytosolic concentration of the sensor is almost impossible to control. But by the logic outlined above, if one can see its fluorescence then the sensor is almost certainly having an impact on cellular Ca2 dynamics. In fact, because of the problem of tissue autofluorescence, in order to image Ca2+ in vivo the cytosolic concentration of the probe must be quite high to see over background. One might argue that the fluorescence of a Ca2+ sensor still reports the electrical activity during behavior, and that the distortions are only relevant to mechanistic studies of Ca2+ dynamics. However, differences in the signal amplitudes between cells may simply reflect different levels of probe expression. Brighter cells might appear less active because their higher sensor concentrations suppress Ca2+ changes. Furthermore, two cells with the same level of sensor but different ke values would also appear different in terms of Ca2+ signaling because the probe would buffer a greater fraction of entering Ca2+ in the low-ke cell compared to the high-ke cell.
Concluding remarks and future perspectives
Researchers will be able to use the techniques of in vivo Ca2+ imaging more effectively if they recognize the artifacts caused by Ca2+ sensors. The direct determination of ke and ks is not practical in many kinds of experiments. Nevertheless, researchers can take some measures to address the buffering action of a Ca2+ sensor. Low affinity dyes are weaker buffers, but also produce smaller fractional fluorescence changes for physiological changes in Ca2+. Calibration of the optical system against test samples of probe, along with assessment of tissue autofluorescence will permit probe brightness within a voxel to be converted to obtain a rough measure of probe concentration. The fluorescence of beads implanted in tissue could be compared to the fluorescence of beads in a transparent medium to assess the impact of tissue light scattering on excitation and light collection. In principle, with an estimate of sensor concentration one could use Eq. 1 to calculate Ks from the Kd and a nominal resting free [Ca2+]i. This can be difficult, though, because free [Ca2+]i is often unknown, probe affinities differ between in vitro and in vivo environments, and the fraction of cytosolic volume accessible to the probe is not precisely known. The accumulated impact of these factors could be quite large. Another approach to evaluating the impact of a Ca2+ sensor is based on decay times. The much longer decay times in virtually all the in vivo reports indicate that the sensor is buffering. One can estimate the decay time of an evoked Ca2+ rise by patch clamping a neuron with a known probe concentration. For a rapidly equilibrating dye, the extrapolation to zero dye concentration will give the biologically relevant decay time for a Ca2+ rise, τobs = τ0(1 +ke) from Eq. 3. he prolongation of this decay time under less well defined loading conditions would then provide an indication of the impact of the sensor, including its increment to k from kd. As in vivo Ca2+ imaging advances, attention to these issues will enhance the power of a technique that is offering unique and remarkable insights into the function of the nervous system.
Outstanding Questions.
How do Ca2+ sensors alter the patterns of observed neural activity?
How much do Ca2+ sensors alter Ca2+ signaling functions of a neuron during sensory input processing and the generation of behavior?
Can technological advances mitigate the artifacts due to Ca2+ buffering by Ca2+ sensors?
Highlights.
Although the buffering actions of Ca2+ sensors are widely known, their magnitude and impact are often not fully appreciated. This buffering action will reduce the amplitude of a change in intracellular Ca2+ and prolong the decay.
The magnitude of this buffering action is reflected in a sensor’s buffering capacity, which depends on the concentration and affinity of the sensor The buffering capacity of a sensor exceeds the buffering capacity of endogenous Ca2+ buffers (Ca2+-binding proteins and metabolites) under the conditions that pertain during most Ca2+ imaging experiments. As a result, the Ca2+ sensor will have a dominant effect on Ca2+ signals.
Recognizing the impact of the Ca2+ sensors used for Ca2+ imaging of intracellular Ca2+ will allow investigators to use these tools more effectively.
Acknowledgements
We thank Dr. Chih-Tien Wang for helpful comments on the manuscript. Supported by NIH grant NS103206.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Neher E (1995) The use of fura-2 for estimating Ca buffers and Ca fluxes. Neuropharmacology 34, 1423–1442. [DOI] [PubMed] [Google Scholar]
- 2.Grienberger C and Konnerth A (2012) Imaging calcium in neurons. Neuron 73 (5), 862–85. [DOI] [PubMed] [Google Scholar]
- 3.Higley MJ and Sabatini BL (2008) Calcium signaling in dendrites and spines: practical and functional considerations. Neuron 59 (6), 902–13. [DOI] [PubMed] [Google Scholar]
- 4.Neher E and Augustine GJ (1992) Calcium gradients and buffers in bovine chromaffin cells. Journal of Physiology 450, 273–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneggenburger R et al. (1993) Fractional contribution of calcium to the cation current through glutamate receptor channels. Neuron 11 (1), 133–43. [DOI] [PubMed] [Google Scholar]
- 6.Helmchen F et al. (1996) Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J 70 (2), 1069–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berlin JR et al. (1994) Intrinsic cytosolic calcium buffering properties of single rat cardiac myocytes. Biophys J 67 (4), 1775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Z and Neher E (1993) Mobile and immobile calcium buffers in bovine adrenal chromaffin cells. J Physiol 469, 245–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu T et al. (1997) Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells Biophys J 73 (1), 532–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jackson MB and Redman SJ (2003) Calcium dynamics, buffering, and buffer saturation in the boutons of the dentate granule-cell axons in the hi l us. Journal of Neuroscience 23, 1612–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMahon SM et al. (2016) Multiple cytosolic calcium buffers in posterior pituitary nerve terminals. J Gen Physiol 147 (3), 243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin KH et al. (2017) Dynamics of volume-averaged intracellular Ca (2+) in a rat CNS nerve terminal during single and repetitive voltage-clamp depolarizations. J Physiol 595 (10), 3219–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabatini BL et al. (2002) The life cycle of Ca (2+) ions in dendritic spines. Neuron 33 (3), 439–52. [DOI] [PubMed] [Google Scholar]
- 14.Tatsumi H and Katayama Y (1993) Regulation of the intracellular free calcium concentration in acutely dissociated neurons from at nucleus basalis. J Physiol 464, 165–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adler EM et al. (1991) Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neuro sci 11 (6), 1496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh M et al. (2018) Presynaptic GCaMP expression decreases vesicle release probability at the Calyx of Held. Synapse [DOI] [PMC free article] [PubMed]
- 17.Steinmetz NA et al. (2017) Aberrant Cortical Activity in Multiple GCaMP6-Expressing Transgenic Mouse Lines. E Neuro 4 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohki K et al. (2005) Functional imaging with cellular resolution reveals precise micro-architecture in visual cortex. Nature 433 (7026), 597–603. [DOI] [PubMed] [Google Scholar]
- 19.Goncalves JT et al. (2013) Circuit level defects in the developing neocortex of Fragile X mice. Nat Neuro sci 16 (7), 903–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sofroniew NJ et al. (2016) A large field of view two-photon meso scope with subcellular resolution for in vivo imaging Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stirman JN et al. (2016) Wide field-of-view, multi-region, two-photon imaging of neuronal activity in the mammalian brain. Nat Bio technol 34 (8), 857–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stosiek C et al. (2003) In vivo two-photon calcium imaging of neuronal networks. Proc Natl A cad Sci U S A 100 (12), 7319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knopfel T et al. (2010) Toward the second generation of optogenetic tools. J Neuro sci 30 (45), 14998–5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akerboom J et al. (2013) Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neuro sci 6, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]