

Abstract
Treatment of 1-(2-bromoarylmethyl)-3,4-dihydroisoquinolines with oxalyl chloride and triethylamine gave 1-(2-bromophenyl)-5,6-dihydropyrrolo[2,1-a]isoquinoline-2,3-dione derivatives, for example, 1-(2-bromophenyl)-5,6-dihydro-8,9-dimethoxypyrrolo[2,1-a]isoquinoline-2,3-dione. Radical cyclisation of these derivatives with tributyltin hydride and 1,1ʹ-azobis(cyclohexanecarbonitrile) afforded telisatin A, telisatin B and lettowianthine.
Keywords: Alkaloid, Dioxoaporphine, Isoquinoline, Radical cyclisation, Synthesis
Introduction
The telisatin-type aporphine alkaloids form a very small sub-group of the aporphine alkaloids in which N-6 and C-7 are fused to an oxalyl function. To date only five members of this type of aporphine alkaloids have been found to occur in Nature. These are telisatin A (1) and telisatin B (2) from Telitoxicum peruvianum Moldenke (Menispermaceae) [1], lettowianthine (3) and 11-methoxy-lettowianthine (4) from Lettowianthus stellatus Diels (Annonaceae) [2], and laurodionine (5) from Phoebe formosana Hayata (Lauraceae) [3]. Annonbraine, isolated from Annona glaba L (Annonaceae), was also assigned the same structure as lettowianthine (3), although there was a big difference in the melting points of the two alkaloids [4]. The structure of telisatin A was elucidated by comparison of spectral data and physical properties with a synthetic compound obtained by Saa and Cava [5], Castedo et al. [6] and Saa et al. [7]. The structures of the remaining alkaloids were assigned based on spectral data analysis.
Figure 1.
Structures of telisatin A (1), telisatin B (2), lettowianthine (annonbraine) (3), 11-methoxylettowianthine (4) and laurodionine (5).
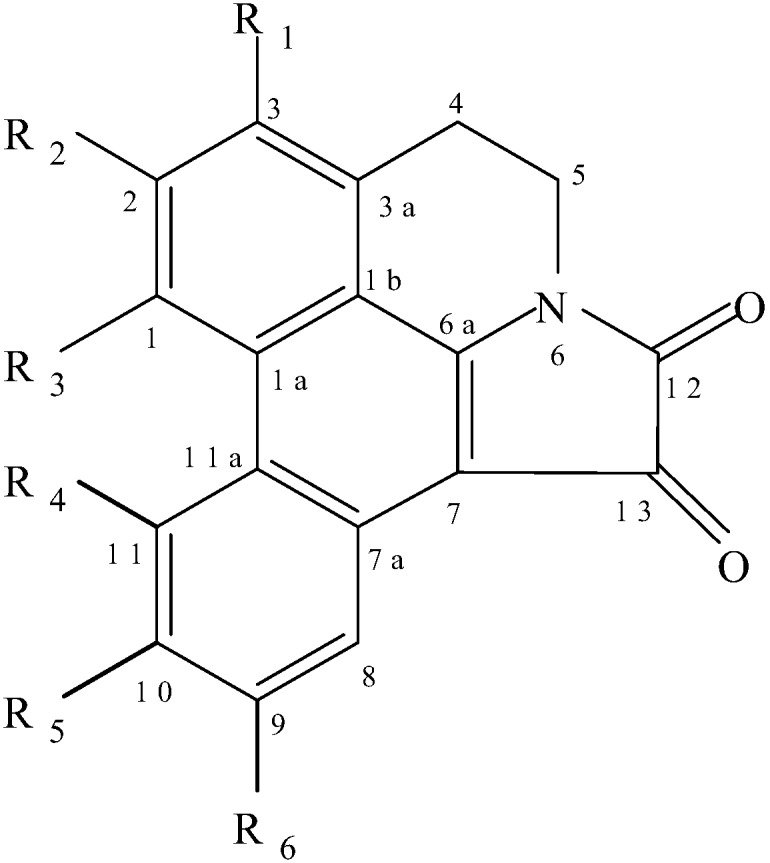
-
1 :
R1 = R4 = R5 = R6 = H, R2 = R3 = OCH3 (telisatin A)
-
2 :
R1 = R2 = R3 = OCH3, R4 = R5 =R6 = H (telisatin B)
-
3 :
R1 = R4 = R5 = R6 = H, R2 + R3 = OCH2O (lettowianthine, annonbraine)
-
4 :
R1 = R5 = R6 = H, R2 + R3 = OCH2O, R4 = OCH3 (11-methoxylettowianthine)
-
5 :
R1 = R4 = H, R2 = R6 = OH, R3 = R5 = OCH3 (laurodionine)
At present there are only two total syntheses of telisatin A reported by Castedo et al. [6] and by Saa et al. [7] The first method involved photochemical cyclisation in reasonably good yield (60%) of a very dilute (0.001 M) solution of 1-(2-bromophenyl)-5,6-dihydro-8,9-dimethoxypyrrolo[2,1-a]isoquinoline-2,3-dione (10a) [6]. This method has obvious inherent limitations with regards to its scalability and convenience. The second method, based on benzyne cycloaddition, gave a low yield (10%) [7]. A partial synthesis reported by Saa and Cava involved acylation of 6a,7-dehydronuciferine with oxalyl chloride [5]. Since 6a,7-dehydroaporphines themselves are not readily accessible, this method therefore lacks generality and convenience.
Results and Discussion
We would like to report herein an extension of the first method described by Castedo et al. [6], shown in Scheme 1. Amides 8a-8c, obtained by conventional methods, were converted by a Bischler–Napieralski reaction to dihydroisoquinolines 9a-9c. We found that 1-(2-bromophenyl)-5,6-dihydropyrrolo[2,1-a]-isoquinoline-2,3-dione derivatives 10a-10c could be more conveniently prepared by the reaction of dihydroisoquinolines 9a-9c with oxalyl chloride in the presence of triethylamine with straightforward workup [8]. The antiplatelet activity of such 1-aryl-5,6-dihydropyrrolo[2,1-a]-isoquinoline-2,3-dione derivatives has been reported [9]. To overcome the limitations of photochemical cyclisation under extreme dilution and based on previous reports on the radical cyclisation of halostilbenes to phenanthrenes [10,11,12,13,14], solutions (0.025 M) of 1-(2-bromophenyl)-5,6-dihydropyrrolo[2,1-a]isoquinoline-2,3-dione derivatives 10a-10c were treated with tributyltin hydride in the presence of 1,1ʹ-azobis(cyclohexanecarbonitrile) (ACCN) to give the corresponding telisatin-type alkaloids in 30-34% yields.
Scheme 1.
Syntheses of telisatin A (1), telisatin B (2) and lettowianthine (3).

Reaction conditions: A) 10 % NaHCO3/ chloroform; B) POCl3/ benzene; C) oxalyl chloride, triethylamine/ chloroform; D) Bu3SnH, ACCN/ dry toluene.
Fortunately, in all cases, it was possible to isolate the pure alkaloids from the crude reaction mixtures by simple crystallization from ethanol. Silica gel chromatography of the residues from the filtrates afforded only minute quantities of the pure alkaloids and was therefore not pursued any further. Comparison of the yields from radical cyclisation using ACCN with those obtained using 2,2ʹ-azobis(isobutyronitrile)(AIBN) was not possible since AIBN is no longer commercially available in Thailand. The spectral data of synthetic telisatin A (1), telisatin B (2) and lettowianthine (3) were in good agreement to those reported for the natural alkaloids.
Conclusions
We have developed an easy and convenient synthesis of the telisatin-type alkaloids. Further applications of the current synthesis to the remaining telisatin-type alkaloids are in progress.
Experimental
General
Melting points were determined on a Stuart Scientific SMP 2 melting point apparatus and are uncorrected. Infrared spectra were recorded on CH2Cl2-films with a Perkin Elmer Spectrum GX FT-IR spectrophotometer. Ultraviolet spectra were recorded on methanol solutions with a Hitachi U-3300 spectrophotometer. 1H- and 13C-NMR spectra were recorded for deuterochloroform solutions at 300 MHz for 1H and 75 MHz for 13C with a Bruker AVANCE 300 spectrometer. Tetramethylsilane was used as the internal standard. High Resolution Mass spectra were recorded with a Bruker Daltonics MicrOTOF mass spectrometer.
N-(3,4-Dimethoxyphenethyl)-2-(2-bromophenyl)acetamide (8a). A mixture of 2-bromophenylacetic acid (15.0 g, 0.07 mol) and thionyl chloride (20.8 g) in benzene (50 mL) was refluxed for 1 h. Removal of the solvent under vacuum gave 2-bromophenylacetyl chloride (7) which was dissolved in ethanol-free chloroform (50 mL) and added to a mixture of 3,4-dimethoxyphenethylamine (6a, 12.7 g, 0.07 mol) in chloroform (100 mL) and 10% sodium hydrogen carbonate (100 mL). The mixture was then stirred for 3 h and the chloroform layer was washed with water (2 × 100 mL), 10% hydrochloric acid (3 × 50 mL), water (100 mL), then dried over anhydrous sodium sulfate. Removal of the solvent under vacuum gave a residue which was recrystallized from ethanol to give amide 8a as a pale yellow solid (22.0 g, 88.1%); m.p. 131-132 °C (Lit. [15] m.p. 127-129 °C); 1H-NMR: δ 7.54 (1H, d, J = 7.8 Hz, Ar-H); 7.30-7.22 (2H, m, Ar-H); 7.18-7.09 (1H, m, Ar-H); 6.72 (1H, d, J = 8.1 Hz, Ar-H); 6.63 (1H, d, J = 1.9 Hz, Ar-H); 6.60 (1H, dd, J = 8.1, 1.9 Hz, Ar-H); 5.58 (1H, s, NH); 3.85 (3H, s, OCH3); 3.82 (3H, s, OCH3); 3.66 (2H, s, CH2); 3.47 (2H, apparent q, J = 6.8 Hz, CH2); 2.71 (2H, apparent t, J = 7.0 Hz, CH2); 13C-NMR: δ 169.48(C), 148.98(C), 147.60(C), 134.79(C), 133.03(CH), 131.63(CH), 131.10(C), 129.04(CH), 127.92(CH), 124.94(C), 120.58(CH), 111.79(CH), 111.32(CH), 55.91(OCH3), 55.82 (OCH3), 43.99(CH2), 40.81(CH2), 34.99(CH2).
N-(2,3,4-Trimethoxyphenethyl)-2-(2-bromophenyl)acetamide (8b). In a similar manner, 8b was obtained in 78.8% yield as a pale yellow solid from ethanol; m.p. 92-93 °C; 1H-NMR: δ 7.57 (1H, d, J = 8.3 Hz, Ar-H); 7.33-7.25 (2H, m, Ar-H); 7.19-7.12 (1H, m, Ar-H); 6.72 (1H, d, J = 8.5 Hz, Ar-H); 6.54 (1H, d, J = 8.5 Hz, Ar-H); 5.83 (1H, br s, NH); 3.88 (3H, s, OCH3); 3.83 (3H, s, OCH3); 3.79 (3H, s, OCH3); 3.67 (2H, s, CH2); 3.43 (2H, apparent q, J = 6.4 Hz, CH2); 2.70 (2H, apparent t, J = 6.7 Hz, CH2); 13C-NMR: δ 169.58(C), 152.59(C), 151.74(C), 142.19(C), 134.91(C), 133.05(CH), 131.78(CH), 128.97(CH), 127.89(CH), 125.10(C), 124.69(C), 124.46(CH), 107.42(CH), 60.92(OCH3), 60.75(OCH3), 56.01(OCH3), 44.05 (CH2), 40.91(CH2), 29.47(CH2).
N-(3,4-Methylenedioxyphenethyl)-2-(2-bromophenyl)acetamide (8c). In a similar manner, 8c was obtained in 82.5% yield as a pale yellow solid from ethanol; m.p. 124-126 °C (Lit. [16] m.p. 128-130 °C); 1H-NMR: δ 7.56 (1H, d, J = 7.8 Hz, Ar-H); 7.33-7.25 (2H, m, Ar-H); 7.19-7.11 (1H, m, Ar-H); 6.65 (1H, d, J = 7.9 Hz, Ar-H); 6.55 (1H, d, J = 1.6 Hz, Ar-H); 6.48 (1H, dd, J = 7.9, 1.6 Hz, Ar-H); 5.91 (2H, s, OCH2O); 5.48 (1H, br s, NH); 3.66 (2H, s, CH2); 3.42 (2H, apparent q, J = 6.7 Hz, CH2); 2.65 (2H, apparent t, J = 6.7 Hz, CH2); 13C-NMR: δ 169.44(C), 147.72(C), 146.11(C), 134.80(C), 133.13(CH), 132.35(C), 131.67(CH), 129.09(CH), 127.97(CH), 124.98(C), 121.57(CH), 109.04(CH), 108.30(CH), 100.85(CH2), 44.04(CH2), 40.87(CH2), 35.15(CH2).
1-(2-Bromobenzyl)-3,4-dihydro-6,7-dimethoxyisoquinoline (9a). A solution of 8a (5.5 g, 14.6 mmol) and phosphorus oxychloride (17.0 g) in benzene (60 mL) was refluxed for 3 h. The excess reagent and solvent were removed under vacuum. The residue was shaken with chloroform (100 mL) and dilute ammonium hydroxide (100 mL). The chloroform layer was washed with water (100 mL), then dried over anhydrous sodium carbonate. Removal of the solvent under vacuum gave dihydroisoquinoline 9a as a pale yellow solid (3.9 g, 75.8%) from ethyl acetate-hexane; m.p. 95-96 °C (Lit. [15] m.p. 93-95 °C). It was found to be unstable and was immediately used in the next step without further purification. 1H-NMR: δ 7.56 (1H, dd, J = 7.9, 1.3 Hz, Ar-H); 7.27 (1H, dd, J = 7.6, 1.7 Hz, Ar-H); 7.18 (1H, dt, J = 7.6, 1.3 Hz, Ar-H); 7.05 (1H, dt, J = 7.6, 1.7 Hz, Ar-H); 6.91 (1H, s, Ar-H); 6.67 (1H, s, Ar-H); 4.20 (2H, s, Ar-CH2); 3.88 (3H, s, OCH3); 3.79 (3H, s, OCH3); 3.73 (2H, t, J = 7.6 Hz, CH2); 2.67 (2H, t, J = 7.6 Hz, CH2); 13C-NMR δ: 165.08(C), 150.76(C), 147.43(C), 137.66(C), 132.78(CH), 131.59(C), 130.21 (CH), 128.18(CH), 127.62(CH), 124.54(C), 121.41(C), 110.25(CH), 109.15(CH), 56.16(OCH3), 55.92(OCH3), 47.29(CH2), 42.53(CH2), 25.73(CH2).
1-(2-Bromobenzyl)-3,4-dihydro-5,6,7-trimethoxyisoquinoline (9b). In a similar manner, 9b was obtained in almost quantitative yield as a yellow viscous oil. 1H-NMR: δ 7.55 (1H, dd, J = 7.9, 1.2 Hz, Ar-H); 7.30-7.26 (1H, m, Ar-H); 7.20-7.15 (1H, m, Ar-H); 7.09-7.01 (1H, m, Ar-H); 6.78 (1H, s, H−8); 4.19 (2H, s, Ar−CH2); 3.88 (3H, s, OCH3); 3.83 (3H, s, OCH3); 3.78 (3H, s, OCH3); 3.71 (2H, t, J = 7.6 Hz, CH2); 2.67 (2H, t, J = 7.6 Hz, CH2); 13C-NMR: δ 164.68(C), 151.69(C), 149.88(C), 144.17 (C), 137.63(C), 132.77(CH), 130.23(CH), 128.19(CH), 127.61(CH), 124.52(C), 124.46(C), 124.09(C), 105.63(CH), 60.88(OCH3), 60.83(OCH3), 56.19(OCH3), 47.09 (CH2), 42.58(CH2), 18.99(CH2).
1-(2-Bromobenzyl)-3,4-dihydro-6,7-methylenedioxyisoquinoline (9c). In a similar manner, 9c was obtained in 42.1% yield from ethanol as a pale yellow solid; m.p. 122-123 °C (Lit. [16] m.p. 121-123 °C); 1H-NMR: δ 7.52 (1H, dd, J = 7.8, 1.0 Hz, Ar-H); 7.22-7.12 (2H, m, Ar-H); 7.06- 6.98 (1H, m, Ar-H); 6.89 (1H, s, Ar-H); 6.61 (1H, s, Ar-H); 5.86 (2H, s, OCH2O); 4.09 (2H, s, Ar-CH2); 3.65 (2H, t, J = 7.6 Hz, CH2); 2.60 (2H, t, J = 7.6 Hz, CH2); 13C-NMR: δ 164.65(C), 149.08(C), 146.44(C), 137.55(C), 133.38(C), 132.79 (CH), 130.30(CH), 128.14(CH), 127.50(CH), 124.79(C), 122.77(C), 107.98(CH), 106.02(CH), 101.31(CH2), 47.09(CH2), 42.55(CH2), 26.33(CH2).
1-(2-Bromophenyl)-5,6-dihydro-8,9-dimethoxypyrrolo[2,1-a]isoquinoline-2,3-dione (10a). Oxalyl chloride (0.2 mL) was added dropwise to a stirred solution of 9a (359 mg, 1 mmol), triethylamine (0.3 mL) in chloroform (10 mL) at room temperature. Stirring was continued for 3 h. Chloroform (20 mL) was added and the chloroform layer was washed with 5% hydrochloric acid (4 × 50 mL), water (50 mL), then dried over anhydrous sodium sulfate. Removal of the solvent under vacuum gave a residue which was recrystallized from ethanol to give 10a as red prisms (247.8 mg, 60.0%); m.p. 195-196 °C. (lit. [6] m.p. 176-178 °C); UV λmax (MeOH) nm (log ε): 203 (4.36), 226sh (4.12), 262 (3.76), 322 (3.65). IR (CH2Cl2-film) νmax cm-1: 2937, 2843, 1744, 1699, 1594, 1575, 1515, 1472, 1428, 1398, 1337, 1312, 1291, 1270, 1225, 1187, 1101, 1034, 987, 865, 798, 735, 683; 1H-NMR: δ 7.70 (1H, dd, J = 8.0, 0.9 Hz, Ar-H); 7.44-7.37 (1H, m, Ar-H); 7.34-7.29 (1H, m, Ar-H); 7.29-7.22 (1H, m, Ar-H); 6.77 (1H, s, Ar-H); 6.66 (1H, s, Ar-H); 3.95 (3H, s, OCH3); 3.89 (2H, t, J = 6.3 Hz, CH2); 3.29 (3H, s, OCH3); 3.10 (2H, t, J = 6.3 Hz, CH2); 13C-NMR: δ 181.59(C), 158.50(C), 158.35(C), 153.80(C), 148.14(C), 133.17(CH), 133.09(C), 132.83(CH), 132.41(C), 129.96(CH), 128.06(CH), 125.82(C), 116.68(C), 111.29(CH), 107.80(C), 56.27(OCH3), 55.17(OCH3), 36.37(CH2), 28.37(CH2). HRMS (ESI-TOF) calcd for C20H16BrNO4 ([M+H+]) = 414.0335, Found 414.0438.
1-(2-Bromophenyl)-5,6-dihydro-7,8,9-trimethoxypyrrolo[2,1-a]isoquinoline-2,3-dione (10b). In a similar manner, 10b was obtained as a deep red solid in 68.2% yield after chromatography over alumina using dichloromethane as eluent; m.p. 69-70 °C; UV (MeOH) λ max nm (log ε): 203 (4.66), 226sh (4.38), 258 (3.95), 332 (3.93); IR (CH2Cl2-film) νmax cm-1: 2939, 2837, 1746, 1702, 1592, 1576, 1467, 1425, 1397, 1342, 1298, 1248, 1182, 1109, 1024, 986, 939, 914, 845, 752; 1H-NMR: δ 7.71 (1H, dd, J = 8.0, 0.9 Hz, Ar-H); 7.44-7.35 (1H, m, Ar-H); 7.32-7.23 (2H, m, Ar-H); 6.53 (1H, s, Ar-H); 3.94 (3H, s, OCH3); 3.88 (3H, s, OCH3); 3.93-3.73 (2H, m, CH2); 3.27 (3H, s, OCH3); 3.20-3.00 (2H, m, CH2); 13C-NMR: δ 182.05(C), 158.07(C), 152.24(C), 150.56(C), 147.02(C), 133.21(CH), 132.70(CH), 132.25(C), 130.03(CH), 128.08(CH), 125.77(C), 125.48(C), 119.38(C), 108.64(C), 108.53(CH), 105.68(C), 61.12(OCH3), 61.08(OCH3), 55.21(OCH3), 36.19(CH2), 21.67(CH2). HRMS (ESI-TOF) calcd for C21H18BrNO5 ([M+H+]) = 444.0441, Found 444.0519.
1-(2-Bromophenyl)-5,6-dihydro-8,9-methylenedioxypyrrolo[2,1-a]isoquinoline-2,3-dione (10c). In a similar manner, 10c was obtained in 47.7% yield from ethanol as a deep red prisms; m.p. 226-227 °C; UV (MeOH) λ max nm (log ε): 203 (4.64), 236sh (4.18), 261sh (3.88), 284 (3.77), 320 (3.67), 388 (3.64); IR (CH2Cl2-film) νmax cm-1: 3056, 2906, 1744, 1698, 1608, 1568, 1505, 1467, 1403, 1378, 1338, 1316, 1286, 1249, 1181, 1036, 938, 868, 748, 736; 1H-NMR: δ 7.71-7.66 (1H, m, Ar-H); 7.42-7.36 (1H, m, Ar-H); 7.31-7.23 (2H, m, Ar-H); 6.79 (1H, s, Ar-H); 6.56 (1H, s, Ar-H); 6.00 (2H, AB q, J = 1.1 Hz, OCH2O); 3.94-3.77 (2H, m, CH2); 3.07 (2H, t, J = 6.3 Hz, CH2); 13C-NMR: δ 181.94(C), 158.10(C), 157.94(C), 152.47(C), 147.37(C), 135.17(C), 133.43(CH), 132.43(CH), 131.68(C), 130.13(CH), 128.14(CH), 125.35(C), 118.23(C), 109.28(CH), 108.50(CH), 108.23(C), 102.25(CH2), 36.20(CH2), 29.24(CH2). HRMS (ESI-TOF) calcd for C19H12BrNO4 ([M+H+]) = 398.0022, Found 397.9895.
Telisatin A (1). A solution of 1,1ʹ-azobis(cyclohexanecarbonitrile) (245.0 mg, 1.0 mmol) and tributyltin hydride (1.2 g, 4.0 mmol) in toluene (20 mL) was added dropwise in four equal portions over 3 h to a refluxing solution of 10a (413.0 mg, 1.0 mmol) in toluene (20 mL) and the resulting mixture was then refluxed for another 8 h. The solvent was then removed under vacuum and the residue was dissolved in acetonitrile (40 mL) and washed with hexane (2 × 30 mL), then dried over anhydrous sodium sulfate. Removal of the solvent gave a brown viscous oil (0.4 g) which was recrystallized with ethanol to give telisatin A (1) as red prisms (109.9 mg, 33.0%); m.p. 234-235 °C (Lit. [1] m.p. 238-239 °C); UV (MeOH) λ max nm (log ε): 207 (4.03), 257 (4.26), 284sh (3.60), 322 (3.70), 336 (3.80), 352sh (3.56); IR (CH2Cl2-film) νmax cm-1: 2925, 1748, 1701, 1605, 1584, 1531, 1462, 1423, 1386, 1306, 1261, 1195, 1149, 1131, 1112, 1037, 969, 924, 802, 759. 1H-NMR δ: 9.41 (1H, br d, J = 8.5 Hz, H−11); 8.63 (1H, dd, J = 8.0, 1.5 Hz, H−8); 7.67-7.60 (1H, m, H−9); 7.54-7.46 (1H, m, H−10); 7.17 (1H, s, H−3); 4.10 (3H, s, OCH3); 3.97 (2H, t, J = 6.5 Hz, CH2); 3.95 (3H, s, OCH3); 3.35 (2H, t, J = 6.5 Hz, CH2); 13C-NMR: δ 179.98(C), 160.34(C), 157.15(C), 153.36(C), 146.65(C), 130.75(C), 129.35(C), 129.21(CH), 128.33(CH), 127.56(C), 125.87(C), 125.62(CH), 123.76(CH), 112.29(CH), 112.18(C), 103.17(C), 59.99(OCH3), 56.62(OCH3), 36.53(CH2), 27.68 (CH2). HRMS (ESI-TOF) calcd for C20H15NO4 ([M+H+]) = 334.1074, Found 334.1125.
Telisatin B (2). In a similar manner, telisatin B (2) was obtained as deep red prisms (30.0%) from ethanol; m.p. 218-219 °C (Lit.[1] m.p. 221-222 °C); UV (MeOH) λ max nm (log ε): 203 (4.33), 223sh (4.26), 257 (4.59), 318sh (3.99), 329 (4.07); IR (CH2Cl2-film) νmax cm-1: 2942, 2864, 1749, 1716, 1702, 1619, 1607, 1579, 1527, 1515, 1452, 1406, 1389, 1323, 1146, 1125, 1071, 1033, 973, 814, 757; 1H-NMR: δ 9.37 (1H, br d, J = 8.5 Hz, H−11), 8.60 (1H, dd, J = 8.0, 1.3 Hz, H−8), 7.64-7.56 (1H, m, H−9), 7.55-7.45 (1H, m, H−10), 4.14 (3H, s, OCH3), 4.03 (3H, s, OCH3), 4.01 (3H, s, OCH3), 3.91 (2H, t, J = 6.5 Hz, CH2), 3.33 (2H, t, J = 6.5 Hz, CH2); 13C-NMR: δ 180.69(C), 159.86(C), 152.58(C), 152.16(C), 152.03(C), 150.16(C), 128.71(CH), 127.53(CH), 126.63(C), 126.38(C), 126.11(C), 125.78(CH), 123.66(CH), 121.18(C), 114.14(C), 104.17(C), 61.47(OCH3), 61.30(OCH3), 60.48 (OCH3), 36.14(CH2), 21.21(CH2); HRMS (ESI-TOF) calcd for C21H17NO5 ([M+H+]) = 364.1179, Found 364.1231.
Lettowianthine (3). In a similar manner, lettowianthine (3) was obtained as red prisms (34.0%); m.p. 294-295 °C (dec.) (Lit. [2] m.p. 314-317 °C (dec.); Lit.[4] m.p. 265-267 °C); UV (MeOH) λ max nm (log ε): 203 (4.37), 212sh (4.29), 247sh (4.07), 257sh (4.04), 287 (3.64), 335 (3.51), 353 (3.28). IR (CH2Cl2-film) νmax cm-1: 2923, 2093, 1737, 1695, 1622, 1610, 1581, 1530, 1506, 1450, 1417, 1301, 1253, 1222, 1176, 1151, 1122, 1050, 927, 867, 749; 1H-NMR: δ 8.80 (1H, br d, J = 8.6 Hz, H−11), 8.55 (1H, br d, J = 7.6 Hz, H−8), 7.61 (1H, t, J = 6.9 Hz, H−9), 7.48 (1H, t, J = 7.2 Hz, H−10), 7.09 (1H, s, H−3), 6.35 (2H, s, OCH2O), 3.93 (2H, t, J = 6.2 Hz, CH2), 3.29 (2H, t, J = 6.2 Hz, CH2); 13C-NMR δ: 179.92(C), 160.33(C), 153.68(C), 151.55(C), 143.08(C), 129.61(C), 129.33(CH), 127.62 (CH), 126.83(C), 125.35(CH), 124.45(C), 123.60(CH), 119.90(C), 112.46(C), 109.23(CH), 103.10 (C), 102.35(CH2), 36.66(CH2), 27.51(CH2); HRMS (ESI-TOF) calcd for C19H11NO4 ([M+H+]) = 318.0761, Found 318.0666.
Footnotes
Sample Availability: All stable products reported in this paper are available from the authors.
References
- 1.Menachery M.D., Blake G.W., Gourley R.C., Freyer A. Telisatin A, telisatin B, and telitoxinone, three nerw aporphinoids from Telitoxicum peruvianum. J. Nat. Prod. 1995;58:1945–1949. doi: 10.1021/np50126a025. [DOI] [Google Scholar]
- 2.Nkunya M.H.H., Jonker S.A., Makangara J.J., Waibel R., Achenbach H. Aporphinoid alkaloids and other constituents from Lettowianthus stellatus. Phytochemistry. 2000;53:1067–1073. doi: 10.1016/S0031-9422(00)00012-1. [DOI] [PubMed] [Google Scholar]
- 3.Chen C.-C., Huang Y.-L., Lee S-S., Ou J.-C. Laurodionine, a new oxalyl-fused aporphine alkaloid from Phoebe formasana. J. Nat. Prod. 1997;60:826–827. doi: 10.1021/np970147c. [DOI] [Google Scholar]
- 4.Chang F.-R., Chen C.-Y., Hsieh T.-J., Cho C-P., Wu Y.-C. Chemical constituents from Annona glabra III. J. Chin. Chem. Soc. 2000;47:913–920. [Google Scholar]
- 5.Saa J.M., Cava M.P. Dehydroaporphines. An acylation study. J. Org. Chem. 1978;43:1096–1099. doi: 10.1021/jo00400a016. [DOI] [Google Scholar]
- 6.Castedo L., Saa C., Saa J.M., Suau R. Synthesis of oxoaporphines. An unusual photocyclization-photoreduction of 2,3-diaryl-Δ2-pyrroline-4,5-diones. J. Org. Chem. 1982;47:513–517. doi: 10.1021/jo00342a028. [DOI] [Google Scholar]
- 7.Saa C., Guitian E., Castedo L., Suau R., Saa J.M. A regioselective entry to 13-substituted 8-oxoprotoberberines. Total synthesis of (±)-corydaline. J. Org. Chem. 1986;51:2781–2784. doi: 10.1021/jo00364a030. [DOI] [Google Scholar]
- 8.Mikhailovskii A.G. Reactions of 2,3-dioxopyrrolo[2,1-a]isoquinolines with sodium borohydride and the properties of its products. Chem. Heterocycl. Comp. 1996;32:590–595. doi: 10.1007/BF01164792. [DOI] [Google Scholar]
- 9.Kuo R.-Y., Wu C.-C., Chang F.-R., Yeh J.-L., Chen I.-J., Wu Y.-C. Antiplatelet activity of synthetic pyrrolo-benzylisoquinolines. Bioorg. Med. Chem. Lett. 2003;13:821–823. doi: 10.1016/S0960-894X(03)00003-9. [DOI] [PubMed] [Google Scholar]
- 10.Harrowven D.C., Nunn M.I.T., Fenwick D. R. Radical cyclisations to arenes for the synthesis of phenanthrenes. Tetrahedron Lett. 2002;43:3185–3187. doi: 10.1016/S0040-4039(02)00505-1. [DOI] [Google Scholar]
- 11.Harrowven D.C., Nunn M.I.T., Fenwick D. R. [5]Helicenes by iterative radical cyclisations to arenes. Tetrahedron Lett. 2002;43:3189–3191. doi: 10.1016/S0040-4039(02)00506-3. [DOI] [Google Scholar]
- 12.Harrowven D.C., Nunn M.I.T., Fenwick D.R. [5]Helicenes by tandem radical cyclisation. Tetrahedron Lett. 2002;43:7345–7347. doi: 10.1016/S0040-4039(02)01720-3. [DOI] [Google Scholar]
- 13.Wang Y.-C., Lin C.-H., Chen C.-M., Liou J.-P. A concise synthesis of denbinobin. Tetrahedron Lett. 2005;46:8103–8104. doi: 10.1016/j.tetlet.2005.09.118. [DOI] [Google Scholar]
- 14.Abbate S., Bazzini C., Caronna T., Fontana F., Gambarotti C., Gangemi F., Longhi G., Mele A., Sora I.N., Panzeri W. Monoaza[5]helicenes. Part 2: synthesis, characterisation and theoretical calculations. Tetrahedron. 2006;62:139–148. doi: 10.1016/j.tet.2005.09.132. [DOI] [Google Scholar]
- 15.Niimi J. Syntheses of biscoclaurine alkaloids. IV. Synthesis of dl-2,4ʹ-bis(2-methyl-6,7-dimethoxy-1,2,3,4-tetrahydro-1-isoquinolylmethyl)diphenyl ether. Yakugaku Zasshi. 1960;80:123–126. [Google Scholar]
- 16.Nimgirawath S., Taylor W.C. Photochemical synthesis of 8H-Benzo[g]-1,3-benzodioxolo-[6, 5,4-de]quinolin-8-one (Liriodenine) via 7-methyl-6,7-dihydro-5H-benzo[g]-1,3-benzodioxolo- [6,5,4-de]quinoline. Aust. J. Chem. 1983;36:1061–1065. doi: 10.1071/CH9831061. [DOI] [Google Scholar]