

Abstract
Secreted proteins are important metabolic regulators in both healthy and disease states. Here, we sought to investigate the mechanism by which the secreted protein complement 1q-like-3 (C1ql3) regulates insulin secretion from pancreatic β-cells, a key process affecting whole-body glucose metabolism. We found that C1ql3 predominantly inhibits exendin-4– and cAMP-stimulated insulin secretion from mouse and human islets. However, to a lesser extent, C1ql3 also reduced insulin secretion in response to KCl, the potassium channel blocker tolbutamide, and high glucose. Strikingly, C1ql3 did not affect insulin secretion stimulated by fatty acids, amino acids, or mitochondrial metabolites, either at low or submaximal glucose concentrations. Additionally, C1ql3 inhibited glucose-stimulated cAMP levels, and insulin secretion stimulated by exchange protein directly activated by cAMP-2 and protein kinase A. These results suggest that C1ql3 inhibits insulin secretion primarily by regulating cAMP signaling. The cell adhesion G protein–coupled receptor, brain angiogenesis inhibitor-3 (BAI3), is a C1ql3 receptor and is expressed in β-cells and in mouse and human islets, but its function in β-cells remained unknown. We found that siRNA-mediated Bai3 knockdown in INS1(832/13) cells increased glucose-stimulated insulin secretion. Furthermore, incubating the soluble C1ql3-binding fragment of the BAI3 protein completely blocked the inhibitory effects of C1ql3 on insulin secretion in response to cAMP. This suggests that BAI3 mediates the inhibitory effects of C1ql3 on insulin secretion from pancreatic β-cells. These findings demonstrate a novel regulatory mechanism by which C1ql3/BAI3 signaling causes an impairment of insulin secretion from β-cells, possibly contributing to the progression of type 2 diabetes in obesity.
Keywords: beta cell (B-cell), insulin secretion, cyclic AMP (cAMP), G protein-coupled receptor (GPCR), second messenger, brain angiogenesis inhibitor 3, complement 1q-like 3, glucagon-like peptide-1, obesity, type 2 diabetes
Introduction
Pancreatic β-cells secrete insulin in response to elevated glucose levels. Glucose enters the β-cells via glucose transporters, where it is oxidized via glycolysis to increase the ATP/ADP ratio and metabolic flux through mitochondria. An increase in the ATP/ADP ratio causes closure of the KATP channel, which leads to membrane depolarization. This stimulates the influx of Ca2+ ions into the cells via l-type calcium channels, resulting in the fusion of insulin granules to the plasma membrane and rapid release of insulin into the plasma (1, 2). A more sustained release of insulin involves activation of cell signaling pathways in β-cells that are derived from the metabolism of nutrients (e.g. glucose, fatty acids, and amino acids), mitochondrial metabolites (e.g. isocitrate, glutamate, glutamine, malate, and α-ketoglutarate), incretin hormones (e.g. glucagon-like peptide-1 and gastric inhibitory peptide), neurotransmitters (e.g. GABA and vasoactive intestinal peptide), and secondary messengers (mono- and diacylglycerol) (3). The mechanisms by which insulin secretagogues regulate the early and sustained phases of insulin secretion are not well-characterized. Specifically, there is a wide gap in our understanding of mechanisms regulating insulin secretion that lead to susceptibility to progress to type 2 diabetes (T2D)2 (4–6).
The complement 1q (C1q)/tumor necrosis factor (Tnf) family is comprised of secreted proteins that are characterized by the presence of a N-terminal signal peptide, a short variable region with the conserved cysteine residue(s), a collagenous domain containing glycine-X-Y repeats, and a C-terminal globular C1q domain (7–9). The C1q domain is characteristic of a target recognition protein that belongs to a classical complement pathway known to function in the innate immune response (10). Moreover, the three-dimensional structure of the C1q domain is almost identical to the TNF homology domain of the Tnfα protein; thus, giving the name “C1q/Tnf family.” The C1q/Tnf-related proteins (CTRP) are evolutionarily conserved from zebrafish to humans, and are similar in sequence and primary structure to adiponectin. Like adiponectin, they form higher order multimers, which can affect their protein function. However, the tissue expression profiles of CTRPs are different from adiponectin, suggesting distinct and unique cellular functions (7–9, 11, 12). The role of CTRPs has been described in lipid and carbohydrate metabolism (8), immunity (10), inflammation (10), and synapse homeostasis (13–15). Overall, the mechanisms of action of CTRPs are not yet well-characterized.
Complement 1q-like-3 (C1ql3, also called CTRP13) is a soluble secreted protein whose primary structure resembles proteins encoded by highly homologous genes: C1ql-1, -2, and -4. It is expressed in the adipose and brain of lean mice (16). Obesity, diet, or homozygosity for the LeptinOb mutation increase the expression of C1ql3 in brain and adipose, whereas caloric restriction decreases its expression (17). C1ql3 levels in serum were increased in LeptinOb mice (17). Wei and colleagues (16) have reported that C1ql3 regulates glucose metabolism. Recombinant C1ql3 protein increases basal- and insulin-stimulated glucose uptake in adipocytes and myocytes by adenosine monophosphate kinase activation. Moreover, in liver H4IIE cells, C1ql3 reduces glucose synthesis and decreases phosphorylation of Jun N-terminal kinase to alleviate fatty acid-induced insulin resistance by improving insulin signaling (16). In the brain, C1ql3 administration decreases food intake, thus acting as an anorexigenic factor (17). Finally, epidemiological studies have reported the association of serum C1ql3 with T2D and metabolic disorders in humans (18, 19). These published studies suggest C1ql3 has an important regulatory role in glucose metabolism in response to nutritional changes.
Brain-specific angiogenesis inhibitor-3 (BAI3; also called Adgrb3) was identified as a cell-surface receptor for C1ql3 (20). It is a cell-adhesion G protein–coupled receptor (GPCR) that binds with high-affinity to C1ql3 in a calcium-dependent manner (14). BAI3 contains a large extracellular N-terminal region, which includes a CUB domain, four thrombospondin type 1 repeats (the C1ql3-binding region) (20), a hormone-binding domain, the GAIN domain, and a GPCR autoproteolysis site (GPS); the characteristic 7-transmembrane repeat of a GPCR; an intracellular α-helical RKR motif; and C-terminal PDZ domain-binding motif. BAI3 is expressed mainly in the brain with relatively low levels in skeletal muscle, lung, and testis (21). In muscle cells, BAI3 is directly involved in myoblast fusion (22). In the brain, BAI3 localizes with the postsynaptic marker, PSD95, and functions to regulate excitatory or maintenance of synapse (13, 20, 23–25). The role of BAI3 in mediating the effects of C1ql3 on excitatory synapse activity has been reported by multiple groups; however, a role for BAI3 in glucose metabolism has not been described.
In screening for obesity-dependent secreted proteins, we identified that C1ql3 was most differentially expressed with obesity in islets of LeptinOb C57B6/J (B6) mice compared with adipose, gastrocnemius, and liver. The role of C1ql3 as a regulator of β-cell function is not known in pancreatic islets. In the present study, we describe the mechanism by which the C1ql3/BAI3 signaling pathway inhibits insulin secretion from pancreatic β-cells.
Results
Effect of C1ql3 on glucose-stimulated insulin secretion
For insulin secretion experiments, we first established that C1ql3 is a secreted protein (14, 16). Hek293FT cells were transiently transfected with a mammalian expression plasmid encoding for C1ql3 along with GFP or GFP alone as control. After 36 h, a significant increase in the relative abundance of C1ql3 mRNA and protein was observed in cells transfected with C1ql3 compared with control plasmid (Fig. 1A). A Western blotting performed using an anti-C1ql3 antibody demonstrate the presence of C1ql3 in the culture media of the Hek293FT cells that were transfected with C1ql3 compared with the control (Fig. 1B). Furthermore, the absence of GFP or actin protein abundance in the conditioned media suggests minimal cross-contamination of proteins due to cell lysis into the conditioned media. Altogether, these results show that C1ql3 is secreted from Hek293FT cells in the culture media. The C1ql3-conditioned media derived from Hek293FT cells was used in subsequent experiments.
Figure 1.
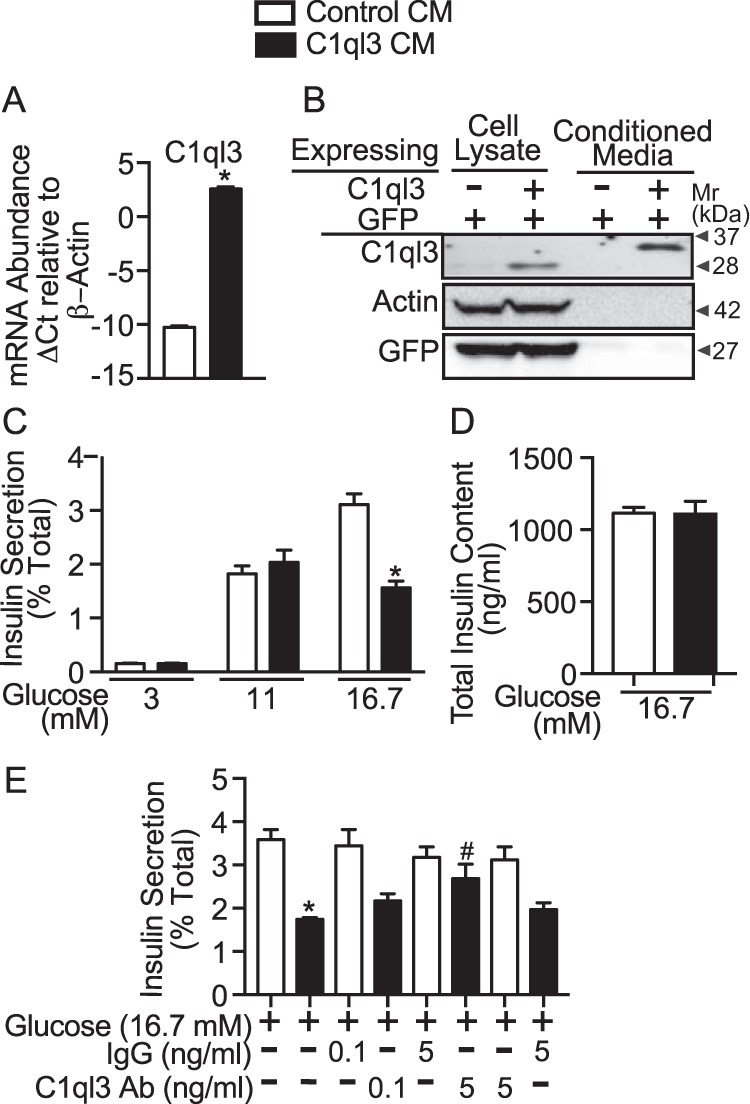
The effect of C1ql3 on glucose-stimulated insulin secretion in mouse islets. Hek293FT cells were plated in a 10-cm tissue culture dishes containing 10 ml of supplemented RPMI 1640 growth media. Approximately, 80% confluent cells were transiently transfected with mammalian expression plasmid that either expressed C1ql3 along with GFP or GFP alone for 36 h. After transfection, cells were harvested, and total RNA and whole cell extracts were prepared. A, quantitative real-time PCR was performed and the mRNA abundance of the C1ql3 was normalized relative to β-actin. The ΔCt was calculated by subtracting the raw Ct of the C1ql3 gene from the raw Ct of the β-actin gene. Values are mean ± S.E. of n ≥ 3. *, p ≤ 0.05 for the mRNA abundance of C1ql3 in cells expressing C1ql3 compared with the control plasmid. B, whole cell extracts (50 μg) and the Hek293 culture media (CM) (60 μl) were subjected to Western blotting. The protein abundance of C1ql3 in whole cell lysates and cell culture media was estimated by using anti-C1ql3 antibody. The protein abundance of actin and GFP served as a control to show that there was no cross-contamination of C1ql3 from cells into the cell culture media. Freshly made C1ql3 and control CM were used for subsequent insulin secretion studies. C, mouse islets were harvest as described under “Experimental procedures.” Islets were cultured overnight at 8 mm glucose. Next day, six size-matched islets were handpicked into each well of a 96-well plate. After preincubating islets with the supplemented RPMI media at 3 mm glucose for 45 min, the cell culture media was replaced with either C1ql3 or control CM with increasing dose of glucose. Insulin secreted in the media and total islet insulin content were determined by using ELISA as described under “Experimental procedures.” The insulin secreted in the media in response to treatments was normalized to the total insulin (secreted plus cellular) and is reported as a percent of total (% Total). The values are mean ± S.E. of n ≥ 3 independent experiments. Moreover, three technical replicates were performed in each experiment. The asterisk (*) indicates that the p value is significantly lower (p ≤ 0.05) for glucose-stimulated insulin secretion in the presence of C1ql3 compared with control CM-treated islets. D, the total insulin content (ng/ml) from islets in response to high glucose treatment was determined by ELISA. The values are the mean ± S.E. of n ≥ 3 independent experiments. E, insulin secretion from mouse islets was determined in response to high glucose treatment in the presence of C1ql3 and control CM. Additionally, the effect of two different doses of anti-C1ql3 or isotype control IgG antibody was determined on insulin secretion from mouse islets incubated with either control CM or C1ql3 CM. The values are mean ± S.E. of n ≥ 3 independent experiments. Each data point in the insulin secretion experiment is an average of three technical replicates. The asterisk (*) indicates that the p value is significantly lower (p ≤ 0.05) for the glucose-stimulated insulin secretion in the presence of C1ql3 compared with the control treated islets. The hash mark (#) indicates that the p value (p ≤ 0.05) is significantly lower for the insulin secreted from cells treated with the C1ql3 antibody and C1ql3 versus C1ql3 alone.
The glucose concentration is the major determinant of β-cell function. Therefore, we determined the effect of C1ql3 on insulin secretion from mouse pancreatic islets at 3, 11, and 16.7 mm glucose concentrations. For these experiments, conditioned media containing native C1ql3 or control was used. Glucose caused a dose-dependent increase in insulin secretion, and C1ql3 inhibited 16.7 mm glucose-stimulated insulin secretion (GSIS) by 40% (p = 0.0027). However, no inhibitory effect of C1ql3 was observed at low (3 mm) or submaximal (11 mm) glucose concentrations (Fig. 1C). Furthermore, this inhibitory effect of C1ql3 on GSIS was not due to the alterations in the total cellular insulin content in the islets (Fig. 1D). To establish the specificity of the inhibitory effects of C1ql3 on GSIS at 16.7 mm, insulin secretion experiments were repeated with and without anti-C1ql3 antibody or isotype-specific IgG control. C1ql3 inhibited high glucose-stimulated insulin secretion, whereas the anti-C1ql3 antibody attenuated this response in a dose-dependent manner compared with the control IgG (Fig. 1E, black bars). As a control, no effect of the C1ql3 antibody or control IgG was observed on the high glucose-stimulated insulin secretion in the absence of C1ql3 (Fig. 1E). This suggests that C1ql3 in the conditioned media modulates pancreatic β-cell function by regulating the secretion of insulin.
Effect of C1ql3 on membrane depolarized stimulated insulin secretion
High glucose can increase GSIS by causing depolarization of the plasma membrane or activation of amplification pathways, or both. Therefore, we performed experiments in mouse islets to determine which of these effects are regulated by C1ql3. Membrane-depolarized insulin secretion can be achieved by treating islets with the sulfonylurea, tolbutamide, or potassium chloride (KCl). Sulfonylureas cause the ATP-independent closure of the KATP channel, which leads to membrane depolarization and influx of calcium in the β-cell, stimulating insulin secretion (26). KCl causes an increase in insulin secretion by directly causing membrane depolarization (1). Both KCl (40 mm) and tolbutamide (500 μm) stimulated insulin secretion at low glucose (3 mm). C1ql3 inhibited KCl- and tolbutamide-stimulated insulin secretion by 25% (p = 0.036) and 33% (p = 0.0018), respectively (Fig. 2, A and B). Moreover, anti-C1ql3 antibody attenuated the inhibition by C1ql3 of KCl and tolbutamide-stimulated insulin secretion (Fig. 2, C and D). These results suggest that the inhibition of insulin secretion by C1ql3 could potentially be mediated by the modulation of KATP-dependent insulin secretion.
Figure 2.
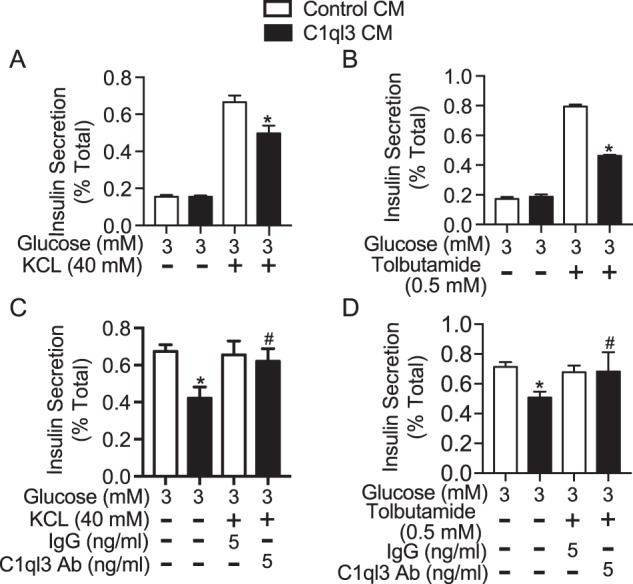
The effect of C1ql3 on membrane depolarized stimulated insulin secretion. Mouse islets were isolated and incubated overnight in the supplemented RPMI 1640 culture media at 8 mm glucose. Next day, the insulin secretion experiment was performed in the presence of C1ql3 or control CM as described under “Experimental procedures” in response to different insulin secretagogues. Insulin secretion in response to (A) 40 mm potassium chloride (KCl) and (B) 0.5 mm tolbutamide at 3 mm glucose was determined in the presence of C1ql3 or control CM. The effect of anti-C1ql3 (5 ng/ml) or IgG (5 ng/ml) antibody on insulin secretion in response to (C) KCl (40 mm) and (D) tolbutamide (0.5 mm) was determined in mouse islets treated with C1ql3 or control. The insulin secreted in response to treatments was normalized to the total insulin and is reported as a percent of total insulin (secreted plus cellular). Values are mean ± S.E. of n ≥ 3. The asterisk (*) indicates that the p value is significantly lower (p ≤ 0.05) for the insulin secreted in the presence of C1ql3 compared with control treated islets. The hash mark (#) indicates that the p value (p ≤ 0.05) is significantly lower for the insulin secreted from cells treated with C1ql3 antibody and C1ql3 versus C1ql3 alone.
Effect of C1ql3 on pathways that are involved in the amplification of insulin secretion
The amplification of GSIS can occur by a number of different pathways, including incretins, fatty acids, amino acids, mitochondrial metabolites, fatty acid oxidation, monoacylglycerol (MAG)- and diacylglycerol (DAG)-activated signaling pathways, among others (1, 3, 27, 28). We systematically determined whether C1ql3 inhibits insulin secretion potentiated by each of these signaling pathways.
Metabolism of glucose increases the flux through mitochondria, which leads to the generation of metabolites such as α-ketoglutarate, malate, ketoisocaproate, glutamate, and isocitrate (see Fig. 3F for a pathway schematic). Treatment of mouse islets with each of these metabolites increased insulin secretion (Fig. 3, A–E, white bars). C1ql3 had no inhibitory effect on GSIS potentiated by any of these metabolites (Fig. 3, A–E, black bars).
Figure 3.

The effect of C1ql3 on mitochondrial metabolite-stimulated insulin secretion. Mouse islets were isolated and incubated overnight in supplemented RPMI 1640 culture media at 8 mm glucose. Next day, the insulin secretion experiment was performed in the presence of C1ql3 or control CM as described under “Experimental procedures” in response to different insulin secretagogues. Insulin secretion experiments were performed in mouse islets treated with cell-permeable (A) 15 mm ketoisocaproate, (B) 100 μm isocitrate, (C) 100 μm α-ketoglutarate, (D) 10 mm malate, or (E) 5 mm glutamate in the presence of control or C1ql3 CM. F, diagram showing mitochondrial metabolites in insulin secretion. The insulin secreted in response to treatments was normalized to total insulin and is reported as a percent of total insulin (secreted plus cellular). Values are mean ± S.E. of n ≥ 3.
Another mechanism that can potentiate GSIS is activation of amino acid signaling pathways (see Fig. 4F for a pathway schematic). Mouse islets were treated with arginine, leucine, ketoisocaproate, l-alanine, and glutamine, all of which potentiated GSIS (Fig. 4, A–D, white bars). Again, C1ql3 had no inhibitory effect on the ability of any of these amino acids to increase GSIS (Fig. 4, A–D, black bars) (3, 29).
Figure 4.
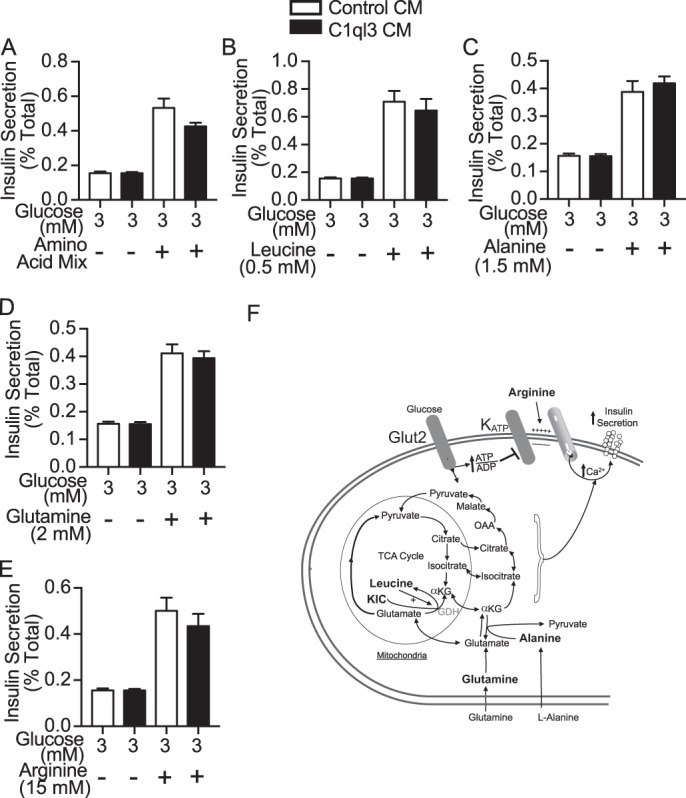
The effect of C1ql3 on amino acid-stimulated insulin secretion. Mouse islets were isolated and incubated overnight in the supplemented RPMI 1640 culture media at 8 mm glucose. Next day, the insulin secretion experiment was performed in the presence of C1ql3 or control CM as described under “Experimental procedures” in response to different insulin secretagogues. Insulin secretion experiments were performed in mouse islets treated with cell-permeable (A) amino acid mixture (0.5 mm leucine, 1.5 mm alanine, and 2 mm glutamine), (B) 0.5 mm leucine, (C) 1.5 mm alanine, (D) 2 mm l-glutamine, and (E) 15 mm l-arginine in the presence of control or C1ql3 CM. F, diagram showing key amino acids that are known to regulate insulin secretion. The insulin secreted in response to treatments was normalized to the total insulin and is reported as a percent of total insulin (secreted plus cellular). Values are mean ± S.E. of n ≥ 3.
A third major mechanism by which GSIS can be potentiated is by fatty acid signaling. Free fatty acids potentiate GSIS by the activating Gq-associated free fatty acid receptors such as GPR40, generating DAG and MAG second messengers. Similarly, inhibition of β-oxidation, by 2-bromo-palmitate potentiates GSIS (see Fig. 5E for a schematic). Palmitate, DAG, and MAG all potentiated GSIS including the inhibitor of β-oxidation, 2-bromo-palmitate, as expected (Fig. 5, A–D, white bars). Addition of C1ql3 had no effect on GSIS potentiated by any of these compounds (Fig. 5, A–E, black bars).
Figure 5.
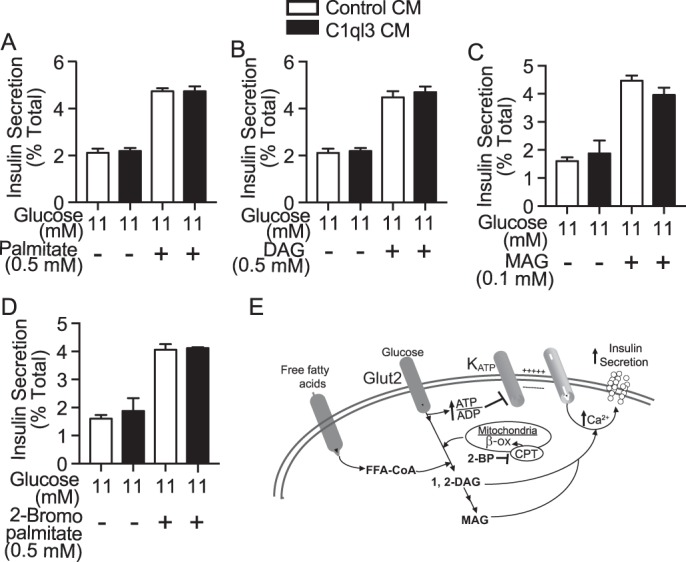
The effect of C1ql3 on intracellular lipid signaling stimulated insulin secretion. Mouse islets were isolated and incubated overnight in the supplemented RPMI 1640 culture media. Next day, the insulin secretion experiment was performed in the presence of C1ql3 or control CM as described under “Experimental procedures” in response to different insulin secretagogues. Insulin secretion experiments were performed in mouse islets treated with cell-permeable (A) 0.5 mm palmitate, (B) 0.5 mm diacylglycerol, (C) 0.1 mm monoacylglycerol, or (D) 0.5 mm 2-Br-palmitate in the presence of control or C1ql3 CM. E, diagram showing fatty acid signaling in insulin secretion. The insulin secreted in response to treatments was normalized to total insulin and is reported as a percent of total insulin (secreted plus cellular). Values are mean ± S.E. of n ≥ 3.
A fourth major mechanism potentiating GSIS involves activation of cAMP signaling. cAMP is a critical second messenger whose levels are modulated by several factors, including receptor-mediated signaling by incretins such as glucagon-like peptide-1 (GLP-1) or gastric inhibitory polypeptide (30). cAMP is required for both basal insulin secretion and is a critical potentiator of GSIS (31–33). We first determined the effect of C1ql3 on the ability of the stable GLP-1 receptor agonist, exendin-4 to potentiate GSIS from mouse islets. Exendin-4 potentiated insulin secretion at both 3 and 11 mm glucose (Fig. 6A, white bars), whereas C1ql3 almost completely inhibited exendin-4-potentiated insulin secretion, independent of glucose concentration (Fig. 6A, black bars; 92%, p = 0.0007). To establish that the observed inhibitory effects of C1ql3 on exendin-4-potentiated GSIS were specific, insulin secretion experiments were repeated with and without an anti-C1ql3 antibody or isotype-specific IgG control. Exendin-4 (10 nm) was added to stimulate cAMP-mediated signaling. C1ql3 inhibited exendin-4-potentiated insulin secretion, whereas the C1ql3 antibody attenuated this response in a dose-dependent manner compared with the control IgG (Fig. 6B, black bars). As controls, there was no effect of the C1ql3 or control IgG antibody of exendin-4-potentiated GSIS (Fig. 6B, white bars).
Figure 6.
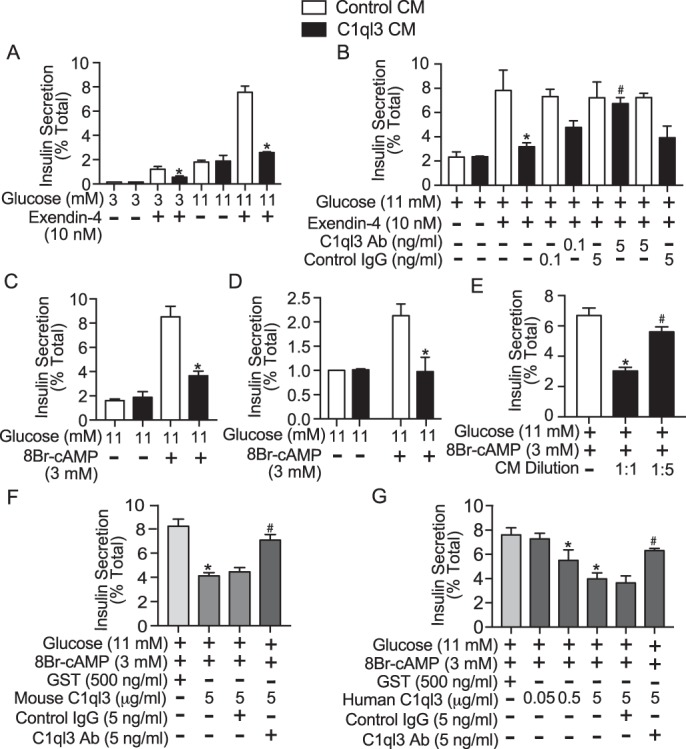
The effect of C1ql3 on exendin-4 incretin signaling stimulated insulin secretion. Mouse islets were isolated and incubated overnight in the supplemented RPMI 1640 culture media at 8 mm glucose. Next day, the insulin secretion experiment was performed in the presence of C1ql3 or control CM as described under “Experimental procedures” in response to different insulin secretagogues. A, insulin secretion experiments were performed in mouse islets treated with 10 nm exendin-4 (a stable analog of GLP-1) in response to 3 and 11 mm glucose, in the presence of C1ql3 or control CM. B, the effect of C1ql3 and IgG isotype control antibody at 0.1 and 5 ng/ml concentrations was determined in mouse islets treated with 10 nm exendin-4 at 11 mm glucose in the presence of C1ql3 CM. The following two control treatments were used in this experiment to show the specificity of C1ql3 and control IgG in this experiment: the effect of C1ql3 IgG in the presence of 10 nm exendin-4 (at 11 mm glucose) along with control CM and isotype control IgG in the presence of 10 nm exendin-4 (at 11 mm glucose) along with C1ql3 CM. The effect of C1ql3 and control CM on insulin secretion was determined in the presence of 3 mm 8Br-cAMP at 11 mm glucose (membrane permeable cAMP) in mouse (C) and human (D) islets. E, the effect of diluting C1ql3 CM five times in the culture media on insulin secretion. F, the effect of mouse C1ql3 recombinant protein compared with the control GST protein (28 kDa, molecular mass similar to C1ql3) on insulin secretion in mouse islets in response to 3 mm 8Br-cAMP at 11 mm glucose. Also, the effect of anti-C1ql3 and IgG isotype control antibodies was determined on insulin secretion from islets treated with C1ql3 recombinant protein in the presence of 3 mm 8Br-cAMP at 11 mm glucose. G, dose-dependent effect of the human C1ql3 recombinant protein compared with the GST protein in mouse islets in response to 3 mm 8Br-cAMP at 11 mm glucose. Also, the effect of anti-C1ql3 and isotype control antibodies was determined on insulin secretion from islets treated with C1ql3 recombinant protein in the presence of 3 mm 8Br-cAMP at 11 mm glucose. The insulin secreted in response to treatments was normalized to the total insulin and is reported as a percent of total insulin (secreted plus cellular). Values are mean ± S.E. of n ≥ 3. The asterisk (*) indicates that the p value is significantly lower (p ≤ 0.05) for the insulin secreted in the presence of C1ql3 compared with control treated islets. The hash mark (#) indicates that the p value (p ≤ 0.05) is significantly lower for the insulin secreted from cells treated with C1ql3 antibody and C1ql3 versus C1ql3 alone.
To look at the effect of C1ql3 on cAMP-potentiated insulin secretion directly, a stable, membrane-permeable cAMP analog, 8Br-cAMP (3 mm), was used to stimulate 11 mm GSIS, with and without the addition of C1ql3. C1ql3 almost completely blocked the cAMP-mediated potentiation of GSIS from both mouse islets (Fig. 6C; 87%, p = 0.0009) and human islets (Fig. 6D, ∼100%, p < 0.05). Moreover, diluting the C1ql3-conditioned media had a dose-dependent effect on inhibiting cAMP-mediated potentiation of GSIS (Fig. 6E). Alternatively, we tested the effect of mouse and human C1ql3 recombinant protein on cAMP-stimulated insulin secretion to corroborate the inhibitory effects of C1ql3 that were obtained by using C1ql3-conditioned media. Treating mouse islets with the mouse C1ql3 recombinant protein (5 μg/ml) inhibited cAMP-stimulated insulin secretion by 50% (Fig. 6F; p = 0.0031 compared with GST treatment). Also, human recombinant C1ql3 protein (0.05, 0.5, and 5 μg/ml) dose-dependently inhibited cAMP-stimulated insulin secretion from mouse islets (Fig. 6G; p = 0.0088 for 5 μg/ml of human C1ql3 compared with GST treatment). The highest dose of 5 μg/ml of the C1ql3 recombinant protein for these experiments was chosen based on the study previously published by Wei et al. (16). The inhibitory effect of mouse and human C1ql3 recombinant proteins on the cAMP-stimulated insulin secretion was significantly attenuated by anti-C1ql3 antibody compared with the control IgG. Altogether, these results confirm that C1ql3 is an inhibitor of cAMP-potentiated GSIS.
Alterations in the ATP/ADP ratio are known to affect insulin secretion. No effect of C1ql3 was observed on the ATP/ADP ratio in response to varying glucose dose (5, 11, and 16.7 mm) and exendin-4 (10 nm) at 11 mm glucose in mouse islets (Fig. 7A). Also, C1ql3 treatment did not affect cell viability (Fig. S1A) or apoptosis determined by the absence of cleaved caspase-3 in mouse islets (Fig. S1B). Glucose is known to increase cAMP content in mouse islets (34). Moreover, elevated cAMP content/signaling contributes to the calcium-independent increase of GSIS (31). Therefore, we determined the effect of C1ql3 on glucose-stimulated cAMP levels in mouse islets. High glucose (16.7 mm) increased cAMP levels by ∼50% compared to 11 or 5 mm glucose treatment in mouse islets. C1ql3 completely inhibited the increase in cAMP levels observed at 16.7 mm glucose (Fig. 7B, p = 0.023). cAMP increases insulin secretion by activating protein kinase A (PKA) and Epac2. We determined the effect of C1ql3 on Epac2 and PKA to stimulate insulin secretion. Epac2 selective agonist 8-pCPT-2′-O-Me-cAMP-AM (8-CPT-AM) increased insulin secretion at 5, 7, and 11 mm glucose from mouse islets. C1ql3 inhibited the 8-CPT-AM potentiated increase in insulin secretion at 5, 7, and 11 mm glucose concentrations by ∼50% (Fig. 7C). Alternatively, PKA selective agonist 6-Bnz also potentiated insulin secretion at 5, 7, and 11 mm glucose concentrations from mouse islets. C1ql3 inhibited the potentiation of insulin secretion by 6-Bnz stimulated at 5, 7, and 11 mm glucose concentrations by ∼40% (Fig. 7D). Moreover, C1ql3 alone did not inhibit insulin secretion at 5, 7, and 11 mm glucose concentrations compared to control (Fig. 7, C and D). Next, we determined whether C1ql3 modulates the activity of PKA by using phospho-PKA substrate antibody, which detects motifs in proteins that are phosphorylated by PKA. cAMP increased the PKA activity in mouse islets at 11 mm glucose. Conditioned media containing C1ql3 and human C1ql3 recombinant protein significantly decreased cAMP-stimulated activity of the PKA by 30–40% (Fig. 7, E and F). These results show that C1ql3 inhibits cAMP levels and also modulates cAMP-stimulated insulin secretion by inhibiting Epac2 and PKA-signaling pathways in mouse islets.
Figure 7.
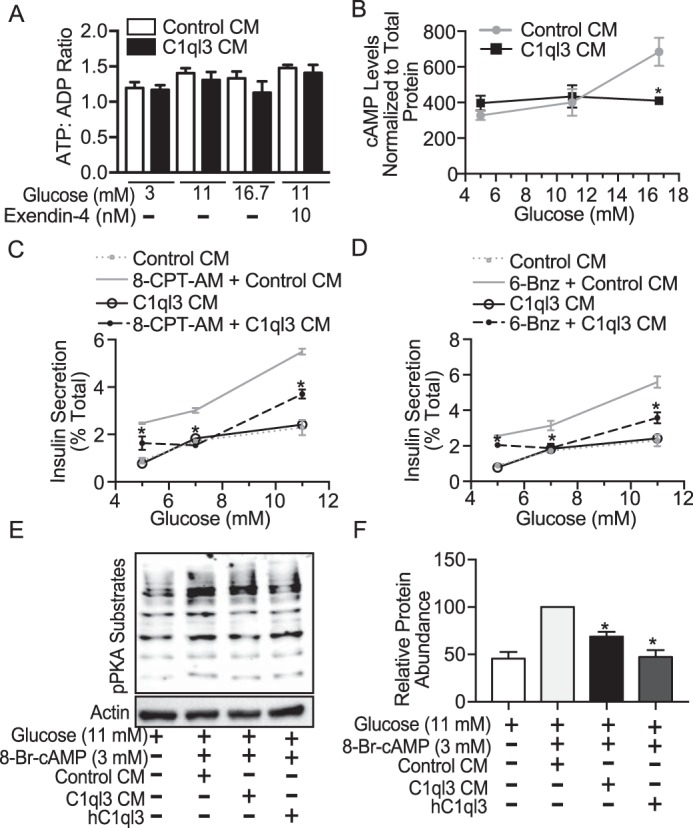
The effect of C1ql3 on cAMP signaling in islets. Mouse islets were isolated and incubated overnight in supplemented RPMI 1640 culture media at 8 mm glucose. Next day, the experiment was performed to determine (A) ATP/ADP ratio as described under “Experimental procedures” in mouse islets treated with insulin secretagogues in the presence of C1ql3 or control CM. B, the effect of C1ql3 and control CM was determined on cAMP levels in mouse islets in response to the increasing dose of glucose as described under “Experimental procedures.” The cAMP levels were normalized to the total protein levels determined by the BCA assay. Values are mean ± S.E. of n ≥ 3. *, p ≤ 0.05 for the cAMP levels in islets treated with C1ql3 CM compared with the control CM in response to high glucose. Insulin secretion was determined in response to (C) Epac2 selective agonist 8-CPT-AM, and (D) PKA selective agonist 6-Bnz in the presence of C1ql3 or control CM at increasing dose of glucose in mouse islets. The insulin secreted in response to treatments was normalized to the total insulin and is reported as a percent of total insulin (secreted plus cellular). Values are mean ± S.E. of n ≥ 3. The asterisk (*) indicates that the p value is significantly lower (p ≤ 0.05) for the insulin secreted in the presence of C1ql3 compared with control treated islets. E, the Western blot shows the relative protein abundance of pPKA substrates in response to C1ql3 CM and human (h) C1ql3 recombinant at 3 mm cAMP at 11 mm glucose in mouse islets. The blot is representative of n = 3 experiments. F, the quantitation for the PKA activity was performed by normalizing the protein abundance of pPKA substrates by the actin. Values are mean ± S.E. of n ≥ 3. *, p ≤ 0.05 for the PKA activity in mouse islets treated with C1ql3 compared with the control CM in response to 3 mm cAMP at 11 mm high glucose.
BAI3 regulates insulin secretion from pancreatic β-cells
BAI3 was reported as a C1ql3 receptor in the brain (14, 35). However, the role for BAI3 in β-cell function has not been described. We first determined the Bai3 mRNA expression pattern in mouse tissues, including pancreatic islets, using quantitative RT-PCR. In tissues from lean C57BL/6J mice, Bai3 is expressed in islets at levels comparable with those of brain (ΔCt = −13 versus ΔCt = −12), whereas the expression in liver, adipose, kidney, or gastrocnemius is relatively low (Fig. 8A). Bai3 is expressed in mouse islets, human islets, and pancreatic β (INS1 (832/13) and MIN6B1) cell lines (Fig. 8B). We performed immunocytochemistry by using confocal imaging to determine the protein expression of BAI3 and Glut2 in INS1 (832/13) cells (Fig. 8C). BAI3 (green) is localized on the plasma membrane of the INS1(832/13) cells. Glut2 (red) is a plasma membrane marker. Merge (orange) shows that BAI3 colocalizes with Glut2. DAPI (blue) stains nucleus of INS1(832/13) cells. No signal was observed from INS1(832/13) cells in the absence of BAI3 primary antibody (Fig. 8C).
Figure 8.
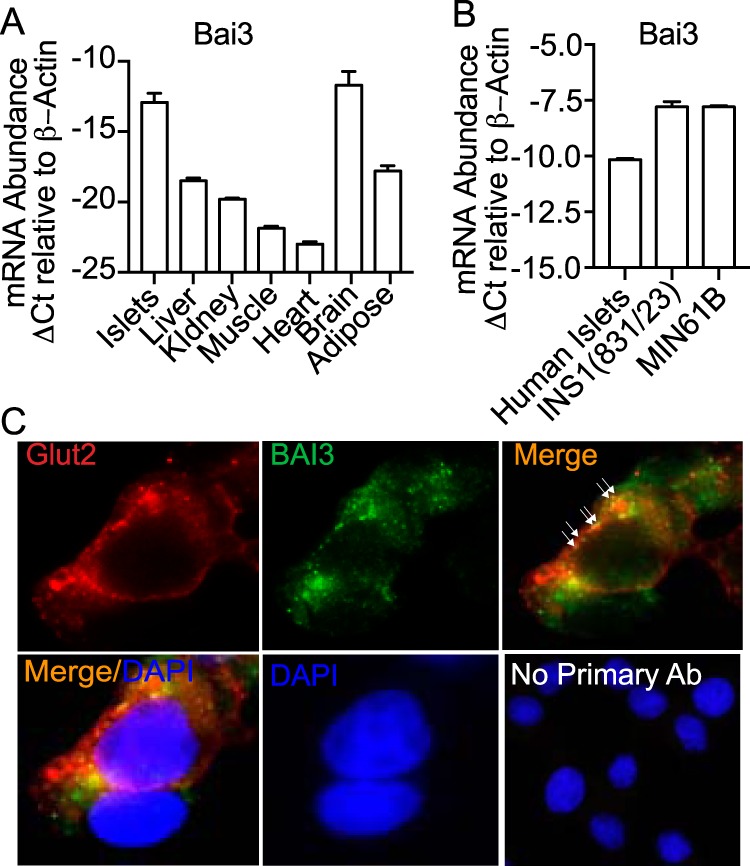
Tissue expression of the BAI3 receptor. A, tissues were isolated from 10-week-old C57BL6/J mice and total mRNA was harvested from islets, liver, adipose, gastrocnemius (gastroc), kidney, heart, and brain. B, human islets, INS1(832/13), and MIN6B1 cells were harvested and total mRNA was prepared. Quantitative real-time PCR was performed to determine the relative mRNA abundance of BAI3. The data were normalized to the β-actin gene. The ΔCt was calculated by subtracting the raw Ct of the BAI3 gene from the raw Ct of the β-actin gene. Values are mean ± S.E. of n ≥ 3. C, immunofluorescence by confocal imaging was performed in INS1(832/13) β-cells using anti-BAI3 antibody (green). The Glut 2 (red) antibody was used as a plasma membrane marker. The localization of BAI3 on the plasma membrane is shown by arrows or as colocalization as a merge in orange. No primary antibody control was used to show that no nonspecific signal was observed from the secondary antibody (scale bar = 10 μm).
The cell-binding assay was performed to determine that C1ql3 in the conditioned media is capable of binding to the BAI3 receptor. For this experiment, Hek293FT cells were transiently transfected with or without Bai3-mVenus expressing plasmid. Immunocytochemistry was performed, and stained cells were imaged by using a fluorescence microscope. BAI3-transfected cells were incubated with either C1ql3 or control conditioned media for 45 min at 37 °C prior to fixing the cells for imaging. Anti-GFP antibody was used to stain for cells expressing BAI3 (green). BAI3-mVenus expressing cells that were incubated with C1ql3 showed far-red staining for C1ql3 on the plasma membrane of Hek293FT cells by using anti-C1ql3 antibody. Merged (yellow) image shows that C1ql3 present in the conditioned media binds to the BAI3 present on the cell membrane (Fig. 9A). No staining for C1ql3 was observed in BAI3-mVenus expressing cells that were incubated with the control conditioned media (Fig. 8B). Additionally, no staining for BAI3 or C1ql3 was observed in untransfected cells that were incubated with C1ql3-conditioned media (Fig. 8C). These results establish that C1ql3 in the conditioned media has the ability to specifically bind to BAI3 G protein–coupled receptor.
Figure 9.
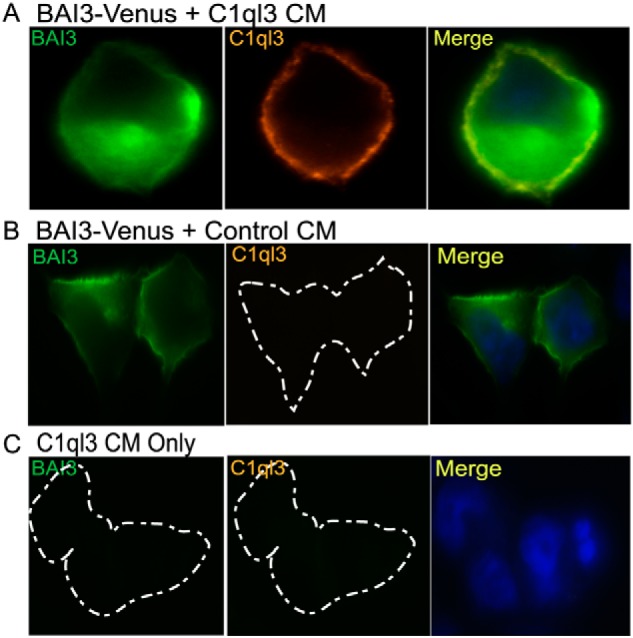
The binding of C1ql3 in the CM to the BAI3 receptor. Cell-surface labeling assay using immunofluorescence was performed in Hek293FT cells that were transiently transfected with BAI3-mVenus or empty vector control. The cells were incubated for 45 min with C1ql3 or control CM at 37 °C in the cell culture incubator. The cells were fixed for staining as described under “Experimental Procedures.” The BAI3 is in green, C1ql3 in far-red, DAPI in blue, and the bound C1ql3 to BAI3 is shown as a merge in yellow color.
BAI3 mediates the inhibitory effects of C1ql3 on insulin secretion
We determined whether BAI3 regulates insulin secretion. For this, insulin secretion experiments were performed in INS1(832/13) cells that were transfected either with small interfering (si) RNA targeting Bai3 or siScramble control. Approximately, 50% reduction in the mRNA abundance of Bai3 was observed in INS1(832/13) cells transfected with siBai3 compared with siScramble (p < 0.05) (Fig. 10A). Insulin secretion experiments were performed at low glucose (3 mm) and high glucose (15 mm) concentrations. A 2-fold (p = 0.0077) increase in insulin secretion was observed in cells treated with siBai3 as compared to siScramble at low glucose (3 mm) (Fig. 10B). Moreover, a 50% increase in insulin secretion (78%, p = 0.0038) was observed in cells treated with siBai3 compared to siScramble at high glucose (15 mm) (Fig. 10C). These outcomes show that BAI3 inhibits insulin secretion from INS1(832/13) cells. C1ql3 treatment inhibited high glucose (15 mm)-stimulated insulin secretion from INS1(832/13) cells by 40% (Fig. 10C). BAI3 knockdown completely attenuated the C1ql3-mediated inhibition of insulin secretion observed in INS1(832/13) (Fig. 10C). Similar to mouse islets, no inhibitory effect of C1ql3 was observed on insulin secretion at low glucose in INS1(832/13) cells (Fig. 10B).
Figure 10.

The effect of BAI3 in mediating the inhibitory effects of C1ql3 on insulin secretion. The INS1(832/13) cells were transiently transfected with siScramble (control) or siBAI3. After 36 h, static insulin secretion in INS1(832/13) cells was performed as described under “Experimental procedures” in response to 3 and 15 mm glucose in the presence of C1ql3 or control CM. A, knockdown of the BAI3 gene in INS1(832/13) β-cells. The relative mRNA abundance of the BAI3 gene was calculated by using the ΔCt method. Values are mean ± S.E. of n ≥ 3. *, p ≤ 0.05 for the mRNA abundance of BAI3 in cells transfected with siBAI3 compared with the siControl. B and C, insulin secretion in response to the BAI3 knockdown in the presence of C1ql3 and control CM at (B) 3 mm and (C) 15 mm glucose. The insulin secreted in response to treatments was normalized to total insulin and is reported as a percent of total insulin (secreted plus cellular). Values are mean ± S.E. of n ≥ 3. The asterisk (*) indicates that the p value is significantly lower (p ≤ 0.05) for insulin secreted in the presence of C1ql3 compared with control treated islets. The hash mark (#) indicates that the p value (p ≤ 0.05) is significantly lower for the insulin secreted from cells treated with siBAI3 versus siControl. D, soluble BAI3 recombinant protein blocks the effect of C1ql3 on insulin secretion. Mouse islets were isolated and incubated overnight in the supplemented RPMI 1640 culture media. Next day, the insulin secretion experiment was performed using C1ql3 or control CM as described under “Experimental procedures” in response to 3 mm 8Br-cAMP at 11 mm glucose in the presence of C1ql3 or control CM. GST or increasing dose of the soluble BAI3 recombinant protein was added to the C1ql3 containing conditioned media. Values are mean ± S.E. of n ≥ 3. The asterisk (*) indicates that the p value is significantly lower (p ≤ 0.05) for insulin secreted in the presence of C1ql3 compared with control treated islets. The hash mark (#) indicates that the p value (p ≤ 0.05) is significantly lower for the insulin secreted from cells treated with the soluble BAI3 fragment along with C1ql3 versus C1ql3 alone.
To further establish that BAI3 mediates the inhibitory effects of C1ql3 on cAMP-stimulated insulin secretion, insulin secretion from mouse islets was stimulated by 11 mm glucose and 3 mm 8Br-cAMP with and without the addition of C1ql3 and a C1ql3 inhibitor, a recombinant soluble N-terminal C1ql3-binding BAI3 fragment. C1ql3 alone inhibited cAMP-potentiated GSIS, repeating the results obtained previously (Fig. 10D). The recombinant soluble N-terminal C1ql3-binding BAI3 fragment dose-dependently blocked the inhibition of cAMP-potentiated GSIS by C1ql3 (Fig. 10D). At the highest concentrations used (0.5 and 2.5 μg/ml sBAI3), the ability of C1ql3 to inhibit insulin secretion was completely attenuated (p = 0.009). As a control, experiments were performed using recombinant GST, chosen for its similar molecular weight to C1ql3, 0.5 μg/ml of recombinant GST was unable to attenuate the inhibitory effects of C1ql3 on cAMP-potentiated insulin secretion (Fig. 10; p = 0.797 as compared with C1ql3 alone). Furthermore, neither 0.5 nor 2.5 μg/ml of sBAI3 inhibited cAMP-potentiated GSIS in the absence of C1ql3 (p = 0.0697 as compared with 0.6987) (Fig. 10). These results confirm that BAI3 has a functional role in regulating cAMP-potentiated insulin secretion and mediates the inhibitory function of C1ql3.
Discussion
Herein, by using biochemical and cell-based approaches, we investigated the effect of C1ql3 protein in regulating insulin secretion from primary isolated islets in response to a variety of different stimulators/potentiators of insulin secretion. The data presented here show that C1ql3 inhibits insulin secretion by attenuating signaling pathways activated in response to high glucose, KCl, tolbutamide, and cAMP signaling. The cell-surface receptor for C1ql3 was identified as BAI3. Our data show that BAI3 is expressed in primary mouse and human pancreatic islets and insulinoma cell lines, and that knockdown of Bai3 stimulates GSIS from β-cell-derived INS1(832/13) cells. Furthermore, by performing BAI3-receptor knockdown and using the soluble N-terminal C1ql3-binding fragment of BAI3 protein we showed that BAI3 is the sole mediator of the inhibitory effects of C1ql3 on insulin secretion. Our data demonstrate that the activation of this novel C1ql3/BAI3 signaling pathway causes impairment in β-cell function. Altered signaling of C1ql3/BAI3 during obesity and T2D might contribute to the β-cell dysfunction observed in these conditions.
The amplification of insulin secretion by various mechanisms requires the triggering of the secretion process itself by closure of the KATP channels, depolarization of the cell membrane, and the influx of Ca2+ from voltage-dependent Ca2+ channels. Many signaling pathways, mitochondrial metabolites, fatty acids, amino acids, and second messengers can potentiate the secretion of insulin that has already been triggered (1, 3). Unique among the second messengers is cAMP. It cannot only amplify GSIS, but is also required for the triggering phase (30, 31, 36–39). The effect of C1ql3 on insulin secretion is completely consistent with its action by regulation of intracellular cAMP. It inhibits insulin secretion triggered and amplified by a stimulatory concentration of glucose (Figs. 1, 2, and 6). In low glucose, it inhibits insulin secretion triggered by direct depolarization of the cell membrane by the sulfonylurea, tolbutamide or KCl (Fig. 2, A and B). C1ql3 has no effect on the potentiation of insulin secretion by any mediator of solely the amplifying pathway tested (Figs. 3–5). The only amplifying pathway C1ql3 inhibited was the cAMP-mediated amplifying pathway. C1ql3 inhibited insulin secretion potentiated by exendin-4 (Fig. 6), a selective ligand of the GLP-1 receptor, known to act by stimulation of cAMP production (40, 41), as well as insulin secretion potentiated by direct activation of the cAMP amplifying pathway by a stable, cell-permeable cAMP analog, 8Br-cAMP (Fig. 6, C and D).
The inhibitory effects of C1ql3 on insulin secretion in islets are mediated by BAI3. The BAI3 receptor is expressed in pancreatic β-cells, and siRNA-mediated knockdown of Bai3 in the INS1(832/13) β-cell line caused an increase in insulin secretion in response to low (1.5 mm) and high (15 mm) glucose treatments (Fig. 10, B and C). These outcomes point to the adaptive role of the BAI3 signaling pathway in response to varying glucose concentrations in β-cell. Moreover, the C1ql3-binding fragment of the BAI3 receptor dose-dependently rescued the inhibitory effects of C1ql3 on cAMP-stimulated insulin secretion from mouse islets (Fig. 10). Taken together, our results indicate that the inhibitory effects of C1ql3/BAI3 signaling on insulin secretion are mediated via negative regulation of the cAMP signaling pathway.
We have confirmed C1ql3 acts through BAI3 to negatively regulate the stimulatory effects of cAMP on insulin secretion. But what are the specific intracellular mechanisms by which this could be occurring? Previously, Tian et al. (36, 37) reported that islets treated with 11–30 mm glucose showed a dose-dependent increase in cAMP oscillations near the plasma membrane, which were not due to changes in the cytoplasmic [Ca2+]i concentrations. Furthermore, Dyachok and co-workers (31, 39) have reported treating islets with high glucose causes cAMP oscillations that contribute to insulin secretion. Glucose, tolbutamide, and KCl have been shown to increase cAMP levels in β-cells via activation of adenylyl cyclase activity (38, 42, 43). Furthermore, tolbutamide was reported to bind and activate Epac2 (44), one of the classical downstream cAMP targets (the other being protein kinase A). Our results show that the inhibitory effects of C1ql3 on insulin secretion observed at 16.7 mm (high) glucose were correlated with the inhibition in the cAMP levels at 16.7 mm glucose (Fig. 7B). Additionally, no effect of C1ql3 was observed on insulin secretion or cAMP levels at 11 or 3 mm in mouse islets. These outcomes along with the complete attenuation of exendin-4/cAMP-stimulated insulin secretion suggest that C1ql3 in β-cells functions by regulating cAMP levels and it's signaling pathway. Determining how C1ql3 inhibits cAMP signaling remains part of the future studies.
The formation of the SNARE complex is required for the fusion of insulin granules to the plasma membrane and secretion of insulin to the plasma (45). This is a major rate-limiting step in the secretion of insulin and is regulated by several SNARE-regulatory proteins. Because C1ql3 does not ubiquitously inhibit stimulation of insulin secretion in response to all insulin secretagogues, it suggests that the effects of the C1ql3-activated signaling pathway in regulating insulin secretion are more distal or upstream to the proteins that are directly involved in the formation of the SNARE complex. Both Epac2 and PKA are known to modulate the activity of the KATP channel to regulate the formation of the SNARE complex: Epac2 (by direct binding) and PKA (by phosphorylation). Incretins such as GLP-1 increase cAMP levels by activating stimulatory GPCRs, causing the adenylate cyclase-mediated conversion of ATP to cAMP (30). Our results show in addition to inhibiting cAMP levels, C1ql3 also inhibits Epac2 and PKA-stimulated insulin secretion. Both PKA and Epac2 regulate insulin secretion by distinct mechanisms. Therefore, it remains to be determined the precise mechanisms by which C1ql3 regulates Epac2 and PKA signaling (e.g. KATP channels, SNARE complex formation) to regulate insulin secretion.
Our data are suggestive of ligand-independent effects of BAI3 on insulin secretion. siRNA-mediated knockdown of the BAI3 receptor in the absence of exogenous ligand increased insulin secretion at low glucose (<2.8 mm) (Fig. 8B). However, the inhibitory effects of C1ql3 on insulin secretion in mouse islets were observed at 16.7 mm, and not at 11 or 3 mm glucose concentration (Fig. 1). The ligand-independent effects of BAI3 could potentially be due to the basal activity of the BAI3 receptor, or it may serve as a receptor for ligands that are similar in structure to C1ql3 (for example, C1ql1, C1ql2, or C1ql4) to regulate insulin secretion. These possibilities are currently being investigated. It is also possible that the islet-derived C1ql3 acts on the β-cell in an autocrine or paracrine fashion; thus, by reducing BAI3 levels we are reducing signaling by endogenous C1ql3. There is precedence for this type of scenario. The classical gut hormone, GLP-1, is now known to have its actions on the β-cell not by its secretion from the gut, but by alternative processing of the prepro-glucagon peptide in the α-cell and paracrine signaling to the β-cell GLP-1 receptor (46, 47).
GWAS studies indicate a SNP (rs2184723) in the human BAI3 gene is associated with T2D in a Mexican American population (p = 4.16E-4) (48, 49). These findings demonstrate that BAI3 expression might be a maladaptation to genetics and the risk allele in the BAI3 may manifest in reducing β-cell function and increasing susceptibility to T2D in the metabolic stress of obesity.
The specificity of C1ql3 for the cAMP signaling pathway has significant clinical relevance. There are already two classes of T2D therapeutics that act through the cAMP pathway: the stable GLP-1 analogs and DPP4 inhibitors (50, 51). These have become well-accepted (52–54). Yet, they are not effective or are incompletely effective in a percentage of T2D patients (55–57). Inhibition of the C1ql3/BAI3 axis may be an alternative target to treat people with diabetes. It has already been demonstrated that dysfunctional cAMP-inhibitory GPCR signaling pathways can at least partially restore glucose-stimulated and incretin-potentiated insulin secretion from islets isolated from T2D mice and humans (58–60). Combined with the potential maladaptive expression of BAI3 in obese and/or diabetic conditions, our data suggest the C1ql3/BAI3 signaling pathway as a novel target for T2D therapeutics.
Experimental procedures
Antibodies and chemicals
Insulin standards and anti-insulin antibodies used in ELISA were purchased from EMD-Millipore and Fitzgerald Industries, respectively. The metabolites used in insulin secretion studies were purchased from Sigma: dimethyl-2-oxoglutarate (αKG, 349631), l-glutamic acid dimethyl ester hydrochloride (glutamate, 49560), 1-monopalmitoleoyl-rac-glycerol (M7890), dimethyl (S)-(−)-malate (374318), sodium 4-methyl-2-oxovalerate (αKIC, K0629), dl-isocitric acid trisodium salt hydrate (I1252), l-glutamine (G3126), l-leucine (L8000), l-alanine (A7627), tolbutamide (T0891), 2-bromohexadecanoic acid (21604), 8-bromoadenosine 3′,5′-cyclic monophosphate sodium salt (B7880), and sodium palmitate (P9767) conjugated to BSA (Fitzgerald, 30-AB79). Exendin-4 was from Med Chem Expres (HY-13443). The anti-FLAG, anti-IgG control, and secondary antibodies were from Cell Signaling Technology (CST). Antibody for C1ql3 (Abcam, ab107006), GFP (Abcam, ab13970), BAI3 (Sigma, SAB4502524), Glut2 (Santa Cruz, sc7580), actin (DSHB, JLA20-s), caspase-3 (Cell Signaling, 9661S), PKA substrate (Cell Signaling, 9624S), and total caspase-3 (Cell Signaling, 14220S) were used in Western blotting or immunofluorescence. Soluble BAI3 and human C1ql3 recombinant proteins were purchased from R & D Systems. Selective Epac2 agonist (Tocris, 4853: 8-pCPT-2-O-Me-cAMP-AM) and PKA agonist (Tocris, 5255: 6-Bnz-cAMP sodium salt) were purchased from Tocris. siRNA oligos for knockdown experiments were from Sigma (MISSION® siRNA Universal, Negative Control #1, and siBAI3 (SIC001)).
Expression constructs
The FLAG-tagged C1ql3 mammalian expression plasmid was purchased from OriGene. The untagged C1ql3 mammalian expression plasmid was generated by cloning C1ql3 cDNA (ATCC) into an Moloney murine leukemia virus-based lentiviral vector (3565) (a gift from Dr. Bill Sugden, University of Wisconsin-Madison, WI). The BAI3-mVenus expression plasmid was a gift from Dr. Thomas Südhof (Stanford University, CA)
Cell culture and transient transfection
INS1(832/13) pancreatic β-cells (gift from Dr. Christopher Newgard, Duke University, NC) were cultured in supplemental RPMI 1640 media containing 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 1 mm sodium pyruvate, 10 mm HEPES, and 100 units/ml of antimycotic-antibiotic along with 50 μm β-mercaptoethanol. Approximately, 500,000 and 100,000 cells were plated in each well of 24- and 96-well plates, respectively. Next day, ∼75–80% confluent cells were transfected with 20 μm siScramble (Control) or siBAI3 using Lipofectamine 2000 (Invitrogen). After 36 h, cells were harvested for RNA or static insulin secretion analysis.
The Hek293FT cells were cultured in Dulbecco's modified Eagle's medium high glucose culture media containing 25 mm glucose, 10% FBS, 0.1 mm nonessential amino acids, 1 mm sodium pyruvate, 6 mm l-glutamine, 100 units/ml of penicillin and streptomycin. The Hek293FT cells were plated at the density of 70–80% confluence before they were transfected with 10 μg of plasmid expressing C1ql3 and GFP by using Lipofectamine 2000. After 6 h, transfection media (Opti-MEM) was removed and replaced with the supplemented RPMI 1640 containing 3 mm glucose without antibiotics. After 36 h, C1ql3 and control conditioned media were used in the experiments.
Mouse islets were isolated from 10–12–week-old C57B6/J male or female mice using a collagenase digestion method (61). Isolated islets were cultured overnight in supplemented RPMI 1640 containing 8 mm glucose. After 16 h, size-matched islets were hand-picked for static insulin secretion assay. Human islets were obtained from our collaborator Dr. Appakalai N. Balamurugan, University of Louisville, KY. Human islets were cultured overnight in supplemented RPMI 1640 culture media containing 8 mm glucose. Next day, the insulin secretion experiment was performed using these islets.
Insulin secretion in INS1(832/13), mouse and human islets
Static insulin secretion in INS1(832/13) pancreatic β-cells was performed in Krebs-Ringer bicarbonate-based buffer (KRB: 118.41 mm NaCl, 4.69 mm KCl, 1.18 mm MgSO4, 1.18 mm KH2PO4, 25 mm NaHCO3, 20 mm HEPES, 2.52 mm CaCl2, pH 7.4, and 0.2% BSA) containing 1.5 mm glucose as described previously (62). Briefly, INS1(832/13) cells were preincubated in 100 μl of KRB-HEPES–based buffer containing 1.5 mm glucose for 2 h. Next, the preincubation buffer was aspirated and replaced with 100 μl of KRB-HEPES–based incubation buffer containing 1.5 or 15 mm glucose. Insulin secreted in the incubation media in response to treatment was quantitated by using in-house insulin ELISA and was expressed as a percent of the total insulin content (secreted plus cellular insulin).
Six size-matched mouse islets were handpicked for secretion assay into each well of a 96-well plate. The procedure was performed as described previously (61). Briefly, islets were preincubated in 100 μl of RPMI 1640 containing 3 mm glucose for 45 min. Next, the preincubation media was removed and replaced with C1ql3 or control conditioned media along with insulin secretagogues for another 45 min. Culture media and islets were processed to determine insulin levels by using in-house ELISA. For the insulin content, islets harvested by using acid-ethanol and the abundance of insulin was estimated by using ELISA. The insulin secreted was normalized to the total insulin (secreted plus content) and the data are represented in this manuscript as a percent of total. Insulin secretion experiments in human islets were performed by using 20 islets.
Isolation and quantitation of RNA
Total mRNA was extracted from mouse tissues, human islets, mouse islets, β-cells, INS1(832/13), and Hek293FT cells by using Qiagen RNeasy Plus Kit. Following extraction, 1 μg of RNA was used for cDNA synthesis (Applied Biosystems). The relative mRNA abundance was determined by quantitative PCR using Fast Start SYBR Green (Applied Biosystems) and gene expression was calculated by the comparative ΔCt method. The data are presented as ΔCt, which is calculated by subtracting the Ct of the test gene from Ct of the β-actin (reference).
Isolation and quantitation of protein
Cells were lysed in 20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm Na2EDTA, 1 mm EGTA, 1% Triton, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture tablet (Roche Applied Science). The extraction of soluble proteins, quantitation, and Western blot analysis was performed as described previously (63).
cAMP production assay
Islets were isolated and preincubated in RPMI 1640 as described above for the insulin secretion assay. For each replicate, 10 islets were picked into each well of a 96-well plate containing 100 μl of C1ql3 or control conditioned media with 200 μm of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (Sigma) and the indicated concentrations of glucose. After 45 min, the islets were pelleted by a centrifugal pulse, and the conditioned media was removed. Islets were immediately lysed for cAMP ELISA according to the manufacturer's protocol (cAMP BiotrakTM enzyme immunoassay system; GE Healthcare). Each sample treatment was measured in triplicates and was averaged to calculate the mean ± S.E.
Cell viability measurement
The CellTiter-Glo Luminescent Cell Viability Assay (Promega Corp., Madison, WI) was used according to the manufacturer's protocol to measure ATP as an indicator of cell viability. Briefly, islets were isolated and preincubated in RPMI 1640 as described above for insulin secretion assays. For each treatment 10 islets were picked into each well of a 96-well plate containing 100 μl of C1ql3 or control conditioned media and the indicated concentrations of glucose. After 45 min, the islets were pelleted by a centrifugal pulse, and the conditioned media was removed. Islets were immediately processed for ATP or ATP/ADP ratio measurements according to the manufacturer's protocol.
ATP/ADP ratio measurement
The ATP to ADP ratio was measured using the EnzyLightTM Assay Kit (ELDT006, BioAssay Systems) for bioluminescent assay according to the manufacturer's protocol. Briefly, islets were isolated and preincubated in RPMI 1640 as described above for insulin secretion assays. For each treatment, 60 islets were picked into each well of a 96-well plate containing 100 μl of C1ql3 or control conditioned media and the indicated concentrations of glucose. After 45 min, the islets were pelleted by a centrifugal pulse, and the conditioned media was removed. Islets were immediately processed for the estimation of the ATP to ADP ratio measurements according to the manufacturer's protocol.
Immunohistochemical analysis
The immunohistochemical study was conducted using INS1 (832/13) cells or human embryonic kidney (Hek293FT) cells. Cells were grown on coverslips coated with poly-d-lysine. The cells are then fixed with a 4% paraformaldehyde in phosphate-buffered saline (PBS) solution, blocked with 10% normal donkey serum for 1 h. Blocking buffer was washed with 0.1% bovine serum albumin (BSA) in PBS with 0.01% sodium azide. A primary antibody was diluted in 2% BSA in PBS containing 0.3% Triton X-100 and 0.01% sodium azide. The antibody was added to the coverslips overnight at 4 °C. Next day, the primary antibody was washed and the secondary antibody from Jackson ImmunoResearch (1:500) was applied to the coverslips for 1 h at the room temperature. Finally, the coverslip was mounted on a microscope slide with a mounting buffer containing DAPI. The slides were analyzed by fluorescent or confocal microscope.
PKA activity assay
Islets were isolated and preincubated in RPMI 1640 as described above for insulin secretion assays. For each treatment, 300 islets were picked into a microcentrifuge tube containing 1 ml of C1ql3 or control conditioned media or human c1ql3 recombinant protein (hC1ql3) with 11 mm glucose in the presence or absence of cAMP. After 30 min, the islets were pelleted by a centrifugal pulse, and the media was removed. Islets were washed quickly using ice-cold PBS and immediately lysed for protein using protein lysis buffer as described above. These samples were then analyzed using Western blotting using phosphor-PKA (pPKA) antibody and the relative abundance of proteins was quantified by ImageJ.
Caspase activity measurement
Islets were isolated and preincubated in RPMI 1640 as described above for insulin secretion assays. For each treatment, 300 islets were picked from a microcentrifuge tube containing 1 ml of C1ql3 or control conditioned media or human C1ql3 recombinant protein (hC1ql3) at 11 mm glucose in the presence or absence of cAMP. After 30 min, the islets were pelleted by a centrifugal pulse, and the media was removed. Islets were washed quickly using ice-cold PBS and immediately lysed for protein using protein lysis buffer as described above. These samples were then analyzed by the Western blotting by using total caspase-3 and cleaved caspase-3 antibody.
Statistical analysis
Data were expressed as mean ± S.E. Statistical comparisons were made using Student's t test at p < 0.05.
Author contributions
R. G., D. C. N., and M. D. S. data curation; R. G. and S. B. investigation; R. G. visualization; R. G., J. E. K., and S. B. methodology; X. L., A. N. B., G. W. W., J.-a. K., and M. E. K. resources; J. E. K., M. E. K., and S. B. writing-review and editing; S. B. conceptualization; S. B. formal analysis; S. B. supervision; S. B. funding acquisition; S. B. validation; S. B. writing-original draft; S. B. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Alan Attie and his laboratory co-workers, Dr. Mark Keller, Mary Rabgalia, and Donnie Stapleton, for their input toward the project and manuscript. We also thank Dr. Sasanka Ramanadham for critical feedback on the project and manuscript.
This work was was supported by National Institutes of Health NLM traineeship Grant T15 LM007359 (to J. E. K.). This work was also supported by National Institutes of Health NIDDK Grant 4 R00 DK95975-03 and Diabetes Research Center (DRC) Grant P30 DK079626 (to S. B.); National Institutes of Health NHLBI Grant R01HL128695 and DRC Grant P30 DK079626 (to J. K.); Postdoctoral fellowship ADA1–18-PDF-103 from the American Diabetes Association (ADA) (to R. G.); and National Institutes of Health NIDDK DK084171 (to G. W. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
- T2D
- type 2 diabetes
- C1q
- complement 1q
- TNF
- tumor necrosis factor
- CTRP
- C1q/TNF-related protein
- C1q13
- complement 1q-like-3
- BAI-3
- brain-specific angiogenesis inhibitor-3
- GPCR
- G protein–coupled receptor
- GSIS
- glucose-stimulated insulin secretion
- MAG
- monoacylglycerol
- DAG
- diacylglycerol
- GLP-1
- glucagon-like peptide-1
- 8-CPT-AM
- 8-pCPT-2′-O-Me-cAMP-AM
- 8-Br-cAMP
- 8-bromo-cyclic AMP
- KRB
- Krebs-Ringer bicarbonate
- PKA
- protein kinase A
- DAPI
- 4′,6-diamidino-2-phenylindole
- 6-Bnz
- 6-Bnz-cAMP-AM.
References
- 1. Henquin J. C. (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49, 1751–1760 10.2337/diabetes.49.11.1751 [DOI] [PubMed] [Google Scholar]
- 2. Seino S., Shibasaki T., and Minami K. (2011) Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J. Clin. Invest. 121, 2118–2125 10.1172/JCI45680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Prentki M., Matschinsky F. M., and Madiraju S. R. (2013) Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 18, 162–185 10.1016/j.cmet.2013.05.018 [DOI] [PubMed] [Google Scholar]
- 4. Cerasi E., and Luft R. (1967) The plasma insulin response to glucose infusion in healthy subjects and in diabetes mellitus. Acta Endocrinol. 55, 278–304 10.1530/acta.0.0550278 [DOI] [PubMed] [Google Scholar]
- 5. Davis S. N., Piatti P. M., Monti L., Brown M. D., Branch W., Hales C. N., and Alberti K. G. (1993) Proinsulin and insulin concentrations following intravenous glucose challenges in normal, obese, and non-insulin-dependent diabetic subjects. Metabolism 42, 30–35 10.1016/0026-0495(93)90168-N [DOI] [PubMed] [Google Scholar]
- 6. Heinitz S., Piaggi P., Bogardus C., and Krakoff J. (2018) Decline in the acute insulin response in relationship to plasma glucose concentrations. Diabetes Metab. Res. Rev. 34, 10.1002/dmrr.2953 10.1002/dmrr.2953 [DOI] [PubMed] [Google Scholar]
- 7. Wong G. W., Krawczyk S. A., Kitidis-Mitrokostas C., Revett T., Gimeno R., and Lodish H. F. (2008) Molecular, biochemical and functional characterizations of C1q/TNF family members: adipose-tissue-selective expression patterns, regulation by PPAR-γ agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem. J. 416, 161–177 10.1042/BJ20081240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Seldin M. M., Tan S. Y., and Wong G. W. (2014) Metabolic function of the CTRP family of hormones. Rev. Endocr. Metab. Disord. 15, 111–123 10.1007/s11154-013-9255-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghai R., Waters P., Roumenina L. T., Gadjeva M., Kojouharova M. S., Reid K. B., Sim R. B., and Kishore U. (2007) C1q and its growing family. Immunobiology 212, 253–266 10.1016/j.imbio.2006.11.001 [DOI] [PubMed] [Google Scholar]
- 10. Schäffler A., and Buechler C. (2012) CTRP family: linking immunity to metabolism. Trends Endocrinol. Metab. 23, 194–204 10.1016/j.tem.2011.12.003 [DOI] [PubMed] [Google Scholar]
- 11. Wong G. W., Krawczyk S. A., Kitidis-Mitrokostas C., Ge G., Spooner E., Hug C., Gimeno R., and Lodish H. F. (2009) Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 23, 241–258 10.1096/fj.08-114991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wei Z., Peterson J. M., Lei X., Cebotaru L., Wolfgang M. J., Baldeviano G. C., and Wong G. W. (2012) C1q/TNF-related protein-12 (CTRP12), a novel adipokine that improves insulin sensitivity and glycemic control in mouse models of obesity and diabetes. J. Biol. Chem. 287, 10301–10315 10.1074/jbc.M111.303651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sigoillot S. M., Iyer K., Binda F., Gonzalez-Calvo I., Talleur M., Vodjdani G., Isope P., and Selimi F. (2015) The secreted protein C1QL1 and its receptor BAI3 control the synaptic connectivity of excitatory inputs converging on cerebellar purkinje cells. Cell Rep. S2211–1247(15)00059–5 10.1016/j.celrep.2015.01.034 [DOI] [PubMed] [Google Scholar]
- 14. Bolliger M. F., Martinelli D. C., and Südhof T. C. (2011) The cell-adhesion G protein-coupled receptor BAI3 is a high-affinity receptor for C1q-like proteins. Proc. Natl. Acad. Sci. U.S.A. 108, 2534–2539 10.1073/pnas.1019577108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bayerdörffer E., Miehlke S., and Mannes G. A. (1994) Helicobacter pylori eradication therapy with omeprazole and amoxicillin: current status. Leber Magen. Darm. 14, 228–232 [PubMed] [Google Scholar]
- 16. Wei Z., Peterson J. M., and Wong G. W. (2011) Metabolic regulation by C1q/TNF-related protein-13 (CTRP13): activation OF AMP-activated protein kinase and suppression of fatty acid-induced JNK signaling. J. Biol. Chem. 286, 15652–15665 10.1074/jbc.M110.201087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Byerly M. S., Swanson R., Wei Z., Seldin M. M., McCulloh P. S., and Wong G. W. (2013) A central role for C1q/TNF-related protein 13 (CTRP13) in modulating food intake and body weight. PLoS ONE 8, e62862 10.1371/journal.pone.0062862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shanaki M., Fadaei R., Moradi N., Emamgholipour S., and Poustchi H. (2016) The circulating CTRP13 in type 2 diabetes and non-alcoholic fatty liver patients. PLoS ONE 11, e0168082 10.1371/journal.pone.0168082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fadaei R., Moradi N., Baratchian M., Aghajani H., Malek M., Fazaeli A. A., and Fallah S. (2016) Association of C1q/TNF-related protein-3 (CTRP3) and CTRP13 serum levels with coronary artery disease in subjects with and without type 2 diabetes mellitus. PLoS ONE 11, e0168773 10.1371/journal.pone.0168773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martinelli D. C., Chew K. S., Rohlmann A., Lum M. Y., Ressl S., Hattar S., Brunger A. T., Missler M., and Südhof T. C. (2016) Expression of C1ql3 in discrete neuronal populations controls efferent synapse numbers and diverse behaviors. Neuron 91, 1034–1051 10.1016/j.neuron.2016.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kee H. J., Ahn K. Y., Choi K. C., Won Song J., Heo T., Jung S., Kim J. K., Bae C. S., and Kim K. K. (2004) Expression of brain-specific angiogenesis inhibitor 3 (BAI3) in normal brain and implications for BAI3 in ischemia-induced brain angiogenesis and malignant glioma. FEBS Lett. 569, 307–316 10.1016/j.febslet.2004.06.011 [DOI] [PubMed] [Google Scholar]
- 22. Hamoud N., Tran V., Croteau L. P., Kania A., and Côté J. F. (2014) G-protein coupled receptor BAI3 promotes myoblast fusion in vertebrates. Proc. Natl. Acad. Sci. U.S.A. 111, 3745–3750 10.1073/pnas.1313886111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Collins M. O., Husi H., Yu L., Brandon J. M., Anderson C. N., Blackstock W. P., Choudhary J. S., and Grant S. G. (2006) Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J. Neurochem. 97, 16–23 10.1111/j.1471-4159.2005.03507.x [DOI] [PubMed] [Google Scholar]
- 24. Kakegawa W., Mitakidis N., Miura E., Abe M., Matsuda K., Takeo Y. H., Kohda K., Motohashi J., Takahashi A., Nagao S., Muramatsu S., Watanabe M., Sakimura K., Aricescu A. R., and Yuzaki M. (2015) Anterograde C1ql1 signaling is required in order to determine and maintain a single-winner climbing fiber in the mouse cerebellum. Neuron 85, 316–329 10.1016/j.neuron.2014.12.020 [DOI] [PubMed] [Google Scholar]
- 25. Duman J. G., Tu Y. K., and Tolias K. F. (2016) Emerging roles of BAI adhesion-GPCRs in synapse development and plasticity. Neural Plast. 2016, 8301737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ashcroft F. M., and Rorsman P. (2013) K(ATP) channels and islet hormone secretion: new insights and controversies. Nat. Rev. Endocrinol. 9, 660–669 10.1038/nrendo.2013.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tomlinson B., Hu M., Zhang Y., Chan P., and Liu Z. M. (2016) An overview of new GLP-1 receptor agonists for type 2 diabetes. Expert Opin. Investig. Drugs 25, 145–158 10.1517/13543784.2016.1123249 [DOI] [PubMed] [Google Scholar]
- 28. Nicholls D. G. (2016) The pancreatic β-cell: a bioenergetic perspective. Physiol. Rev. 96, 1385–1447 10.1152/physrev.00009.2016 [DOI] [PubMed] [Google Scholar]
- 29. Rutter G. A., Pullen T. J., Hodson D. J., and Martinez-Sanchez A. (2015) Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem. J. 466, 203–218 10.1042/BJ20141384 [DOI] [PubMed] [Google Scholar]
- 30. Ramos L. S., Zippin J. H., Kamenetsky M., Buck J., and Levin L. R. (2008) Glucose and GLP-1 stimulate cAMP production via distinct adenylyl cyclases in INS-1E insulinoma cells. J. Gen. Physiol. 132, 329–338 10.1085/jgp.200810044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tengholm A. (2012) Cyclic AMP dynamics in the pancreatic β-cell. Ups J. Med. Sci. 117, 355–369 10.3109/03009734.2012.724732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seino S., and Shibasaki T. (2005) PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol. Rev. 85, 1303–1342 10.1152/physrev.00001.2005 [DOI] [PubMed] [Google Scholar]
- 33. Furman B., Ong W. K., and Pyne N. J. (2010) Cyclic AMP signaling in pancreatic islets. Adv. Exp. Med. Biol. 654, 281–304 10.1007/978-90-481-3271-3_13 [DOI] [PubMed] [Google Scholar]
- 34. Seino S. (2012) Cell signalling in insulin secretion: the molecular targets of ATP, cAMP and sulfonylurea. Diabetologia 55, 2096–2108 10.1007/s00125-012-2562-9 [DOI] [PubMed] [Google Scholar]
- 35. Ressl S., Vu B. K., Vivona S., Martinelli D. C., Südhof T. C., and Brunger A. T. (2015) Structures of C1q-like proteins reveal unique features among the C1q/TNF superfamily. Structure 23, 688–699 10.1016/j.str.2015.01.019 [DOI] [PubMed] [Google Scholar]
- 36. Tian G., Sandler S., Gylfe E., and Tengholm A. (2011) Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes 60, 1535–1543 10.2337/db10-1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim J. W., Roberts C. D., Berg S. A., Caicedo A., Roper S. D., and Chaudhari N. (2008) Imaging cyclic AMP changes in pancreatic islets of transgenic reporter mice. PLoS ONE 3, e2127 10.1371/journal.pone.0002127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Landa L. R. Jr, Harbeck M., Kaihara K., Chepurny O., Kitiphongspattana K., Graf O., Nikolaev V. O., Lohse M. J., Holz G. G., and Roe M. W. (2005) Interplay of Ca2+ and cAMP signaling in the insulin-secreting MIN6 beta-cell line. J. Biol. Chem. 280, 31294–31302 10.1074/jbc.M505657200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dyachok O., Idevall-Hagren O., Sågetorp J., Tian G., Wuttke A., Arrieumerlou C., Akusjärvi G., Gylfe E., and Tengholm A. (2008) Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab. 8, 26–37 10.1016/j.cmet.2008.06.003 [DOI] [PubMed] [Google Scholar]
- 40. Doyle M. E., and Egan J. M. (2007) Mechanisms of action of glucagon-like peptide 1 in the pancreas. Pharmacol. Ther. 113, 546–593 10.1016/j.pharmthera.2006.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miura Y., and Matsui H. (2003) Glucagon-like peptide-1 induces a cAMP-dependent increase of [Na+]i associated with insulin secretion in pancreatic β-cells. Am. J. Physiol. Endocrinol. Metab. 285, E1001–E1009 10.1152/ajpendo.00005.2003 [DOI] [PubMed] [Google Scholar]
- 42. Nenquin M., and Henquin J. C. (2016) Sulphonylurea receptor-1, sulphonylureas and amplification of insulin secretion by Epac activation in β cells. Diabetes Obes. Metab. 18, 698–701 10.1111/dom.12607 [DOI] [PubMed] [Google Scholar]
- 43. Zhang C. L., Katoh M., Shibasaki T., Minami K., Sunaga Y., Takahashi H., Yokoi N., Iwasaki M., Miki T., and Seino S. (2009) The cAMP sensor Epac2 is a direct target of antidiabetic sulfonylurea drugs. Science 325, 607–610 10.1126/science.1172256 [DOI] [PubMed] [Google Scholar]
- 44. Herbst K. J., Coltharp C., Amzel L. M., and Zhang J. (2011) Direct activation of Epac by sulfonylurea is isoform selective. Chem. Biol. 18, 243–251 10.1016/j.chembiol.2010.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jewell J. L., Oh E., and Thurmond D. C. (2010) Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R517–R531 10.1152/ajpregu.00597.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Traub S., Meier D. T., Schulze F., Dror E., Nordmann T. M., Goetz N., Koch N., Dalmas E., Stawiski M., Makshana V., Thorel F., Herrera P. L., Böni-Schnetzler M., and Donath M. Y. (2017) Pancreatic α cell-derived glucagon-related peptides are required for β cell adaptation and glucose homeostasis. Cell Rep. 18, 3192–3203 10.1016/j.celrep.2017.03.005 [DOI] [PubMed] [Google Scholar]
- 47. Vasu S., Moffett R. C., Thorens B., and Flatt P. R. (2014) Role of endogenous GLP-1 and GIP in β cell compensatory responses to insulin resistance and cellular stress. PLoS ONE 9, e101005 10.1371/journal.pone.0101005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Johnson A. D., and O'Donnell C. J. (2009) An open access database of genome-wide association results. BMC Med. Genet. 10, 6 10.1186/1471-2350-10-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hayes M. G., Pluzhnikov A., Miyake K., Sun Y., Ng M. C., Roe C. A., Below J. E., Nicolae R. I., Konkashbaev A., Bell G. I., Cox N. J., and Hanis C. L. (2007) Identification of type 2 diabetes genes in Mexican Americans through genome-wide association studies. Diabetes 56, 3033–3044 10.2337/db07-0482 [DOI] [PubMed] [Google Scholar]
- 50. Brunton S. (2014) GLP-1 receptor agonists vs. DPP-4 inhibitors for type 2 diabetes: is one approach more successful or preferable than the other? Int. J. Clin. Pract. 68, 557–567 10.1111/ijcp.12361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Halimi S. (2008) DPP-4 inhibitors and GLP-1 analogues: for whom? which place for incretins in the management of type 2 diabetic patients? Diabetes Metab. 34, S91–95 10.1016/S1262-3636(08)73400-1 [DOI] [PubMed] [Google Scholar]
- 52. Ross S. A., and Ballantine J. (2013) Early use of glucagon-like peptide-1 receptor agonists (GLP-1 RAs) in type 2 diabetes. Curr. Med. Res. Opin. 29, 1617–1626 10.1185/03007995.2013.837817 [DOI] [PubMed] [Google Scholar]
- 53. Pratley R. E., and Gilbert M. (2008) Targeting incretins in type 2 diabetes: role of GLP-1 receptor agonists and DPP-4 inhibitors. Rev. Diabet. Stud. 5, 73–94 10.1900/RDS.2008.5.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pinkney J., Fox T., and Ranganath L. (2010) Selecting GLP-1 agonists in the management of type 2 diabetes: differential pharmacology and therapeutic benefits of liraglutide and exenatide. Ther. Clin. Risk Manag. 6, 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aschner P., Kipnes M. S., Lunceford J. K., Sanchez M., Mickel C., Williams-Herman D. E., and Sitagliptin Study 021 Group (2006) Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 29, 2632–2637 10.2337/dc06-0703 [DOI] [PubMed] [Google Scholar]
- 56. Raz I., Hanefeld M., Xu L., Caria C., Williams-Herman D., Khatami H., and Sitagliptin Study 023 Group (2006) Efficacy and safety of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy in patients with type 2 diabetes mellitus. Diabetologia 49, 2564–2571 10.1007/s00125-006-0416-z [DOI] [PubMed] [Google Scholar]
- 57. Collet-Gaudillat C., Petit-Aubert G., Desforges-Bullet V., and Beressi J. P. (2012) Exenatide efficacy in unselected patients: comparison with clinical trials. J. Diabet. Mellitus 2, 118–121 10.4236/jdm.2012.21019 [DOI] [Google Scholar]
- 58. Kimple M. E., Keller M. P., Rabaglia M. R., Pasker R. L., Neuman J. C., Truchan N. A., Brar H. K., and Attie A. D. (2013) Prostaglandin E2 receptor, EP3, is induced in diabetic islets and negatively regulates glucose- and hormone-stimulated insulin secretion. Diabetes 62, 1904–1912 10.2337/db12-0769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Neuman J. C., Schaid M. D., Brill A. L., Fenske R. J., Kibbe C. R., Fontaine D. A., Sdao S. M., Brar H. K., Connors K. M., Wienkes H. N., Eliceiri K. W., Merrins M. J., Davis D. B., and Kimple M. E. (2017) Enriching islet phospholipids with eicosapentaenoic acid reduces prostaglandin E2 signaling and enhances diabetic β-cell function. Diabetes 66, 1572–1585 10.2337/db16-1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kimple M. E., Neuman J. C., Linnemann A. K., and Casey P. J. (2014) Inhibitory G proteins and their receptors: emerging therapeutic targets for obesity and diabetes. Exp. Mol. Med. 46, e102 10.1038/emm.2014.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bhatnagar S., Oler A. T., Rabaglia M. E., Stapleton D. S., Schueler K. L., Truchan N. A., Worzella S. L., Stoehr J. P., Clee S. M., Yandell B. S., Keller M. P., Thurmond D. C., and Attie A. D. (2011) Positional cloning of a type 2 diabetes quantitative trait locus; tomosyn-2, a negative regulator of insulin secretion. PLoS Genet. 7, e1002323 10.1371/journal.pgen.1002323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bhatnagar S., Soni M. S., Wrighton L. S., Hebert A. S., Zhou A. S., Paul P. K., Gregg T., Rabaglia M. E., Keller M. P., Coon J. J., and Attie A. D. (2014) Phosphorylation and degradation of tomosyn-2 de-represses insulin secretion. J. Biol. Chem. 289, 25276–25286 10.1074/jbc.M114.575985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bhatnagar S., Damron H. A., and Hillgartner F. B. (2009) Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J. Biol. Chem. 284, 10023–10033 10.1074/jbc.M808818200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.