

Abstract
The Hippo pathway controls cell proliferation, differentiation, and survival by regulating the Yes-associated protein (YAP) transcriptional coactivator in response to various stimuli, including the mechanical environment. The major YAP regulators are the LATS1/2 kinases, which phosphorylate and inhibit YAP. LATS1/2 are activated by phosphorylation on a hydrophobic motif (HM) outside of the kinase domain by MST1/2 and other kinases. Phosphorylation of the HM motif then triggers autophosphorylation of the kinase in the activation loop to fully activate the kinase, a process facilitated by MOB1. The angiomotin family of proteins (AMOT, AMOTL1, and AMOTL2) bind LATS1/2 and promote its kinase activity and YAP phosphorylation through an unknown mechanism. Here we show that angiomotins increase Hippo signaling through multiple mechanisms. We found that, by binding LATS1/2, SAV1, and YAP, angiomotins function as a scaffold that connects LATS1/2 to both its activator SAV1–MST1 and its target YAP. Deletion of all three angiomotins reduced the association of LATS1 with SAV1–MST1 and decreased MST1/2-mediated LATS1/2-HM phosphorylation. Angiomotin deletion also reduced LATS1/2's ability to associate with and phosphorylate YAP. In addition, we found that angiomotins have an unexpected function along with MOB1 to promote autophosphorylation of LATS1/2 on the activation loop motif independent of HM phosphorylation. These results indicate that angiomotins enhance Hippo signaling by stimulating LATS1/2 autophosphorylation and by connecting LATS1/2 with both its activator SAV1–MST1/2 and its substrate YAP.
Keywords: Hippo pathway; LATS (Warts, Wts); mechanotransduction; cell signaling; Yes-associated protein (YAP); protein kinase; mammalian sterile 20-like kinase 1 (MST1); mammalian sterile 20-like kinase 2 (MST2); Salvador (Sav); scaffold protein; AMOT; angiomotin; MOB1; SAV1
Introduction
The Hippo pathway regulates the transcriptional coactivators YAP2 and TAZ (hereafter referred to as YAP) to control cell fate decisions regarding cell proliferation, survival, and differentiation (1). Misregulation of the Hippo–YAP pathway is commonly associated with cancer (2). The LATS1 and LATS2 kinases (LATS1/2) are major regulators of YAP. LATS1/2 phosphorylate YAP to trigger its cytoplasmic retention and degradation (3–5). In the canonical Hippo pathway, the MST1 and MST2 kinases (MST1/2) activate LATS1/2 (6). LATS1/2 can also be activated by MAP4K family and TAO kinases (7–9). All upstream kinases phosphorylate LATS1/2 on a conserved hydrophobic motif (HM; Thr1079 and Thr1041 in LATS1 and LATS2, respectively). LATS1/2-HM phosphorylation stimulates it to autophosphorylate in the activation loop (AL: Ser909 and Ser872 in LATS1 and LATS2, respectively) and become fully active (6, 10, 11). This process is facilitated by the MOB1 protein, which enhances LATS1/2-HM phosphorylation by MST1/2 and stimulates LATS1/2-AL autophosphorylation (10–12). MST1/2 phosphorylation of LATS1/2 is also stimulated by colocalization of both proteins on the membrane, which is promoted by SAV1 and NF2, respectively (13). In addition, SAV1 can function as a scaffold because it can bind both MST1/2 and LATS1/2 (14, 15). Although there are numerous other proteins that can promote Hippo signaling, their mechanism of action is generally not well-understood.
A major regulator of Hippo signaling is the mechanical environment, which includes factors such as cell density, substrate stiffness, and cell stretch or mechanical tension. How these mechanical stimuli act through the core Hippo regulators to control YAP activity is not clear. Several studies have shown that the actin cytoskeleton, which responds dynamically to mechanical changes, is a major regulator of Hippo signaling (16–20). Reduction in F-actin activates LATS1/2. How F-actin controls LATS1/2 activity is not certain but may involve angiomotins (AMOT, AMOTL1, and AMOTL2) (21–23). Angiomotins bind to both LATS1/2 and YAP and can inhibit YAP through two mechanisms: binding and retention of YAP in the cytoplasm/plasma membrane and activation of LATS1/2 (21, 24–27). Other studies have shown that AMOT can control YAP nuclear/cytoplasmic localization, depending on its phosphorylation state (28). Angiomotins also bind the LATS1/2 activator NF2 (29, 30), and a study using proximity labeling found that SAV1 and MOB1 may interact with AMOT (31). We and others have proposed that angiomotins may act as scaffolds to promote LATS1/2 activation. A conserved N-terminal region in the long form of AMOT (AMOT130, hereafter referred to as AMOT) contains an F-actin binding motif and flanking L/PPXY sites that bind WW-containing proteins (such as YAP). We have shown previously that F-actin and YAP compete for binding to angiomotins (22), which may provide one mechanism for how F-actin can influence YAP activity; when F-actin levels go down, angiomotins are free to bind and inhibit YAP in the cytoplasm and possibly activate LATS1/2. Angiomotins are required for relocalization of YAP from the nucleus to the cytoplasm when F-actin is disrupted (22). In addition, elimination of angiomotin regulation of YAP causes patterning defects in early mouse embryos (26, 27), transformation of Madin-Darby canine kidney cells (25), and epithelial to mesenchymal transition in MCF10A cells (32). How angiomotins activate LATS1/2 is not known. Here we show that the angiomotin protein AMOT interacts with multiple core Hippo pathway regulators to stimulate LATS1/2 to phosphorylate YAP via three distinct mechanisms. First, AMOT promotes interaction between LATS1/2 and its activators MST1/2 and SAV1. Second, AMOT enhances LATS1/2 phosphorylation of YAP by promoting interaction between the two proteins. And third, AMOT collaborates with MOB1 to enhance LATS1/2-AL autophosphorylation, even in the absence LATS-HM site phosphorylation.
Results
Angiomotins promote LATS1/2 activation
Phosphorylation at the LATS2-HM site is presumed to be the primary regulatory site for LATS1/2 because, when phosphorylated, it promotes autophosphorylation in the AL of the kinase to make it fully active (6, 10, 11), allowing it to phosphorylate YAP and other substrates. We tested the effect of angiomotins on phosphorylation at the LATS1/2-HM site. Overexpression of all three angiomotins (AMOT, AMOTL1, and AMOTL2) promoted LATS2-HM phosphorylation (Fig. 1A). (Note that in this study all experiments utilize the long form of AMOT (AMOT130)). Consistent with these observations, deletion of all three angiomotin genes (Amot-3KO) in HEK293 cells using CRISPR (Fig. S1, A and B) resulted in reduced activating phosphorylation of LATS1/2 at the HM and AL sites after F-actin disruption by Latrunculin B (Lat B) treatment (Fig. 1B). Similar results were observed in HeLa cells with all three angiomotins knocked down using siRNA (Fig. S1C). Consistent with our findings, other studies showed that knockdown of angiomotins reduces LATS1/2 activity, as judged by reduced YAP Ser127 phosphorylation in HEK293T cells, Madin-Darby canine kidney cells, and MCF10A cells (21, 25, 32). Together, these findings show that angiomotins can activate LATS1/2 by promoting phosphorylation at the HM site in response to F-actin perturbation.
Figure 1.

Angiomotins require MST1/2, SAV1, and NF2 to activate LATS1/2. A, HEK293 (WT) or HEK293 cells with MST1 and MST2 inactivated using CRISPR (MST1/2-KO) were transfected with a LATS2-FLAG–expressing plasmid and either a control plasmid or a plasmid for expressing Myc-tagged versions of each of the three angiomotin genes (AMOT, AMOTL1, and AMOTL2). Cell lysates were analyzed by Western blotting using antibodies against the LATS1/2-HM phosphorylation site (pLATS2-HM), Myc (Angiomotins-Myc), and FLAG (LATS2-FLAG). Tubulin levels in cell lysates are also shown. B, HEK293 (WT) or HEK293 cells with AMOT, AMOTL1, and AMOTL2 inactivated using CRISPR (Amot-3KO) were treated with or without Lat B, and cell lysates were analyzed by Western blotting using antibodies against LATS1/2-HM phosphorylation, LATS1/2-AL phosphorylation, and LATS1. Tubulin levels in cell lysates are also shown. Quantification of LATS1/2-HM and LATS1/2-AL phosphorylation relative to untreated WT HEK293 cells is shown. Mean ± S.D.; n = 3; NS, p ≥ 0.05; *, p ≤ 0.05; **, p ≤ 0.01; t test. C, HEK293 (WT) or HEK293 cells with NF2 (NF2-KO) or SAV1 (SAV1-KO) inactivated using CRISPR were transfected with a control plasmid or a plasmid for expressing Myc-tagged versions of AMOT or the AMOT-S175E mutant. Cell lysates were analyzed by Western blotting using antibodies against the LATS1/2-HM phosphorylation site (pLATS1/2-HM) and Myc (AMOT-Myc). Tubulin levels in cell lysates are also shown. D, HEK293 (WT) or MST1/2-KO HEK293 cells were transfected with LATS2-FLAG– and AMOT-Myc–expressing plasmids. Myc or control (IgG) antibodies were used for immunoprecipitation (IP) from cell lysates, and immune complexes and cell lysates were analyzed by Western blotting for LATS2-FLAG and AMOT-Myc levels. Quantification of LATS2 levels in AMOT immune complexes is shown. Mean ± S.D.; n = 3; ***, p ≤ 0.001; t test. E, HEK293 (WT), NF2-KO, or SAV1-KO HEK293 cells were transfected with LATS2-FLAG and AMOT-Myc expressing plasmids. Myc or control (IgG) antibodies were used for immunoprecipitations from cell lysates, and immune complexes and cell lysates were analyzed by Western blotting for LATS2-FLAG and AMOT-Myc levels. Quantification of LATS2 levels in AMOT immune complexes is shown. Mean ± S.D.; n = 3; **, p ≤ 0.01; NS, p ≥ 0.05; t test.
To better understand how angiomotins activate LATS1/2, we tested which core Hippo components are required for angiomotins to promote HM phosphorylation. We found that, in HEK293 cells, MST1/2 are the primary LATS1/2-HM kinases because deletion of MST1/2 (Fig. S1D) causes a major decrease in HM phosphorylation in unperturbed and Lat B– and okadaic acid–treated cells (Fig. S1, E and F). Consistent with this observation, the increase LATS2-HM phosphorylation after angiomotin overexpression was largely lost in MST1/2-deleted cells (Fig. 1A), suggesting that angiomotin-stimulated LATS2-HM phosphorylation works primarily through MST1/2 in HEK293 cells. Similar results were observed in the absence of SAV1 and NF2 (Fig. 1C and Figure S1D), the MST1/2 and LATS1/2 binding partners, respectively. Note that not even the phosphomimetic (active) form of AMOT (AMOT-175E) is able to activate LATS1/2 in the absence of SAV1 or NF2. These results are consistent with the idea that angiomotins may act together with the two kinase modules (MST-SAV1 and LATS1/2-NF2) to promote LATS1/2 activation.
MST1/2 and SAV1 stimulate AMOT-LATS2 binding
While examining how MST1/2, SAV1, and NF2 contribute to angiomotin activation of LATS1/2, we discovered a potential positive feedback loop whereby MST1/2 and SAV1 promote AMOT-LATS2 binding. When we tested whether MST1/2, SAV1, and NF2 affect AMOT-LATS2 binding, we observed that deletion of MST1/2 or SAV1 greatly reduced binding of AMOT to LATS2 (NF2 deletion reduced binding, but the effect did not reach the level of significance) (Fig. 1, D and E; see Fig. S1G for further antibody controls). One explanation for these results could be that AMOT-LATS2 binding requires LATS2 activity and that MST1/2 and SAV1 are required for LATS2 activation. This would be consistent with our previous study showing that AMOTL2 bound better to WT than kinase-dead LATS2 or LATS2 with both sites of activating phosphorylation mutated to alanine residues (21). We tested whether AMOT had similar binding preferences. This experiment showed that AMOT, like AMOTL2, bound preferentially to WT LATS2 compared with LATS2 with both sites of activating phosphorylation mutated (Fig. 2A). Because LATS2 is known to phosphorylate AMOT on Ser175 (22, 26, 33–35), these results could be explained if either LATS2 phosphorylation of AMOT promotes AMOT-LATS2 binding or if AMOT preferentially binds to the active form of LATS2. Consistent with the first model, we observed, as seen previously (26), that a phosphomimetic version of AMOT (AMOT-175E) binds to (Fig. 2B) and activates LATS2 (Fig. S2A) better than the nonphosphorylatable form of AMOT (AMOT-175A). However, because expression of AMOT-175E did not rescue the AMOT-LATS2 binding defect in MST1/2-KO cells, additional factors must contribute to the AMOT-LATS2 binding defect in MST1/2-KO cells (Fig. 2C). Therefore, we tested the second model; namely, that angiomotins bind preferentially to active LATS2. If correct, then expression of a phosphomimetic version of LATS2 (LATS2–1041E) should rescue the defect in AMOT-LATS2 interaction in MST1/2-deleted cells. Indeed, this turned out to be the case (Fig. 2D). These results show that MST1/2 and SAV1 activation of LATS1/2 stimulates a positive feedback mechanism by promoting AMOT-LATS1/2 binding.
Figure 2.

AMOT binds to active forms of LATS2. A, HEK293 cells were transfected with plasmids for expressing AMOT-Myc and either LATS2 or LATS2 with the AL and HM phosphorylation sites (Ser872 and Thr1041) mutated to alanine (LATS2–2A). Myc or control (IgG) antibodies were used for immunoprecipitation (IP) from cell lysates, and immune complexes and cell lysates were analyzed by Western blotting for LATS2-FLAG and AMOT-Myc levels. Quantification of AMOT levels in LATS2 immune complexes is shown. Mean ± S.D.; n = 3; **, p ≤ 0.01; t test). B, HEK293 cells were transfected with plasmids for expressing LATS2-FLAG and either AMOT-Myc, AMOT-175A-Myc, or AMOT-175E-Myc. Immunoprecipitations were performed on each cell lysate with either FLAG (LATS2-FLAG) or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for LATS2-FLAG and AMOT-Myc levels. Quantification of AMOT levels in LATS2 immune complexes is shown. Mean ± S.D.; n = 3; **, p ≤ 0.01; ***, p ≤ 0.001; t test. C, HEK293 cells (WT) or MST1/2-KO cells were transfected with plasmids for expressing LATS2-FLAG and AMOT-175E-Myc. Immunoprecipitations were performed on each cell lysate with either FLAG (LATS2-FLAG) or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for LATS2-FLAG and AMOT-Myc levels. Quantification of AMOT levels in LATS2 immune complexes is shown. Mean ± S.D.; n = 3; ***, p ≤ 0.001; t test. D, HEK293 cells (WT) or MST1/2-KO cells were transfected with plasmids for expressing LATS2-T1041E-FLAG and AMOT-Myc. Immunoprecipitations were performed on cell lysates with either Myc (AMOT-Myc) or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for LATS2-FLAG and AMOT-Myc levels. Quantification of LATS2 levels in AMOT immune complexes is shown. Mean ± S.D.; n = 3; NS, p ≥ 0.05; t test.
AMOT is a Hippo pathway scaffold protein
AMOT could activate LATS1/2 either by stimulating MST1/2 activation or by promoting the ability of MST1/2 to phosphorylate LATS1/2. Our results support the later possibility because AMOT overexpression did not affect the levels of MST1/2-activating phosphorylation (Fig. S2B). (Note that, to remove potential issues with feedback from LATS1/2, we conducted these and subsequent experiments with AMOT-175E.) We next investigated whether AMOT might function as a scaffold to bring LATS1/2 together with its activator MST1/2 and substrate YAP. This and previous studies showed that AMOT binds LATS1/2 (21, 36). Thus, one potential mechanism of action for AMOT could be that it connects LATS1/2 with the MST1/2-SAV1 complex and YAP. Indeed, when we analyzed endogenous AMOT complexes using chromosomally tagged sfGFP-MAP-AMOT (37) in HEK293 cells (created using CRISPR-mediated genome modification), we could detect LATS1, SAV1, MST1, and YAP coming down with sfGFP-MAP-AMOT isolated using streptavidin beads (note that part of the sfGFP-MAP tag is a streptavidin-binding peptide) (Fig. 3A), consistent with endogenous AMOT associating with core Hippo pathway proteins. We next examined whether AMOT might contribute to association of core Hippo pathway proteins by overexpressing AMOT with different combinations of Hippo pathway proteins followed by immunoprecipitation. Because AMOT bound LATS1/2, we tested whether AMOT could stimulate LATS2 activation by also binding MST1. Although we were unable to observe binding between AMOT and MST1 when both proteins were co-expressed (Fig. 3B), AMOT did bind to SAV1 in a manner that depended on the SAV1 WW domains and the 3 L/PPXY motifs (WW domain ligands) in AMOT (Fig. 3C). SAV1 expression allowed MST1 to come down in AMOT immune complexes (Fig. 3B), indicating that SAV1 could bridge interaction between AMOT and MST1. These results support the notion that AMOT could connect LATS2 to SAV1-MST1. However, as observed previously in Drosophila (14), SAV1 was also able to bind LATS2 (Fig. 3D) (presumably by interaction of its WW domain with the PPXY motif in LATS2), showing that it can act as a scaffold for MST1 and LATS2 without AMOT (at least when overexpressed). Interestingly, the amount of SAV1 that came down in LATS2 immune complexes was enhanced by expression of AMOT (Fig. 3D). Similar experiments were also done to examine how the interaction of MST1 with LATS2 might depend on SAV1 and AMOT (Fig. 3E). MST1 and LATS2 were found to not interact unless SAV1 was co-expressed (Fig. 3E). As with the SAV1–LATS2 interaction (Fig. 3D), AMOT alone could not promote interaction between LATS2 and MST1 but was able to enhance the interaction of MST1 and SAV1 with LATS2 (Fig. 3E). Together, these results suggest that AMOT may enhance the assembly/stabilization of a complex between SAV1-MST1 and LATS2. In addition, AMOT may also act as a scaffold to bring LATS1/2 in proximity with its substrate YAP. We observed that, when YAP and LATS2 were co-expressed, YAP was only poorly recovered in LATS2 immune complexes (Fig. 3F) unless AMOT was also expressed. The ability of AMOT to enhance LATS2-YAP binding depended on the L/PPXY motifs in AMOT (Fig. 3F). This result suggested that YAP binding by AMOT might not just inhibit YAP by sequestering it in the cytoplasm as reported previously but might also bring it together with its inhibitory kinase LATS1/2. Consistent with this model, Amot-3KO cells show defects in YAP phosphorylation (Fig. S3A) and are defective in keeping YAP out of the nucleus after growth to high density or serum deprivation (Fig. S3B). We also saw defects in association of endogenous core Hippo pathway proteins in the absence of angiomotins. Although immunoprecipitates of endogenous LATS1 from HEK293 cells contained SAV1 and YAP, both proteins were almost completely absent in LATS1 immunoprecipitates from Amot-3KO cells (Fig. 4A). Similarly, when we immunoprecipitated endogenous SAV1 from HEK293 cells, we could detect MST1, LATS1, AMOT, and YAP in the immune complexes. However, SAV1 immune complexes from Amot-3KO cells contained MST1 but almost no YAP and LATS1 (Fig. 4B). This indicates that, at endogenous protein levels, SAV1 alone cannot efficiently scaffold LATS1/2 with MST1/2. Collectively, these results show that AMOT may regulate LATS1/2 and YAP at least in part by acting as a scaffold to connect LATS1/2 to both its activator SAV1-MST1/2 as well as its target YAP.
Figure 3.

AMOT promotes assembly of LATS2-SAV1-MST1 complexes. A, HEK293 cells expressing sfGFP-AMOT from the chromosomal locus were created using CRISPR-mediated genome modification (see “Experimental procedures”). Streptavidin beads were used to pull down (PD) sfGFP-MAP-AMOT protein (GFP-AMOT) (WT HEK293 cells where the endogenous AMOT locus has not been tagged with the sfGFP-MAP tag were used as a control). Protein complexes on beads were analyzed by Western blotting using antibodies against the indicated proteins. The levels of these proteins in cell lysates are shown. B, HEK293 cells were transfected with plasmids for expressing HA-MST1, HA-SAV1, and AMOT-Myc as indicated. Immunoprecipitation (IP) was performed cell lysates with either Myc (AMOT-Myc) or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for HA-MST1, HA-SAV1, and AMOT-Myc levels. C, HEK293 cells were transfected with plasmids for expressing HA-SAV1 or HA-SAV1 with both WW domains mutated (HA-SAV1–2ww) and either AMOT-Myc or AMOT-3PxY-Myc (eliminates all three L/PPXY motifs in AMOT). Immunoprecipitations were performed on each cell lysate with either Myc (AMOT-Myc) or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for HA-SAV1 and AMOT-Myc levels. D, HEK293 cells were transfected with plasmids for expressing HA-SAV1, LATS2-FLAG, and AMOT-Myc as indicated. Immunoprecipitations were performed on each cell lysate with either FLAG (LATS2-FLAG) or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for LATS2-FLAG, HA-SAV1, and AMOT-Myc levels. Quantification of SAV/LATS2 ratios in LATS2 immune complexes for cells expressing LATS2 and SAV1 (LATS2+S) or LATS2, SAV1, and AMOT (LATS2+A+S) is shown. Mean ± S.D.; n = 3; *, p ≤ 0.05; t test). E, HEK293 cells were transfected with combinations of plasmids for expressing LATS2-FLAG, HA-MST1, HA-SAV1, and AMOT-Myc as indicated. Immunoprecipitations were performed on each cell lysate with either FLAG (LATS2-FLAG) or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for LATS2-FLAG, HA-MST1, HA-SAV1, and AMOT-Myc levels. Quantification of MST1/LATS2 and SAV1/LATS2 ratios in LATS2 immune complexes for cells co-expressing LATS2 and MST1 (L + M); LATS2, MST1, and SAV (L + M + S); LATS2, MST1, and AMOT (L + M + A); and LATS2, MST1, AMOT, and SAV1 (L + M + A + S) is shown. Mean ± S.D.; n = 3; *, p ≤ 0.05; **, p ≤ 0.01; ****, p ≤ 0.0001; t test. NS, not significant. F, HEK293 cells were transfected with combinations of plasmids for expressing GFP-LATS2, FLAG-YAP2, and AMOT-Myc as indicated. Immunoprecipitations were performed on each cell lysate with either GFP (LATS2-GFP) or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for LATS2-GFP, FLAG-YAP2, and AMOT-Myc levels.
Figure 4.
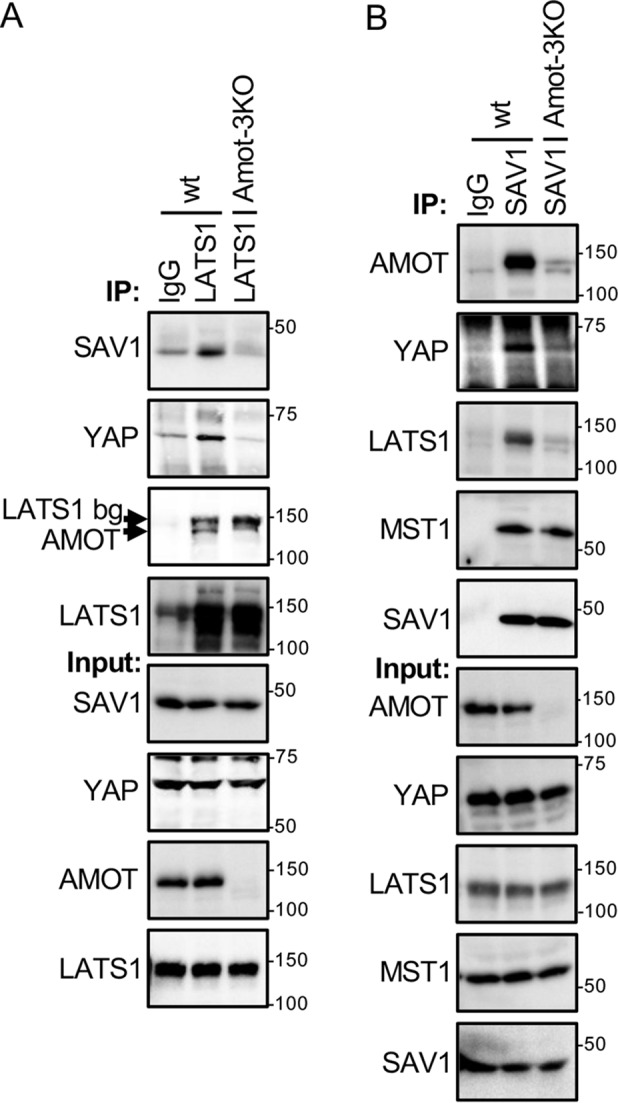
Angiomotins function as scaffolds for the Hippo pathway in vivo. A, HEK293 (WT) and Amot-3KO HEK293 cells were grown to high density and serum-starved for 4 h. Immunoprecipitation (IP) was performed on cell lysates with either anti-LATS1 or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for AMOT, SAV1, LATS1, and YAP levels. LATS1 bg, residual LATS1 signal after stripping. B, HEK293 (WT) and Amot-3KO HEK293 cells were grown as in A. Immunoprecipitations were performed on cell lysates with either anti-SAV1 or control (IgG) antibodies, and immune complexes and cell lysates were analyzed by Western blotting for AMOT, MST1, SAV1, LATS1, and YAP levels.
Angiomotins promote LATS1/2-AL autophosphorylation
The ability of AMOT to promote a complex containing SAV1, MST1, and LATS2 could explain why LATS1/2-activating phosphorylation at the HM site (the site phosphorylated by MST1/2) is reduced in Amot-3KO cells' response to Lat B treatment (Fig. 1B). Similarly, we observed that LATS1/2-HM phosphorylation is reduced in Amot-3KO cells compared with WT when MST1 is overexpressed (Fig. 5A). The defect in LATS2-HM phosphorylation in Amot-3KO cells in response to MST1 overexpression could be rescued by co-overexpression of SAV1 (Fig. 5B), consistent with SAV1 being able to replace the scaffolding function of angiomotins, at least when overexpressed. Interestingly, although Amot-3KO cells overexpressing MST1 and SAV1 had HM phosphorylation levels similar to WT controls expressing MST1 and SAV1, they still had a defect in LATS2 phosphorylation at the AL site (Fig. 5B). These results raised the possibility that angiomotins may promote LATS2 autophosphorylation at the LATS2-AL site, independent of their effect on MST1/2 phosphorylation at the LATS2-HM site. Note that the defect in LATS2-AL phosphorylation in Amot-3KO cells could be rescued by re-expression of the three angiomotin proteins (Fig. S1B). To distinguish between direct effects of AMOT on LATS2-AL phosphorylation and indirect effects via LATS2-HM phosphorylation, we tested whether AMOT could promote AL phosphorylation of a version of LATS2 that mimicked the HM-phosphorylated form (LATS2–1041E) but could not be regulated by MST1/2 (Fig. S4A) or other upstream HM kinases. We used LATS2–1041E because we assumed that AMOT alone might not be able promote LATS2-AL phosphorylation without HM phosphorylation. We found that AMOT expression enhanced AL phosphorylation of LATS2–1041E (Fig. 5C). Thus, AMOT can promote LATS2 autophosphorylation at the AL site independent of any effects it has on LATS2-HM phosphorylation. The only other protein known to stimulate LATS1/2 autophosphorylation is MOB1 (10, 12). We observed that, when co-expressed (Fig. S4B), MOB1A could be detected in AMOT immune complexes, showing that the two proteins might interact. Therefore, we investigated the relationship between MOB1 and AMOT in promoting LATS2-AL phosphorylation. Several lines of evidence suggested that MOB1 may act independent of angiomotins and that the effects of AMOT on LATS1/2-AL phosphorylation may depend on MOB1. For example, MOB1A binding to LATS2 was not affected by deletion of all three angiomotins (Fig. S4C), and overexpression of AMOT was not able to significantly enhance AL phosphorylation of LATS2–1041E, which had a point mutation in the MOB-binding domain (MBD, R657A) (11), rendering it unable to bind MOB1 (Fig. 5D). In addition, strong overexpression of MOB1A could stimulate similar levels of AL phosphorylation of LATS2–1041E in angiomotin-deleted cells compared with WT cells (Figs. 5E). Interestingly, despite having the same levels of LATS-AL phosphorylation, the angiomotin-deleted cells had a significant defect in YAP phosphorylation (Fig. 5E), consistent with our earlier results suggesting a scaffolding role for angiomotins to promote LATS1/2 phosphorylation of YAP. Other evidence suggests that MOB1A and AMOT may have additive or synergistic effects on LATS1/2-AL phosphorylation. When both MOB1A and AMOT were moderately overexpressed (at levels where each alone barely enhanced LATS2-AL phosphorylation), we observed that co-expression of both proteins significantly increased AL phosphorylation of LATS2 (Fig. 5F) beyond that observed for either protein alone. These results prompted us to test whether AMOT and MOB1A could promote AL phosphorylation even in the absence of HM phosphorylation (using LATS2–1041A). We found that, although moderately expressed AMOT and MOB1A alone did not increase AL phosphorylation, expression of both proteins significantly increased AL phosphorylation of LATS2–1041A, albeit to lower levels than with WT LATS2 (Fig. 5F). We think that the AL phosphorylation of LATS2–1041A was due to autophosphorylation and not phosphorylation by another kinase or endogenous LATS1/2 because a LATS2 kinase-dead mutant did not show AL phosphorylation after MOB1A and AMOT expression (Fig. 5F). Together, these experiments indicate that AMOT and MOB1A can promote LATS2 autophosphorylation on the AL site independent of HM phosphorylation by upstream kinases.
Figure 5.

AMOT promotes LATS2-AL activation loop phosphorylation independent of LATS2-HM phosphorylation. A, HEK293 (WT) or Amot-3KO HEK293 cells were transfected with a control plasmid or a plasmid for expressing HA-MST1. Cell lysates were analyzed by Western blotting using antibodies against the LATS1/2-HM phosphorylation site (pLATS1/2-HM), HA (HA-MST1), and LATS1. Tubulin levels in cell lysates are also shown. Quantification of LATS1/2-HM phosphorylation is shown. Mean ± S.D.; n = 3; NS, p ≥ 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ****, p ≤ 0.0001; t test. B, HEK293 (WT) or Amot-3KO KO HEK293 cells were transfected with a control plasmid or a combination of plasmids (L + M + S) for expressing LATS2-FLAG, HA-MST1, and HA-SAV1. Cell lysates were analyzed by Western blotting using antibodies for LATS2-HM and LATS2-AL phosphorylation, FLAG (LATS2-FLAG), and HA (HA-MST1 and HA-SAV1). Tubulin levels in cell lysates are also shown. Quantification of LATS1/2-HM phosphorylation is shown. Mean ± S.D.; n = 3; NS, p ≥ 0.05; **, p ≤ 0.001; t test. C, HEK293 cells were transfected with a control plasmid, LATS2–1041E-FLAG, or AMOT-175E-Myc as indicated. Cell lysates were analyzed by Western blotting using antibodies for LATS2-AL phosphorylation (pLATS2-AL), FLAG (LATS2–1041E-FLAG), and Myc (AMOT-175E-Myc). Tubulin levels in cell lysates are also shown. Quantification of LATS1/2-AL phosphorylation relative to LATS2–1041E is shown. L-1041E, LATS2–1041E; A-175E, AMOT-175E. Mean ± S.D.; n = 3; ****, p ≤ 0.0001; t test. D, HEK293 cells were transfected with a control plasmid or LATS2–1041E-FLAG, LATS2–1041E-MBD-FLAG (MBD is a LATS2 mutant defective for binding MOB1), or AMOT-175E-Myc as indicated. Cell lysates were analyzed by Western blotting using antibodies for LATS2-AL phosphorylation (pLATS2-AL), FLAG (LATS2–1041E-FLAG or LATS2–1041E-MBD-FLAG), and Myc (AMOT-Myc). Tubulin levels in cell lysates are also shown. Quantification of LATS1/2-AL phosphorylation relative to LATS2–1041E-FLAG levels is shown. L-1041E, LATS2–1041E; A, AMOT-175E. Mean ± S.D.; n = 3; NS, p ≥ 0.05; **, p ≤ 0.01; t test). E, HEK293 (WT) or Amot-3KO HEK293 cells were transfected with a plasmid for expressing LATS2–1041E-FLAG and either a control plasmid or a plasmid for expressing Myc-MOB1A. Cell lysates were analyzed by Western blotting using antibodies for LATS2-AL phosphorylation (pLATS2-AL), YAP phosphorylation on Ser127 (pYAP-S127), FLAG (LATS2–1041E-FLAG), and Myc (Myc-MOB1A). Tubulin levels in cell lysates are also shown. Quantification of LATS1/2-AL and pYAP-Ser127 phosphorylation is shown. Mean ± S.D.; n = 3; NS, p ≥ 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001; t test). F, HEK293 cells were transfected with a control plasmid, LATS2-FLAG, LATS2–1041E-FLAG, kinase-dead LATS2 (LATS2-KD-FLAG), AMOT-Myc, or Myc-MOB1A as indicated. Cell lysates were analyzed by Western blotting using antibodies for LATS2-AL phosphorylation (pLATS2-AL), FLAG (LATS2-FLAG), or Myc (AMOT-Myc and Myc-MOB1A). Tubulin levels in cell lysates are also shown. L, LATS2; L-1041A, LATS2–1041A; L-KD, LATS2-kinase dead; A, AMOT-175E; M, MOB1A. Quantification of LATS1/2-AL phosphorylation is shown. Mean ± S.D.; n = 3; NS, p ≥ 0.05; *, p ≤ 0.05; **, p ≤ 0.01; ****, p ≤ 0.0001; t test).
Discussion
Previous studies showed that angiomotins promote LATS1/2 activity and YAP phosphorylation, but the mechanism was not known (21, 25, 32). We show here that angiomotins carry out these functions through multiple modes of action (Fig. 6). Several lines of evidence are consistent with AMOT acting as a scaffolding protein. Previous studies have shown that angiomotins bind LATS1/2, NF2, and YAP (21, 22, 24–27, 29, 30), and here we show that they can also bind SAV1-MST1/2. Thus, they have the ability to bring LATS1/2 together with both their activator and substrate. Interestingly, the SAV1 protein has also been proposed to have a scaffolding function because it binds LATS1/2 (15) and MST1 (38–41). However, we found that cells lacking angiomotins show severe defects in binding between endogenous SAV1-MST1 and LATS1/2, suggesting that, at physiological levels, angiomotins are important for SAV/MST–LATS complex stability. This raises the question of how Hippo signaling complexes are assembled in animals like Drosophila that lack angiomotins. One possible explanation is a rearrangement in the PPXY motif ligands for the SAV WW domains. In Drosophila, SAV binds to the LATS1/2 homolog WARTS through interaction between its WW domains and the five PPXY motifs in WARTS (14). Although a similar mechanism could operate in humans, it may not be as robust because LATS1 and LATS2 contain fewer PPXY motifs than Drosophila WARTS (LATS1 and LATS2 have two and one PPXY motifs, respectively). AMOT could act to recruit SAV1-MST1 to AMOT-LATS2 complexes via its three L/PPXY motifs (two PPXY motifs and one LPXY motif), which could supply additional binding sites for the WW domains of SAV1. Thus, Drosophila may not need angiomotins to recruit WW domain proteins to WARTS because WARTS has five PPXY motifs instead of the two and one in LATS1 and LATS2, respectively. Utilization of the PPXY domains of AMOT to recruit WW domain proteins to an AMOT–LATS1/2 complex could provide another level of regulation because we have previously shown that the L/PPXY motifs of AMOT are masked in the presence of F-actin. Here we showed that the phosphomimetic point mutant (AMOT-175E) that disrupts F-actin binding by AMOT (22, 26, 33, 34) (thereby making the L/PPXY motifs of AMOT available for binding to WW domains) is better than the WT at promoting LATS2-HM phosphorylation. Together, this would create a mechanism whereby a reduction in F-actin frees AMOT to bind the WW domains of both SAV1 and YAP, allowing it to promote LATS1/2 activation and YAP phosphorylation by bringing LATS1/2 together with its activator SAV1-MST1/2 and its target YAP.
Figure 6.

Model for AMOT function in Hippo signaling. A, when F-actin levels are high, AMOT is bound to F-actin and interacts poorly with its binding partners. B, when F-actin levels are reduced, AMOT becomes phosphorylated by LATS and is free to bind YAP, SAV-MST, and LATS. Assembly of this complex allows MST to phosphorylate LATS on the HM site. Both AMOT phosphorylation and LATS HM phosphorylation enhance AMOT-LATS binding. C, to become fully active, LATS must autophosphorylate on the AL site. HM phosphorylation enhances LATS autophosphorylation. In addition, we show that both MOB1 and AMOT enhance LATS-AL phosphorylation independent of any effects they have on HM phosphorylation. D, the ability of fully active LATS to phosphorylate YAP may be enhanced by formation of a LATS–AMOT–YAP complex. Note that the model is speculative and its purpose is to illustrate the primary steps involved in AMOT activation of Hippo signaling. The Hippo pathway is regulated at many other levels, which could be occurring at the same time but are not shown for simplicity. It is presently not clear whether complexes exist in cells as pictured, with all components simultaneously bound to each other.
This model may also be relevant to our observation showing weak co-immunoprecipitation between YAP and LATS2, except for when AMOT was co-expressed, and reduced interaction between endogenous LATS1 and YAP in Amot-3KO cells. Here again, AMOT might be acting to provide additional L/PPXY sites for the WW domains of YAP to bind. Previous reports have given differing results regarding interaction between LATS2 and YAP, with some showing that LATS2 interacts with YAP via the first WW domain of YAP (42–44), whereas another report failed to observe co-immunoprecipitation between YAP and LATS2 (45). These studies could be consistent with the interaction between YAP and LATS2 being weak but observable when high enough levels of each protein are expressed. We showed previously that a competition for binding to AMOT between F-actin and YAP caused a reduction in F-actin levels to trigger AMOT to bind YAP and sequester it in the cytoplasm (22). Our current results suggest that the increased binding of AMOT to YAP caused by reduction in F-actin levels may help connect LATS1/2–AMOT complexes to YAP to promote its phosphorylation and cytoplasmic sequestration.
The other major discovery of this study was that AMOT can stimulate LATS2 to autophosphorylate on its activation loop. Previously, MOB1 was the only protein known to enhance LATS1/2-AL phosphorylation (10, 12). We observed that co-expression of AMOT with MOB1A greatly enhanced LATS2-AL phosphorylation more than expression of either protein alone. Because both AMOT and MOB1A promote LATS1/2-HM site phosphorylation, and this phosphorylation can trigger LATS1/2 to autophosphorylate at the AL site, we needed to be able to analyze LATS2-AL phosphorylation in isolation. To do this, we used LATS2 mutants that had the HM phosphorylation site mutated to either nonphosphorylatable alanine or phosphomimetic glutamate. These results showed that activation or increased levels of AMOT and MOB1 can activate LATS2 even without activation by upstream kinases. The physiological relevance is at this point not clear. It is possible that AMOT and MOB1 always act in conjunction with LATS1/2-HM phosphorylation to promote activation of LATS1/2 through AL phosphorylation, or, alternatively, they might function under some circumstances to activate LATS2 independent of HM phosphorylation. Further studies will be required to address these possibilities.
Experimental procedures
Cell culture and drug and siRNA treatments
HEK293 cells were grown in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (Gibco) and 1% (v/v) penicillin/streptomycin (Gibco) in a humidified incubator at 37 °C with 5% CO2. Cells were incubated with Lat B for 45 min at concentrations of 1 μm (Enzo Life Sciences) or 0.2 μm (Sigma). Okadaic acid was used at 100 μm for 2 h. Angiomotins were stabilized with XAV-939 (Selleckchem) overnight at 10 μM. Knockdowns with siRNA in HeLa cells were performed using 75 nm control (GL2) siRNA or 25 nm each AMOT, AMOTL1, and AMOTL2 SMARTpool siRNA (Dharmacon M-015417, M-017595, and M-013232, respectively) and 18 μl of Lipofectamine RNAiMAX (Invitrogen) in 6-well plates following the manufacturer's directions. After 48 h, cells were collected following Latrunculin B treatment as indicated.
Plasmids and mutagenesis
The term “well” below refers to a single well from a 12-well plate. AMOT (WT, S175E, and S175A (22)), AMOTL1, and AMOTL2 were expressed from pCDNA4-Myc-His at 400 ng/well for all assays except the for the experiments in Fig. S2A, where 750 ng was used. In Fig. S1B, all three angiomotins were expressed together from 500 ng of plasmid/10-cm plate. SAV1 was expressed from pcDNA3–3HA, LATS2 (WT and phosphomimetic LATS2-T1041E (21)) from pcDNA3.1-FLAG, and MST1 from pcDNA-HA, all at 400 ng/well. MOB1A was expressed from pcDNA3.1-Myc at 400 ng/well and pEGFP at 200 ng/well. AMOT-3PxY-Myc was described previously (22). The LATS2-R657A MOB1 binding site mutant (MBD) was made on the 1041E background using the QuikChange II site mutagenesis kit (Agilent), and primers were designed using the manufacturer's web tool (CTACCAGAAAGAGTCTAATTACAACGCGTTAAAGAGGGCCAAGATG and CATCTTGGCCCTCTTTAACGCGTTGTAATTAGACTCTTTCTGGTAG). A similar procedure was followed to make SAV1-ww but using QuikChange Lightning Multi (Agilent) and primers AACACAAATACAACTCACGCGAGCCATGCTCTTGAGCGAGAAGGAC (for ww1) and CACAAATAAGAAGGCCCAAGCCAGGCATGCCTGTGCTCCTAGTGTA (for ww2).
Antibodies
The following dilutions were used for antibodies in Western blots. The mouse anti-MST1 (Proteintech, 66663) was used at 1:2000. Mouse anti-tubulin (Proteintech, 66031), mouse anti-FLAG (Sigma, F3165), mouse anti-GFP (Santa Cruz, 9996), rabbit anti-Myc (Santa Cruz, 789), and mouse anti-Myc (Santa Cruz, 40) were used at 1:1000. Rabbit anti-YAP (Proteintech, 13584), rabbit anti pYAP Ser127 (Cell Signaling, 4911), rabbit anti LATS1 (Cell Signaling, 3477), rabbit anti LATS2 (Cell Signaling, 5888), rabbit anti pLATS1-S909 (Cell Signaling, 9157), rabbit anti MST1/2 (Bethyl, A300-466A), and rabbit anti pLATS1-T1079 (Cell Signaling, 8654) were used at 1:500. Mouse anti-SAV1 (Santa Cruz, 374366) and mouse anti-YAP (Santa Cruz, 101199) were used at 1:100. The rabbit anti-AMOT antibody was generated by Maria Fernandes (Université Laval) and used at 1:2000. Rabbit anti-AMOTL1 was provided by Anthony Schmitt (Pennsylvania State University) and used at 1:1000. Rabbit anti AMOTL2 was generated by Wenqi Wang (University of California Irvine) and used at 1:1000. Immunoprecipitation of endogenous proteins was carried out with rabbit anti-SAV1 (Cell Signaling, 13301) and rabbit anti LATS1 (Cell Signaling, 3477)
Cell transfection, immunoprecipitation, and Western blotting
For the experiment in Fig. S1B, AMOT, AMOTL1, and AMOTL2 were transfected at low levels into HEK293 Amot-3KO cells with FuGENE® HD (Promega) according to the manufacturer's protocol. Rescued A3KO cells were trypsinized 8 h after transfection, replated, and incubated overnight. The next morning, cells were treated with Lat B as indicated. Lipofectamine 2000 (Invitrogen) was the transfection reagent of choice for all other experiments. Because the Hippo pathway is activated by cell resuspension, all protein was collected from cells snap-frozen with liquid nitrogen in the plates on which they were grown. For the experiments shown in Figs. 3A and 4, A and B, a lysis buffer containing glycerol was used (10% glycerol, 20 μm Tris HCl (pH 7), 137 mm NaCl, 2 mm EDTA, and 1% NP40). The lysis buffer for all other experiments contained 1% NP-40, 150 mm NaCl, 2 mm EDTA, 6 mm Na2HPO4, and 4 mm NaH2PO4. Both lysis buffers contained phosphatase and protease inhibitors (1 mm phenylmethylsulfonyl fluoride, 1 mm Na3VO4, and mammalian protease inhibitor mixture (Sigma)). Lysis buffers were added to frozen cells, and cells were solubilized with the help of a cell scraper and back-and-forth pipetting. Lysates were cleared by centrifugation at 15,000 × g for 5 min at 4 °C. Dynabeads (Invitrogen) were used for all immunoprecipitation experiments according to the manufacturer's protocol. Nonspecific IgG antibodies were typically used as a control for immunoprecipitation experiments. For experiments involving coimmunoprecipitation of proteins with different epitope tags, control experiments were done to show that antibodies against the tag on the protein being immunoprecipitated did not bind to the co-expressed protein being tested for co-immunoprecipitation (Fig. S1G).
CRISPR-mediated KO cell line development
Target sequences were selected using the web tool developed by the Zhang lab at the Massachusetts Institute of Technology (http://crispr.mit.edu).3 Complementary oligos (below) containing the target sequence (uppercase) and appropriate overhangs (lowercase) were annealed and cloned into a variant of the px330 plasmid with puromycin resistance (46): AMOT, caccGCCATACACCAGCAAGCCAC and aaacGTGGCTTGCTGGTGTATGGC; AMOTL1, caccgCAAGTTCATGTTCTCGGTTG and aaacCAACCGAGAACATGAACTTGc; AMOTL2, caccGCAGCGTGCGCGTCTCAGTC and aaacGACTGAGACGCGCACGCTGC; MST1, caccGGATCGTTATGGAGTACTGT and aaacACAGTACTCCATAACGATCC; MST2, caccGTTATGGAGTACTGTGGCGC and aaacGCGCCACAGTACTCCATAAC; SAV1, caccGTTGGAATTGTTGGACCATGc and aaacGCATGGTCCAACAATTCCAAc; NF2, caccgGAACTCCATCTCGGCGTCCA and aaacTGGACGCCGAGATGGAGTTC. KO cell lines were generated by transfecting 500 ng of the px330-based plasmid containing the CRISPR target sequence into HEK293 cells plated in 12-well plates using Lipofectamine 2000 (Invitrogen). The next day, cells were placed under selection for 48 h with 1 μg/ml of puromycin (Gibco). Puromycin-resistant cells were then heavily diluted and plated on 10-cm plates for colony isolation. Clonal lines were then expanded, and the expression of the target gene was determined by Western blotting. Clonal lines that lacked expression of the target gene were reisolated from single cells and tested by Western blotting to ensure clonality. The sfGFP-MAP-AMOT–expressing cells were created similarly to the KO cell lines, but instead of px330 we used MLM3636 (Addgene, 43860) to generate the guide RNA (oligo sequences acaccGAATTTCTCATCTCTATTGCg and aaaacGCAATAGAGATGAGAAATTCc), which cuts near the start codon for AMOT; JDS246 (Addgene, 43861) to express Cas9; and a rescue plasmid containing sfGFP-MAP (37) flanked by ∼700 bp of genomic sequence immediately upstream and downstream of the AMOT start codon.
Pooled puromycin-resistant cells were used for further analysis. Western blotting using AMOT and GFP antibodies was used to confirm that the cells expressed full-length sfGFP-MAP-AMOT.
Quantification and statistical analysis
Student's t test (*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001; ****, p ≤ 0.0001) was performed with the GraphPad web tool) and represents mean ± S.D. for experiments done in triplicates. Western blotting densitometry quantifications were either performed with LI-COR Odyssey (when fluorescent probes were used) or with ImageJ (47) on images obtained with a Bio-Rad Chemi-Touch when horseradish peroxidase–conjugated antibodies were used.
Author contributions
S. M.-C. and D. M. conceptualization; S. M.-C. data curation; S. M.-C. formal analysis; S. M.-C. and D. M. investigation; S. M.-C. methodology; S. M.-C. and D. M. writing-original draft; S. M.-C. and D. M. writing-review and editing; D. M. funding acquisition; D. M. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Anthony Schmitt (Pennsylvania State University) and Dr. Wenqi Wang (University of California Irvine) for antibodies against AMOTL1 and AMOTL2, respectively.
This work was supported by National Institutes of Health grant RO1 GM058406 (to D. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party–hosted site.
- YAP
- Yes-associated protein
- HM
- hydrophobic motif
- AL
- activation loop
- AMOT
- angiomotin
- KO
- knockout
- HEK
- human embryonic kidney
- Lat B
- Latrunculin B
- MAP
- multifunctional affinity tandem purification
- MBD
- MOB-binding domain
- HA
- hemagglutinin
- sfGFP
- superfolder GFP.
References
- 1. Meng Z., Moroishi T., and Guan K. L. (2016) Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17 10.1101/gad.274027.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zanconato F., Cordenonsi M., and Piccolo S. (2016) YAP/TAZ at the roots of cancer. Cancer Cell 29, 783–803 10.1016/j.ccell.2016.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dong J., Feldmann G., Huang J., Wu S., Zhang N., Comerford S. A., Gayyed M. F., Anders R. A., Maitra A., and Pan D. (2007) Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133 10.1016/j.cell.2007.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao B., Li L., Tumaneng K., Wang C. Y., and Guan K. L. (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(β-TRCP). Genes Dev. 24, 72–85 10.1101/gad.1843810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu C. Y., Zha Z. Y., Zhou X., Zhang H., Huang W., Zhao D., Li T., Chan S. W., Lim C. J., Hong W., Zhao S., Xiong Y., Lei Q. Y., and Guan K. L. (2010) The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFβ-TrCP E3 ligase. J. Biol. Chem. 285, 37159–37169 10.1074/jbc.M110.152942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chan E. H., Nousiainen M., Chalamalasetty R. B., Schäfer A., Nigg E. A., and Silljé H. H. (2005) The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 24, 2076–2086 10.1038/sj.onc.1208445 [DOI] [PubMed] [Google Scholar]
- 7. Meng Z., Moroishi T., Mottier-Pavie V., Plouffe S. W., Hansen C. G., Hong A. W., Park H. W., Mo J. S., Lu W., Lu S., Flores F., Yu F. X., Halder G., and Guan K. L. (2015) MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 6, 8357 10.1038/ncomms9357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zheng Y., Wang W., Liu B., Deng H., Uster E., and Pan D. (2015) Identification of Happyhour/MAP4K as alternative Hpo/Mst-like kinases in the Hippo kinase cascade. Dev. Cell 34, 642–655 10.1016/j.devcel.2015.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Plouffe S. W., Meng Z., Lin K. C., Lin B., Hong A. W., Chun J. V., and Guan K. L. (2016) Characterization of Hippo pathway components by gene inactivation. Mol. Cell 64, 993–1008 10.1016/j.molcel.2016.10.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hergovich A., Schmitz D., and Hemmings B. A. (2006) The human tumour suppressor LATS1 is activated by human MOB1 at the membrane. Biochem. Biophys. Res. Commun. 345, 50–58 10.1016/j.bbrc.2006.03.244 [DOI] [PubMed] [Google Scholar]
- 11. Hoa L., Kulaberoglu Y., Gundogdu R., Cook D., Mavis M., Gomez M., Gomez V., and Hergovich A. (2016) The characterisation of LATS2 kinase regulation in Hippo-YAP signalling. Cell Signal. 28, 488–497 10.1016/j.cellsig.2016.02.012 [DOI] [PubMed] [Google Scholar]
- 12. Ni L., Zheng Y., Hara M., Pan D., and Luo X. (2015) Structural basis for Mob1-dependent activation of the core Mst-Lats kinase cascade in Hippo signaling. Genes Dev. 29, 1416–1431 10.1101/gad.264929.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yin F., Yu J., Zheng Y., Chen Q., Zhang N., and Pan D. (2013) Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell 154, 1342–1355 10.1016/j.cell.2013.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tapon N., Harvey K. F., Bell D. W., Wahrer D. C., Schiripo T. A., Haber D., and Hariharan I. K. (2002) Salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 110, 467–478 10.1016/S0092-8674(02)00824-3 [DOI] [PubMed] [Google Scholar]
- 15. Guo C., Tommasi S., Liu L., Yee J. K., Dammann R., and Pfeifer G. P. (2007) RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr. Biol. 17, 700–705 10.1016/j.cub.2007.02.055 [DOI] [PubMed] [Google Scholar]
- 16. Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S., Elvassore N., and Piccolo S. (2011) Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183 10.1038/nature10137 [DOI] [PubMed] [Google Scholar]
- 17. Fernández B. G., Gaspar P., Brás-Pereira C., Jezowska B., Rebelo S. R., and Janody F. (2011) Actin-capping protein and the Hippo pathway regulate F-actin and tissue growth in Drosophila. Development 138, 2337–2346 10.1242/dev.063545 [DOI] [PubMed] [Google Scholar]
- 18. Sansores-Garcia L., Bossuyt W., Wada K., Yonemura S., Tao C., Sasaki H., and Halder G. (2011) Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 30, 2325–2335 10.1038/emboj.2011.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wada K., Itoga K., Okano T., Yonemura S., and Sasaki H. (2011) Hippo pathway regulation by cell morphology and stress fibers. Development 138, 3907–3914 10.1242/dev.070987 [DOI] [PubMed] [Google Scholar]
- 20. Zhao B., Li L., Wang L., Wang C. Y., Yu J., and Guan K. L. (2012) Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 26, 54–68 10.1101/gad.173435.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paramasivam M., Sarkeshik A., Yates J. R. 3rd, Fernandes M. J., and McCollum D. (2011) Angiomotin family proteins are novel activators of the LATS2 kinase tumor suppressor. Mol. Biol. Cell 22, 3725–3733 10.1091/mbc.e11-04-0300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mana-Capelli S., Paramasivam M., Dutta S., and McCollum D. (2014) Angiomotins link F-actin architecture to Hippo pathway signaling. Mol. Biol. Cell 25, 1676–1685 10.1091/mbc.e13-11-0701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feng X., Degese M. S., Iglesias-Bartolome R., Vaque J. P., Molinolo A. A., Rodrigues M., Zaidi M. R., Ksander B. R., Merlino G., Sodhi A., Chen Q., and Gutkind J. S. (2014) Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 25, 831–845 10.1016/j.ccr.2014.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan S. W., Lim C. J., Chong Y. F., Pobbati A. V., Huang C., and Hong W. (2011) Hippo pathway-independent restriction of TAZ and YAP by angiomotin. J. Biol. Chem. 286, 7018–7026 10.1074/jbc.C110.212621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhao B., Li L., Lu Q., Wang L. H., Liu C. Y., Lei Q., and Guan K. L. (2011) Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 25, 51–63 10.1101/gad.2000111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hirate Y., Hirahara S., Inoue K., Suzuki A., Alarcon V. B., Akimoto K., Hirai T., Hara T., Adachi M., Chida K., Ohno S., Marikawa Y., Nakao K., Shimono A., and Sasaki H. (2013) Polarity-dependent distribution of angiomotin localizes Hippo signaling in preimplantation embryos. Curr. Biol. 23, 1181–1194 10.1016/j.cub.2013.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leung C. Y., and Zernicka-Goetz M. (2013) Angiomotin prevents pluripotent lineage differentiation in mouse embryos via Hippo pathway-dependent and -independent mechanisms. Nat. Commun. 4, 2251 10.1038/ncomms3251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moleirinho S., Hoxha S., Mandati V., Curtale G., Troutman S., Ehmer U., and Kissil J. L. (2017) Regulation of localization and function of the transcriptional co-activator YAP by angiomotin. eLife 6, e23966 10.7554/eLife.23966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yi C., Troutman S., Fera D., Stemmer-Rachamimov A., Avila J. L., Christian N., Persson N. L., Shimono A., Speicher D. W., Marmorstein R., Holmgren L., and Kissil J. L. (2011) A tight junction-associated Merlin-angiomotin complex mediates Merlin's regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell 19, 527–540 10.1016/j.ccr.2011.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Y., Zhou H., Li F., Chan S. W., Lin Z., Wei Z., Yang Z., Guo F., Lim C. J., Xing W., Shen Y., Hong W., Long J., and Zhang M. (2015) Angiomotin binding-induced activation of Merlin/NF2 in the Hippo pathway. Cell Res. 25, 801–817 10.1038/cr.2015.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Couzens A. L., Knight J. D., Kean M. J., Teo G., Weiss A., Dunham W. H., Lin Z. Y., Bagshaw R. D., Sicheri F., Pawson T., Wrana J. L., Choi H., and Gingras A. C. (2013) Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 6, rs15 10.1126/scisignal.2004712 [DOI] [PubMed] [Google Scholar]
- 32. Wang W., Huang J., and Chen J. (2011) Angiomotin-like proteins associate with and negatively regulate YAP1. J. Biol. Chem. 286, 4364–4370 10.1074/jbc.C110.205401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chan S. W., Lim C. J., Guo F., Tan I., Leung T., and Hong W. (2013) Actin-binding and cell proliferation activities of angiomotin family members are regulated by Hippo pathway-mediated phosphorylation. J. Biol. Chem. 288, 37296–37307 10.1074/jbc.M113.527598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dai X., She P., Chi F., Feng Y., Liu H., Jin D., Zhao Y., Guo X., Jiang D., Guan K. L., Zhong T. P., and Zhao B. (2013) Phosphorylation of angiomotin by Lats1/2 kinases inhibits F-actin binding, cell migration, and angiogenesis. J. Biol. Chem. 288, 34041–34051 10.1074/jbc.M113.518019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Adler J. J., Johnson D. E., Heller B. L., Bringman L. R., Ranahan W. P., Conwell M. D., Sun Y., Hudmon A., and Wells C. D. (2013) Serum deprivation inhibits the transcriptional co-activator YAP and cell growth via phosphorylation of the 130-kDa isoform of Angiomotin by the LATS1/2 protein kinases. Proc. Natl. Acad. Sci. U.S.A. 110, 17368–17373 10.1073/pnas.1308236110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim M., Kim M., Park S. J., Lee C., and Lim D. S. (2016) Role of Angiomotin-like 2 mono-ubiquitination on YAP inhibition. EMBO Rep. 17, 64–78 10.15252/embr.201540809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ma H., McLean J. R., Chao L. F., Mana-Capelli S., Paramasivam M., Hagstrom K. A., Gould K. L., and McCollum D. (2012) A highly efficient multifunctional tandem affinity purification approach applicable to diverse organisms. Mol. Cell. Proteomics 11, 501–511 10.1074/mcp.O111.016246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Callus B. A., Verhagen A. M., and Vaux D. L. (2006) Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS J. 273, 4264–4276 10.1111/j.1742-4658.2006.05427.x [DOI] [PubMed] [Google Scholar]
- 39. Cairns L., Tran T., Fowl B. H., Patterson A., Kim Y. J., Bothner B., and Kavran J. M. (2018) Salvador has an extended SARAH domain that mediates binding to Hippo kinase. J. Biol. Chem. 293, 5532–5543 10.1074/jbc.RA117.000923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bae S. J., Ni L., Osinski A., Tomchick D. R., Brautigam C. A., and Luo X. (2017) SAV1 promotes Hippo kinase activation through antagonizing the PP2A phosphatase STRIPAK. eLife 6, e30278 10.7554/eLife.30278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hwang E., Ryu K. S., Pääkkönen K., Güntert P., Cheong H. K., Lim D. S., Lee J. O., Jeon Y. H., and Cheong C. (2007) Structural insight into dimeric interaction of the SARAH domains from Mst1 and RASSF family proteins in the apoptosis pathway. Proc. Natl. Acad. Sci. U.S.A. 104, 9236–9241 10.1073/pnas.0610716104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kawahara M., Hori T., Chonabayashi K., Oka T., Sudol M., and Uchiyama T. (2008) Kpm/Lats2 is linked to chemosensitivity of leukemic cells through the stabilization of p73. Blood 112, 3856–3866 10.1182/blood-2007-09-111773 [DOI] [PubMed] [Google Scholar]
- 43. Zhang J., Smolen G. A., and Haber D. A. (2008) Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res. 68, 2789–2794 10.1158/0008-5472.CAN-07-6205 [DOI] [PubMed] [Google Scholar]
- 44. Oka T., Mazack V., and Sudol M. (2008) Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J. Biol. Chem. 283, 27534–27546 10.1074/jbc.M804380200 [DOI] [PubMed] [Google Scholar]
- 45. Jagannathan R., Schimizzi G. V., Zhang K., Loza A. J., Yabuta N., Nojima H., and Longmore G. D. (2016) AJUBA LIM proteins limit Hippo activity in proliferating cells by sequestering the Hippo core kinase complex in the cytosol. Mol. Cell. Biol. 36, 2526–2542 10.1128/MCB.00136-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hainer S. J., Gu W., Carone B. R., Landry B. D., Rando O. J., Mello C. C., and Fazzio T. G. (2015) Suppression of pervasive noncoding transcription in embryonic stem cells by esBAF. Genes Dev. 29, 362–378 10.1101/gad.253534.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schneider C. A., Rasband W. S., and Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.