

Abstract
Members of the G protein–coupled receptor and TMEM16 (transmembrane protein 16) protein families are phospholipid scramblases that facilitate rapid, bidirectional movement of phospholipids across a membrane bilayer in an ATP-independent manner. On reconstitution into large unilamellar vesicles, these proteins scramble more than 10,000 lipids/protein/s as measured with co-reconstituted fluorescent nitrobenzoxadiazole (NBD)-labeled phospholipids. Although NBD-labeled phospholipids are ubiquitously used as reporters of scramblase activity, it remains unclear whether the NBD modification influences the quantitative outcomes of the scramblase assay. We now report a refined biochemical approach for measuring the activity of scramblase proteins with radiolabeled natural phosphatidylinositol ([3H]PI) and exploiting the hydrolytic activity of bacterial PI-specific phospholipase C (PI-PLC) to detect the transbilayer movement of PI. PI-PLC rapidly hydrolyzed 50% of [3H]PI in large symmetric, unilamellar liposomes, corresponding to the lipid pool in the outer leaflet. On reconstitution of a crude preparation of yeast endoplasmic reticulum scramblase, purified bovine opsin, or purified Nectria haematococca TMEM16, the extent of [3H]PI hydrolysis increased, indicating that [3H]PI from the inner leaflet had been scrambled to the outer leaflet. Using transphosphatidylation, we synthesized acyl-NBD-PI and used it to compare our PI-PLC–based assay with conventional fluorescence-based methods. Our results revealed quantitative differences between the two assays that we attribute to the specific features of the assays themselves rather than to the nature of the phospholipid. In summary, we have developed an assay that measures scrambling of a chemically unmodified phospholipid by a reconstituted scramblase.
Keywords: G protein–coupled receptor (GPCR), liposome, phosphatidylinositol, Phospholipase C, glycerophospholipid, membrane transport, scramblase, TMEM16
Introduction
Glycerophospholipids are essential components of all eukaryotic membranes. The most abundant classes, phosphatidylcholine (PC)3 and phosphatidylethanolamine (PE), are synthesized via the Kennedy pathways (1) on the cytoplasmic face of the endoplasmic reticulum (ER) (2–7). There are no parallel pathways to synthesize phospholipids in the luminal leaflet of the ER. Thus, de novo synthesized glycerophospholipids are deposited exclusively into the cytoplasmic leaflet of the ER membrane bilayer, creating an imbalance between cytoplasmic and luminal leaflets. To restore or maintain the stability of the bilayer, newly synthesized lipids have to be transported from the cytoplasmic to the luminal side of the membrane. However, because spontaneous transbilayer movement of glycerophospholipids is energetically unfavorable, occurring with half-times of up to 100 h in a fluid PC bilayer, specialized proteins are needed to catalyze this process (reviewed in Ref. 8). Some of these transporters couple lipid translocation to ATP hydrolysis to transport lipids in a unidirectional way, whereas others function as ATP-independent scramblases and transport lipids bidirectionally (reviewed in Refs. 5 and 8–12).
Glycerophospholipid scramblase activity in the ER was demonstrated over three decades ago (13, 14), but the molecular identity of the protein(s) responsible for this activity has remained elusive (8, 15). However, in recent years other phospholipid scramblases have been identified and characterized. Members of the Xk-related family of proteins, including Xkr8, were found to be involved in exposure of phosphatidylserine (PS) on the surface of mammalian cells during apoptosis (16). Likewise, TMEM16F, a member of the TMEM16 family of ion channels and scramblases, is implicated in the exposure of PS on activated blood platelets that is needed for blood coagulation (17, 18). Although it is unclear whether the Xkr8 proteins are scramblases themselves or only indirectly contribute to PS exposure, several TMEM16 proteins, including TMEM16F, have been purified and reconstituted into phospholipid bilayers and explicitly shown to have Ca2+-dependent scramblase activity (19–22). Unexpectedly, a number of G protein–coupled receptors including rhodopsin, the β1- and β2-adrenergic receptors, and the adenosine A2A receptor, were also shown to have scramblase activity after reconstitution into phospholipid bilayers (23–27). The phospholipid scramblase activity of rhodopsin provides the molecular basis for previous observations of rapid lipid scrambling across intact bovine photoreceptor discs (28–30), including the transient, light-dependent change in their transbilayer lipid asymmetry (30), and has been implicated in disc membrane lipid homeostasis (reviewed in Ref. 31). A hallmark of the ER scramblase, as well as the TMEM16 and G protein–coupled receptor scramblases, is their lack of specificity. Reconstitution-based assays show that these proteins are able to scramble all common phospholipids (8). Thus, exposure of PS at the surface of activated platelets is a consequence of general scrambling of all phospholipids by TMEM16F rather than specific transport of PS.
Transbilayer organization and movement of glycerophospholipids in the plasma membrane of eukaryotic cells has historically been determined using lipid-modifying enzymes, including phospholipases (reviewed in Ref. 32). Differential accessibility of membrane lipids to phospholipases A2 or C added to the outside of cells demonstrated that phospholipids in the plasma membrane are arranged asymmetrically (33, 34). These findings were subsequently corroborated in assays in which the distribution of fluorescent or spin-labeled phospholipid analogs inserted into the outer leaflet of the plasma membrane was determined (35–37). Labeled lipid probes have also been used to measure transbilayer distribution and movement of glycerophospholipids in artificial systems (38). In particular, glycerophospholipid analogs carrying fluorescent nitrobenzoxadiazole (NBD) groups in their hydrophilic heads or hydrophobic tails have proven useful to identify and characterize phospholipid scramblases in membrane preparations and reconstituted proteoliposomes (16, 19, 20, 23, 39, 40). Although these lipid probes appear generally to reflect the behavior of natural glycerophospholipids, it cannot be excluded that quantitative aspects of scramblase activity measurements are affected by the presence of the NBD tag (41). Because precise measurements of scrambling rates are important for mechanistic understanding, it is important to quantify the rate at which natural lipids are scrambled and compare these results with those obtained with NBD phospholipids.
We now report a novel biochemical assay to measure the activity of scramblases using radiolabeled natural phosphatidylinositol (PI) as reporter (42, 43). Given the reported lack of lipid specificity of phospholipid scramblases, it seemed reasonable to assume that PI would also be scrambled by these proteins. We exploited the ability of bacterial PI-specific phospholipase C (PI-PLC) to hydrolyze PI located in the outer leaflet of liposomes, whereas PI in the inner leaflet is protected from the action of the enzyme. In this approach, an increase in the extent of PI-PLC–mediated PI hydrolysis in proteoliposomes compared with protein-free liposomes reflects the presence of scramblase activity in the former. We also synthesized acyl-NBD-PI from acyl-NBD-PC by using a novel phospholipase D enzyme with positional specificity on myo-inositol (44) and used this novel lipid probe in a side-by-side comparison with natural PI in activity assays. As sources of scramblase activity, we used an extract of yeast microsomes that was previously shown to contain scramblase activity, as well as two purified scramblases (bovine opsin and Nectria haematococca TMEM16 (nhTMEM16)). Comparison of the new approach with NBD phospholipid–based measurements revealed unexpected quantitative differences in the deduced rate of scrambling rate. We discuss the possible basis of these differences.
Results
PI-PLC–mediated hydrolysis of [3H]PI in liposomes
Bacterial PI-PLC enzymes catalyze the cleavage of PI into diacylglycerol and inositol 1,2-cyclic phosphate (45, 46), which can be subsequently hydrolyzed to inositol 1-phosphate (45, 47). If [3H-inositol]PI is used as substrate, the extent of hydrolysis by PI-PLC is conveniently determined by partitioning the reaction between aqueous and organic phases and measuring the appearance of [3H]inositol 1,2-cyclic phosphate/[3H]inositol 1-phosphate in the aqueous phase and unreacted [3H-inositol]PI in the organic phase.
The addition of PI-PLC to symmetric, large unilamellar liposomes containing a trace quantity of [3H]PI should result in hydrolysis of ∼50% of [3H]PI, i.e. the portion of total PI that is present in the outer leaflet of the bilayer. [3H]PI in the inner leaflet is protected from PI-PLC action because the enzyme cannot cross the membrane bilayer, and the rate at which [3H]PI translocates spontaneously to the outer leaflet is too slow to be detected on the time scale of our experiments. In contrast, if the liposomes are reconstituted with a scramblase capable of translocating [3H]PI across the bilayer, all [3H]PI should be available for hydrolysis by PI-PLC over a short time period (Fig. 1).
Figure 1.

Schematic of PI-PLC assay. Incubation of unilamellar liposomes composed of egg PC and egg PA (9:1, molar ratio; green symbols) and trace amounts of [3H]PI (red symbols) with PI-PLC results in hydrolysis of [3H]PI (to inositol cyclic phosphate and diacylglycerol (DAG)) in the outer leaflet of the bilayer, whereas [3H]PI in the inner leaflet is protected from the enzyme. Because [3H]PI distributes equally between the two leaflets during preparation of liposomes, ∼50% is expected to be hydrolyzed by PI-PLC. In unilamellar proteoliposomes reconstituted with scramblase protein(s), [3H]PI from the inner leaflet is translocated to the outer leaflet, where it becomes accessible to PI-PLC. The extent of [3H]PI hydrolysis in scramblase-containing proteoliposomes is expected to reach 100% on a relatively short time scale.
To test the principle of the assay, we first prepared [3H]PI-containing liposomes composed of egg PC and egg PA (9:1, mol/mol). After adding PI-PLC, aliquots of the liposome suspension were removed and added to chloroform:methanol (1:2, v/v) to stop the reaction immediately. Two phases were induced by adding additional chloroform, methanol, and water, and aliquots of the aqueous and organic phases were taken for liquid scintillation counting to determine the extent of [3H]PI hydrolysis. The results show that on adding PI-PLC (0.5–3.0 μl from a stock solution of 0.01 units/μl) to liposomes (3 mm phospholipid, final concentration), hydrolysis of [3H]PI occurred rapidly, reaching a maximum of ∼45, ∼47, and ∼52% of total [3H]PI for samples treated with 0.5, 1.0, and 3.0 μl of PI-PLC, respectively (Fig. 2A). On destruction of the membrane barrier with Triton X-100 (0.9% (w/v), final concentration), >95% of [3H]PI was hydrolyzed as expected. The extent of [3H]PI hydrolysis was also determined by an alternative procedure in which TCA precipitation was used to separate [3H]PI from 1,2-cyclic [3H]inositol phosphate/[3H]inositol 1-phosphate (see “Experimental procedures”); the results were similar to those obtained with organic solvent extraction. We conclude that [3H]PI is symmetrically reconstituted in the vesicles and that PI-PLC is a valid topological probe that detects only [3H]PI molecules in the outer leaflet.
Figure 2.
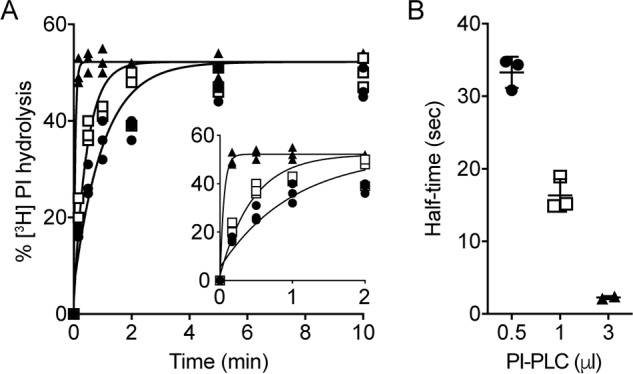
PI-PLC–mediated hydrolysis of [3H]PI in liposomes. A, liposomes composed of egg PC, egg PA (9:1, molar ratio), and trace amounts of [3H]PI were treated with different amounts of PI-PLC at room temperature. At the indicated times, aliquots of the suspensions were removed, and lipids were extracted using a two-phase extraction system. Radioactivity in the upper aqueous and lower organic phases, containing 1,2-cyclic [3H]inositol phosphate/[3H]inositol 1-phosphate and ([3H]PI), respectively, was used to calculate the fraction of [3H]PI hydrolyzed by PI-PLC. The data points are from three independent experiments done in the presence of 0.5 μl (filled circles), 1.0 μl (open squares), or 3.0 μl (filled triangles) PI-PLC. In control incubations in the absence of PI-PLC, [3H]PI hydrolysis was <3%. For better visibility, the inset shows the curves during the first 2 min of incubation. The data were fitted to a one-phase exponential function. B, half-times of [3H]PI hydrolysis in the presence of different amounts of PI-PLC calculated from the curves shown in A. The data are mean values ± standard deviations from three independent experiments.
The time taken to reach maximal [3H]PI hydrolysis depended on the amount of PI-PLC (Fig. 2A). The hydrolysis kinetics were well described by a single-phase exponential function (Fig. 2A), revealing half-times of ∼21, ∼13, and ∼2 s for samples treated with 0.5, 1.0, and 3.0 μl of PI-PLC, respectively (Fig. 2B). A better estimate of the half-time of hydrolysis when using 3.0 μl of PI-PLC was not possible because it was not practical to sample the reaction sufficiently soon (the earliest time point taken was 10 s after PI-PLC addition). In all subsequent experiments we used 3.0 μl of PI-PLC to have the best chance of kinetically resolving transbilayer scrambling from the process of detecting outer leaflet [3H]PI via PI-PLC–mediated hydrolysis.
PI-PLC–mediated hydrolysis of [3H]PI in proteoliposomes reconstituted with ER membrane proteins
It has been shown previously that a detergent extract of crude microsomes from Saccharomyces cerevisiae (termed Triton extract (TE), enriched in ER membrane proteins) contains specific protein(s) with phospholipid scrambling activity (40, 48–50). Treatment of [3H]PI-containing proteoliposomes reconstituted with TE (protein to phospholipid ratio (PPR) of ∼36 mg/mmol) with 3.0 μl of PI-PLC resulted in biphasic hydrolysis of >80% of [3H]PI (Fig. 3A), indicating that the [3H]PI located in the inner leaflet of the proteoliposomes scrambles to the outer leaflet, thereby becoming accessible to cleavage by PI-PLC during the experiment. The initial rapid phase of [3H]PI hydrolysis, half-time ∼2 s, accounts for PI in the outer leaflet of the vesicles, whereas the slower second phase, half-time ∼20 s, indicates the rate of scrambling.
Figure 3.
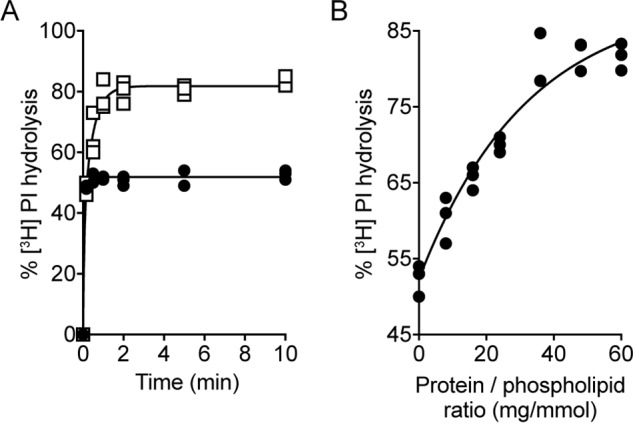
PI-PLC–mediated hydrolysis of [3H]PI in proteoliposomes reconstituted with yeast Triton extract. Preformed liposomes composed of egg PC and egg PA (9:1, molar ratio) were reconstituted with trace amounts of [3H]PI and a Triton extract from S. cerevisiae containing phospholipid scrambling activity. A, proteoliposomes (open squares) and control liposomes (mock reconstituted; filled circles) were treated with 3 μl of PI-PLC at room temperature, and the extent of [3H]PI hydrolysis was determined as described in Fig. 2A. The data points are from three independent experiments. The time course for [3H]PI hydrolysis in proteoliposomes was fitted to a double-exponential function. To display all data points, the graph of mock-treated liposomes was shifted by +0.3 min on the x axis. B, dependence of [3H]PI hydrolysis on the protein to phospholipid ratio. Maximal [3H]PI hydrolysis after 10 min of PI-PLC treatment was determined in proteoliposomes reconstituted with different amounts of yeast Triton extract and plotted against the protein to phospholipid ratio. The data points are from three independent experiments and fitted into a one-phase exponential function.
When the amount of TE used for reconstitution was varied systematically to obtain vesicles with PPRs in the range 0 to ∼60 mg/mmol, the extent of [3H]PI hydrolysis measured after a 10-min incubation increased from ∼50% to 82% (Fig. 3B) (in control experiments, >95% of [3H]PI was hydrolyzed on adding PI-PLC to liposomes that had been disrupted with Triton X-100). Analysis of these data (Fig. 3B) by fitting to a monoexponential function, yielded a fit constant of 32 ± 6 mg/mmol. This value is ∼4-fold greater than reported previously with the NBD phospholipid–based fluorescence assay for scrambling in proteoliposomes reconstituted with a yeast ER extract (48). As shown and discussed later, this apparent discrepancy is because scrambling is not complete after a 10-min incubation in samples prepared with lower PPR values.
PI-PLC–mediated [3H]PI hydrolysis in proteoliposomes reconstituted with purified scramblases
We next applied the PI-PLC assay to measure phospholipid scramblase activity of purified bovine opsin and nhTMEM16 (Fig. S1). We reconstituted a high amount of these proteins to ensure maximum occupancy of vesicles with a scramblase and measured the extent of [3H]PI hydrolysis on adding PI-PLC. Compared with ∼50% hydrolysis seen for protein-free liposomes that had been treated with 3 μl of PI-PLC for 10 min, 80 ± 2% and 82 ± 2% (mean ± S.D., n = 3) hydrolysis was observed with opsin-containing proteoliposomes (PPR ∼6 mg/mmol) and nhTMEM16-containing proteoliposomes (PPR =∼7 mg/mmol, in the presence of 250 μm Ca2+), respectively. Despite the high amount of protein used for reconstitution in each case, 100% hydrolysis was not achieved because, as noted previously, a significant fraction of vesicles is refractory to reconstitution for reasons that are not well understood (19, 26, 51). These results indicate that both opsin and nhTMEM16 are able to scramble natural PI.
Quantitative analysis of opsin-mediated scrambling measured via PI-PLC–mediated [3H]PI hydrolysis
Using various NBD phospholipid reporters, e.g. NBD-PC, opsin-mediated lipid scrambling was reported to occur with a half-time of <10 s (23, 24, 26, 27). To determine whether the PI-PLC–based assay revealed a similar scrambling rate, we assayed opsin-containing proteoliposomes prepared with a high PPR of ∼6 mg/mmol (corresponding to ∼25 opsin dimers per ∼175-nm diameter vesicle). As in the case of proteoliposomes reconstituted with a microsomal proteins (Fig. 3A), we found [3H]PI hydrolysis in opsin proteoliposomes to be a biphasic process: a first phase with the expected half-time of ∼2 s, followed by a slow phase (half-time ∼50 s) (Fig. 4A). This result suggests that PI scrambling may occur more slowly than seen with NBD-PC (Fig. 4B).
Figure 4.

Opsin-mediated scrambling of [3H]PI and NBD-PC. A, proteoliposomes composed of egg PC and egg PA (9:1, molar ratio) were reconstituted with [3H]PI and purified opsin using protein to phospholipid ratios of 0 (filled circles) or ∼6 (open squares) mg/mmol. The extent of PI-PLC–mediated hydrolysis of [3H]PI was determined at each time point as described in Fig. 2A. The data are from two independent experiments. The time course for [3H]PI hydrolysis in proteoliposomes was fitted to a double-exponential function. To display all data points, the graphs of opsin-liposomes at ∼3 mg/mmol and mock-treated liposomes were shifted by +0.3 and +0.6 min, respectively, on the x axis. B, liposomes composed of egg PC and egg PA (9:1 molar ratio) were reconstituted with trace amounts of NBD-PC in the absence or presence of purified opsin using a protein to phospholipid ratio of ∼0.4 mg/mmol. Dithionite (to reduce the fluorescent signal of NBD-PC) was added after 60 s, and fluorescence was recorded at room temperature with constant stirring. The traces are representative of three independent experiments and reflect opsin liposomes (solid line) and mock-reconstituted liposomes (dashed line). C, proteoliposomes composed of egg PC and egg PA (9:1, molar ratio) were reconstituted with [3H]PI and NBD-PC and purified opsin using protein to phospholipid ratios as indicated. The extent of PI-PLC–mediated hydrolysis of [3H]PI (white bars) and reduction of NBD-PC fluorescence by dithionite (gray bars) was determined as described above. The data represent mean values ± standard deviations from three independent experiments.
To explore this point further, we carried out a side-by-side comparison of the two activity assays using proteoliposomes prepared with different PPRs, ∼0.4, ∼4.0, and ∼6.0 mg/mmol (corresponding to ∼1–2, ∼15, and ∼23 opsin dimers/vesicle, respectively). We used a 7-min time point to assess activity via the fluorescence-based assay and a 10-min time point for the PI-PLC–based assay. The fluorescence-based assay showed a reduction of >80% fluorescence for the proteoliposome preparations, as expected (Fig. 4C, gray bars). However, the PI-PLC–based assay showed only ∼60 and ∼70% hydrolysis of [3H]PI in vesicles prepared with PPR values of ∼0.4 and ∼4.0, respectively, reaching ∼80% hydrolysis only at a PPR of ∼6 mg/mmol (Fig. 4, C, white bars, and A, end point of time course). This result indicates that the PI-PLC–based assay reports a lower scrambling rate than that observed with the fluorescence-based assay.
PI-PLC–based versus fluorescence-based assays of the scramblase activity of nhTMEM16
To determine whether the slow rate of scrambling as revealed by the PI-PLC–based assay is unique to opsin-mediated scrambling, we analyzed the scramblase activity of purified nhTMEM16. In these experiments, [3H]PI and NBD-PC were co-reconstituted into nhTMEM16-containing proteoliposomes, composed of egg PC and dioleoylphosphatidylglycerol (DOPG) (9:1, molar ratio), and hydrolysis of [3H]PI by PI-PLC and reduction of NBD-PC by dithionite were measured using aliquots from the same preparation. A time course of [3H]PI hydrolysis for nhTMEM16 proteoliposomes prepared with PPR ∼7 mg/mmol, revealed biphasic kinetics with half-times for fast and slow phases similar to those obtained for opsin, ∼2 and ∼40 s, respectively (Fig. 5A). Measurements taken 10 min after adding PI-PLC revealed that as the PPR was increased from ∼1–7 mg/mmol, hydrolysis increased from ∼60% to ∼80% (Fig. 5C). In contrast, >90% of NBD-PC was reduced by dithionite (Fig. 5, B and C) at the lowest PPR tested (∼1 mg/mmol). Thus, as seen for opsin, the nhTMEM16 analyses reveal that the rate of scrambling reported by the PI-PLC assay is substantially lower than that seen in the fluorescence assay.
Figure 5.

nhTMEM16-mediated scrambling of [3H]PI and NBD-PC. A, proteoliposomes composed of egg PC and DOPG (9:1, molar ratio) were reconstituted with [3H]PI and purified nhTMEM16 using protein to phospholipid ratios of 0 (filled circles) or ∼7 (open squares) mg/mmol in the presence of 250 μm Ca2+. The extent of PI-PLC–mediated hydrolysis of [3H]PI was determined at each time point as described in Fig. 2A. The data are from two independent experiments. The time course for [3H]PI hydrolysis in proteoliposomes was fitted to a double-exponential function. To display all data points, the graph of mock-treated liposomes was shifted by +0.3 min on the x axis. B, liposomes and nhTMEM16 proteoliposomes at a PPR of ∼1 mg/mmol containing trace amount of NBD-PC were reconstituted as in Fig. 4B, except for the addition of 250 μm Ca2+ in the reconstitution solution. Dithionite was added after 1 min, and fluorescence was recorded for 8 min. Traces of liposomes (gray) and nhTMEM16 proteoliposomes (black) are representative of three independent experiments. C, liposomes and nhTMEM16 proteoliposomes at PPRs of ∼1, ∼2, and ∼4 mg/mmol containing trace amounts of [3H]PI and NBD-PC were prepared as above in the presence of 250 μm Ca2+. The extent of PI-PLC–mediated hydrolysis of [3H]PI (white bars) and reduction of NBD-PC fluorescence by dithionite (gray bars) was determined as described above. The data represent mean values ± standard deviations from three independent experiments.
We quantified the scrambling/hydrolysis rate in nhTMEM16 proteoliposomes prepared with a PPR ∼1.0 mg/mmol. Measurements taken over a 4-h period revealed that whereas the extent of [3H]PI hydrolysis in protein-free liposomes remained constant after an initial burst phase (Fig. 6A, filled circles), indicating undetectable scrambling over 4 h as expected, the extent of [3H]PI that was hydrolyzed in proteoliposomes increased sharply above that of the protein-free sample within 10 min and then continued to increase more slowly over the remainder of the time course with a t½ of ∼350 min (Fig. 6A). We next carried out a similar experiment with proteoliposomes reconstituted with yeast TE at a PPR of ∼10 mg/mmol where only ∼60% [3H]PI hydrolysis was observed after a 10-min incubation (Fig. 3B). As in the case of nhTMEM16 proteoliposomes, the extent of [3H]PI that was hydrolyzed increased sharply above that of the protein-free sample within 10 min and then continued to increase more slowly (t½ ∼50 min) over the remainder of the time course (Fig. 6B). We conclude that for both nhTMEM16 and yeast ER proteins, the PI-PLC–based assay yields a more complex readout in which scrambling-coupled [3H]PI hydrolysis in both nhTMEM16 proteoliposomes and TE proteoliposomes occurs quite slowly (over tens of minutes) after an initial burst.
Figure 6.

Time-course of PI-PLC–mediated hydrolysis of [3H]PI in nhTMEM16 and TE-containing proteoliposomes. A, liposomes (filled circles) and nhTMEM16 proteoliposomes (open squares) at PPR of ∼1 mg/mmol containing trace amounts of [3H]PI were prepared as Fig. 5. The extent of PI-PLC–mediated hydrolysis of [3H]PI was determined at each time point as described in Fig. 2A. The data are from two independent experiments. B, liposomes (filled circles) and TE-containing proteoliposomes (open squares) at PPR of ∼10 mg/mmol containing trace amounts of [3H]PI were prepared as Fig. 3. The extent of PI-PLC–mediated hydrolysis of [3H]PI was determined at each time point as described in Fig. 2A. The data are from two independent experiments.
Probing the basis for the different readouts in the PI-PLC– versus fluorescence-based assays
Effect of the structure of the lipid reporter
Our results thus far indicate significant differences in the quantitative readout of scramblase activity obtained via the fluorescence-based versus PI-PLC–based assays. Because both assays use topological probes that detect lipids only in the outer leaflet of the liposomes (dithionite does not enter vesicles containing opsin or nhTMEM16 as evinced by protection of trapped NBD-glucose (19, 24, 27), and PI-PLC is a large protein that cannot enter vesicles), they report on the scrambling of lipids from the inner to the outer leaflet, albeit with different characteristics. We considered the possibility that the structure of the lipid reporter, i.e. an NBD-labeled phospholipid versus natural PI, may account for the observed difference. However, we note (i) that the position of the NBD fluorophore on the phospholipid does not detectably impact scrambling, i.e. N-NBD-PE and acyl-NBD-PE are both scrambled faster than the rate at which dithionite reduces the NBD group (23), indicating that lipids with either a natural head group (acyl-NBD-PE) or a natural diacylglycerol moiety (N-NBD-PE) are similarly scrambled by ER scramblases, opsin and nhTMEM16; (ii) that the scrambling rate measured in the fluorescence-based assay does not depend on the concentration of NBD phospholipids in the membrane, indicating that NBD phospholipids are not exclusively or preferentially scrambled compared with natural lipids in the vesicles (19); and (iii) that acyl-NBD-PI and acyl-NBD-PC appear to be scrambled equally effectively in proteoliposomes reconstituted with Triton X-100–solubilized rat liver ER membrane proteins (39). These points suggest that the structure of the lipid reporter per se may not account for the difference between the two assays.
Nevertheless, to investigate this point definitively, we assayed the scramblase activity of nhTMEM16 using NBD-PI as reporter. We synthesized acyl-NBD-PI from acyl-NBD-PC and myo-inositol via phospholipase D-catalyzed transphosphatidylation using a variant of the enzyme with positional selectivity toward the 1-OH group of myo-inositol (Fig. S2, A–D). Fig. 7A shows that acyl-NBD-PI is scrambled effectively in nhTMEM16-containing proteoliposomes (PPR = ∼1 mg/mmol), with >80% of the probe being bleached within 1 min, similar to results obtained with acyl-NBD-PC (Fig. 5B). However, when an aliquot of the same vesicles was treated with PI-PLC for a similar time period (10 min), the extent of NBD-PI hydrolysis was low, ∼50% (Fig. 7B; note that in this assay the extent of NBD-PI hydrolysis in protein-free liposomes was only 46%), increasing to ∼60% after 4 h (Fig. 7B). In proteoliposomes prepared at a higher PPR (4 mg/mmol), NBD-PI hydrolysis was ∼55% after 10 min of PI-PLC treatment, increasing to ∼80% after 4 h of treatment. This behavior resembles that of [3H]PI shown in Fig. 5C. Thus, the PI-PLC–based assay reports a slower rate of NBD-PI scrambling than the dithionite-based assay.
Figure 7.
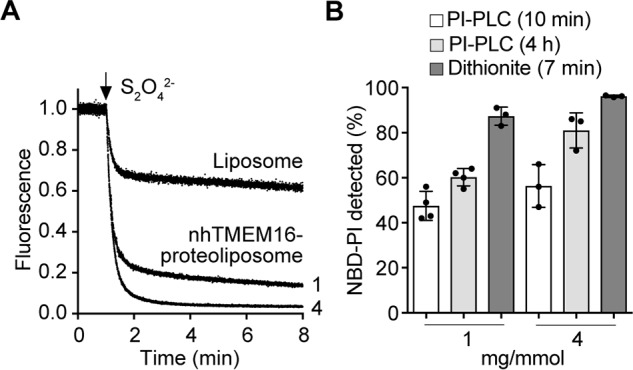
nhTMEM16-mediated scrambling of NBD-PI probed by dithionite and PI-PLC. A, liposomes and nhTMEM16 proteoliposomes at a PPR of ∼1 or 4 mg/mmol containing trace amounts of NBD-PI (Fig. S2) were reconstituted as in Fig. 4B, except for the addition of 250 μm Ca2+ in the reconstitution solution. Dithionite was added after 1 min, and fluorescence was recorded for 8 min. Traces of liposomes and nhTMEM16 proteoliposomes (PPR of ∼1 or ∼4 mg/mmol, indicated next to the corresponding trace) are representative of three independent experiments. B, nhTMEM16 proteoliposomes at PPRs of ∼1 and ∼4 mg/mmol containing trace amounts of NBD-PI were prepared as above in the presence of 250 μm Ca2+. The extent of PI-PLC–mediated hydrolysis of NBD-PI after 10 min (white bars) and 4 h (light gray bars) was determined as described under “Experimental procedures,” and reduction of NBD-PI fluorescence by dithionite (dark gray bars) was determined as described above. The data represent mean values ± standard deviations from three or four measurements.
Effect of PI and/or diacylglycerol
Because the two assays with NBD-PI yielded different quantitative outcomes, we considered other possibilities to account for the differences: (i) PI has an inhibitory effect on scramblase activity, and (ii) diacylglycerol, the product of PI hydrolysis, is inhibitory for scrambling.
Vesicle reconstitution is intrinsically heterogeneous (52), and so even with a fixed average ratio of PI:PC:PA(DOPG) in our reconstituted samples, individual vesicles could contain higher or lower amounts of PI. We considered whether the scramblase activity of opsin or TMEM16 vesicles with a higher proportion of PI would be silenced, whereas those with less PI would display activity albeit at a lower rate. To test whether PI inhibits scramblase activity, liposomes and nhTMEM16-containing proteoliposomes (at PPR of ∼2 mg/mmol) were reconstituted with [3H]PI in the absence or presence of 2% (relative to total phospholipid) nonlabeled PI, incubated with 3 μl of PI-PLC for 10 min, and processed to determine the extent of [3H]PI hydrolysis. As shown in Fig. 8A, the extent of hydrolysis after a 10-min incubation period was ∼65% for both types of proteoliposomes, demonstrating that the presence of 2% PI in proteoliposomes did not affect scramblase activity.
Figure 8.
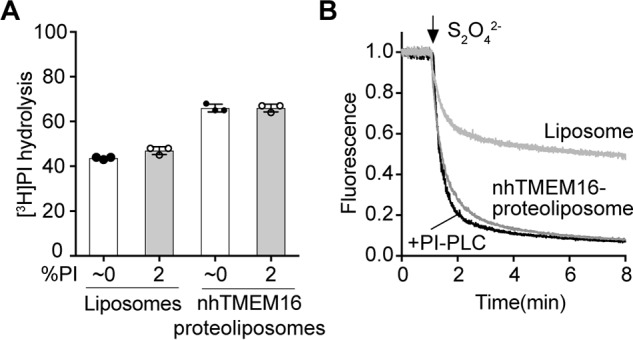
The effect of PI and diacylglycerol on nhTMEM16-mediated scrambling of [3H]PI and NBD-PC. A, liposomes and nhTMEM16-containing proteoliposomes (PPR = ∼2 mg/mmol) composed of egg PC and DOPG (9:1) were reconstituted with trace amounts of [3H]PI in the absence or presence of 2% (relative to total phospholipid) nonlabeled PI and in the presence of 250 μm Ca2+. Subsequently, the samples were treated with PI-PLC for 10 min, and the extent of [3H]PI hydrolysis was determined after TCA precipitation as described under “Experimental procedures.” The data represent mean values ± standard deviations from three independent experiments. B, liposomes and nhTMEM16-containing proteoliposomes (PPR = ∼1 mg/mmol) were reconstituted in the presence of [3H]PI, 2 mol % nonlabeled PI and NBD-PC and incubated for 10 min in the absence or presence of PI-PLC. Subsequently, the samples were taken for the fluorescence-based scramblase assay: dithionite was added as indicated, and fluorescence was recorded at room temperature with constant stirring. The traces are representative of two independent experiments.
A possible effect of diacylglycerol on NBD phospholipid scrambling was tested by treating nhTMEM16 proteoliposomes containing [3H]PI, 2% PI, and NBD-PC with PI-PLC for 10 min before measuring NBD-PC scrambling using the dithionite assay. The results showed that ∼80% of NBD-PC was reduced in both mock- and PI-PLC–treated proteoliposomes, demonstrating that the presence of the product of the PI-PLC reaction, diacylglycerol, had no effect on NBD-PC scrambling (Fig. 8B).
Discussion
We report a new assay of phospholipid scramblase activity that exploits the ability of PI-PLC to hydrolyze [3H]PI located in the outer leaflet of (proteo)liposomes, whereas the fraction of [3H]PI located in the inner leaflet is protected from the action of the enzyme unless it is exposed by scramblase activity. Although most currently used scramblase assays make use of synthetic head- or tail-labeled phospholipid analogs bearing covalently linked fluorescent or spin-labeled functional groups (see e.g. (19, 20, 41, 53, 54)), our assay measures the transbilayer movement of a natural phospholipid. We demonstrate that PI is scrambled in proteoliposomes reconstituted with yeast ER phospholipid scramblases, purified bovine opsin, or purified fungal nhTMEM16. Under our assay conditions, spontaneous scrambling of PI is undetectable—in fact, protein-free liposomes did not expose inner leaflet PI for a period of at least 4 h at room temperature.
Interestingly, the readout from the PI-PLC–based assay is quantitatively different from that of the conventional fluorescence-based assay that uses dithionite to probe NBD phospholipids in the outer leaflet of vesicles. [3H]PI hydrolysis resulting from scrambling, i.e. hydrolysis in excess of the initial hydrolysis of PI already in the outer leaflet, appears to occur significantly more slowly compared with results from the fluorescence-based assay. For proteoliposomes prepared at relatively low PPR, the PI-PLC–based assay showed only 60–70% [3H]PI hydrolysis after a standard 10-min incubation, whereas the fluorescence-based assay showed a reduction of 80–90% fluorescence of NBD-PC and NBD-PI in the same time frame. Only at higher PPR did the extent of [3H]PI hydrolysis reach values >80% within 10 min. This observation was independent of the scramblase used for reconstitution. Extended time courses using proteoliposomes reconstituted with crude ER scramblases or purified nhTMEM16 at relatively low PPR revealed that scrambling-coupled [3H]PI hydrolysis mediated by PI-PLC occurred slowly, on the time scale of tens of minutes, after an initial burst. Experiments with NBD-PI, a lipid amenable to analysis by both fluorescence- and PI-PLC–based assays, indicated that the difference in assay readout was due to an aspect of the assay itself rather than a difference between an NBD-labeled lipid and a natural lipid. Thus, when using dithionite, NBD-PI scrambling was seen to be rapid, resembling that of other NBD phospholipids such as NBD-PC, whereas when using the PI-PLC–based assay, NBD-PI scrambling was slow, resembling that of [3H]PI.
Our results are enigmatic and do not lend themselves to a straightforward explanation. Although the burst kinetics seen with PI-PLC–mediated hydrolysis of [3H]PI in nhTMEM16- and TE-containing proteoliposomes (Fig. 6) indicate that there is pool of PI that is scrambled rapidly as expected from the fluorescence-based assay, the slow continued hydrolysis of [3H]PI indicates the additional presence of a slowly released pool. This pool can be converted into the fast scrambling pool by reconstituting additional scramblases per vesicle, i.e. by increasing the PPR of the sample. We ruled out that this effect could be due to the presence of PI or its hydrolysis product, diacylglycerol. Although it has been previously suggested that diacylglycerol may enhance PI-PLC activity through effects on the membrane (55), we cannot explicitly eliminate the possibility that diacylglycerol produced in the initial stages of the assay, e.g. during PI-PLC–mediated hydrolysis of outer leaflet PI, would have an adverse effect on the ability of the enzyme to hydrolyze PI efficiently, giving rise to the observed slow rate. More work needs to be done to elucidate the mechanistic basis for the quantitative differences that we have identified.
Experimental procedures
Unless otherwise stated, all reagents were of analytical grade and purchased from Sigma–Aldrich or Merck. Phosphatidylcholine and phosphatidic acid from egg yolk (egg PC and egg PA, respectively, 99% purity) DOPG and NBD-PC were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). l-α-[myo-Inositol-2-3H(N)] PI ([3H]PI; 11.8 Ci/mmol) was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO). PI-PLC from Bacillus cereus was purchased from Thermo Fisher Scientific. SM-2 Bio-Beads adsorbents were from Bio-Rad. Triton X-100 was from Roche Applied Science. n-Dodecyl-β-d-maltopyranoside (DDM) was purchased from Anatrace (Berkshire, UK).
Preparation of acyl-NBD-PI
Acyl-NBD-PI (henceforth NBD-PI) was synthesized from acyl-NBD-PC and myo-inositol by phospholipase D (PLD)-mediated transphosphatidylation (Fig. S2A). We used an engineered variant of a microbial PLD having amino acid replacement of G186T/W187N/Y385R. This enzyme is capable of transferring the phosphatidyl group with positional selectivity toward the 1-OH group of myo-inositol, thereby synthesizing 1-PI, a naturally occurring isomer (56). 1-ml chloroform solution of 1 mg/ml NBD-PC was dried under nitrogen gas and redissolved in 100 μl of ethyl acetate. To this lipid solution was added 80 μl of NaCl-saturated 50 mm acetate buffer (pH 5.6) containing 18 mg of myo-inositol and 20 μl of 0.5 mg/ml of the variant PLD. This biphasic mixture was vigorously mixed at 20 °C for 24 h. The use of NaCl-saturated buffer is important to increase the product yield by localizing the enzyme toward the solvent–water interface (57). 10 μl of 1 m HCl was added to stop the reaction, and the lipid was extracted with 200 μl of chloroform:methanol (2:1). After the removal of the solvent under nitrogen gas, the lipid was redissolved in 50 μl of chloroform and loaded onto a small (100 mg) silica gel column. The lipids were eluted with 2 ml of chloroform:methanol (9:1, by volume) to elute NBD-phosphatidic acid (NBD-PA) and then with 5 ml of chloroform:methanol (7:3, by volume) to elute NBD-PI. Eluents were collected into 200-μl fractions, each of which was analyzed by thin layer chromatography (TLC). Because the fractions containing NBD-PI were contaminated with NBD-PA, they were combined and rechromatographed. As shown in Fig. S2B, the purified lipid contained mainly NBD-PI with small amount of NBD-PA. Liquid chromatography–MS analysis gave a peak of NBD-PI at 24 min having m/z value of 847.2 (M − H)− and a small (∼6% of total area) peak of NBD-PA at 23 min having m/z value of 685.15 (M − H)− (Fig. S2, C and D).
Preparation of TE containing yeast ER membrane proteins
A crude preparation of S. cerevisiae ER membrane proteins was obtained as previously described (49) by solubilizing salt-washed yeast microsomes with 1% (w/v) ice-cold Triton X-100.
Bovine opsin
Thermostable FLAG-tagged bovine opsin was prepared as described before (24, 58) using HEK293S GnTI− cells and FLAG affinity chromatography. A standard preparation yielded 100 ng/μl of opsin in buffer containing 0.1% (w/v) DDM.
nhTMEM16
His10-tagged nhTMEM16 was expressed in S. cerevisiae FGY217 cells and purified exactly as described (21) using affinity chromatography on nickel–nitrilotriacetic acid–resin followed by gel filtration chromatography on a Superdex-200 column.
Preparation of scramblase-containing proteoliposomes
Opsin-containing proteoliposomes were reconstituted as described previously, by destabilizing preformed liposomes composed of egg PC and egg PA at a molar ratio of 9:1 (58). Protein-free liposomes were prepared in parallel. nhTMEM16-containing proteoliposomes were reconstituted in a similar way as opsin proteoliposomes, except that the preformed liposomes were made using egg PC and DOPG (9:1, molar ratio) and destabilized using DDM in a volume of 840 μl (2.8 mm phospholipid and 4.5 mm DDM) in the presence of 250 μm Ca2+. Reconstitution of proteoliposomes with yeast Triton extract was done as previously described (48).
Quantification of phospholipids
Aliquots of liposomes and proteoliposomes were extracted according to (59) (see below), and lipid phosphorus was quantified as described (60).
Scramblase activity assay using [3H]PI as probe
The assay was initiated by adding 3 μl of PI-PLC to (proteo)liposomes (100 μl; 2.4 mm phospholipid) and rapidly mixing. After incubation at room temperature for 0–4 h, the reaction was terminated by removing an aliquot (103 μl) and adding it to a glass tube containing 0.5 ml of chloroform and 1.0 ml of methanol. Lipids were extracted by adding 397 μl of distilled water and frequent vigorous vortexing for 10 min at room temperature. After addition of a further 0.5 ml of chloroform and 0.5 ml of methanol (to reach a final ratio of water:chloroform:methanol = 1:1:1, v/v/v), the resulting two phase extract was vortexed and then centrifuged for 5 min at 2500 rpm using a Universal 2S centrifuge (Hettich, Tuttlingen, Germany). The lower organic phase, containing nonhydrolyzed [3H]PI, was transferred to a scintillation tube using a long, blunt-tip needle syringe, dried under a stream of nitrogen, and counted in a liquid scintillation counter. The upper aqueous phase, containing [3H]inositol-cyclic-phosphate, was transferred to a microcentrifuge tube, dried in a SpeedVac to reduce the volume, transferred to scintillation tubes, and counted.
Alternatively, aliquots (100 μl) were removed, and the reaction was stopped by adding 5 μl of cytochrome c (4 mg/ml in water) followed by 13 μl of 100% TCA and placed on ice for 1 h. Subsequently, samples were centrifuged for 15 min at top speed in an Eppendorf centrifuge to collect precipitated phospholipids including nonhydrolyzed [3H]PI. The resulting supernatant (100 μl), containing [3H]inositol 1,2-cyclic phosphate/[3H]inositol 1-phosphate, was transferred to scintillation tubes and counted. To determine the extent of [3H]PI hydrolysis, TCA samples prepared before the addition of PI-PLC were taken as background, and TCA samples taken after PI-PLC treatment for 10 min in the presence of 0.9% Triton X-100 (added from a stock solution of 10% in buffer A) were taken as 100%.
Scramblase activity assay using NBD-labeled PC as probe
Scrambling of NBD-PC was measured exactly as described before (58). Briefly, liposomes or proteoliposomes composed of egg PC, egg PA, and trace amounts of NBD-PC were diluted with buffer A, and NBD fluorescence was monitored in a fluorimeter until a stable signal was obtained. Bleaching of the fluorescence in the outer leaflet of liposomes was achieved by addition of dithionite.
Scrambling activity assays using multiple probes
In some experiments, [3H]PI and NBD-PC or NBD-PI were added simultaneously during reconstitution of liposomes. Determination of the extent of [3H]PI and NBD-PI hydrolysis by PI-PLC and NBD-PC or NBD-PI reduction by dithionite allowed direct comparison of scrambling activity using the different probes.
Scrambling of NBD-PI using PI-PLC as a probe
Liposomes or proteoliposomes containing NBD-PI were reconstituted in the same way as for NBD-PC mentioned above. The scrambling of NBD-PI was initiated by adding 3 μl of PI-PLC to (proteo)liposomes (200 μl; 2.4 mm phospholipid) and rapidly mixing. After incubation at room temperature for 10 min or 4 h, the reaction was terminated by adding it to a glass tube containing 0.5 ml of chloroform and 1.0 ml of methanol. The lipids were extracted as described under “Scramblase activity assay using [3H]PI as probe” and then resolved on a silica TLC plate using chloroform:petroleum ether:methanol:acetic acid (4:3:2:1, v/v/v/v) as the solvent system. NBD-PI and NBD-diacylglycerol spots on TLC were visualized under blue light and quantified by ImageJ or scraped off and extracted before quantification of NBD fluorescence in a fluorimeter (470-nm excitation; 530-nm emission).
Author contributions
L. W., A. K. M., and P. B. conceptualization; L. W., A. K. M., and P. B. data curation; L. W., Y. I., K. K. A., A. K. M., and P. B. formal analysis; L. W., Y. I., K. K. A., K.P., A. K. M., and P. B. investigation; L. W., Y. I., K. K. A., K.P., A. K. M., and P. B. methodology; L. W. writing-original draft; A. K. M. and P. B. supervision; A. K. M. and P. B. funding acquisition; A. K. M. and P. B. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Birgit Ploier for preparing purified opsin and for instructing L. W. on the opsin reconstitution protocol. P. B. thanks A. Niederer, M. Bütikofer, and D. Reynolds for advice and stimulation.
This work was supported by Grant 881 from the Velux Foundation (to P. B. and A. K. M.) and in part by Swiss National Science Foundation Grant 31003A_169355 (to P. B.) and National Institutes of Health Grants EY028314 and GM106717 (to A. K. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- PC
- phosphatidylcholine
- DDM
- n-dodecyl-β-d-maltopyranoside
- DOPG
- dioleoylphosphatidylglycerol
- ER
- endoplasmic reticulum
- NBD
- nitrobenzoxadiazole
- NBD-PC
- 1-palmitoyl-2-{6-[7-nitro-2–1,3-benzoxadiazole-4-yl)amino]hexanoyl}-sn-glycero-3-phosphocholine
- nhTMEM16
- N. haematococca TMEM16
- PA
- phosphatidic acid
- PE
- phosphatidylethanolamine
- PI
- phosphatidylinositol
- PI–PLC
- phosphatidylinositol-specific phospholipase C
- PLD
- phospholipase D
- PPR
- protein to phospholipid ratio
- TE
- Triton extract, enriched in yeast ER membrane proteins.
References
- 1. Kennedy E. P., and Weiss S. B. (1956) The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 222, 193–214 [PubMed] [Google Scholar]
- 2. Ballas L. M., and Bell R. M. (1980) Topography of phosphatidylcholine, phosphatidylethanolamine and triacylgycerol biosynthetic enzymes in rat liver microsomes. Biochim. Biophys. Acta 602, 578–590 10.1016/0005-2736(80)90336-3 [DOI] [PubMed] [Google Scholar]
- 3. Kuchler K., Daum G., and Paltauf F. (1986) Subcellular and submitochondrial localization of phospholipid-synthesizing enzyme in Saccharomyces cerevisiae. J. Bacteriol. 165, 901–910 10.1128/jb.165.3.901-910.1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Henneberry A. L., Wright M. M., and McMaster C. R. (2002) The major sites of cellular phospholipid synthesis and molecular determinants of fatty acid and lipid head group specificity. Mol. Biol. Cell. 13, 3148–3161 10.1091/mbc.01-11-0540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Coleman J. A., Quazi F., and Molday R. S. (2013) Mammalian P4-ATPases and ABC transporters and their role in phospholipid transport. Biochim. Biophys. Acta 1831, 555–574 10.1016/j.bbalip.2012.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coleman R., and Bell R. M. (1978) Evidence that biosynthesis of phosphatidylethanolamine, phosphatidylcholine, and triacylglycerol occurs on the cytoplasmic side of microsomal vesicles. J. Cell Biol. 76, 245–253 10.1083/jcb.76.1.245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chauhan N., Farine L., Pandey K., Menon A. K., and Bütikofer P. (2016) Lipid topogenesis: 35 years on. Biochim. Biophys. Acta 1861, 757–766 10.1016/j.bbalip.2016.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pomorski T. G., and Menon A. K. (2016) Lipid somersaults: uncovering the mechanisms of protein-mediated lipid flipping. Prog. Lipid Res. 64, 69–84 10.1016/j.plipres.2016.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sebastian T. T., Baldridge R. D., Xu P., and Graham T. R. (2012) Phospholipid flippases: building asymmetric membranes and transport vesicles. Biochim. Biophys. Acta 1821, 1068–1077 10.1016/j.bbalip.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Montigny C., Lyons J., Champeil P., Nissen P., and Lenoir G. (2016) On the molecular mechanism of flippase- and scramblase-mediated phospholipid transport. Biochim. Biophys. Acta 1861, 767–783 10.1016/j.bbalip.2015.12.020 [DOI] [PubMed] [Google Scholar]
- 11. Sanyal S., and Menon A. K. (2009) Flipping lipids: why an' what's the reason for? ACS Chem. Biol. 4, 895–909 10.1021/cb900163d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pomorski T., and Menon A. K. (2006) Lipid flippases and their biological functions. Cell. Mol. Life Sci. 63, 2908–2921 10.1007/s00018-006-6167-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bishop W. R., and Bell R. M. (1985) Assembly of the endoplasmic reticulum phospholipid bilayer: the phosphatidylcholine transporter. Cell 42, 51–60 10.1016/S0092-8674(85)80100-8 [DOI] [PubMed] [Google Scholar]
- 14. Backer J. M., and Dawidowicz E. A. (1987) Reconstitution of a phospholipid flippase from rat liver microsomes. Nature 327, 341–343 10.1038/327341a0 [DOI] [PubMed] [Google Scholar]
- 15. Menon A. K., Watkins W. E. 3rd, and Hrafnsdóttir S. (2000) Specific proteins are required to translocate phosphatidylcholine bidirectionally across the endoplasmic reticulum. Curr. Biol. 10, 241–252 10.1016/S0960-9822(00)00356-0 [DOI] [PubMed] [Google Scholar]
- 16. Suzuki J., Denning D. P., Imanishi E., Horvitz H. R., and Nagata S. (2013) Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406 10.1126/science.1236758 [DOI] [PubMed] [Google Scholar]
- 17. Suzuki J., Fujii T., Imao T., Ishihara K., Kuba H., and Nagata S. (2013) Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem. 288, 13305–13316 10.1074/jbc.M113.457937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki J., Umeda M., Sims P. J., and Nagata S. (2010) Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838 10.1038/nature09583 [DOI] [PubMed] [Google Scholar]
- 19. Malvezzi M., Chalat M., Janjusevic R., Picollo A., Terashima H., Menon A. K., Accardi A., Janjusevic R., Chalat M. N., Terashima H., Menon A. K., and Accardi A. (2013) Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat. Commun. 4, 2367 10.1038/ncomms3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brunner J. D., Lim N. K., Schenck S., Duerst A., and Dutzler R. (2014) X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature 516, 207–212 10.1038/nature13984 [DOI] [PubMed] [Google Scholar]
- 21. Lee B. C., Menon A. K., and Accardi A. (2016) The nhTMEM16 scramblase is also a nonselective ion channel. Biophys. J. 111, 1919–1924 10.1016/j.bpj.2016.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Watanabe R., Sakuragi T., Noji H., and Nagata S. (2018) Single-molecule analysis of phospholipid scrambling by TMEM16F. Proc. Natl. Acad. Sci. U.S.A. 115, 3066–3071 10.1073/pnas.1717956115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Menon I., Huber T., Sanyal S., Banerjee S., Barré P., Canis S., Warren J. D., Hwa J., Sakmar T. P., and Menon A. K. (2011) Opsin is a phospholipid flippase. Curr. Biol. 21, 149–153 10.1016/j.cub.2010.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goren M. A., Morizumi T., Menon I., Joseph J. S., Dittman J. S., Cherezov V., Stevens R. C., Ernst O. P., and Menon A. K. (2014) Constitutive phospholipid scramblase activity of a G protein–coupled receptor. Nat. Commun. 5, 5115 10.1038/ncomms6115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morra G., Razavi A. M., Pandey K., Weinstein H., Menon A. K., and Khelashvili G. (2018) Mechanisms of lipid scrambling by the G protein–coupled receptor opsin. Structure 26, 356–367 10.1016/j.str.2017.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ploier B., Caro L. N., Morizumi T., Pandey K., Pearring J. N., Goren M. A., Finnemann S. C., Graumann J., Arshavsky V. Y., Dittman J. S., Ernst O. P., and Menon A. K. (2016) Dimerization deficiency of enigmatic retinitis pigmentosa-linked rhodopsin mutants. Nat. Commun. 7, 12832 10.1038/ncomms12832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pandey K., Ploier B., Goren M. A., Levitz J., Khelashvili G., and Menon A. K. (2017) An engineered opsin monomer scrambles phospholipids. Sci. Rep. 7, 16741 10.1038/s41598-017-16842-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wu G., and Hubbell W. L. (1993) Phospholipid asymmetry and transmembrane diffusion in photoreceptor disc membranes. Biochemistry 32, 879–888 10.1021/bi00054a020 [DOI] [PubMed] [Google Scholar]
- 29. Hessel E., Herrmann A., Müller P., Schnetkamp P. P., and Hofmann K. P. (2000) The transbilayer distribution of phospholipids in disc membranes is a dynamic equilibrium: evidence for rapid flip and flop movement. Eur. J. Biochem. 267, 1473–1483 10.1046/j.1432-1327.2000.01147.x [DOI] [PubMed] [Google Scholar]
- 30. Hessel E., Müller P., Herrmann A., and Hofmann K. P. (2001) Light-induced reorganization of phospholipids in rod disc membranes. J. Biol. Chem. 276, 2538–2543 10.1074/jbc.M009061200 [DOI] [PubMed] [Google Scholar]
- 31. Ernst O. P., and Menon A. K. (2015) Phospholipid scrambling by rhodopsin. Photochem. Photobiol. Sci. 14, 1922–1931 10.1039/C5PP00195A [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kobayashi T., and Menon K. A. (2018) Transbilayer lipid asymmetry. Curr. Biol. [DOI] [PubMed] [Google Scholar]
- 33. Chap H. J., Zwaal R. F., and van Deenen L. L. M. (1977) Action of highly purified phospholipases on blood platelets. Evidence for an asymmetric distribution of phospholipids in the surface membrane. Biochim. Biophys. Acta 467, 146–164 10.1016/0005-2736(77)90192-4 [DOI] [PubMed] [Google Scholar]
- 34. Tsai K. H., and Lenard J. (1975) Asymmetry of influenza virus membrane bilayer demonstrated with phospholipase C. Nature 253, 554–555 10.1038/253554a0 [DOI] [PubMed] [Google Scholar]
- 35. Seigneuret M., and Devaux P. F. (1984) ATP-dependent asymmetric distribution of spin-labeled phospholipids in the erythrocyte membrane: relation to shape changes. Proc. Natl. Acad. Sci. U.S.A. 81, 3751–3755 10.1073/pnas.81.12.3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McIntyre J. C., and Sleight R. G. (1991) Fluorescence assay for phospholipid membrane asymmetry. Biochemistry 30, 11819–11827 10.1021/bi00115a012 [DOI] [PubMed] [Google Scholar]
- 37. Pomorski T., Muller P., Zimmermann B., Burger K., Devaux P. F., and Herrmann A. (1996) Transbilayer movement of fluorescent and spin-labeled phospholipids in the plasma membrane of human fibroblasts: a quantitative approach. J. Cell Sci. 109, 687–698 [DOI] [PubMed] [Google Scholar]
- 38. Wolf D. E., Winiski A. P., Ting A. E., Bocian K. M., and Pagano R. E. (1992) Determination of the transbilayer distribution of fluorescent lipid analogues by nonradiative fluorescence resonance energy transfer. Biochemistry 31, 2865–2873 10.1021/bi00126a004 [DOI] [PubMed] [Google Scholar]
- 39. Vishwakarma R. A., Vehring S., Mehta A., Sinha A., Pomorski T., Herrmann A., and Menon A. K. (2005) New fluorescent probes reveal that flippase-mediated flip-flop of phosphatidylinositol across the endoplasmic reticulum membrane does not depend on the stereochemistry of the lipid. Org. Biomol. Chem. 3, 1275–1283 10.1039/b500300h [DOI] [PubMed] [Google Scholar]
- 40. Chang Q. L., Gummadi S. N., and Menon A. K. (2004) Chemical modification identifies two populations of glycerophospholipid flippase in rat liver ER. Biochemistry 43, 10710–10718 10.1021/bi049063a [DOI] [PubMed] [Google Scholar]
- 41. Devaux P. F., Fellmann P., and Hervé P. (2002) Investigation on lipid asymmetry using lipid probes: comparison between spin-labeled lipids and fluorescent lipids. Chem. Phys. Lipids 116, 115–134 10.1016/S0009-3084(02)00023-3 [DOI] [PubMed] [Google Scholar]
- 42. Higgins J. A., Hitchin B. W., and Low M. G. (1989) Phosphatidylinositol-specific phospholipase C of Bacillus thuringiensis as a probe for the distribution of phosphatidylinositol in hepatocyte membranes. Biochem. J. 259, 913–916 10.1042/bj2590913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bütikofer P., Lin Z. W., Chiu D. T., Lubin B., and Kuypers F. A. (1990) Transbilayer distribution and mobility of phosphatidylinositol in human red blood cells. J. Biol. Chem. 265, 16035–16038 [PubMed] [Google Scholar]
- 44. Masayama A., Tsukada K., Ikeda C., Nakano H., and Iwasaki Y. (2009) Isolation of phospholipase D mutants having phosphatidylinositol-synthesizing activity with positional specificity on myo-inositol. Chembiochem. 8601, 559–564 [DOI] [PubMed] [Google Scholar]
- 45. Volwerk J. J., Shashidhar M. S., Kuppe A., and Griffith O. H. (1990) Phosphatidylinositol-specific phospholipase C from Bacillus cereus combines intrinsic phosphotransferase and cyclic phosphodiesterase activities: a 31P NMR study. Biochemistry 29, 8056–8062 10.1021/bi00487a010 [DOI] [PubMed] [Google Scholar]
- 46. Griffith O. H., and Ryan M. (1999) Bacterial phosphatidylinositol-specific phospholipase C: structure, function, and interaction with lipids. Biochim. Biophys. Acta 1441, 237–254 10.1016/S1388-1981(99)00153-5 [DOI] [PubMed] [Google Scholar]
- 47. Zhou C., Wu Y., and Roberts M. F. (1997) Activation of phosphatidylinositol-specific phospholipase C toward inositol. Biochemistry 36, 347–355 10.1021/bi960601w [DOI] [PubMed] [Google Scholar]
- 48. Chalat M., Menon I., Turan Z., and Menon A. K. (2012) Reconstitution of glucosylceramide flip-flop across endoplasmic reticulum: implications for mechanism of glycosphingolipid biosynthesis. J. Biol. Chem. 287, 15523–15532 10.1074/jbc.M112.343038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sanyal S., Frank C. G., and Menon A. K. (2008) Distinct flippases translocate glycerophospholipids and oligosaccharide diphosphate dolichols across the endoplasmic reticulum. Biochemistry 47, 7937–7946 10.1021/bi800723n [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Vehring S., Pakkiri L., Schröer A., Alder-Baerens N., Herrmann A., Menon A. K., and Pomorski T. (2007) Flip-flop of fluorescently labeled phospholipids in proteoliposomes reconstituted with Saccharomyces cerevisiae microsomal proteins. Eukaryot. Cell 6, 1625–1634 10.1128/EC.00198-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee S. Y., Letts J. A., and MacKinnon R. (2009) Functional reconstitution of purified human Hv1 H+ channels. J. Mol. Biol. 387, 1055–1060 10.1016/j.jmb.2009.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Larsen J., Hatzakis N. S., and Stamou D. (2011) Observation of inhomogeneity in the lipid composition of individual nanoscale liposomes. J. Am. Chem. Soc. 133, 10685–10687 10.1021/ja203984j [DOI] [PubMed] [Google Scholar]
- 53. McNamee M. G., and McConnell H. M. (1973) Transmembrane potentials and phospholipid flip-flop in excitable membrane vesicles. Biochemistry 12, 2951–2958 10.1021/bi00740a001 [DOI] [PubMed] [Google Scholar]
- 54. Marx U., Lassmann G., Holzhütter H. G., Wüstner D., Müller P., Höhlig A., Kubelt J., and Herrmann A. (2000) Rapid flip-flop of phospholipids in endoplasmic reticulum membranes studied by a stopped-flow approach. Biophys. J. 78, 2628–2640 10.1016/S0006-3495(00)76807-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ahyayauch H., Sot J., Collado M. I., Huarte N., Requejo-Isidro J., Alonso A., and Goñi F. M. (2015) End-product diacylglycerol enhances the activity of PI-PLC through changes in membrane domain structure. Biophys. J. 108, 1672–1682 10.1016/j.bpj.2015.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Damnjanović J., Kuroiwa C., Tanaka H., Ishida K., and Nakano H. (2016) Directing positional specificity in enzymatic synthesis of bioactive 1-phosphatidylinositol by protein engineering of a phospholipase D. Biotechnol. Bioeng. 113, 62–71 10.1002/bit.25697 [DOI] [PubMed] [Google Scholar]
- 57. Muraki M., Damnjanovi J., Nakano H., and Iwasaki Y. (2016) Salt-induced increase in the yield of enzymatically synthesized phosphatidylinositol and the underlying mechanism. J. Biosci. Bioenerg. 122, 276–282 10.1016/j.jbiosc.2016.02.011 [DOI] [PubMed] [Google Scholar]
- 58. Ploier B., and Menon A. K. (2016) A fluorescence-based assay of phospholipid scramblase activity. J. Vis. Exp. 10.3791/54635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 10.1139/y59-099,10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 60. Rouser G., Fleischer S., and Yamamoto A. (1970) Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 5, 494–496 10.1007/BF02531316 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.