

Abstract
Deregulation of the HECT ubiquitin ligase UBE3A/E6AP has been implicated in Angelman syndrome as well as autism spectrum disorders. We and others have previously identified the 26S proteasome as one of the major UBE3A-interacting protein complexes. Here, we characterize the interaction of UBE3A and the proteasomal subunit PSMD4 (Rpn10/S5a). We map the interaction to the highly conserved Zn2+-binding N-terminal (AZUL) domain of UBE3A, the integrity of which is crucial for binding to PSMD4. Interestingly, two Angelman syndrome point mutations that affect the AZUL domain show an impaired ability to bind PSMD4. Although not affecting the ubiquitin ligase or the estrogen receptor α–mediated transcriptional regulation activities, these AZUL domain mutations prevent UBE3A from stimulating the Wnt/β-catenin signaling pathway. Taken together, our data indicate that impaired binding to the 26S proteasome and consequential deregulation of Wnt/β-catenin signaling might contribute to the functional defect of these mutants in Angelman syndrome.
Keywords: autism, proteasome, estrogen receptor, Wnt signaling, E3 ubiquitin ligase, Angelman syndrome, AZUL domain, E6AP, PSMD4, UBE3A
Introduction
UBE3A/E6AP3 is a HECT (homologous to E6AP C terminus) ubiquitin ligase that has been implicated in human papillomavirus (HPV)-positive cancers, Angelman syndrome (AS), and some cases of autism spectrum disorder. The UBE3A gene is located on chromosome 15 (15q11.2–13). Differential splicing results in the expression of at least three UBE3A isoforms that differ only in their N termini, although no different functions for those three isoforms have yet been described (1). UBE3A was originally named E6-associated protein (E6AP) after its initial identification as an interaction partner of the E6 protein expressed by a subset of HPVs that are associated with cervical cancer, other anogenital cancers, and oropharyngeal cancer (2–4). The E6 proteins encoded by this group of cancer-associated HPVs, referred to as high-risk HPVs (hrHPVs), hijack UBE3A to form a ternary complex with the tumor suppressor p53 protein, resulting in p53 ubiquitylation and proteasome-mediated degradation (5). The functional inactivation of the p53 pathway is a crucial step in HPV-mediated carcinogenesis. Subsequent studies revealed that UBE3A has important neurological roles and that alterations in UBE3A expression levels are associated with specific neurogenetic disorders. Loss of UBE3A function in the brain has been identified as the cause of Angelman syndrome, whereas an increase in UBE3A gene dosage has been linked to autism spectrum disorders (6–10). In addition to and independently of its ubiquitin ligase activity, UBE3A is also involved in the transcriptional regulation of steroid hormone receptors (11–13). Interestingly, both stimulating and repressing roles for these signaling pathways have been described for UBE3A, indicating that the function of UBE3A might depend on the cellular context and the responsive gene (11–13).
Ubiquitin ligases are involved in substrate recognition specificity as part of an enzymatic cascade resulting in the covalent attachment of ubiquitin to its target proteins (14, 15). HECT ubiquitin ligases are characterized by the presence of a C-terminal HECT domain that contains a catalytic cysteine residue. This active-site cysteine forms a thioester bond with the C-terminal glycine residue of ubiquitin in a step preceding the final transfer of ubiquitin to the substrate protein (15, 16). Mutation of the active-site cysteine inactivates the ability of HECT ubiquitin ligases to transfer ubiquitin to its substrates (16). UBE3A has been shown to catalyze the assembly of polyubiquitin chains through the Lys-48 residue of the ubiquitin moiety, providing a signal for proteasomal degradation (17, 18).
The 26S proteasome is a large cellular protein complex that contains a 20S barrel structure with a 19S lid structure on both sides of the barrel (19). Although the proteins of the lid complex are responsible for the recruitment of ubiquitylated proteins, the barrel and its intrinsic proteolytic activities mediate the breakdown of proteins (20). UBE3A associates with the proteasome (21–29). In that context, the UBE3A-interacting protein PMSD4 (also known as Rpn10/S5a) is part of the regulatory lid complex and known to bind to polyubiquitin chains of ubiquitylated proteins via its ubiquitin-interacting motif (UIM), resulting in their recruitment to the proteasome (30–32). PSMD4, however, also exists unbound to the proteasome in the cytosol, and its proteasome-independent functions are still unclear (23, 29).
Although UBE3A is biallelically expressed in most tissues, expression of the paternal allele is silenced in specific brain regions, including the hippocampus. Loss of a functional maternal UBE3A allele and therefore absence of UBE3A in these brain regions causes Angelman syndrome (33). AS patients are characterized by a severe neurodevelopmental phenotype. Symptoms include behavioral uniqueness, severe impairment of speech, developmental delay, and movement and balance disorder (34–37). The genetic mechanisms resulting in Angelman syndrome are varied. The majority of cases are caused by deletions of the maternal 15q11.2–13 region, but some cases show paternal uniparental disomy or imprinting defects, all of which result in a complete loss of functional UBE3A expression (38). Some patients carry intragenic UBE3A mutations, including frameshift or missense mutations that are thought to result in the expression of truncated, nonfunctional proteins. A number of UBE3A point mutations leading to single amino acid substitutions have also been described (38). Most of these point mutations are located in the C-terminal catalytic domain of UBE3A, affecting its ubiquitin ligase activity, whereas others along the length of the protein destabilize the protein, resulting in a functional loss of UBE3A (39, 40). Analysis of such point mutations could potentially provide important insights into whether additional functions of UBE3A or other cellular protein interactions could contribute to the AS phenotype.
A number of UBE3A-interacting proteins and potential substrates, including RAD23A, UBQLN1/2, and estrogen receptor α (ERα) have been described (41–43). UBE3A has also been recently shown to interact with a large protein complex consisting of HERC2, UBE3A, and NEURL4 (HUN complex) (21–25, 27–29, 44). However, the cellular functions of UBE3A and the physiological relevance of those UBE3A interactions remain largely uncertain.
UBE3A has also been shown to associate with the 26S proteasome. The physiological consequences of this interaction are not fully understood; however, conflicting studies have reported both a stimulating and an inhibitory role for UBE3A in proteasome function, which might be explained by differences in the physiological conditions used in these studies (10, 28, 45). A recent and interesting finding of the inhibitory effect of UBE3A on the proteasome is that interaction of UBE3A with the proteasome results in the ubiquitylation and degradation of several proteasomal subunits. It was suggested that as a consequence of this proteasome inhibition, the Wnt pathway signaling protein β-catenin becomes stabilized, resulting in an up-regulation of Wnt signaling (45). A recently described autism-linked UBE3A mutation (T485A) has been shown to have increased ubiquitin ligase activity and thus results in a deregulation of the Wnt/β-catenin signaling pathway. Wnt signaling plays an important role in developmental processes, including those of neuronal cells, and thus this finding might provide a direct link between a deregulation of UBE3A/proteasome interaction and autism development (10, 45).
Although there is not yet a structure for the full-length UBE3A protein, structures for several UBE3A domains have been reported. The structures of the C-terminal HECT domain alone and in complex with the ubiquitin-conjugating enzyme UbcH7 as well as the structure of the E6-binding motif of UBE3A in complex with the HPV16 E6 protein and p53 have been determined (46–48). Also, a solution NMR structure of the N terminus of UBE3A revealed that this conserved domain, named the AZUL domain (N-terminal Zn2+ finger of UBE3A ligase), folds into a novel Zn2+-binding fold in which four conserved cysteine residues coordinate the binding of one Zn2+ ion (49).
In this study, we characterize the UBE3A interaction with the proteasomal subunit PSMD4. Using immunoprecipitation followed by MS (IP/MS), we were able to map the PSMD4/26S proteasome-binding site to the N-terminal AZUL domain of UBE3A. We were also able to show that an intact AZUL domain is necessary and sufficient for the binding of UBE3A to PSMD4. Additionally, two Angelman syndrome mutations localized within the AZUL domain are impaired in binding to PSDM4. UBE3A mutant proteins containing these AS mutations retain their E3 ligase activity and the ability to repress ERα-responsive genes. Previous studies have implicated the UBE3A/proteasome interaction in Wnt/β-catenin signaling and deregulation of its function in autism development (45). We were able to show that both ubiquitin ligase function and interaction with PSMD4 are necessary for UBE3A to activate Wnt/β-catenin signaling, raising the possibility that a disruption of the UBE3A/PSMD4 interaction might have physiological consequences that contribute to the development of Angelman syndrome.
Results
The 26S proteasome binds to the N terminus of UBE3A
The 26S proteasome is one of the major cellular complexes interacting with UBE3A (21–29). Although immunoprecipitation and MS cannot distinguish which specific protein subunit within the 26S proteasome directly binds UBE3A, PSMD4 was the only proteasomal subunit identified as a direct UBE3A-binding partner in a yeast two-hybrid screen we recently reported, suggesting that PSMD4 likely bridges the interaction of UBE3A with the 26S proteasome (24).
To map the regions of UBE3A that mediate its various protein-binding interactions, we used IP/MS with HA-tagged full-length UBE3A and a variety of UBE3A fragments (shown in Fig. 1) expressed in 293 T-REx cells. To separate nonspecific background proteins from high-confidence interacting proteins (HCIPs), the data from the IP/MS experiments were analyzed using the Comparative Proteomic Analysis Software Suite (CompPASS), which allowed us to assign a specific normalized weight D (NWD) score for each identified interacting protein (see “Experimental procedures”) (50). PSMD4 as well as other proteasomal subunits bound to all UBE3A truncation mutants that contained the UBE3A N terminus. UBE3A mutants lacking the N terminus coprecipitated neither PSMD4 nor any other proteasomal subunits. These experiments map the binding site for the proteasome to the first 64 amino acids of UBE3A (amino acid numbers reflect UBE3A Isoform 1) (Fig. 1). A summary of the mass spectrometry data obtained for the different proteasomal subunits that were identified as HCIPs for at least one UBE3A bait can be found in Table S1. Immunoprecipitation of the isolated N-terminal 64 amino acids alone coprecipitated both PSMD4 and other proteasomal subunits, whereas a mutant depleted of these 64 amino acids did not bind PSMD4 but still bound to HERC2, indicating that this truncated protein folds correctly and can still bind to other UBE3A interactors. The first 64 amino acids of UBE3A are therefore both necessary and sufficient to bind to PSMD4 and the 26S proteasome.
Figure 1.

UBE3A associates with the proteasome through its N terminus. Upper left corner, schematic representation of the UBE3A protein (Isoform 1), showing the positions of the AZUL domain, the HERC2- and hrHPVE6-binding sites, and the HECT domain. Numbers represent amino acids (aa) at a given position (UBE3A Isoform 1). Lower left corner, schematic representation of the UBE3A fragments used in the IP/MS mapping experiment. Right, heat map indicating whether or not a UBE3A fragment was associated with proteasomal subunits or the HUN complex (HERC2 and NEURL4). The map is based on the NWD scores (95% cutoff) obtained for several preys identified in several IP/MS experiments using the N-terminally HA-tagged UBE3A or UBE3A fragments shown on the left. The different intensities of blue reflect the range of NWD scores obtained for each identified interactor.
The N terminus of each of the three UBE3A isoforms mediates binding to PSMD4
The IP/MS experiment was performed with Isoform 1 of UBE3A. The three different splice variants of UBE3A differ only in their very N termini. Isoforms 2 and 3 display 23 and 20 additional amino acids, respectively. We therefore next examined whether the elongated N termini would affect binding to PSMD4. To test this, the HA-tagged N termini of the three UBE3A isoforms were cotransfected with a V5-tagged version of PSMD4 in HEK293T cells, and binding was assessed by HA pulldowns. We used ectopically expressed V5-tagged PSMD4 in all immunoprecipitation experiments because endogenous PSMD4 migrates at the same size as the heavy chain of the Igs used for immunoprecipitation experiments. HA pulldown of the N-terminal fragments of each of the three UBE3A isoforms showed binding to PSMD4 but not to HERC2 (Fig. 2), the binding site of which is localized to amino acids 150–200 of UBE3A (44). The N-terminal deletion mutant lacking the first 64 amino acids (Isoform 1) was not able to precipitate V5-tagged PSMD4 but was still able to bind endogenous HERC2, confirming our finding from the IP/MS experiments (Fig. 1). These results indicate that all three isoforms of UBE3A interact with PSMD4 through their N-terminal AZUL domain.
Figure 2.
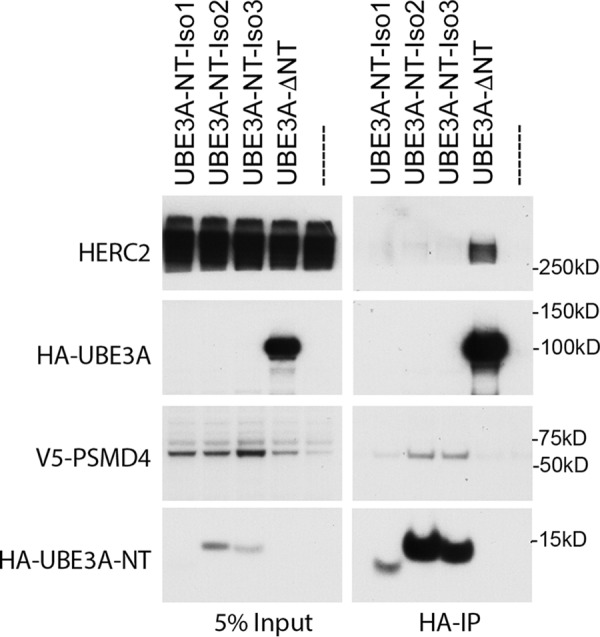
The AZUL domain of all three UBE3A isoforms mediates binding to PSMD4. HEK293T cells were transfected with V5-tagged PSMD4 and either HA-tagged N-terminal expression constructs of the three UBE3A variants (Isoform (Iso) 1, aa 1–64; Isoform 2, aa 1–87; Isoform 3, aa 1–84), an N-terminal deletion mutant of E6AP Isoform 1 (Δaa 1–64), or empty vector. Cells were then harvested and lysed 24 h after transfection, and HA-tagged proteins were precipitated using HA-agarose beads. 5% of the lysate used for immunoprecipitation was used as input. Precipitated proteins were detected by Western blot analysis using anti-HA and anti-V5 antibodies, respectively.
An intact N-terminal AZUL domain is necessary for binding to PSMD4
The NMR structure of the N-terminal AZUL domain (amino acids 1–64) of UBE3A has been determined and shows that it folds into a Zn2+ finger in which several cysteine residues coordinate a Zn2+ ion (49). Interestingly, two Angelman syndrome patients with point mutations affecting the N terminus of UBE3A have been described previously (51, 52). One of these mutations directly affects a cysteine residue shown to be involved in the coordination of Zn2+ (C21Y), whereas the second mutation affects an adjacent glycine residue (G20V). We next asked whether an intact AZUL domain is necessary for UBE3A to bind to PSMD4 and whether the two mutations described in AS patients affect this interaction.
We examined the ability of several different UBE3A mutants (Fig. 3A) to bind to V5-tagged PSMD4. We generated a double mutant in which the first two cysteine residues of the AZUL domain were substituted by alanine to affect the coordination of the Zn2+ ion within the Zn2+ finger motif (AZUL domain mutant (AZUL-mut)). We also tested the G20V and C21Y AS mutants as well as an additional mutant containing the AZUL domain substitution R39H described in another AS patient. Of note, the R39H mutation was found in a UBE3A gene that additionally contained a frameshift mutation that resulted in a C-terminal truncated protein (51). The NMR structure reveals that the Gly-20 and Cys-21 residues are located in close proximity to the bound Zn2+, whereas the Arg-39 residue is located on the surface of the AZUL domain with its side chain exposed to the solvent and therefore most likely not involved in the coordination of Zn2+ (Fig. 3B). Indeed, it has been previously noted that the R39H mutation might not affect the function of UBE3A or have a causal role in this Angelman syndrome patient (49). Nonetheless, because the amino acid substitution could potentially affect the interaction surface between UBE3A and binding partners, we included the R39H mutant in this analysis. To compensate for different protein stabilities of the various tested UBE3A variants, the transfected cells were treated with the proteasome inhibitor bortezomib for 4 h prior to harvesting. As shown in Fig. 3C, WT UBE3A as well as the ubiquitin ligase-inactive UBE3A variant (catalytically inactive (CA)), in which the active-site cysteine is substituted by alanine, pulled down PSMD4. The AZUL-mut and the G20V and C21Y mutants were strongly impaired in binding PSMD4 but still capable of binding HERC2. The finding that AZUL-mut shows a highly reduced interaction with PSMD4 indicates that a correctly folded AZUL domain is necessary for this interaction. Additionally, the G20V and C21Y AS mutants, which presumably also affect the structure of the AZUL domain, were also impaired in their interaction with PSMD4. In contrast, the R39H substitution mutant was able to bind to V5-PSMD4 comparably with UBE3A-WT or UBE3A-CA (Fig. 3D). This indicates that the R39H mutation does not affect the binding of UBE3A to PSMD4 and likely does not affect the structure of the AZUL domain.
Figure 3.

Mutations in the AZUL domain of UBE3A affect binding to PSMD4. A, sequence alignment of UBE3A-WT and AZUL domain mutants. G20V and C21Y represent point mutations identified in Angelman patients. In the AZUL mutant, two Zn2+-coordinating cysteine residues were exchanged to alanine to affect AZUL structure (note that this mutant additionally carries the C820A mutation in the HECT domain, rendering it ligase-inactive). R39H represents a presumably benign point mutation identified in an Angelman patient with an additional frameshift mutation. B, NMR structure of the UBE3A AZUL domain shows localization of Zn2+-coordinating cysteine residues (pink), the glycine mutated in the G20V AS mutant (green), and the arginine mutated in the putative benign R39H mutation (orange). C and D, HEK293T cells were transfected with V5-tagged PSMD4 and HA-tagged UBE3A of the different N-terminal UBE3A mutants or empty vector. Prior to harvesting, cells were treated for 4 h with 300 nm bortezomib. Cells were lysed 24 h after transfection, and HA-tagged proteins were precipitated using HA-agarose beads. 5% of the lysate used for immunoprecipitation was used as input. Precipitated proteins were detected by Western blot analysis using anti-HA and anti-V5 antibodies, respectively. E, GST pulldown assay using recombinant bacterially expressed GST-PSMD4 and GST-HPVE7 (negative control) and recombinant bacterially expressed Trx-tagged AZUL domain (WT, G20V, and C21Y variants). GST-proteins were precipitated using GSH-Sepharose. The ratio of pulled down Trx-AZUL domain proteins to 5% input is indicated. AZUL-WT directly interacts with PSMD4, whereas the AZUL(G20V) and AZUL(C21Y) variants showed significantly impaired binding to PSMD4.
We recently identified PSMD4 as a UBE3A-interacting protein in a yeast two-hybrid screen, indicating that PSMD4 is likely a direct interaction partner of UBE3A (24). To examine this possibility further, we tested binding of the isolated UBE3A AZUL domain to PSMD4 with recombinant proteins expressed in bacteria. As shown in Fig. 3E, GST-PSMD4 was indeed able to coprecipitate the AZUL domain of UBE3A-WT (AZUL-WT), indicating that the binding of PSMD4 to the AZUL domain is direct and furthermore that PSMD4 does not need to be incorporated into the proteasome for this interaction. Additionally, we also tested the effect of the G20V and C21Y Angelman syndrome mutations in this experiment. Although residual binding could be detected, both mutants were significantly impaired in their binding to PSMD4 (Fig. 3E).
The N-terminal UBE3A Angelman syndrome mutants display decreased expression levels but still exhibit ubiquitin ligase activity toward substrate proteins
Point mutations in UBE3A associated with Angelman syndrome have thus far been reported to result either in destabilization of the protein or inactivation of its ubiquitin ligase activity (39). We know from earlier experiments in our laboratory using an N-terminal truncated form of UBE3A that the AZUL domain is not required for its ubiquitin ligase activity (53). In addition, UBE3A(C21Y) has been shown to have ligase activity in vitro (39). Because UBE3A(G20V) and UBE3A(C21Y) are expressed at lower levels than UBE3A-WT, we next examined whether these two mutants can still target UBE3A substrates for proteasomal degradation. To answer this question, we first established a HeLa (HPV-positive) cell line stably expressing mRuby-p53(R273C), a dominant-negative p53 mutant that can be targeted by UBE3A/E6 for ubiquitin-mediated proteolysis (54). These cells were then used to generate a HeLa cell line with a CRISPR/CAS9-induced knockout of UBE3A. Note that knockout of UBE3A in HPV-positive cells stabilizes p53, resulting in the induction of apoptosis. The stable expression of the dominant-negative p53(R273C) mutant, however, prevents p53-mediated apoptosis, permitting us to successfully establish a UBE3A−/− HeLa cell line. Due to the stabilization of mRuby-p53(R273C) upon loss of endogenous UBE3A, we were able to analyze the cells for mRuby fluorescence. After two rounds of FACS for mRuby fluorescence to enrich for UBE3A−/− cells, we performed single-cell cloning (Fig. 4A). In the last step, we tested several single-cell clones (SSC15, SSC16, SSC19, and SSC26) for their p53 response to the reintroduction of UBE3A-WT or UBE3A-CA (Fig. S1). We selected SSC19 for our further experiments (Fig. 4B).
Figure 4.

The G20V and C21Y AS mutants display ubiquitin ligase activity. A, HeLa cells stably expressing mRuby-p53(R723C) were used to establish a cell line in which CRISPR/Cas9-mediated knockout of endogenous UBE3A was established. Cells were sorted twice for stabilization of p53 (as seen by increased mRuby fluorescence), and the levels of UBE3A and actin were detected by Western blot analysis after each sorting event and compared with cells without induction of UBE3A knockout (no Cas9). The twice-sorted population of cells was then used to generate SSCs. Comparison of UBE3A and actin (loading control) levels in these cells identified SSCs with different knockout efficiencies. SSCs 15, 19, 26, and 31 were considered UBE3A−/− cell lines. B, HeLa UBE3A−/− mRuby-p53(R273C) SSCs were compared in their p53 levels after expression of WT or CA UBE3A. The various SSCs were transduced with lentiviruses carrying the expression constructs for HA-tagged UBE3A-WT and UBE3A-CA. Cells were harvested 72 h after infection and lysed, and protein extracts were adjusted for equal protein amounts. Samples were then analyzed by SDS-PAGE and subsequent Western blotting. Endogenous p53 and mRuby-p53(R273C) were detected using anti-p53 antibody. Anti-actin antibody was used to detect actin as a loading control. The depicted Western blot represents HeLa UBE3A−/− mRuby-p53(R273C) SSC19. See Fig. S1 for additional SSCs. not inf., not infected. C, HeLa UBE3A−/− mRuby-p53(R273C) SSC19 cells were transfected with the indicated expression constructs for HA-tagged UBE3A variants. Cells were harvested 48 h after transfection and lysed, and protein extracts were adjusted for equal protein amounts. Samples were then analyzed by SDS-PAGE and subsequent Western blotting. Endogenous p53 and mRuby-p53(R273C) were detected using anti-p53 antibody. UBE3A was detected using anti-HA antibody. Actin (detected by anti-actin antibody) served as a loading control. Numbers reflect the quantified values of the above Western blot signals (endogenous p53 or mRuby-p53) normalized to mock-transfected cells. See Fig. S2 for an uncropped version of the Western blot. D, HEK293T cells were transfected with the indicated HA-tagged constructs of UBE3A variants and an HA-GFP expression construct as a transfection control. Cells were harvested 48 h after transfection and lysed, and protein extracts were adjusted for equal protein amounts. Samples were then analyzed by SDS-PAGE and subsequent Western blotting. HA-UBE3A and HA-GFP were detected by Western blot analysis using anti-HA antibody. HA-GFP served as a transfection efficiency control.
To test the G20V and C21Y UBE3A mutants for ubiquitin ligase activity, we transfected UBE3A-WT, UBE3A-CA, UBE3A-ΔNT, UBE3A(G20V), or UBE3A(C21Y) into HeLa UBE3A−/− mRuby-p53(R273C) SSC19 cells. Cell extracts were then examined for p53 protein levels (endogenous as well as mRuby-p53(R273C)). Although UBE3A-CA did not reduce endogenous p53 or mRuby-p53(R273C) levels, a UBE3A mutant lacking the AZUL domain (UBE3A-ΔNT) was able to induce degradation of both endogenous and mRuby-p53(R273C) (Fig. 4C), confirming previously published results that the AZUL domain is not necessary for UBE3A ubiquitin ligase activity (53). Additionally, both UBE3A(G20V) and UBE3A(C21Y) were able to induce p53 degradation when compared with mock-infected cells (Fig. 4C). UBE3A-WT induced p53 degradation more efficiently than the two mutants, but its expression levels were significantly higher than those of UBE3A(G20V) and UBE3A(C21Y). (Fig. 4C). This is in agreement with previous reports that the G20V and C21Y mutants show lower expression levels than WT UBE3A, an attribute that could contribute to the observed phenotype of Angelman syndrome (10, 39). Based on this observation, it was previously suggested that, compared with WT UBE3A, these point mutations cause UBE3A to be less stable. To examine the stability of the mutant UBE3A proteins, we transfected HA-tagged WT UBE3A, UBE3A(G20V), and UBE3A-C21, as well as the CA versions of each and examined their protein expression levels in 293T cells. Whereas UBE3A-CA showed significantly higher expression levels than WT UBE3A, the substitution of the catalytic cysteine with alanine in either of the two AS mutant proteins did not result in an increase in protein levels, indicating that the lower expression levels of the AS mutants are not a consequence of decreased stability due to UBE3A autoubiquitylation (Fig. 4D). We can instead hypothesize that G20V and C21Y are not expressed as well as the WT protein, perhaps due to folding problems. Expression levels of AZUL domain variants (AZUL-WT, AZUL(G20V), and AZUL(C21Y)) in bacteria also support this hypothesis. The AS mutant variants of the AZUL domain showed significantly lower levels of soluble protein in bacteria. However, there was no detectable difference in the induction efficiency (Fig. S3A) or the expression levels of WT and mutant AZUL domain in whole bacterial cell lysate (Fig. S3B). Comparison of soluble and insoluble fractions revealed that the majority of AZUL-WT was found in the soluble fraction, whereas the majority of AZUL(G20V) and AZUL(C21Y) was found in the insoluble fraction (Fig. S3B), suggesting that the G20V and C21Y mutants may have a folding defect. Nevertheless, these two mutants are still able to induce degradation of p53, indicating that even at very low levels those two UBE3A mutants still function as ubiquitin ligases (Fig. 4C).
The N-terminal Angelman syndrome mutants do not affect UBE3A transcriptional repressor activity toward ERα-responsive genes
In addition to being a ubiquitin ligase, UBE3A has also been shown to have a role in the transcriptional regulation of steroid hormone receptors (11, 12). Although previous studies showed that the ubiquitin ligase activity of UBE3A is not necessary for its function as a transcriptional regulator, it has been speculated that both functions might contribute to the development of Angelman syndrome (11–13). Therefore, we next examined whether the G20V and C21Y mutants were able to repress ERα-mediated transcription of an ER-responsive luciferase reporter. In these experiments, we induced the ERα-responsive reporter construct by expressing ERα and then tested the effect of coexpressing increasing amounts of UBE3A-WT, UBE3A-CA, UBE3A(G20V), or UBE3A(C21Y) (Fig. 5). ERα expression induced luciferase activity, and cotransfection of high amounts (2.4 μg) of UBE3A-WT as well as the UBE3A variants repressed ER signaling. Notably, at lower DNA amounts of the UBE3A variants (1.4 μg), the repressor effect was only visible for UBE3A-WT and catalytically inactive UBE3A, likely reflecting the lower expression levels of UBE3A(G20V) and UBE3A(C21Y) (Fig. 5). Thus, both UBE3A(G20V) and UBE3A(C21Y) can function as repressors of estrogen receptor signaling. However, the lower protein levels observed for these mutants also raise the possibility that reduced expression levels might preclude their proper function as transcriptional regulators and contribute to the Angelman syndrome phenotype.
Figure 5.
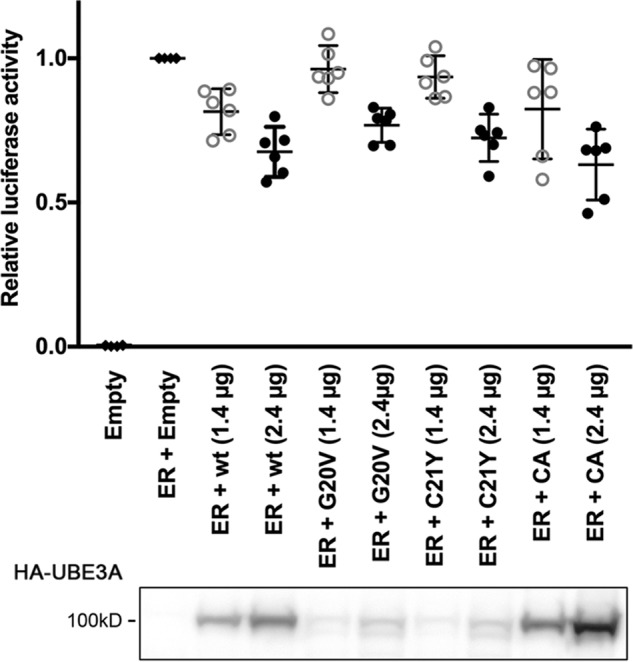
The UBE3A G20V and C21Y AS mutants act as repressors of estrogen receptor signaling. HEK293T cells were transfected with an ER-responsive firefly luciferase reporter construct and a construct encoding Renilla luciferase to adjust for transfection efficiency either in the absence or presence of an expression construct for ERα. To determine the effect of different UBE3A variants on ER signaling, increasing amounts of the different HA-tagged UBE3A variants were expressed. The effect of ERα on the reporter construct was measured using a Dual-Luciferase assay. The values of firefly luciferase were normalized to those of Renilla luciferase and then expressed as relative values to induced (only ERα-expressing) cells. The depicted graph shows data points collected in six independent experiments. Bars represent means and 95% confidence intervals. Open gray circles represent 1.4 μg of transfected UBE3A plasmid; closed black circles represent 2.4 μg of transfected UBE3A plasmid. A Western blot showing expression levels of transfected HA-UBE3A variants of one representative experiment is shown below; loaded lysate amounts have been adjusted for Renilla luciferase activity. UBE3A was detected using anti-HA antibody.
Proteasome binding is necessary for UBE3A to stimulate Wnt/β-catenin signaling
UBE3A has been previously reported to affect the Wnt/β-catenin signaling pathway via the 26S proteasome (55, 56). More recently, an autism-linked de novo UBE3A mutation (UBE3A(T485A)) has been reported to have increased ubiquitin ligase activity due to the loss of an inhibitory phosphorylation site on Thr-485 (10). This results in an increased degradation of PSMD4 and other proteasomal subunits and consequently in the inhibition of proteasome function, stabilization of β-catenin, and activation of Wnt signaling. Thus, the UBE3A(T485A) mutant resulted in a deregulation of the Wnt/β-catenin signaling pathway, implicating a deregulation of UBE3A/proteasome interaction in autism (45). This link among an autism-causing UBE3A mutation, proteasome function, and Wnt/β-catenin signaling raises the question of whether the reduction in the UBE3A/proteasome interaction observed for the G20V and C21Y AS mutants could also lead to deregulation of Wnt/β-catenin signaling. To test this hypothesis, we analyzed the effects of expressing different UBE3A variants in a Wnt/β-catenin reporter assay. Because previous studies showed that UBE3A stimulates Wnt/β-catenin signaling independently of Wnt ligand, we performed our experiments in the absence of stimulation by Wnt ligand (45). As reported previously, expression of UBE3A-WT resulted in ∼4-fold induction of Wnt/β-catenin signaling (Fig. 6, A and B, and Ref. 45). This induction depends on the ubiquitin ligase activity of UBE3A and is not observed with UBE3A-CA (45) (Fig. 6, A and B). Additionally, deletion of the AZUL domain (UBE3A-ΔNT) abolished the Wnt/β-catenin stimulating effect of UBE3A, indicating that both the ubiquitin ligase activity and proteasome binding are necessary for this effect (Fig. 6A). We also tested the effect of UBE3A(G20V) and UBE3A(C21Y) AS mutants in this assay. Compared with WT UBE3A, the two AS mutants were unable to stimulate Wnt/β-catenin signaling (Fig. 6B). Although lower expression levels of G20V and C21Y might contribute to this effect, the protein levels of UBE3A-ΔNT are comparable with those of WT UBE3A, and this deletion mutant is also unable to stimulate the Wnt/β-catenin signaling pathway (Fig. 6A). We therefore hypothesize that in addition to its ubiquitin ligase activity, UBE3A needs to associate through its AZUL domain with the proteasome via PSMD4 to stimulate the Wnt/β-catenin signaling pathway. Knockout of either function impairs UBE3A's ability to activate Wnt/β-catenin signaling, consistent with a potential role in the development of Angelman syndrome.
Figure 6.

The ability of UBE3A to bind the proteasome is required to stimulate Wnt/β-catenin signaling. A and B, HEK293T cells were transfected with a Wnt/β-catenin firefly luciferase reporter construct and a construct encoding Renilla luciferase to adjust for transfection efficiency. To determine their effects on Wnt/β-catenin signaling, different HA-tagged UBE3A variants were concomitantly expressed with the reporter. Wnt/β-catenin signaling was measured using a Dual-Luciferase assay. The values of firefly luciferase were normalized to those of Renilla luciferase and then expressed as relative values compared with cells transfected with the Wnt/β-catenin reporter construct only. The depicted graph shows data points collected in four independent experiments. Bars represent means and 95% confidence intervals. A Western blot showing expression levels of transfected HA-UBE3A variants of one representative experiment is shown below; loaded lysate amounts were adjusted for Renilla luciferase activity. UBE3A was detected using anti-HA antibody.
Discussion
The ubiquitin-proteasome system (UPS) plays a critical role in maintaining proteostasis, a process crucial for all cells, including neurons. The UPS has been shown to modulate the function and plasticity of synapses, and the proteasome itself has been shown to play an important role in synaptic plasticity (57–61). The proteasome is important in protecting cells, including neurons, under conditions of cellular stress that induce protein aggregation (62). Protein aggregation of misfolded proteins is a commonality of a number of neurodegenerative diseases like Parkinson's and Alzheimer's diseases (63). The deregulation of the UPS has also been described in other neurological pathologies. UBE3A, among others, is one of the proteasome-associated ubiquitin ligases and has been shown to associate with proteasomes in a variety of different systems and tissues (22, 28, 64, 65). This includes the brain where it interacts with both cytosolic and synaptic proteasomes (26). The impact of the 26S proteasome in neurophysiology and the fact that deregulation of UBE3A has severe neuropathological consequences highlight the importance of studies on the role of the UBE3A/proteasome interaction in Angelman syndrome and UBE3A-related autism spectrum disorders.
Previous studies have established the association of UBE3A with the proteasome and with PSMD4. Here we show, using MS, that the proteasome associates with the N-terminal Zn2+-coordinating AZUL domain of UBE3A. Although MS allows identification of interacting protein complexes, yeast two-hybrid interactions are thought mainly to be direct binary protein/protein interactions. The fact that PSMD4 was the only proteasomal subunit found to interact with UBE3A in a yeast two-hybrid screen suggests that PSMD4 likely links UBE3A to the proteasome (24). Now, using recombinant proteins, we were able to confirm that PSMD4 directly binds UBE3A in a proteasome-independent manner.
Interestingly, the interaction of UBE3A and PSMD4 seems to be highly conserved throughout evolution. Although other parts of UBE3A changed during evolution, the N-terminal AZUL domain and the C-terminal HECT domain necessary for its ubiquitin ligase activity are well-conserved, indicating that those domains are crucial for the function of UBE3A. In this context, an Angelman syndrome model has been described in Drosophila melanogaster, and Drosophila Ube3a (Dube3a) was shown to interact with and ubiquitylate the Drosophila ortholog of PSMD4 (Rpn10) (22, 66–68). Dube3a and Rpn10 synergistically act in the same pathway, implying that this evolutionarily conserved interaction might play an important role in the neurological function of UBE3A (22, 66–68).
PSMD4 functions as a ubiquitin receptor in the 19S lid of the 26S proteasome to help recruit polyubiquitylated proteins to the proteasome for their subsequent proteolytic degradation. However, PSMD4 also exists in a proteasome-unbound manner in the cytosol (23, 29). As other proteasomal subunits are copurified with PSMD4 in UBE3A immunoprecipitation experiments, we can assume that UBE3A binds to the proteasome-associated PSMD4. Additionally, combined gel-filtration and UBE3A immunoprecipitation experiments show both high- and low-molecular-weight complexes of PSMD4, indicating that UBE3A is associated with PMSD4 in both proteasome-bound and unbound states in mammalian cells (23). Lastly, GST pulldown experiments using recombinant PSMD4 and UBE3A AZUL domain further confirmed that UBE3A can interact with proteasome-unbound PSMD4. We are only beginning to understand the physiological relevance of these different UBE3A/PSMD4 interactions. Although it has been suggested that UBE3A directly binds to and targets PSMD4 (Rpn10) for degradation in Drosophila (22), another study has suggested that PSMD4 is a promiscuous substrate that is targeted for degradation by a variety of ubiquitin ligases. The UIM of PSMD4 binds to growing ubiquitin chains, and in this process PSMD4 becomes ubiquitylated itself (27). We and others have found that PSMD4 interacts with both catalytically active and inactive UBE3A (Fig. 3, C and D, and Ref. 28). Catalytically inactive UBE3A cannot form ubiquitin chains, but because UBE3A has been suggested to form trimers (69), it is possible that the ectopically expressed catalytically inactive UBE3A forms mixed trimers with endogenous WT UBE3A, producing active trimers with ubiquitylation activity that are able to bind to PSMD4. However, we also show that PSMD4 interacts with the isolated N terminus of UBE3A, which does not contain the HECT domain and thus neither has catalytic activity nor the ability to form oligomers with endogenous UBE3A. Therefore, it seems that PSMD4 might interact with UBE3A via two distinct mechanisms: first, indirectly via binding of the UIM domain to growing ubiquitin chains on the HECT domain of UBE3A, and second, via a direct interaction with the N-terminal AZUL domain of UBE3A. Although it has been suggested that PSMD4 is subsequently degraded when bound via the UIM domain, it is unclear whether both interaction mechanisms will lead to PSMD4 degradation (27). Note that to stabilize the G20V and C21Y AS mutants that consistently showed lower expression levels than WT UBE3A in our binding assays, we performed our coexpression and coimmunoprecipitation experiments after treating the transfected cells for a few hours with a proteasome inhibitor. Therefore, degradation of PSMD4 is not expected to be observed under those conditions.
The G20V and C21Y UBE3A mutations belong to the small subgroup of AS point mutations. The majority of UBE3A point mutations either result in frameshift or nonsense mutations leading to the expression of truncated and likely nonfunctional UBE3A protein variants. Most reported point mutations that result in single amino acid substitution mutants of UBE3A have been linked to the loss of UBE3A ubiquitin ligase activity or to destabilization of the protein. Notably, the severity of symptoms observed in Angelman syndrome patients seems to correlate with the molecular mechanism causing the disease (70, 71). A C21Y patient, for example, has been described to present a mild AS phenotype, whereas a full loss of UBE3A results in the most severe phenotype (51). It can be assumed that a complete loss of UBE3A leads to the loss of all UBE3A functions, including its ubiquitin ligase and transcriptional regulation activities, as well as the loss of its interaction with all binding partners. One possible explanation as to why UBE3A point mutants like C21Y display a milder AS phenotype is that these mutants are expressed at much lower protein levels than WT UBE3A and might not reach the optimal threshold to efficiently perform all its cellular functions but still prevent the severe effects of complete loss of UBE3A. However, another possibility is that the severe UBE3A phenotype reflects an additive effect of the loss of different UBE3A functions and that certain point mutations of UBE3A cause the loss of a single UBE3A function, leading to a less severe phenotype. Detailed physiological studies of UBE3A mutations like proteasome binding mutations and catalytically inactive UBE3A mutations in animal model systems should provide better insights into how loss of the different functions of UBE3A contribute to UBE3A-related pathologies. Another argument supporting the hypothesis that loss of certain UBE3A interactions can result in Angelman syndrome–like phenotypes is the finding that a point mutation in one of the major UBE3A-interacting proteins, HERC2, which results in HERC2 destabilization, is associated with a phenotype that closely resembles that of AS, demonstrating that the loss of binding to a major UBE3A-interacting protein can result in Angelman syndrome–like symptoms (72, 73). Although the AS mutations G20V and C21Y seem to affect UBE3A expression levels, these mutants are still able to target p53 for degradation in the presence of hrHPV16 E6 protein and to inhibit ERα-mediated transcription in a luciferase reporter assay. Nevertheless, it is still unclear what levels of protein are needed for UBE3A to properly fulfil its functions. Thus, we cannot conclude that the low expression levels of the G20V and C21Y AS mutants do not cause or at least contribute to AS development. However, these mutants also exhibit a second deficiency, namely a decrease in the ability to bind to one of the major UBE3A-binding partners, the 26S proteasome. Together, our data suggest that either the lower expression levels, the impairment of PSMD4 binding, or a combination of both may contribute to the observed Angelman syndrome phenotype. Although the interaction between UBE3A and the 26S proteasome has been previously established, different physiological consequences of this interaction have been described in the literature. In one publication, a stimulating function of UBE3A on the 26S proteasome that depends on its ubiquitin ligase activity has been reported (28). We thus wanted to test the behavior of the G20V and C21Y mutants in a proteasome activity assay. Unfortunately, we were unable to reproduce the reported stimulating effect of WT UBE3A and did not observe an inhibitory effect of catalytically inactive UBE3A expression on proteasome activity and thus could not test the effects of the G20V and C21Y AS mutants in this assay (data not shown).
Interestingly, UBE3A has also been reported to inhibit proteasome function (10, 45). This inhibition is a consequence of UBE3A-mediated degradation of various proteasomal subunits, including PSMD4, and is thus dependent on the presence of its ubiquitin ligase activity. One downstream effect of proteasome inhibition by UBE3A is the stabilization of β-catenin, which can then translocate to the nucleus and act as a coactivator for Wnt signaling pathway genes (45). Yi et al. (10, 45) reported that the expression of an autism-linked UBE3A mutant, T485A, results in the disruption of an inhibitory phosphorylation site and consequent expression of a constitutively active UBE3A, increased proteasome inhibition, and overstimulation of the Wnt/β-catenin signaling pathway. As UBE3A-related autism cases are thought to be caused by an excess of UBE3A activity, whereas Angelman syndrome is caused by a loss of UBE3A function, we wondered whether a deregulation of Wnt/β-catenin signaling could also be involved in AS development. We thus compared the effect of UBE3A mutants impaired in proteasome binding (UBE3A(G20V), UBE3A(C21Y), and UBE3A-ΔNT) with that of WT and catalytically inactive UBE3A. We found that to stimulate Wnt/β-catenin signaling, UBE3A must be able to both bind to PSMD4/proteasome and, in agreement with previous reports, retain ubiquitin ligase activity. The G20V and C21Y AS mutations seem to affect Wnt/β-catenin signaling in an opposite way to the autism-linked UBE3A(T485A) mutation, and it can be speculated that expression of the AS mutants would result in a lack of UBE3A-mediated activation of Wnt/β-catenin signaling. The Wnt pathway plays a crucial role in neurodevelopment and has been linked to neurological diseases, including autism and other intellectual disabilities (74). Because proteasome binding and ubiquitin ligase activity are necessary for UBE3A to function in Wnt/β-catenin signaling, it is likely that most AS mutations, whether leading to a complete loss of UBE3A expression, the loss of ubiquitin ligase activity, or the loss of binding to the proteasome, would similarly result in a deregulation of the Wnt/β-catenin pathway. Thus, further investigation of the role of UBE3A and various UBE3A mutant proteins in Wnt/β-catenin signaling using AS animal models would help us to understand the role of Wnt/β-catenin deregulation in the development of Angelman syndrome.
In summary, here we describe that the proteasomal subunit PSMD4 binds to UBE3A via the N-terminal Zn2+ coordinating AZUL domain of UBE3A and that an intact and properly folded AZUL domain is necessary for the interaction of UBE3A with the proteasome. We also provide evidence that two previously described Angelman syndrome mutations, G20V and C21Y, which are localized in the AZUL domain, show reduced binding to PSMD4. To our knowledge, this is the first report that a mutation found in AS patients affects the interaction of UBE3A with one of its major binding partners. Our findings suggest that the impairment of those mutants to bind to the proteasome and activate the Wnt/β-catenin signaling pathway may contribute to the symptoms observed in Angelman syndrome. This also highlights the possibility that different UBE3A mutations affecting different functions of UBE3A (i.e. proteasome binding and ubiquitin ligase activity) might result in comparable physiological or pathological outcomes.
Experimental procedures
Plasmids
Expression constructs for UBE3A variants and fragments and PSMD4 were generated using the Gateway system (Invitrogen). UBE3A and UBE3A fragments were PCR-amplified and recombined into pDONR223 to generate entry vectors. The PSMD4 entry vector in pDONR223 was obtained from the Center for Cancer Systems Biology (CCSB) Human ORFeome collection (http://horfdb.dfci.harvard.edu/index.php?page=home)4 (85). Open reading frames contained in the entry vectors were then recombined into the pHAGE expression vectors pHAGE-P CMVt N-HA GAW (23) and pHAGE-P CMVt N-V5 GAW (24) or the bacterial expression vectors Gateway Nova pET-60-DEST and Gateway Nova pET-59-DEST using the Gateway LR II Clonase Enzyme Mix (Invitrogen) according to the manufacturer's instructions.
The estrogen receptor–responsive reporter construct 3X ERE TATA Luc and the estrogen receptor expression construct VP16-ERα gifts from Donald McDonnell (Addgene plasmids 11345 and 11351, respectively) (75, 76). The β-catenin–responsive Wnt luciferase reporter construct pTOPflash was described previously (77). The Renilla luciferase construct pRL-TK (Promega) used for normalization in Dual-Luciferase reporter assays was described previously (78).
The reporter vector pHAGE-N CMVt N-RIG3 p53(R273C), which expresses a bicistronic mRNA encoding the fusion proteins mRuby-p53(R273C) and H2B-SGFP2 separated by a CMV internal ribosome entry site, was made from a p53(R273C) entry vector by recombining the ORF of p53(R273C) into the destination vector pHAGE-N CMVt RIG3 GAW as described above. The fluorescent proteins mRuby and SGFP2 have been described before (79, 80). The bacterial expression construct pGex-2T-E7 has been described previously (81). Cas9 was expressed from the vector lentiCas9-Blast, a gift from Feng Zhang (Addgene plasmid 52962) (82).
Antibodies
The various HA-tagged E6AP variants were detected by Western blot analysis using a mouse monoclonal anti-HA antibody conjugated to HRP (Roche Applied Science). V5-tagged PSMD4 was detected using a mouse monoclonal anti-V5 antibody conjugated to HRP (Invitrogen). HERC2 was detected using a mouse monoclonal anti-HERC2 antibody (BD Transduction Laboratories). Actin was detected using an HRP-coupled mouse monoclonal anti-actin antibody (Abcam). Trx-tagged proteins were detected using Trx tag mouse mAb 71542-3 (Novagen). GST-tagged proteins were detected using anti-GST-HRP ab 58626 (Abcam). p53 was detected using an HRP-coupled anti-p53 antibody (R&D Systems). If necessary, primary antibodies were detected using sheep anti-mouse IgG HRP-linked antibodies (Amersham Biosciences/GE Healthcare). Quantification of the intensity of Western blotting signals was performed with Image StudioTM Lite (LI-COR Biosciences).
IP/MS experiments
IP/MS experiments using HA-tagged UBE3A Isoform 1 or UBE3A fragments as baits were performed T-REx 293 cells were performed as described previously (23). The IP/MS data were analyzed by CompPASS, which relies on an unbiased comparative approach to identify HCIPs (50, 83). For the analysis of these data, we used a library from 171 HA coimmunoprecipitations, also performed in T-REx 293 cells, generated by the Harper laboratory (83). CompPASS calculates weight D (WD) scores for each protein. In this case, a threshold was calculated in a way that 95% of the data are under it. The NWD scores result from dividing each WD score by the calculated threshold. Proteins were considered HCIPs when their NWD score was equal to or greater than 1 (WD score equal to or greater than the calculated threshold). The complete set of data for IP/MS experiments is presented in Tables S2–S9. The heat map (Fig. 1) was made using Orange, an open-source data visualization, machine learning, and data mining toolkit (84).
Immunoprecipitation
For coimmunoprecipitation experiments, HEK293T cells were transfected with the indicated HA-E6AP expression constructs and V5-PSMD4. Where indicated, cells were treated with 300 nm VELCADE® for 4 h prior to harvesting. Protein extracts were prepared 24 h after transfection in lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5% Nonidet P-40, 1 mm EDTA) supplemented with a protease inhibitor mixture (Roche Applied Science). 30 μl of a 50:50 slurry of anti-HA–agarose resin (Sigma) were added. Cell extracts were incubated at 4 °C overnight. Precipitated proteins were detected by Western blot analysis using anti-HA and anti-V5 antibodies, respectively.
Cell culture
HEK293T cells, HeLa mRuby-p53(R723C), and UBE3A−/− HeLa mRuby-p53(R273C) cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal bovine serum. To create the HeLa N-RIG3 p53(R273C) cells, HeLa cells were transduced with pHAGE-N CMVt N-RIG3 p53(R273C) and then selected with neomycin. Cells were then sorted by flow cytometry, selecting cells with a strong green fluorescence but not red fluorescence, indicating a robust expression of the transduced genes. To obtain HeLa UBE3A−/− N-RIG3 mRuby-p53(R273C) cells, the HeLa mRuby-p53(R273C) cells were cotransfected with UBE3A single guide RNA (target sequence ATTAATCTTATAAAGCTCGA) and the CAS9 expression construct. Cells were then sorted twice for mRuby fluorescence, used as an indicator of p53 stabilization. As low levels of UBE3A were still detectable by Western blotting in protein extracts of sorted cells, single-cell cloning was performed, and single-cell clones were tested for UBE3A protein levels by Western blot analysis. Single-cell clones either displayed normal (+/+), low (−/+ or +/−), or no (−/−) signal for UBE3A. One clone (SSC19) with undetectable levels for UBE3A was chosen for further experiments.
GST pulldown assay
GST- and Trx-tagged proteins were expressed in Escherichia coli BL21 RIL (Agilent Technologies). Expression was induced for 4 h with 400 μm isopropyl 1-thio-β-d-galactopyranoside at 37 °C. Bacterial pellets were resuspended in PBS, 1% Triton, and cells were lysed on ice using sonification. Bacterial lysates containing GST-tagged proteins (PSMD4 or HPVE7) were mixed with lysates containing Trx-tagged AZUL domains (AZUL-WT, AZUL(G20V), or AZUL(C21Y)) and incubated with GSH-Sepharose 4B (GE Healthcare) at 4 °C for 3 h. Precipitated proteins were detected by Western blot analysis using anti-Trx and anti-GST antibodies, respectively.
Luciferase assays
ERα reporter assays
HEK293T cells were transfected in 6-well plates with the indicated luciferase and expression constructs. Twenty-four hours after transfection, cells were harvested and lysed on the plate in 300 μl of 1× Passive Lysis Buffer (Promega). Luciferase activity was determined using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Luciferase readings were measured using a SpectraMax L luminescence microplate reader (Molecular Devices). The firefly luciferase readings were normalized to the respective Renilla luciferase readings.
Wnt/β-catenin reporter assays
HEK293T cells were transfected in 6-well plates with the indicated luciferase and expression constructs. Twenty-four hours after transfection, the medium was changed to starvation medium (0.2% fetal calf serum). After 6 h of starvation, cells were switched back to regular medium for 16 h. Cells were then harvested and lysed on the plate in 300 μl of 1× Passive Lysis Buffer. Luciferase activity was determined using the Dual-Luciferase Reporter Assay System according to the manufacturer's instructions. Luciferase readings were measured using a SpectraMax L luminescence microplate reader. The firefly luciferase readings were normalized to the respective Renilla luciferase readings.
Author contributions
S. K., G. M.-N., J. W. H., and P. M. H. conceptualization; S. K., G. M.-N., F. L., and S. D. H. data curation; S. K., G. M.-N., J. W. H., and P. M. H. formal analysis; S. K. and G. M.-N. validation; S. K., G. M.-N., and F. L. investigation; S. K., G. M.-N., and J. W. H. methodology; S. K. writing-original draft; S. K., G. M.-N., S. D. H., J. W. H., and P. M. H. writing-review and editing; P. M. H. resources; P. M. H. supervision; P. M. H. funding acquisition; P. M. H. visualization; P. M. H. project administration.
Supplementary Material
Acknowledgment
We thank Patricia Szajner for comments and helpful discussions.
This work was supported by National Institutes of Health Grants R35CA197262 (to P. M. H) and AG011085 (to J. W. H.) and German Research Foundation (Deutsche Forschungsgemeinschaft) Scholarship Grant KU 3196/2-1 (to S. K.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S3 and Tables S1–S9.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party-hosted site.
- E6AP
- E6-associated protein
- HECT
- homologous to E6AP C terminus
- HPV
- human papillomavirus
- AS
- Angelman syndrome
- hrHPV
- high-risk HPV
- AZUL domain
- N-terminal Zn2+ finger of UBE3A ligase
- ER
- estrogen receptor
- UIM
- ubiquitin-interacting motif
- HUN complex
- HERC2, UBE3A, and NEURL4
- IP/MS
- immunoprecipitation followed by MS
- HA
- hemagglutinin
- GST
- glutathione S-transferase
- HCIP
- high-confidence interacting protein
- CompPASS
- Comparative Proteomic Analysis Software Suite
- WD
- weight D
- NWD
- normalized WD
- AZUL-mut
- AZUL domain mutant
- CA
- catalytically inactive
- SSC
- single-cell clone
- UPS
- ubiquitin-proteasome system
- Dube3a
- Drosophila Ube3a
- CMV
- cytomegalovirus
- HRP
- horseradish peroxidase
- Trx
- thioredoxin
- aa
- amino acids.
References
- 1. Yamamoto Y., Huibregtse J. M., and Howley P. M. (1997) The human E6-AP gene (UBE3A) encodes three potential protein isoforms generated by differential splicing. Genomics 41, 263–266 10.1006/geno.1997.4617 [DOI] [PubMed] [Google Scholar]
- 2. Schiffman M., Herrero R., Desalle R., Hildesheim A., Wacholder S., Rodriguez A. C., Bratti M. C., Sherman M. E., Morales J., Guillen D., Alfaro M., Hutchinson M., Wright T. C., Solomon D., Chen Z., et al. (2005) The carcinogenicity of human papillomavirus types reflects viral evolution. Virology 337, 76–84 10.1016/j.virol.2005.04.002 [DOI] [PubMed] [Google Scholar]
- 3. Muñoz N., Bosch F. X., de Sanjosé S., Herrero R., Castellsagué X., Shah K. V., Snijders P. J., Meijer C. J., and International Agency for Research on Cancer Multicenter Cervical Cancer Study Group (2003) Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 348, 518–527 10.1056/NEJMoa021641 [DOI] [PubMed] [Google Scholar]
- 4. Huibregtse J. M., Scheffner M., and Howley P. M. (1991) A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 10, 4129–4135 10.1002/j.1460-2075.1991.tb04990.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scheffner M., Huibregtse J. M., Vierstra R. D., and Howley P. M. (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505 10.1016/0092-8674(93)90384-3 [DOI] [PubMed] [Google Scholar]
- 6. Kishino T., Lalande M., and Wagstaff J. (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15, 70–73 10.1038/ng0197-70 [DOI] [PubMed] [Google Scholar]
- 7. Glessner J. T., Wang K., Cai G., Korvatska O., Kim C. E., Wood S., Zhang H., Estes A., Brune C. W., Bradfield J. P., Imielinski M., Frackelton E. C., Reichert J., Crawford E. L., Munson J., et al. (2009) Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature 459, 569–573 10.1038/nature07953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hogart A., Wu D., LaSalle J. M., and Schanen N. C. (2010) The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol. Dis. 38, 181–191 10.1016/j.nbd.2008.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Noor A., Dupuis L., Mittal K., Lionel A. C., Marshall C. R., Scherer S. W., Stockley T., Vincent J. B., Mendoza-Londono R., and Stavropoulos D. J. (2015) 15q11.2 duplication encompassing only the UBE3A gene is associated with developmental delay and neuropsychiatric phenotypes. Hum. Mutat. 36, 689–693 10.1002/humu.22800 [DOI] [PubMed] [Google Scholar]
- 10. Yi J. J., Berrios J., Newbern J. M., Snider W. D., Philpot B. D., Hahn K. M., and Zylka M. J. (2015) An autism-linked mutation disables phosphorylation control of UBE3A. Cell 162, 795–807 10.1016/j.cell.2015.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nawaz Z., Lonard D. M., Smith C. L., Lev-Lehman E., Tsai S. Y., Tsai M. J., and O'Malley B. W. (1999) The Angelman syndrome-associated protein, E6-AP, is a coactivator for the nuclear hormone receptor superfamily. Mol. Cell. Biol. 19, 1182–1189 10.1128/MCB.19.2.1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ramamoorthy S., and Nawaz Z. (2008) E6-associated protein (E6-AP) is a dual function coactivator of steroid hormone receptors. Nucl. Recept. Signal. 6, e006. 10.1621/nrs.06006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kühnle S., Mothes B., Matentzoglu K., and Scheffner M. (2013) Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein Arc. Proc. Natl. Acad. Sci. U.S.A. 110, 8888–8893 10.1073/pnas.1302792110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hershko A., and Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 10.1146/annurev.biochem.67.1.425 [DOI] [PubMed] [Google Scholar]
- 15. Hershko A., Heller H., Elias S., and Ciechanover A. (1983) Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 258, 8206–8214 [PubMed] [Google Scholar]
- 16. Huibregtse J. M., Scheffner M., Beaudenon S., and Howley P. M. (1995) A family of proteins structurally and functionally related to the E6-AP ubiquitin-protein ligase. Proc. Natl. Acad. Sci. U.S.A. 92, 2563–2567 10.1073/pnas.92.7.2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chau V., Tobias J. W., Bachmair A., Marriott D., Ecker D. J., Gonda D. K., and Varshavsky A. (1989) A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science 243, 1576–1583 10.1126/science.2538923 [DOI] [PubMed] [Google Scholar]
- 18. Thrower J. S., Hoffman L., Rechsteiner M., and Pickart C. M. (2000) Recognition of the polyubiquitin proteolytic signal. EMBO J. 19, 94–102 10.1093/emboj/19.1.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kish-Trier E., and Hill C. P. (2013) Structural biology of the proteasome. Annu. Rev. Biophys. 42, 29–49 10.1146/annurev-biophys-083012-130417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fu H., Sadis S., Rubin D. M., Glickman M., van Nocker S., Finley D., and Vierstra R. D. (1998) Multiubiquitin chain binding and protein degradation are mediated by distinct domains within the 26 S proteasome subunit Mcb1. J. Biol. Chem. 273, 1970–1981 10.1074/jbc.273.4.1970 [DOI] [PubMed] [Google Scholar]
- 21. Altun M., Besche H. C., Overkleeft H. S., Piccirillo R., Edelmann M. J., Kessler B. M., Goldberg A. L., and Ulfhake B. (2010) Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. J. Biol. Chem. 285, 39597–39608 10.1074/jbc.M110.129718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee S. Y., Ramirez J., Franco M., Lectez B., Gonzalez M., Barrio R., and Mayor U. (2014) Ube3a, the E3 ubiquitin ligase causing Angelman syndrome and linked to autism, regulates protein homeostasis through the proteasomal shuttle Rpn10. Cell. Mol. Life Sci. 71, 2747–2758 10.1007/s00018-013-1526-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martínez-Noël G., Galligan J. T., Sowa M. E., Arndt V., Overton T. M., Harper J. W., and Howley P. M. (2012) Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes. Mol. Cell. Biol. 32, 3095–3106 10.1128/MCB.00201-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Martínez-Noël G., Luck K., Kühnle S., Desbuleux A., Szajner P., Galligan J. T., Rodriguez D., Zheng L., Boyland K., Leclere F., Zhong Q., Hill D. E., Vidal M., and Howley P. M. (2018) Network analysis of UBE3A/E6AP-associated proteins provides connections to several distinct cellular processes. J. Mol. Biol. 430, 1024–1050 10.1016/j.jmb.2018.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Subbaiah V. K., Zhang Y., Rajagopalan D., Abdullah L. N., Yeo-Teh N. S., Tomaić V., Banks L., Myers M. P., Chow E. K., and Jha S. (2016) E3 ligase EDD1/UBR5 is utilized by the HPV E6 oncogene to destabilize tumor suppressor TIP60. Oncogene 35, 2062–2074 10.1038/onc.2015.268 [DOI] [PubMed] [Google Scholar]
- 26. Tai H. C., Besche H., Goldberg A. L., and Schuman E. M. (2010) Characterization of the brain 26S proteasome and its interacting proteins. Front. Mol. Neurosci. 3, 12 10.3389/fnmol.2010.00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Uchiki T., Kim H. T., Zhai B., Gygi S. P., Johnston J. A., O'Bryan J. P., and Goldberg A. L. (2009) The ubiquitin-interacting motif protein, S5a, is ubiquitinated by all types of ubiquitin ligases by a mechanism different from typical substrate recognition. J. Biol. Chem. 284, 12622–12632 10.1074/jbc.M900556200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tomaić V., and Banks L. (2015) Angelman syndrome-associated ubiquitin ligase UBE3A/E6AP mutants interfere with the proteolytic activity of the proteasome. Cell Death Dis. 6, e1625 10.1038/cddis.2014.572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tomaić V., Ganti K., Pim D., Bauer C., Blattner C., and Banks L. (2013) Interaction of HPV E6 oncoproteins with specific proteasomal subunits. Virology 446, 389–396 10.1016/j.virol.2013.08.016 [DOI] [PubMed] [Google Scholar]
- 30. Wang Q., Young P., and Walters K. J. (2005) Structure of S5a bound to monoubiquitin provides a model for polyubiquitin recognition. J. Mol. Biol. 348, 727–739 10.1016/j.jmb.2005.03.007 [DOI] [PubMed] [Google Scholar]
- 31. Elsasser S., Chandler-Militello D., Müller B., Hanna J., and Finley D. (2004) Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 279, 26817–26822 10.1074/jbc.M404020200 [DOI] [PubMed] [Google Scholar]
- 32. Elsasser S., and Finley D. (2005) Delivery of ubiquitinated substrates to protein-unfolding machines. Nat. Cell Biol. 7, 742–749 10.1038/ncb0805-742 [DOI] [PubMed] [Google Scholar]
- 33. Rougeulle C., Glatt H., and Lalande M. (1997) The Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in brain. Nat. Genet. 17, 14–15 10.1038/ng0997-14 [DOI] [PubMed] [Google Scholar]
- 34. Williams C. A., Angelman H., Clayton-Smith J., Driscoll D. J., Hendrickson J. E., Knoll J. H., Magenis R. E., Schinzel A., Wagstaff J., Whidden E. M., and Zori R. T. (1995) Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation. Am. J. Med. Genet. 56, 237–238 10.1002/ajmg.1320560224 [DOI] [PubMed] [Google Scholar]
- 35. Williams C. A., Beaudet A. L., Clayton-Smith J., Knoll J. H., Kyllerman M., Laan L. A., Magenis R. E., Moncla A., Schinzel A. A., Summers J. A., and Wagstaff J. (2006) Angelman syndrome 2005: updated consensus for diagnostic criteria. Am. J. Med. Genet. A 140, 413–418 10.1002/ajmg.a.31074 [DOI] [PubMed] [Google Scholar]
- 36. Angelman H. (1965) 'Puppet' children. A report on three cases. Dev. Med. Child Neurol. 7, 681–688 [DOI] [PubMed] [Google Scholar]
- 37. Mabb A. M., Judson M. C., Zylka M. J., and Philpot B. D. (2011) Angelman syndrome: insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34, 293–303 10.1016/j.tins.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fang P., Lev-Lehman E., Tsai T. F., Matsuura T., Benton C. S., Sutcliffe J. S., Christian S. L., Kubota T., Halley D. J., Meijers-Heijboer H., Langlois S., Graham J. M. Jr., Beuten J., Willems P. J., Ledbetter D. H., et al. (1999) The spectrum of mutations in UBE3A causing Angelman syndrome. Hum. Mol. Genet. 8, 129–135 10.1093/hmg/8.1.129 [DOI] [PubMed] [Google Scholar]
- 39. Cooper E. M., Hudson A. W., Amos J., Wagstaff J., and Howley P. M. (2004) Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein. J. Biol. Chem. 279, 41208–41217 10.1074/jbc.M401302200 [DOI] [PubMed] [Google Scholar]
- 40. Sadikovic B., Fernandes P., Zhang V. W., Ward P. A., Miloslavskaya I., Rhead W., Rosenbaum R., Gin R., Roa B., and Fang P. (2014) Mutation update for UBE3A variants in Angelman syndrome. Hum. Mutat. 35, 1407–1417 10.1002/humu.22687 [DOI] [PubMed] [Google Scholar]
- 41. Kleijnen M. F., Shih A. H., Zhou P., Kumar S., Soccio R. E., Kedersha N. L., Gill G., and Howley P. M. (2000) The hPLIC proteins may provide a link between the ubiquitination machinery and the proteasome. Mol. Cell 6, 409–419 10.1016/S1097-2765(00)00040-X [DOI] [PubMed] [Google Scholar]
- 42. Kumar S., Talis A. L., and Howley P. M. (1999) Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J. Biol. Chem. 274, 18785–18792 10.1074/jbc.274.26.18785 [DOI] [PubMed] [Google Scholar]
- 43. Li L., Li Z., Howley P. M., and Sacks D. B. (2006) E6AP and calmodulin reciprocally regulate estrogen receptor stability. J. Biol. Chem. 281, 1978–1985 10.1074/jbc.M508545200 [DOI] [PubMed] [Google Scholar]
- 44. Kühnle S., Kogel U., Glockzin S., Marquardt A., Ciechanover A., Matentzoglu K., and Scheffner M. (2011) Physical and functional interaction of the HECT ubiquitin-protein ligases E6AP and HERC2. J. Biol. Chem. 286, 19410–19416 10.1074/jbc.M110.205211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yi J. J., Paranjape S. R., Walker M. P., Choudhury R., Wolter J. M., Fragola G., Emanuele M. J., Major M. B., and Zylka M. J. (2017) The autism-linked UBE3A T485A mutant E3 ubiquitin ligase activates the Wnt/β-catenin pathway by inhibiting the proteasome. J. Biol. Chem. 292, 12503–12515 10.1074/jbc.M117.788448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martinez-Zapien D., Ruiz F. X., Poirson J., Mitschler A., Ramirez J., Forster A., Cousido-Siah A., Masson M., Vande Pol S., Podjarny A., Travé G., and Zanier K. (2016) Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 529, 541–545 10.1038/nature16481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zanier K., Charbonnier S., Sidi A. O., McEwen A. G., Ferrario M. G., Poussin-Courmontagne P., Cura V., Brimer N., Babah K. O., Ansari T., Muller I., Stote R. H., Cavarelli J., Vande Pol S., and Travé G. (2013) Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science 339, 694–698 10.1126/science.1229934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Huang L., Kinnucan E., Wang G., Beaudenon S., Howley P. M., Huibregtse J. M., and Pavletich N. P. (1999) Structure of an E6AP-UbcH7 complex: insights into ubiquitination by the E2-E3 enzyme cascade. Science 286, 1321–1326 10.1126/science.286.5443.1321 [DOI] [PubMed] [Google Scholar]
- 49. Lemak A., Yee A., Bezsonova I., Dhe-Paganon S., and Arrowsmith C. H. (2011) Zn-binding AZUL domain of human ubiquitin protein ligase Ube3A. J. Biomol. NMR 51, 185–190 10.1007/s10858-011-9552-y [DOI] [PubMed] [Google Scholar]
- 50. Sowa M. E., Bennett E. J., Gygi S. P., and Harper J. W. (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138, 389–403 10.1016/j.cell.2009.04.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Malzac P., Webber H., Moncla A., Graham J. M., Kukolich M., Williams C., Pagon R. A., Ramsdell L. A., Kishino T., and Wagstaff J. (1998) Mutation analysis of UBE3A in Angelman syndrome patients. Am. J. Hum. Genet. 62, 1353–1360 10.1086/301877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mueller O. T., and Coovadia A. (2008) Gene symbol: UBE3A. Disease: Angelman syndrome. Hum. Genet. 124, 304 [PubMed] [Google Scholar]
- 53. Huibregtse J. M., Scheffner M., and Howley P. M. (1993) Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol. Cell. Biol. 13, 4918–4927 10.1128/MCB.13.8.4918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scheffner M., Takahashi T., Huibregtse J. M., Minna J. D., and Howley P. M. (1992) Interaction of the human papillomavirus type 16 E6 oncoprotein with wild-type and mutant human p53 proteins. J. Virol. 66, 5100–5105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lichtig H., Gilboa D. A., Jackman A., Gonen P., Levav-Cohen Y., Haupt Y., and Sherman L. (2010) HPV16 E6 augments Wnt signaling in an E6AP-dependent manner. Virology 396, 47–58 10.1016/j.virol.2009.10.011 [DOI] [PubMed] [Google Scholar]
- 56. Sominsky S., Kuslansky Y., Shapiro B., Jackman A., Haupt Y., Rosin-Arbesfeld R., and Sherman L. (2014) HPV16 E6 and E6AP differentially cooperate to stimulate or augment Wnt signaling. Virology 468–470, 510–523 10.1016/j.virol.2014.09.007 [DOI] [PubMed] [Google Scholar]
- 57. Karpova A., Mikhaylova M., Thomas U., Knöpfel T., and Behnisch T. (2006) Involvement of protein synthesis and degradation in long-term potentiation of Schaffer collateral CA1 synapses. J. Neurosci. 26, 4949–4955 10.1523/JNEUROSCI.4573-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fonseca R., Vabulas R. M., Hartl F. U., Bonhoeffer T., and Nägerl U. V. (2006) A balance of protein synthesis and proteasome-dependent degradation determines the maintenance of LTP. Neuron 52, 239–245 10.1016/j.neuron.2006.08.015 [DOI] [PubMed] [Google Scholar]
- 59. DiAntonio A., and Hicke L. (2004) Ubiquitin-dependent regulation of the synapse. Annu. Rev. Neurosci. 27, 223–246 10.1146/annurev.neuro.27.070203.144317 [DOI] [PubMed] [Google Scholar]
- 60. Yi J. J., and Ehlers M. D. (2007) Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol. Rev. 59, 14–39 10.1124/pr.59.1.4 [DOI] [PubMed] [Google Scholar]
- 61. Tai H. C., and Schuman E. M. (2008) Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat. Rev. Neurosci. 9, 826–838 10.1038/nrn2499 [DOI] [PubMed] [Google Scholar]
- 62. Goldberg A. L. (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899 10.1038/nature02263 [DOI] [PubMed] [Google Scholar]
- 63. Sherman M. Y., and Goldberg A. L. (2001) Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29, 15–32 10.1016/S0896-6273(01)00177-5 [DOI] [PubMed] [Google Scholar]
- 64. Besche H. C., Sha Z., Kukushkin N. V., Peth A., Hock E. M., Kim W., Gygi S., Gutierrez J. A., Liao H., Dick L., and Goldberg A. L. (2014) Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. EMBO J. 33, 1159–1176 10.1002/embj.201386906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jacobson A. D., MacFadden A., Wu Z., Peng J., and Liu C. W. (2014) Autoregulation of the 26S proteasome by in situ ubiquitination. Mol. Biol. Cell 25, 1824–1835 10.1091/mbc.e13-10-0585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu Y., Bolduc F. V., Bell K., Tully T., Fang Y., Sehgal A., and Fischer J. A. (2008) A Drosophila model for Angelman syndrome. Proc. Natl. Acad. Sci. U.S.A. 105, 12399–12404 10.1073/pnas.0805291105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Miura K., Kishino T., Li E., Webber H., Dikkes P., Holmes G. L., and Wagstaff J. (2002) Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiol. Dis. 9, 149–159 10.1006/nbdi.2001.0463 [DOI] [PubMed] [Google Scholar]
- 68. Cheron G., Servais L., Wagstaff J., and Dan B. (2005) Fast cerebellar oscillation associated with ataxia in a mouse model of Angelman syndrome. Neuroscience 130, 631–637 10.1016/j.neuroscience.2004.09.013 [DOI] [PubMed] [Google Scholar]
- 69. Ronchi V. P., Klein J. M., Edwards D. J., and Haas A. L. (2014) The active form of E6-associated protein (E6AP)/UBE3A ubiquitin ligase is an oligomer. J. Biol. Chem. 289, 1033–1048 10.1074/jbc.M113.517805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lossie A. C., Whitney M. M., Amidon D., Dong H. J., Chen P., Theriaque D., Hutson A., Nicholls R. D., Zori R. T., Williams C. A., and Driscoll D. J. (2001) Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet. 38, 834–845 10.1136/jmg.38.12.834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Moncla A., Malzac P., Voelckel M. A., Auquier P., Girardot L., Mattei M. G., Philip N., Mattei J. F., Lalande M., and Livet M. O. (1999) Phenotype-genotype correlation in 20 deletion and 20 non-deletion Angelman syndrome patients. Eur. J. Hum. Genet. 7, 131–139 10.1038/sj.ejhg.5200258 [DOI] [PubMed] [Google Scholar]
- 72. Harlalka G. V., Baple E. L., Cross H., Kühnle S., Cubillos-Rojas M., Matentzoglu K., Patton M. A., Wagner K., Coblentz R., Ford D. L., Mackay D. J., Chioza B. A., Scheffner M., Rosa J. L., and Crosby A. H. (2013) Mutation of HERC2 causes developmental delay with Angelman-like features. J. Med. Genet. 50, 65–73 10.1136/jmedgenet-2012-101367 [DOI] [PubMed] [Google Scholar]
- 73. Puffenberger E. G., Jinks R. N., Wang H., Xin B., Fiorentini C., Sherman E. A., Degrazio D., Shaw C., Sougnez C., Cibulskis K., Gabriel S., Kelley R. I., Morton D. H., and Strauss K. A. (2012) A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum. Mutat. 33, 1639–1646 10.1002/humu.22237 [DOI] [PubMed] [Google Scholar]
- 74. Kwan V., Unda B. K., and Singh K. K. (2016) Wnt signaling networks in autism spectrum disorder and intellectual disability. J. Neurodev. Disord. 8, 45 10.1186/s11689-016-9176-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hall J. M., and McDonnell D. P. (1999) The estrogen receptor β-isoform (ERβ) of the human estrogen receptor modulates ERα transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology 140, 5566–5578 10.1210/endo.140.12.7179 [DOI] [PubMed] [Google Scholar]
- 76. Chang C. y., Norris J. D., Grøn H., Paige L. A., Hamilton P. T., Kenan D. J., Fowlkes D., and McDonnell D. P. (1999) Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors α and β. Mol. Cell. Biol. 19, 8226–8239 10.1128/MCB.19.12.8226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Korinek V., Barker N., Morin P. J., van Wichen D., de Weger R., Kinzler K. W., Vogelstein B., and Clevers H. (1997) Constitutive transcriptional activation by a β-catenin-Tcf complex in APC−/− colon carcinoma. Science 275, 1784–1787 10.1126/science.275.5307.1784 [DOI] [PubMed] [Google Scholar]
- 78. Tan M. J., White E. A., Sowa M. E., Harper J. W., Aster J. C., and Howley P. M. (2012) Cutaneous β-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. U.S.A. 109, E1473–E1480 10.1073/pnas.1205991109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kredel S., Oswald F., Nienhaus K., Deuschle K., Röcker C., Wolff M., Heilker R., Nienhaus G. U., and Wiedenmann J. (2009) mRuby, a bright monomeric red fluorescent protein for labeling of subcellular structures. PLoS One 4, e4391 10.1371/journal.pone.0004391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Kremers G. J., Goedhart J., van den Heuvel D. J., Gerritsen H. C., and Gadella T. W. Jr. (2007) Improved green and blue fluorescent proteins for expression in bacteria and mammalian cells. Biochemistry 46, 3775–3783 10.1021/bi0622874 [DOI] [PubMed] [Google Scholar]
- 81. Phelps W. C., Bagchi S., Barnes J. A., Raychaudhuri P., Kraus V., Münger K., Howley P. M., and Nevins J. R. (1991) Analysis of trans activation by human papillomavirus type 16 E7 and adenovirus 12S E1A suggests a common mechanism. J. Virol. 65, 6922–6930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Behrends C., Sowa M. E., Gygi S. P., and Harper J. W. (2010) Network organization of the human autophagy system. Nature 466, 68–76 10.1038/nature09204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Demsar J., Curk T., Erjavec A., Gorup Č., Hočevar. T, Milutinovič M., Možina M., Toplak M., Starič A., Štajdohar M., Umek L., Žagar L., Žbontar J., Žitnik M., and Zupan B. (2013) Orange: data mining toolbox in Python. J. Mach. Learn. Res. 14, 2349–2353 [Google Scholar]
- 85. Lamesch P., Li N., Milstein S., Fan C., Hao T., Szabo G., Hu Z., Venkatesan K., Bethel G., Martin P., Rogers J., Lawlor S., McLaren S., Dricot A., Borick H., et al. (2007) hORFeome 3.1: a resource of human open reading frames covering over 10,000 human genes. Genomics 89, 307–315 10.1016/j.ygeno.2006.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.