

Abstract
Correlation between transcriptional regulation and positioning of genes at the nuclear envelope is well established in eukaryotes, but the mechanisms involved are not well understood. We show that brr6-1, a mutant of the essential yeast envelope transmembrane protein Brr6p, impairs normal positioning and expression of the PAB1 and FUR4-GAL1,10,7 loci. Similarly, expression of a dominant negative nucleoplasmic Brr6 fragment in wild-type cells reproduced many of the brr6-1 effects. Histone chromatin immunoprecipitation (ChIP) experiments showed decreased acetylation at the key histone H4K16 residue in the FUR4-GAL1,10,7 region in brr6-1. Importantly, blocking deacetylation significantly suppressed selected brr6-1 phenotypes. ChIPseq with FLAG-tagged Brr6 fragments showed enrichment at FUR4 and several other genes that showed striking changes in brr6-1 RNAseq data. These associations depended on a Brr6 putative zinc finger domain. Importantly, artificially tethering the GAL1 locus to the envelope suppressed the brr6-1 effects on GAL1 and FUR4 expression and increased H4K16 acetylation between GAL1 and FUR4 in the mutant. Together these results argue that Brr6 interacts with chromatin, helping to maintain normal chromatin architecture and transcriptional regulation of certain loci at the nuclear envelope.
INTRODUCTION
Transcriptional regulation is intimately linked to dynamic spatial organization of genes within the nucleus (reviewed in Rajapakse and Groudine [2011], Zimmer and Fabre [2011], and Taddei and Gasser [2012]) and the nuclear envelope has emerged as an important organizing entity in chromatin architecture and regulation (reviewed in Steglich et al. [2013], Stancheva and Schirmer [2014], and Czapiewski et al. [2016]). Mutations in nuclear envelope transmembrane (NET) protein genes are linked to numerous human genetic diseases and certain cancers (reviewed in Stancheva and Schirmer [2014], Wong et al. [2014], Janin et al. [2017]), underscoring the importance of understanding the role of NET proteins in transcriptional regulation.
Correlation between localization of certain genes to the nuclear periphery and either activation or silencing has been demonstrated from yeast to mammals and artificially breaking or creating a tether to the nuclear envelope affects gene activity in some cases (e.g., Andrulis et al. [1998], Galy et al. [2000], Feuerbach et al. [2002], Taddei et al. [2006]). Work in the yeast system has been instrumental in identifying various mechanisms by which genes are targeted to the nuclear envelope including DNA zip codes and transcription factor binding (reviewed in Brickner [2017]). However, in spite of recent progress, the mechanistic relationship between envelope association and gene regulation is not well understood. The complexities of this problem are well exemplified by the yeast GAL1,10,7 gene cluster required for galactose utilization in budding yeast. The GAL1-10 locus relocates to the envelope upon galactose induction and transcriptional activation is necessary though not sufficient for localization (Cabal et al., 2006). However, localization to the envelope is not required for activation and has been proposed instead to allow for rapid repression following inactivation (Green et al., 2012). Two DNA zip codes, GRS4 and GRS5, present upstream of GAL1 have been shown to target the GAL locus to the envelope, but only GRS4 affects activation, raising further questions regarding the function of envelope association (Brickner et al., 2017).
Positioning genes in subcompartments such as the nuclear periphery, rich in chromatin modifying, transcription and processing factors, is thought to contribute to regulation (reviewed in Rajapakse and Groudine [2011], Zimmer and Fabre [2011], Taddei and Gasser [2012]). However, genes are often closely spaced in yeast such that recruitment of one locus may expose an adjacent gene with different regulatory requirements to the same general environment. Consistent with this idea, a recent study of GAL locus repositioning upon galactose induction showed that other loci distantly located on the same chromosome were also peripheralized (Dultz et al., 2016). Even within the same locus, differential regulation is required to curtail the generation of deleterious noncoding RNA (ncRNA) transcription from bidirectional promoters (reviewed in Wei et al. [2011]). How these requirements intersect with recruitment of loci at the nuclear rim is not well understood.
At a mechanistic level, gene activity is regulated by various chromatin modifications, such as acetylation on histone tails, that determine access of transcription factors to the DNA (reviewed in Grunstein and Gasser [2013]). In particular, acetylation status of the histone H4K16 residue has emerged as an important determinant of chromatin compaction (Dorigo et al., 2003; Shogren-Knaak et al., 2006; Robinson et al., 2008; Allahverdi et al., 2011; Liu et al., 2011; Zhang et al., 2017), and deacetylation/acetylation at this residue has been proposed to act as a switch that controls the binding of regulatory proteins and chromatin remodelers (Millar et al., 2004).
Chromatin modifications are also believed to play a part in targeting genes to the nuclear envelope in yeast and higher eukaryotes through various mechanisms (reviewed in Harr et al. [2016] and Brickner [2017]). For example, the yeast SAGA histone acetyl transferase (Cabal et al., 2006; Luthra et al., 2007) and the Rpd3(L) deacetylase (Randise-Hinchliff et al., 2016) affect gene recruitment to the envelope by regulating association of transcription factors that interact with the nuclear pore complex (NPC). In addition, deacetylation at H4K16 provides a physical link between yeast telomeres and the envelope by promoting association of Sir silencing proteins that in turn bind to the NET protein Esc1 (Andrulis et al., 2002; Taddei et al., 2004; Oppikofer et al., 2011; Laporte et al., 2016).
A clearer understanding of gene regulation at the nuclear envelope will require greater insight into the role of chromatin modifications as well as the identification of the membrane components that mediate gene recruitment. Most attention has focused on NPC components in this regard (reviewed in Ptak et al. [2014] and Sood and Brickner [2014]); however, several yeast NETs besides Esc1 (Scs2 Mps3, Scr1, and Nur1) have also been found to interact with chromatin (Brickner and Walter, 2004; Bupp et al., 2007; Grund et al., 2008; Mekhail et al., 2008). Here we demonstrate a previously unknown role for another NET protein, Brr6, in the recruitment of specific genes to the nuclear envelope, maintenance of appropriate H4K16 acetylation, and transcriptional regulation. We originally identified BRR6 via isolation of the brr6-1 allele in a dT50 in situ hybridization screen for cold-sensitive mRNA export mutants in Saccharomyces cerevisiae. We showed that Brr6 is a c-terminally anchored nuclear envelope integral membrane protein that is required for normal nuclear pore distribution but is not itself a nucleoporin (de Bruyn Kops and Guthrie [2001] and unpublished data). Brr6 also affects lipid homeostasis and NPC assembly in S. cerevisiae (Scarcelli et al., 2007; Hodge et al., 2010; Lone et al., 2015) and spindle pole body insertion in Schizosaccharomyces pombe (Tamm et al., 2011).
We show here that the brr6-1 mutant 1) impairs positioning of the PAB1 and GAL1,10,7 loci to the nuclear envelope; 2) associates physically with specific genes, including FUR4 located adjacent to GAL1,10,7; and 3) alters expression of the GAL1,10,7 and FUR4 genes as well as noncoding transcripts. We reproduce many of these effects in wild-type cells expressing a dominant-negative non–membrane-bound form of Brr6 in the nucleoplasm. Importantly, we link misregulation at FUR4-GAL7 to hypoacetylation at H4K16 and show that artificial recruitment of the GAL1 locus to the envelope overcomes GAL1 and FUR4 expression defects, concomitant with increased H4K16 acetylation in the region. Our results suggest that Brr6 helps recruit specific genes to the nuclear envelope, promoting appropriate differential regulation by enabling acetylation at H4K16.
RESULTS
We previously identified brr6-1 in a dT50 in situ hybridization screen for mutants in S. cerevisiae that accumulated bulk mRNA in the nucleus (de Bruyn Kops and Guthrie, 2001). Our subsequent characterization showed that cells in which BRR6 expression was shut off also accumulated mRNA in the nucleus (Supplemental Figure 1A). Notably, a nucleoplasmic form of Brr6 (the galactose-controlled PGAL_NLS-Brr6N fragment lacking the membrane anchor and luminal portion) did not rescue a ∆brr6 strain; instead, expression of the NLS-Brr6N fragment in wild-type cells was dominant negative and caused a bulk mRNA export defect, consistent with the fragment competing with the endogenous protein (Supplemental Figure 1, B–F). The brr6-1 mutation is located in a putative zinc finger and a Brr6 fragment in which the zinc finger domain was deleted (PGAL_NLS-Brr6∆C4N) showed no effects on growth or export, pointing to the importance of this domain for Brr6 function.
Our earlier work showed that Brr6 is a c-terminally anchored nuclear envelope integral membrane protein that is required for normal nuclear pore distribution but is not itself a nucleoporin (de Bruyn Kops and Guthrie, 2001). The brr6-1 mutant affects localization of nucleoporins comprising the cytoplasmic fibrils of the NPC (Scarcelli et al., 2007; Hodge et al., 2010) but most core nucleoporins and those comprising the nuclear basket structure important in RNA export are not strongly affected (Scarcelli et al. [2007], Hodge et al. [2010], and de Bruyn Kops [unpublished data]). Evidence suggests that the mutant affects NPC assembly but not stability (Scarcelli et al., 2007) and brr6-1 showed no effect on protein trafficking (de Bruyn Kops and Guthrie, 2001), consistent with a functional NPC. Thus, it has not been clear how Brr6 impacts mRNA export.
The brr6-1 mutant impairs PAB1 transcript levels, indirectly causing the bulk mRNA export defect
To better understand the nature of the brr6-1 bulk mRNA export defect seen by dT50 in situ hybridization, we wanted to examine the localization of a specific mRNA. We chose the PAB1 transcript because it is abundant and decreased Pab1GFP signal seen by fluorescence microscopy (Figure 1A) was consistent with a possible PAB1 mRNA export defect. Interestingly, Pab1GFP signal in brr6-1 showed high cell–cell variability in keeping with the incomplete penetrance of the bulk mRNA export defect (de Bruyn Kops and Guthrie, 2001). Flow cytometry experiments confirmed both the decreased Pab1GFP levels and increased cell–cell variance in brr6-1 (Figure 1A). Decreased Pab1GFP levels were also observed on expression of the NLS-Brr6N but not the NLS-Brr6∆C4N fragment (Figure 1B).
FIGURE 1:

The brr6-1 mutant and the NLS-Brr6N fragment impair PAB1 expression. (A) Fluorescence microscopy localizing Pab1GFP in live isogenic WT (wild-type, yDBK398) and b6 (brr6-1, yDBK399) cells. Arrowheads indicate cells with little Pab1GFP protein signal. Plots show quantitation of Pab1GFP levels by flow cytometry. (B) Pab1GFP in wild-type cells (W303) carrying empty vector (pJL602), B6N (pPGAL_NLS-BRR6N-FLAG), or ∆C4N (pPGAL_NLS-brr6∆C4N-FLAG) grown in raffinose media then induced with galactose O/N.
To determine whether brr6-1 affected PAB1 mRNA export, we used single-molecule fluorescence in situ hybridization (smFISH) to localize individual PAB1GFP mRNA molecules using an established probe mix against the GFP sequence (Abruzzi et al., 2006) and methods developed by the Singer lab (Zenklusen and Singer, 2010). Wild-type cells grown at 30°C showed numerous PAB1GFP mRNA molecules throughout the cell (Figure 2A). Staining of untagged cells showed no signal (Supplemental Figure S2), confirming that the probe detected PAB1GFP mRNA. PAB1 mRNAs were also detected throughout the cell in the mutant. Interestingly, the PAB1GFP mRNA data mirrored the protein localization results with brr6-1 cells showing reduced numbers of mRNAs relative to wild-type and high cell–cell variance (Figure 2A). Thus, we did not observe a defect in export of PAB1GFP mRNA in brr6-1 even though nuclear accumulation of bulk mRNA had been detected in brr6-1 by dT50 in situ hybridization. It is possible that the nuclear dT50 signal reflected mild decreases in export of many transcripts not detectable at the specific mRNA level.
FIGURE 2:
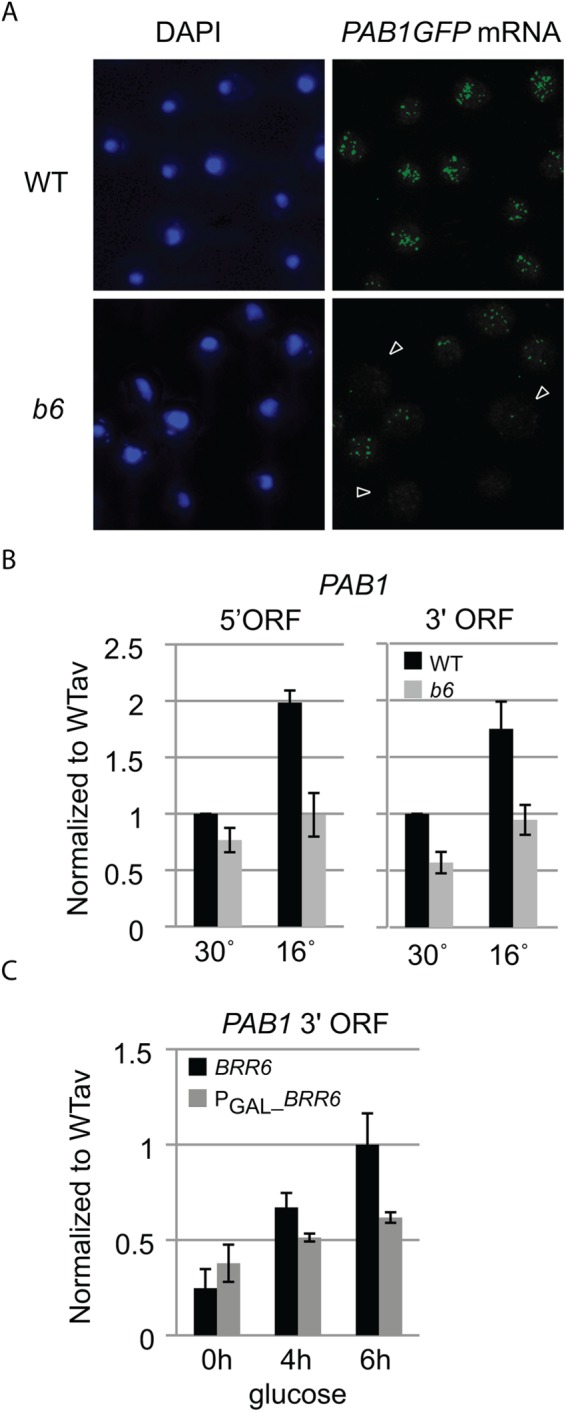
brr6-1 decreases PAB1 transcript levels. (A) Single-molecule FISH detecting PAB1GFP mRNA in fixed isogenic WT (wild-type, yDBK165) and b6 (brr6-1, yDBK166) cells. Nucleus is detected by DAPI staining. Arrows indicate cells with little PAB1GFP RNA signal. Untagged control is shown in Supplemental Figure S2. (B) DN9 primed RT-qPCR detection of PAB1 5′ and 3′ ORF transcripts in total RNA prepared from WT (wild type, yDBK165) and b6 (brr6-1, yDBK166) cells grown at 30°C or shifted for 3 h to 16°C. (C) RT-qPCR detection of PAB1 3′ transcripts from BRR6 (yDBK155) and PGAL_BRR6 (yDBK192) cells following a time course of glucose repression. RT-qPCR data were normalized against a Cryptococcus RNA control (see Materials and Methods) and expressed relative to an averaged wild-type sample. Error bars (SEM) reflect four biological replicates.
The smFISH results suggested that reduced PAB1 mRNA levels, rather than an export defect, are responsible for decreased Pab1 protein expression. To confirm the decrease in PAB1 mRNA levels, we used quantitative reverse transcription PCR (RT-qPCR) to quantify PAB1 transcripts in wild type and brr6-1. cDNAs were synthesized using a DN9 primer and quantified by qPCR using primers specific for the PAB1 5′ and 3′ open reading frame (ORF) regions. Results were normalized using a Cryptococcus RNA control added to each RT reaction (see Materials and Methods). The brr6-1 cells grown at 30° C showed lower levels of PAB1 transcript than wild type with both primer sets, and cells shifted to 16°C for 3 h showed stronger effects (Figure 2B). A similar effect was observed during a BRR6 shutoff using a PGAL_BRR6 strain switched from galactose to glucose media (Figure 2C). These results confirmed that BRR6 is required for normal PAB1 transcript levels.
brr6-1 impairs positioning of PAB1 and GAL1-10 loci at the nuclear rim
The effects of the brr6-1 mutation, the BRR6 shut-off, and the NLS-Brr6N fragment on PAB1 RNA and protein levels suggest a role for Brr6 in PAB1 regulation. Because Brr6 is a nuclear envelope transmembrane protein and the regulation of some genes correlates with their recruitment to the nuclear envelope, we wondered whether the PAB1 locus is recruited to the envelope. To examine this, we employed a commonly used method for visualizing the position of specific gene loci in individual cells in which a LAC operon tag (an approximately 14-kilobase-pair insert consisting of LAC O repeats and a marker [Rohner et al., 2013]) is inserted near the gene of interest and localized in living cells by binding of LacI GFP to the operon repeats. Comparison of PAB1 locus position in isogenic wild-type and brr6-1 strains with a locus tag just upstream of the PAB1 promoter (LAC O:PAB1), showed preferential positioning of the PAB1 locus at the envelope in wild-type but not brr6-1 cells (Figure 3A), indicating that PAB1 is recruited to the envelope in a Brr6-dependent manner. The magnitude of this effect was comparable to GAL1-10 locus positioning defects reported previously in various mRNA biogenesis mutants (Cabal et al., 2006; Green et al., 2012). Expression of the NLS-Brr6N fragment also caused decreased rim association of the PAB1 locus compared with the NLS-Brr6∆C4N fragment in cells grown in 2% raffinose/0.04% sucrose followed by induction with 2% galactose for 2 h (Figure 3B).
FIGURE 3:
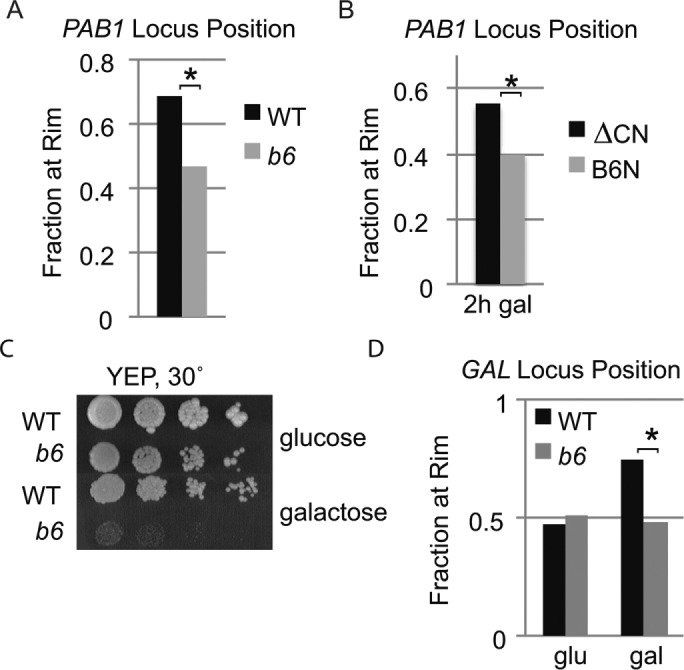
brr6-1 and the NLS-Brr6N fragment impair PAB1 and GAL1-10 locus positioning. (A, B) Locus positioning assay showing fraction of PAB1 locus at the nuclear rim in homozygous diploid LAC O:PAB1 cells (A) WT (wild type, yDBK523) vs. b6 (brr6-1, yDBK524) and (B) wild-type (W303) cells carrying B6N (pPGAL_NLS-BRR6N) or ∆C4 (pPGAL_NLS-brr6∆C4N) fragment constructs. (C) Growth of WT (wild-type, yDBK165) and b6 (brr6-1, yDBK166) strains on YEP media containing glucose or galactose. (D) Locus positioning assay for GAL1-10 locus in homozygous diploid LAC O:GAL1 cells (WT [wild type, yDBK535] vs. b6 [brr6-1, yDBK536]). Asterisks indicate p value ≤6 × 10–5.
Interestingly, the LAC O cassette adjacent to the PAB1 promoter in a LAC O:PAB1 strain both restored Pab1GFP protein levels and overcame the bulk mRNA export defect (Supplemental Figure S3, A and B). We speculate that insertion of the large, repeat-rich tag substantially alters chromatin architecture in the region, affecting PAB1 expression. Elimination of the export defect in brr6-1 LAC O:PAB1 argues strongly that the bulk mRNA export phenotype in brr6-1 stems from perturbed Pab1 protein levels, consistent with the known requirement for Pab1 in mRNA export (Brune et al., 2005; Dunn et al., 2005). Because the LAC O:PAB1 insertion was identical in wild type and brr6-1, the decreased rim association seen in the mutant was not the result of the effects of the tag on PAB1 expression. The brr6-1 effects on PAB1 expression were also independent of the tag as they were observed in untagged strains.
The GAL1,10,7 gene cluster required for galactose utilization is among the most studied loci regulated at the envelope in budding yeast. Poor growth of the brr6-1 mutant on galactose media (Figure 3C) suggested that Brr6 might also play a role in GAL gene expression. Therefore, we asked whether brr6-1 also affected GAL1-10 locus positioning using isogenic wild-type and brr6-1 strains derived from crosses with a strain containing a LAC O tag inserted in the GAL1-10 intergenic region (Schmid et al., 2006). We saw no difference in GAL1-10 locus position between wild-type and brr6-1 cells in glucose. However, envelope recruitment in cells grown in 2% raffinose/0.04% sucrose and then induced with 2% galactose for 5 h was impaired in brr6-1 (Figure 3D). These results show that the brr6-1 mutation interferes with recruitment of both PAB1 and GAL1-10 loci to the nuclear envelope.
brr6-1 alters expression at the GAL locus
To examine the effect of brr6-1 on expression of genes in the GAL1,10,7 cluster, we compared transcripts produced in wild-type and brr6-1 cells using whole genome RNA deep sequencing analysis (RNAseq). The mutant showed decreased transcript levels across the GAL1,10,7 gene cluster compared with wild type (Figure 4A and Supplemental Table S3). Notably, the expression changes for the GAL transcripts were on the order of twofold decreases, similar to that observed for PAB1 (Figure 2 and Supplemental Table S3); yet the brr6-1 mutant showed a dramatic growth defect on galactose media in the absence of other carbon sources (Figure 3). We think that the explanation for this can be found in the PAB1 mRNA and protein localization experiments as well as the flow cytometry, each of which indicates high cell-to-cell variance. In both the mRNA and protein localization experiments, some cells appear mostly normal, while others are severely impacted. Such nonpenetrant phenotypes can indicate stochasticity in underlying processes (e.g., Raj et al. [2010] and reviewed in Kærn et al. [2005] and Neems and Kosak [2010]). The brr6-1 mutation is a conservative Arg to Lys change at the tip of a putative zinc finger. This mutation is unlikely to disrupt the structure of the zinc finger but could make its interactions less robust. Stochastic, transient protein binding events involved in gene expression can be stabilized by additional interactions during the formation of functional entities (reviewed in Misteli [2001]). Weak binding of the Brr6-1 mutant protein may decrease the opportunity for stable associations necessary for GAL and PAB1 expression. A complete inhibition of GAL expression in 50% of cells would give a modest twofold effect in bulk assays such as RT-qPCR and RNAseq yet would represent an important disruption of function. Loss of GAL function in an additional 50% of cells during each subsequent cell cycle could result in the dramatic growth defect observed on galactose plates.
FIGURE 4:

brr6-1 and the NLS-Brr6N fragment perturb coding and noncoding transcription at the FUR4-GAL1,10,7 gene region. (A) RNAseq results comparing transcript levels (read density [transcripts per million, TPM]) for GAL7, GAL10, GAL1, and FUR4 in WT (wild type, yPH399) vs. b6 (brr6-1, yDBK168). (B) RT-qPCR measurement of FUR4 ORF transcripts in wild-type cells (W303) carrying vector (pJL602) vs. the B6N (pPGAL_NLS-BRR6N) construct. (C) Bar plot of RPM-normalized aligned sense (blue) reads for GAL1 and FUR4 coding and intergenic regions (red bracket) and sense and antisense (red) reads for GAL7 (representative replicates). (D) DN9 primed RT-qPCR of ncRNA (nc1, 2, and 3) transcripts in WT (wild-type, yDBK165) and b6 (brr6-1, yDBK166) cells (left) and the nc2 transcript in wild-type cells (W303) carrying vector (pJL602) vs. the B6N (pPGAL_NLS-BRR6N) construct (middle). Amplicons (GAL nc1-3) are indicated by red bars in the region from KAP104 to GAL7 (right). qPCR data were normalized and expressed as in Figure 2. Error bars (SEM) reflect ≥3 biological replicates.
Interestingly, regulation of the FUR4 gene immediately adjacent to GAL1 was also altered in the RNAseq data but showed increased read density. The increase in FUR4 transcript levels was also observed by RT-qPCR following expression of the GAL promoter-driven NLS-Brr6N fragment in wild-type cells (Figure 4B). We did not see changes in GAL gene transcript levels in the NLS-Brr6N samples (unpublished data). It may be that twofold decreases in transcription are difficult to detect on top of high levels of GAL transcripts transcribed before the NLS-Brr6N protein fragment accumulated. Although GAL transcript half-lives are known to be short in wild-type cells (Bennett et al., 2008; Munchel et al., 2011), we do not know whether NLS-Brr6N induction affects RNA turnover rates. Alternatively, the effect of Brr6 on FUR4 and GAL gene expression may be inherently different in ways we do not currently understand.
Interestingly, the RNAseq data also showed increased transcription in the GAL1-FUR4 intergenic region (Figure 4C, bracket), indicating that there is misregulation in noncoding as well as coding regions. Similarly, increased antisense reads in the GAL7-KAP104 region suggested that aberrant noncoding transcription may also occur on the other side of the GAL1,10,7 locus (Figure 4C). The antisense read density was too low to be included in the DESeq2 statistical analyses (50 reads per kilobase cutoff); therefore we carried out RT-qPCR to confirm whether aberrant ncRNA is detectable downstream of GAL7 using a primer set (nc1) located in the KAP104-GAL7 intervening sequence. We detected antisense RNA at low levels in both mutant and wild-type cells following but not prior to a 2 h galactose induction, with levels in brr6-1 being slightly higher than in wild type (Supplemental Figure 4). To ask whether this RNA extended further towards GAL7, we increased the amplicon size by moving the GAL7 proximal primer to the middle (primer nc2) and beginning (primer nc3) of the GAL7 3′ untranslated region (UTR), respectively. With both of these primers, we detected >2× higher levels of antisense transcript in RNA from brr6-1 than from wild type (Figure 4D). Thus, it appears that an ncRNA transcript is produced on galactose induction that is restricted more effectively in wild type than in mutant. Increased levels of the nc2 ncRNA were also detected following NLS-Brr6N fragment induction (Figure 4D). Together, these results show that Brr6 is required for normal transcript levels at and around the GAL1,10,7 locus.
Histone H4 hypoacetylation underlies the brr6-1 transcription defects
Transcription of both sense and antisense RNAs is regulated by various histone modifications such as acetylation on histone tails (reviewed Grunstein and Gasser [2013]). Hence, we wondered whether histone acetylation patterns in the GAL7 region were altered in brr6-1. We examined histone H3 and H4 acetylation in the GAL7 region using chromatin immunoprecipitation (ChIP). Acetylation relative to total H3 and H4 was determined using qPCR primer sets in the region spanning the GAL10 3′ ORF to the GAL7 3′UTR. Multiple lysine residues on both H3 and H4 are known to be acetylated; therefore we initially used pan-acetyl antibodies recognizing all of these marks. In addition, we included samples from a brr6-1/∆hda1 double mutant because the HDA1 histone deacetylase (HDAC) has been linked to regulation of the GAL gene cluster (Wu et al., 2001; Houseley et al., 2008). We observed hypoacetylation of H4 with all four primer sets in brr6-1 (Figure 5A). In contrast, we saw no difference in H3 acetylation between wild type and brr6-1. Pan H4 acetylation returned to wild-type levels in the brr6-1/∆hda1 double mutant; this was somewhat surprising given the reported specificity of Hda1 for histones H2A, H2B, and H3 (Wu et al., 2001). However, HDACs show significant functional overlap and also deacetylate many nonhistone proteins in various cell processes, including chromatin dynamics (reviewed in Ekwall [2005] and Glozak et al. [2005]), that could affect activity of either histone acetylases (HATs) or HDACs. Detailed studies of the four acetylated lysines in H4 (K5, 8, 12, and 16) have shown that H4K5, 8 and 12 behave similarly in gene regulation while the functions of K16 appears to be distinct from those of the other residues (Millar et al., 2004). Therefore, we also carried out ChIP with antibodies specific for acetylated H4K16. In this case, brr6-1 showed marked hypoacetylation of the GAL7 ORF and 3′ UTR but not the GAL10 3′ ORF (Figure 5B); this effect was reversed in the brr6-1/∆hda1 double mutant. Similarly, we observed a decrease in H4K16 acetylation in the GAL1-FUR4 intergenic region where increased noncoding transcription was also evident (Figure 5C).
FIGURE 5:

Aberrant transcription at FUR4 and GAL7 correlates with hypoacetylation at histone H4K16. (A, B) ChIP ratios of (A) pan-acetylated histone H4/total H4 pan-acetylated histone H3/total H3, and (B) lysine 16–acetylated H4 in WT (wild type, yDBK165), b6 (brr6-1, yDBK166), and b6/∆hda1 (brr6-1/∆hda1, yDBK169) cells following 2 h galactose induction. DNA was amplified using primer sets #1-4: #1-GAL10 3′ ORF, #2-GAL7 5′ ORF, #3-GAL7 3′ ORF, and #4-GAL7 3′ UTR. Total H4 and H3 values were normalized against WT prior to calculation of ratios. All antibody ChIP levels were >10× above mock ChIP. Error bars (SEM) reflect average of four biological replicates. (C) ChIP ratios of lysine 16–acetylated H4 amplified with primers for the GAL1-FUR4 intergenic region. (D) Growth of WT (wild type, yDBK165), b6 (brr6-1, yDBK166), and b6/∆hda1 (brr6-1/∆hda1, yDBK169) double mutants on YEP +2% galactose, 0.04% sucrose at 30°C. (E) DN9 RT-qPCR detection of GAL ncRNA (nc2 and nc3) transcripts in WT, b6, and b6/∆hda1 following galactose induction for 2 h at 30°C. qPCR data were normalized as in Figure 1. Error bars (SEM) reflect ≥3 biological replicates.
Aberrant H4K16 deacetylation could readily explain the altered transcript levels in the FUR4-GAL1,10,7 region seen in both brr6-1 and following NLS-Brr6N fragment expression. Deletion of HDA1 restored acetylation of H4K16 in brr6-1 (Figure 5B); therefore, we examined the impact of ∆hda1 on selected brr6-1 phenotypes (impaired growth on galactose media and the aberrant ncRNA downstream of GAL7). To examine the growth of the double mutant, cells were grown in 2% raffinose/0.04% sucrose and plated on 2% galactose/0.04% sucrose media. The small amount of sucrose improves growth of wild type and mutant in raffinose and galactose media, decreasing the extreme sickness of brr6-1 seen on galactose alone (Figure 3) while still allowing detection of a significant growth defect. The brr6-1/∆hda1 mutant showed substantial suppression of the brr6-1 growth defect at 30°C (Figure 5D), consistent with restored GAL gene expression. The brr6-1/∆hda1 strain also showed much less of the extended GAL7 ncRNA than brr6-1 alone (Figure 5E); thus, these brr6-1 effects are significantly decreased when H4K16 acetylation is restored in brr6-1/∆hda1. These results indicate that the misregulation in the GAL7 region is caused in part by altered H4K16 acetylation.
RNAseq analysis revealed genomewide transcriptional changes and CHIII disomy in brr6-1
In addition to changes in the FUR4-GAL1,10,7 region, analysis of the RNAseq data showed that expression at 809 ORFs and 168 antisense transcripts was significantly (p < 0.01) decreased or increased ≥1.5× in brr6-1 grown in galactose (Figure 6A and Supplemental Table S3; see Supplemental Table S4 for gene expression changes in glucose). To see how these changes were distributed throughout the genome, we mapped the Log2-fold changes to the midpoint of each affected gene on the chromosomes (Figure 6B and Supplemental Figure S5). This showed that expression increased approximately twofold for many genes across CHIII but not other chromosomes (e.g., CHV), suggesting disomy at CHIII. Comparison of genomic DNA from wild-type cells and 2 separate brr6-1 integrants confirmed stable disomy exclusively at CHIII (Supplemental Results and Supplemental Figure S6A).
FIGURE 6:
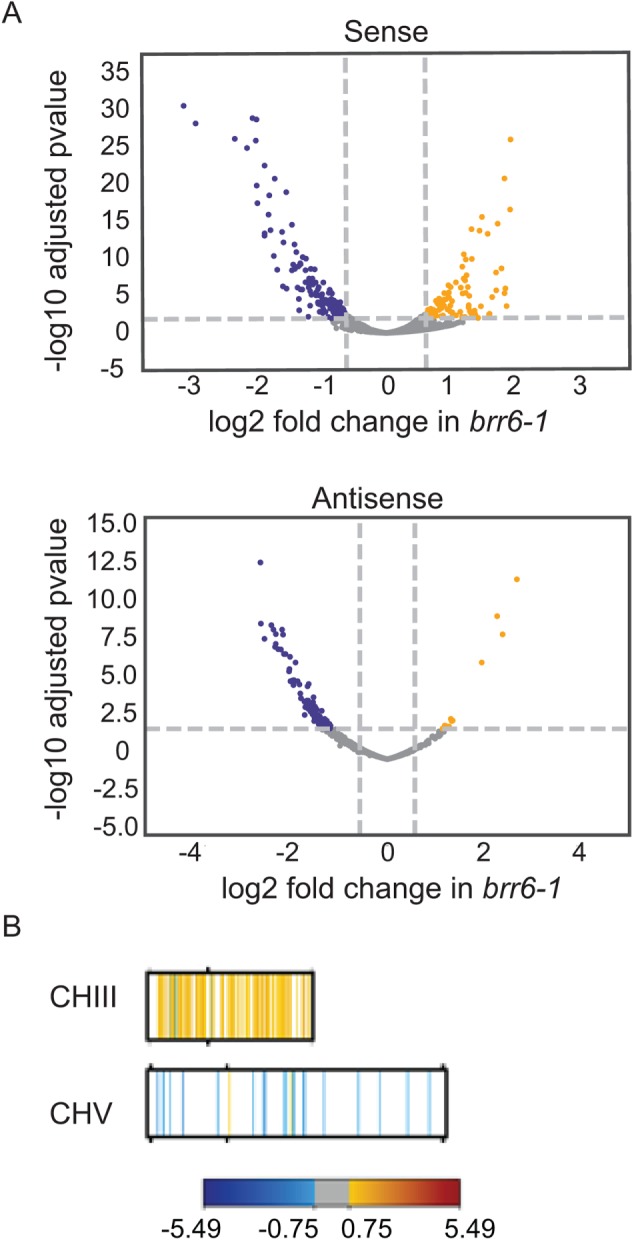
RNAseq analysis detects genomewide expression changes and CHIII disomy. (A) Volcano plots showing distribution of fold sense and anti-sense changes and adjusted p values in brr6-1(yDBK168) relative to wild-type (yPH399) cells following 2 h galactose induction (blue, fold decreases; yellow, fold increases; gray, changes between ± 0.75 log2 fold). Genes showing significant changes in galactose and glucose are listed in Supplemental Tables 3 and 4. (B) Mapping of genes showing altered sense reads in brr6-1 (galactose) along CHIII and CHV (for other chromosomes and glucose results see Supplemental Figure S5, A and B).
The disomy raises the possibility that some of the expression changes result from altered CHIII gene copy number. However, GO term analysis of the genes affected in brr6-1 (Supplemental Results and Supplemental Figure S6B) showed different enrichment patterns from those typically associated with disomy (Torres et al., 2007), suggesting that some of the expression effects may relate to Brr6 function. We know that this is the case for the misregulation across the FUR4-GAL1,10,7 region seen in brr6-1 because increases in FUR4 ORF and GAL7 ncRNA transcript levels also occurred when the NLS-Brr6N fragment was transiently expressed in wild-type cells (Figure 4, B and D). In these experiments, the NLS-Brr6N fragment was transiently expressed following galactose induction for 2–12 h, making it unlikely that a large fraction of cells would have had time to generate a stable disomy. In fact, we have confirmed that CHIII is present in a single copy following overnight (O/N) induction of the NLS-Brr6N fragment (see below). The NLS-Brr6N fragment also duplicated the brr6-1 effects on PAB1 locus positioning and expression and the resulting mRNA export defect (Figures 1 and 3 and Supplemental Figure S1). Thus, the experiments with the NLS-Brr6N fragment confirm that Brr6 plays a role in regulation of both the FUR4-GAL1,10,7, and PAB1 loci at the nuclear envelope.
The NLS-Brr6 fragment interacts physically with specific genes
Because brr6-1 and the NLS-Brr6N fragment affect both recruitment of loci to the envelope and transcriptional regulation, we wondered whether Brr6 associates with chromatin. We attempted to carry out ChIP with Brr6 but encountered prohibitively low immunoprecipitation efficiencies, as is frequently the case with integral membrane proteins. The FLAG-tagged nucleoplasmic versions of the Brr6 fragments provided an alternative means of testing for physical interactions between Brr6 and chromatin. We carried out ChIPseq experiments using W303α cells carrying the pPGAL_NLS-Brr6N-FLAG or pPGAL_NLSBrr6∆C4N-FLAG constructs or the empty vector grown O/N in galactose. qPCR of the DNA libraries confirmed the absence of CHIII disomy in the PGAL_NLS-Brr6N-FLAG fragment strain (Supplemental Figure 7). Using a sliding window analysis (see Materials and Methods), we identified a small set of genes showing >2x enrichment in the NLS-Brr6N ChIP sample compared with vector (Figure 7A).
FIGURE 7:

The NLS-Brr6N fragment associates with chromatin at FUR4 and several glucose- and heatshock-responsive genes. (A) Right: Heatmap of the average read density (RPKM, ChIP normalized to whole cell extract) of Brr6 binding sites in ChIP samples from W303 cells carrying vector (pJL602), B6N (pPGAL_NLS-BRR6N-FLAG), or ∆C4N (pPGAL_NLS-brr6∆C4N-FLAG) prepared following O/N galactose induction (two biological replicates). Regions of Brr6 binding were identified by sliding window (see Materials and Methods). Only regions with at least 1.5-fold ChIP/WCE are shown. Values are shown in Supplemental Table S5. qPCR confirming absence of CHIII disomy following B6N expression is shown in Supplemental Figure S7. (A) Left, changes in expression in brr6-1 obtained by RNAseq analysis are shown for associated genes (*genes on CHIII not affected by brr6-1 are predicted to show ∆log2 ≈ 1 expression changes due to disomy). (B) Bar plot of RPKM-normalized reads enriched in vector (pJL602), B6N (pPGAL_NLS-BRR6N-FLAG), or ∆C4N (pPGAL_NLS-brr6∆C4N-FLAG) samples showing GAL1-FUR4, PAB1, and HXT7-HXT6 and HSP30. (C) Left: RT-qPCR analyses of GAL1 5′ ORF and FUR4 mid-ORF transcript levels in cells carrying a LAC O tag upstream of GAL1 (see schematic) and either Nup2 and Nup2-Lac I URA3-marked constructs. (C) Right, comparison of H4K16 acetylation in the FUR4-GAL1 intergenic region brr6-1 cells carrying the LAC O tag and the Nup2 vs. Nup2-Lac I construct. Error bars (SEM) reflect three biological replicates.
Of particular interest, the set included the FUR4 gene located adjacent to the GAL1,7,10 locus. The NLS-Brr6∆C4N fragment showed no enrichment at FUR4 (Figure 7A), indicating that this effect was dependent on the Brr6 putative zinc finger. We were unable to determine whether the GAL1,10,7 genes interacted with Brr6 because the nonspecific background (signal in vector sample) over the GAL1,10,7 ORFs was very high (Figure 7B). A similar high background was observed over the PAB1 ORF as well as the ORFs of the adjacent high-affinity glucose importer genes HXT6 and HXT7. NLS-Brr6N-specific association evident in the HXT6-7 intergenic region indicated a physical link between these genes and Brr6; however, the data did not reveal whether PAB1 also associates. The high background over these ORFs was not a general feature of the ChIP data set and may reflect chromatin characteristics stemming from intense transcriptional activity as the GAL1,7,10 and HXT6,7 are induced in galactose and low glucose conditions respectively and PAB1 is constituitively highly expressed.
The ability of the NLS-Brr6N fragment to both associate with FUR4 and alter its expression (Figure 4) suggests that physical association with Brr6 may play a role in FUR4 regulation. Several other gene regions showing NLS-Brr6N zinc finger-dependent enrichment also exhibited significant expression changes in brr6-1 in galactose, suggesting that association with Brr6 may also contribute to their regulation. These included the HXT7 and HXT6 genes and the heat shock gene HSP30 that are induced under conditions of limited glucose; notably, different expression effects were observed for these genes in galactose versus glucose (Figure 7A). Another heatshock protein, HSP26, also showed both association and expression effects. BRR6 itself was enriched in the fragment samples relative to the vector control; however, this reflected increased copy number from the plasmids encoding the Brr6 fragments, as indicated by the fact that the transmembrane and luminal portions of the sequence that are absent in the fragment constructs were not enriched (unpublished data). The ChIP results show that the NLS-Brr6N fragment associates physically with a small set of genes, including FUR4.
Together, the effect of brr6-1 and the NLS-Brr6N fragment on recruitment of the GAL locus to the envelope and the FUR4 ChIP result, along with the H4K16 acetylation and expression defects (Figures 4 and 5), raise the possibility that Brr6 performs a gene tethering function necessary for appropriate acetylation and regulation in this region. To test this idea, we asked whether artificially tethering the GAL1 locus to the NPC could overcome the acetylation and expression defects. We made use of an established tethering approach in which wild-type and brr6-1 cells carried a LAC O tag upstream of GAL1 and a URA+ construct expressing a Nup2-Lac I fusion protein or Nup2 alone. Cells were grown in 2% raffinose/0.04% sucrose media lacking uracil to retain the plasmids and assayed following a 2 h galactose induction. In the presence of untagged Nup2, brr6-1 showed decreased expression of both GAL1 and FUR4 relative to wild type (Figure 7C). FUR4 is a uracil importer that is expressed under low uracil growth conditions and repressed in rich media (Séron et al., 1999); hence, the decreased expression shows that brr6-1 impairs optimal expression in low uracil as well as preventing appropriate repression in rich media (Figure 4). Importantly, the Nup2-Lac I construct largely overcame the expression defects for both GAL1 and FUR4 (Figure 7C). This argues strongly that these effects stem from failed envelope recruitment of the GAL1-FUR4 region in brr6-1. Comparison of H4K16 acetylation in brr6-1 cells carrying the Nup2-Lac I construct versus Nup2 alone showed greatly increased acetylation in the presence of the Nup2-Lac I construct (Figure 7C). Together, these results suggest that association of the GAL-FUR4 region with the NPC promotes H4K16 acetylation and that artificial recruitment substitutes for a Brr6 tethering function needed for appropriate GAL1 and FUR4 regulation.
DISCUSSION
Our results showed that brr6-1 impairs PAB1 transcript levels (Figure 2) and disrupts PAB1 and GAL1-10 locus positioning (Figure 3), suggesting a role for Brr6 in controlling gene expression at the nuclear envelope. RNAseq analysis revealed changes in both coding and noncoding transcript levels across the FUR4-GAL1,10,7 region (Figure 4 and Supplemental Table S3) and at numerous other genes (Figure 6). The discovery that brr6-1 is disomic for CHIII raised the possibility that some of these effects stemmed from increased copy number of CHIII genes. However, disomy was not present in wild-type cells where the NLS-Brr6N fragment was expressed (Supplemental Figure S7). Therefore, we were able to use the NLS-Brr6N fragment to link many of the brr6-1 effects at the PAB1 and FUR4-GAL1,10,7 loci to Brr6 function, including the following: 1) decreased PAB1 expression (Figure 1), 2) defective PAB1 locus positioning (Figure 3), and 3) expression changes across the FUR4-GAL1-10 region (Figure 4). Together with our ChIPseq results showing zinc finger-dependent association of the NLS-Brr6N fragment with the FUR4 gene and the ability of an artificial NPC tether to overcome GAL1 and FUR4 expression effects in brr6-1 (Figure 7), these results argue that Brr6 functions as a tether, helping to recruit the FUR4 gene region to the envelope to ensure appropriate regulation.
Tethering via Brr6 promotes appropriate H4K16 acetylation and expression in the FUR4-GAL1,10,7 region
We observed altered expression of the GAL1,10,7 and FUR4 ORFs and detected aberrant noncoding transcripts between GAL1 and FUR4 and between GAL7 and KAP104 (Figure 4) in brr6-1, consistent with general disruption of gene expression in the region. While it is possible that the expression changes reflect defects in other aspects of mRNA metabolism, the Histone H4K16 hypoacetylation seen in both of these locations in brr6-1 (Figure 5) is consistent with effects on transcriptional regulation. Importantly, the brr6-1 growth and GAL7 ncRNA defects were substantially suppressed when H4K16 acetylation was restored in the ∆hda1/brr6-1 double mutant (Figure 5), showing that these defects were caused at least in part by histone deacetylation at H4K16.
The ectopically expressed PGAL_NLS-Brr6N fragment caused a dominant negative growth defect (Supplemental Figure S1) and recapitulated brr6-1 expression changes across the FUR4-GAL1,7,10 region, increasing both FUR4 ORF and GAL7 ncRNA transcript levels in rich media (Figure 4). Although the presence of the GAL promoter in the expression construct precluded assaying the effect of the NLS-Brr6N fragment on GAL locus positioning, the effect of the fragment on PAB1 locus positioning duplicated the brr6-1 effect. The similarity between brr6-1 and the nucleoplasmic Brr6N fragment phenotypes, suggests that the NLS-Brr6N fragment competes with membrane bound Brr6. The NLS-Brr6N fragment associated physically with the FUR4 locus (Figure 7) and GAL locus positioning was disrupted in brr6-1 (Figure 3), making it likely that membrane-bound Brr6 also interacts with the FUR4 region. Together, the expression changes, positioning defects and ChIP results raised the possibility that tethering of this region to the envelope by Brr6 may affect expression near FUR4. Importantly, the expression of an artificial NPC-Lac I tether largely eliminated GAL1 and FUR4 expression defects in brr6-1 and strikingly increased H4K16 acetylation in the GAL1-FUR4 region. This result strongly suggests that the NPC-Lac I substituted for a Brr6 tethering function necessary for optimal GAL1 and FUR4 regulation.
H4K16 acetylation promoted by Brr6 may optimize expression of certain inducible genes
Acetylation/deacetylation of the histone H4K16 residue has been proposed to act as a switch for chromatin architecture by promoting binding of different effector complexes and determining chromatin compaction (Millar et al., 2004). Numerous in vitro studies have provided evidence that H4K16 acetylation decreases compaction by reducing internucleosome interactions (Dorigo et al., 2003; Shogren-Knaak et al., 2006; Robinson et al., 2008; Allahverdi et al., 2011; Liu et al., 2011; Zhang et al., 2017). Although the role of H4K16 in chromatin compaction in vivo is not fully understood, H4K16 acetylation is correlated with polycomb chromatin puffs in Drosophila (Bone et al., 1994) and deacetylation plays a key role in heterochromatin formation in yeast (reviewed in Millar et al. [2004]). In addition, a H4K16 histone acetyltransferase functions in X chromosome decondensation in Drosophila (Lau et al., 2016). However, studies in mammalian cells failed to demonstrate a connection with linear chromatin compaction (Taylor et al., 2013). Similarly, linear chromatin compaction near the GAL locus was not perturbed in living yeast following deletion of the SAGA histone acetylase GCN5 or in the presence of the histone-deacetylase inhibitor trichostatin A (TSA) (Dultz et al., 2018); however, mutants in the TSA-resistant deacetylase Sir2 (Bernstein et al., 2000) and the SAS histone acetylase complex responsible for the H4K16 acetylation in vivo (Shia et al., 2005) were not examined in these studies.
Based on the effects of H4K16 acetylation on chromatin compaction in vitro and in some in vivo systems, an attractive model is that the tethering function of Brr6 promotes access to regulatory proteins by helping maintain appropriate H4K16-mediated compaction in the FUR4-GAL gene cluster and other Brr6-interacting genes. Alternatively, H4K16 acetylation could alter interaction of regulators independent of chromatin compaction. In either scenario, we predict that gene-specific transcriptional outcomes of tethering would depend on both the presence of binding sites for specific regulators in different genes as well as the levels of regulators under different environmental conditions.
The fact that FUR4 locus is differentially regulated from the immediately adjacent GAL1 gene is consistent with gene-specific consequences of gene recruitment. Both the GAL1 and the FUR4 promoters carry the Gal4-responsive activating sequence (UASGAL), yet the normal increase in GAL1 expression in wild-type cells following galactose induction is approximately two orders of magnitude greater (Figure 4A). Differences in response to galactose among genes containing the UASGAL, are thought to be determined both by the number and affinity of UASGAL sequences that vary among genes (Lohr et al., 1995) and by differences in chromosome architecture in genes carrying the UASGAL (reviewed in Traven et al. [2006]). In brr6-1, FUR4 expression increases 3.5× relative to wild type in rich media while transcript levels from the adjacent GAL1 gene decrease 1.7×, indicating opposing misregulation of the adjacent genes. FUR4 expression is also tightly controlled by uracil levels (Séron et al., 1999), leading to increased and decreased transcripts in conditions of low uracil and rich media, respectively. The opposite effects of brr6-1 on FUR4 transcript levels in uracil dropout (Figure 7) versus rich media (Figure 4) reinforces the idea that failed Brr6 tethering results in misregulation rather than consistently either aberrant activation or repression.
It is interesting that we detected interactions between the NLS-Brr6N fragment and only a few genes. This may reflect an inability of the fragment to consistently reproduce interactions of the native membrane-bound protein. However, it is striking that most of the regions detected in the NLS-Brr6N ChIP carry genes that are regulated in a particular context such as heatshock (HSP26, HSP30, SSA3), mating type a cells (STE2, BAR1, MFA2) or carbon source (HXT7, HXT6). It may be that Brr6 functions to recruit a very small set of highly regulated genes that require tight transcriptional regulation under specific inducing conditions.
Conclusion
A large body of information links nuclear envelope components with chromatin organization and transcriptional regulation. Histone modifications govern chromatin architecture, providing the mechanistic basis for eukaryotic transcriptional regulation. The current work argues that the Brr6 nuclear membrane protein aids in the recruitment of specific genes to the envelope and that gene tethering may promote a H4K16 chromatin architectural switch that is key for fine-tuning regulation of inducible genes as well as for allowing individual control of adjacent genes with different regulatory requirements.
This work establishes Brr6 as a valuable model for understanding the relationship of the nuclear envelope to chromatin architecture and gene regulation.
MATERIALS AND METHODS
Strains and plasmids
The pPGAL_NLS-BRR6N (pDBK101) and pPGAL_NLSbrr6-1N (pDBK105) constructs were made by inserting a PCR-generated NLS(MAPKKKRKV)-BRR6 or -brr6-1(codons 2–158) sequence (lacking the c-terminal transmembrane domain and luminal portion) into the pJL602 CEN/ARS vector containing the GAL10 promoter and a LEU2 marker (gift from Joachim Li, University of California, San Francisco). The pPGAL_NLS-brr6∆C4N zinc finger deletion construct (pDBK103) was generated by replacing BRR6 sequence encoding amino acids 96–124 with an in-frame ClaI/SpH1 cassette (ATCGATGC). The FLAG-tagged versions of the (pDBK102, pDBK104, and pDBK106) constructs were generated by inserting a FLAG tag (DYKDDDDK) fragment with stop codon with Spe1/SacII ends at the C-termini of the fragment coding sequences. The tagged and untagged constructs and pJL602 vector were transformed into a W303 wild-type strain. The NUP2 pRS316 construct was a gift from Jonathan Loeb and Gerald Fink, Massachusetts Institute of Technology. Sequence encoding the Lac I tag was inserted into the plasmid in frame to give the NUP2-LACI pRS316 construct.
Strains used in this study are listed in Supplemental Table S1. GFP:HIS3 tagged strains (PAB1, NUP60, POM34) used in strain generation were obtained from the O’Shea library (Huh et al., 2003). PAB1GFP strains were generated by crossing PAB1GFP:HIS3 with wild-type and mutant BRR6 strains (yDBK164 and yDBK165). LAC O:PAB1 strains were made by inserting LAC O repeats at nucleotide –260 with respect to the PAB1 ORF in a wild-type s288C strain background (ATC20089) using a previously described LAC O:LEU cassette and marker replacement method (Rohner et al., 2008). Haploid wild-type and mutant strains were obtained from a cross with a strain carrying unmarked brr6-1 and Pom34GFP:HIS3 (s288C background). These were subsequently crossed with BRR6:HIS:LEU,LACIGFP:HIS3 and brr6-1:HIS3:LEU2,LACIGFP:HIS3 strains (W303 background) to obtain diploids used in positioning assays. LAC O:GAL1 strains were made by crossing a wild-type strain carrying the GAL1 locus tag and LACIGFP:HIS3 (YGA133 [Schmid et al., 2006]) with unmarked wild-type and brr6-1 strains (W303 background). These were subsequently crossed against ∆brr6::HIS3 strains covered by BRR6 and brr6-1 on TRP1 plasmids and carrying NUP60:GFP:HIS3 to give diploids used in positioning assays. Diploid LAC O:GAL1 strains were sporulated to yield the haploid LAC O:GAL1, BRR6 and brr6-1-unmarked strains without LACIGFP:HIS3 used in Nup2-Lac I tethering experiments. A YPH399 strain carrying the brr6-1 mutation (yDBK168) was made by targeted insertion of brr6-1 into the BRR6 downstream sequence in the ∆brr6::HIS3 deletion strain yDBK123 (de Bruyn Kops and Guthrie, 2001). The resulting strain carries the brr6-1 allele and LEU2 marker flanked by ∆brr6::HIS3.
Growth assays
Strains were grown to saturation and then diluted identically prior to pinning on appropriate media: BRR6 mutant and wild-type cells were grown in yeast extract peptone (YEP) raffinose media and tested for growth on plates containing 2% glucose or 2% galactose (Figure 3C and Supplemental Figure S1E) or 2% galactose/0.04% sucrose (Figure 5D) at 30°C. For experiments with wild-type cells carrying the Brr6 fragment constructs or empty vector, YEP was replaced with synthetic minimal media (SD–Leu) media (Supplemental Figure S1E).
Fluorescence microscopy, locus positioning assays, and in situ hybridization
Fluorescence microscopy was carried out using an Olympus BX60 microscope equipped with a 100× UPlanApo oil-immersion objective and Hi-Q DAPI, Hi-Q FITC, and Edow long-pass GFP and CY3 filters (Chroma Technologies) and z-axis controller and 24-bit black-and-white Photometrics Sensys charge-coupled device (CCD) camera. Images were collected and processed using iVision (v.4.0.0), BioVision Technologies. Cells for all microscopy experiments were grown to 0.25–0.5 OD600.
PabGFP localization and locus positioning assays.
Diploid cultures (5 ml) were grown in glucose media (YEPD) at 30°C, harvested by brief centrifugation and resuspended in 10–20 μl synthetic complete (SC) media prior to live imaging to decrease media fluorescence. Diploid cells carried the LAC O repeat tag integrated adjacent to the PAB1 or GAL1 locus and LacIGFP as well as either Nup60GFP or Pom34GFP to locate the nuclear envelope. Loci were detected as a bright spot of LacI GFP signal that was readily distinguishable from the dimmer nucleoporin signal. Having both locators tagged with GFP allowed scoring of Loci positions without filter registration concerns. Cells with in-focus loci were scored as rim or internal depending on whether they contacted the envelope signal. Fields were photographed sufficient to give p ≤ 0.00006 using a two-tailed, pooled two-proportions Z-test appropriate for binomial data.
In situ hybridization.
Localization of bulk poly A mRNA in fixed cells was carried out as previously described (de Bruyn Kops and Guthrie, 2001) using a digoxygenin-labeled dT50 probe. Staining with 4’,6-diamidine-2’-phenylindole dihydrochloride (DAPI) was used to identify the nucleus. Single-molecule FISH Localization of PAB1GFP mRNA was carried out according to methods developed by the Singer lab (Zenklusen and Singer, 2010). The GFP probe consisted of 4 AMC6-dT–modified 56mer oligonucleotides (OGM191-194[KD209-212]) identical to those used to detect GFP RNA sequences previously (Abruzzi et al., 2006). The probe was conjugated to CY3 dye (GE Healthcare) according to manufacturer’s instructions. Images are pseudo-colored maximal projections of z-series.
Flow cytometry
GFP tagged and untagged cells were grown at 30°C to 0.3–0.5 OD600 in SC media with 2% glucose. Signals in the FITC channel were recorded from 40,000 cells/sample without media change using a LSRII flow cytometer (BD Biosiences). Data were analyzed using FlowJo single-cell analysis software. Coefficient of variation is defined CV = SD/mean and displayed in percent.
RT-qPCR detection of transcripts
Cells were grown to mid–log phase in YEP media containing glucose or 2% raffinose/0.04% sucrose as appropriate. For GAL gene induction, galactose was added to a final concentration of 2% at designated times prior to harvesting. RNA was extracted using an established hot acid phenol protocol. Total 20 μg RNA was treated with RNase-free DNase (10 U; NEB), phenol-chloroform extracted, ethanol-precipitated, and measured by Nano-Drop (Thermo Scientific). DNased RNA (0.5 µg) was reverse transcribed using SuperscriptIII (Invitrogen) with a DN9 random primer. Because the full range of BRR6 transcription targets is unknown, it was not possible to select a gene for use as an internal control for normalization. Instead reactions were doped with 0.5 μg DNased Cryptococcus neoformins grubii H99 RNA to control for variabilities in RT efficiency and recovery and transcript levels were normalized to the level of Crypto PAB (CNAG_04441). Control experiments established complete absence of cross-detection with Saccharomyces and Cryptococcus probes (unpublished data). qPCR was carried out with four technical replicates (see Supplemental Table S2 for primer sets). Transcript levels from a minimum of three cryptonormalized biological replicates were normalized to the average of a wild-type sample.
qPCR determination of chromosome number
Genomic DNA from cells grown to mid–log phase was prepared by bead-beating in phenol. DNA was treated with RNase, quantified by nanodrop, and diluted to 3 ng/ul. DNA (5 μl) was assayed by qPCR (three biological replicates) using primers specific for selected genes on each chromosome (Supplemental Table S2).
RNA deep sequencing (RNAseq)
Sequencing experiments were done in the YPH399 (S288C derivative) background rather than W303, because a completely assembled and annotated reference genome is available for S288C. Cells (brr6-1 [yDBK168] and wild-type parent [YPH399], two biological replicates) were grown at 30°C in 2% glucose or in raffinose/0.04% sucrose YEP media followed by a 2-h galactose (2%) induction prior to harvesting in mid–log phase. RNA was isolated as for RT-qPCR (see above) and rRNA was removed using a Ribominus kit for yeast and concentration module (Invitrogen). RNA was cleaned and DNase treated using the RNA Clean and Concentrator Kit (Zymo Research). RNA quality was checked by bioanalyzer using the Agilent RNA 6000 Pico Kit. RNA libraries were prepared using a NEBNext Ultra Directional RNA Library Prep kit for Illumina (E7420) and standard NEB protocol for ribosome-depleted RNA. Libraries were size selected (200–400 base pairs) and quality checked by bioanalyzer using the Agilent High Sensitivity DNA Kit prior to multiplexing for Illumina sequencing (single end, 50-base-pair reads) on Hiseq 4000. Genes showing significant changes in expression in brr6-1 are listed in Supplemental Tables S3 (galactose) and S4 (glucose).
RNAseq data analysis.
Sequencing data (two biological replicates per genotype) were aligned to the S288C (parental strain for YPH399) genome (Fisk et al., 2006) using TopHat1 (Trapnell et al., 2009) using the following parameters: tophat1 –min-intron-length 20 –max-intron-length 2000 –max-multihits 2 –library-type fr-firststrand –segment-mismatches 3 –no-coverage-search –segment-length 20 –min-coverage-intron 10 –bowtie1. Alignments were sorted and indexed with SAMtools (Li et al., 2009), and bedgraph files were created with BEDTools (Quinlan, 2014). Reads in annotated transcripts were counted using HTseq-count (Anders et al., 2015) and differential expression was determined using DESeq2 (Love et al., 2014). Transcripts with a log2-fold change of greater than 0.75 or less than –0.75 and an adjusted p value of at most 0.01 were considered significantly changed. The log2-fold change threshold was chosen based on the differential expression of GAL7 in the brr6-1 mutant, which is deficient for growth on galactose. Antisense regions were defined by the boundaries of each annotated transcript plus 300 base pairs downstream of the stop codon. A cutoff of 50 reads per kilobase was used for antisense transcripts. The differential expression of RNA in the antisense region was analyzed as described above. Data from RNAseq and ChIPseq (see below) experiments are available in a GEO record (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE113746). Expression changes for the affected transcripts were mapped to the midpoint position for each gene using chromosomal location data from the Yeast Genome Database (www.yeastgenome.org/).
Chromatin immunoprecipitation
Histone ChIP.
Cells (DBK165 and DBK166) were grown to mid–log phase in YEP media containing 2% raffinose/0.04% sucrose and induced for 2 h by addition of 2% galactose. Cells (80 ml OD600 = 0.5 equivalents) were cross-linked for 15 min in 1% formaldehyde, quenched 15 min with 125 mM glycine, washed 2× in TBS, and resuspended in ChIP buffer (50 mM Tris [pH 7.4], 125 mM KCl, and 0.1% NP40) plus protease inhibitors (100 μM PMSF), Sigma and Complete protease tablet (Roche). Cells lysates generated by bead beating were bath sonicated 8 × 7.5 min and clarified by centrifugation 2 × 10 min at 2000 relative centrifugal force. Cell lysate (200 μl/sample) was incubated O/N at 4°C with 2–5 μl antibodies against histones: H3(Thermo PA5-16183), H4 (Millipore 04-858), acetylated H3(Millipore 06-599), pan-acetylated H4 (Millipore 06-866), and acetylated H4K16 (Millipore 07-329). Anti-histone and mock ChIP samples were incubated at 4°C for 3 h with 30 μl equilibrated protein A sepharose CL-4B beads (GE Healthcare) and then washed twice with 1 ml lysis buffer, once with 1 ml lysis buffer + 500 mM NaCl at 4°C, and once with Tris-EDTA buffer (TE) at room temperature. Beads were dissociated by heating 15 min at 65°C in 300 μl TE +1% SDS. Cross-links were reversed O/N at 65°C prior to proteinase K digestion and phenol chloroform extraction.
Brr6 fragment ChIPseq.
Two biological replicates of wild-type cells carrying empty vector(pJL602), pPGAL_NLS-BRR6N-FLAG, or pPGAL_NLS-brr6∆C4N-FLAG were grown to mid–log phase in YEP media containing 2% raffinose/0.04% sucrose, diluted into media containing 2% galactose/0.04% sucrose, and grown O/N. Cells (80 ml OD600 = 1.0) were cross-linked and lysates were generated as above. Extracts were sonicated 4 × 7.5 min in a bath sonicator, clarified, and incubated (350 μl/sample) O/N with 40 μl EZview Red anti-FLAG M2 affinity gel (Sigma) and washed as above. Cross-links were reversed and DNA libraries prepared as described in Inada et al. (2016). Library quality was confirmed by bioanalyzer using the Agilent High Sensitivity DNA Kit prior to multiplexing for Illumina sequencing (single end, 50-base-pair reads). Genes showing association with the NLS-Brr6N fragment are listed in Supplemental Table S5.
ChIP analysis methods.
Adaptor was trimmed from the 3′ end of reads using Cutadapt (Martin, 2011). Trimmed reads (two biological replicates per genotype) were then aligned to the S288C genome (R64-2-1_20150113) using Bowtie (Langmead et al., 2009) with the following parameters: bowtie -p8 -v2 -M1 –best –un B6N_a_multi_ un.fastq –max B6N_a_multi.fastq S288C_genome -q B6N_a_trim. fastq –sam B6N_a_multi.sam. Sequence alignment map (SAM) files were converted to binary alignment map (BAM) files, sorted, and indexed using SAMtools (Li et al., 2009). Alignment was done against S288C because the W303 genome is not fully assembled. Sorted and indexed BAM files were converted to bedgraph files using BEDTools (Quinlan, 2014). Bedgraph files were smoothed with a rolling mean of 100 base pairs using the Pandas Python package.
To identify regions of Brr6 binding, smoothed ChIP signal from the tagged samples (B6N) were divided by the signal from the untagged sample (PJL) after normalizing to the total number of aligned reads. A sliding window of 200 base pairs moving in increments of 20 base pairs was used to detect regions that had at least twofold enrichment of signal in the tagged sample over the untagged sample. Regions that had at least 50% overlap in both replicates were selected. Only the overlapping portion of the region was considered for further analysis steps. Finally, the average read density (in RPKM) was calculated in each region of Brr6 binding for tagged and untagged and then normalized to the signal from the corresponding whole cell extract from that sample.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by National Institutes of Health–National Institute of General Medical Sciences grant 5R01GM021119 to C.G. C.G. was also supported by an American Cancer Society Professorship in Molecular Genetics. J.B. is supported by an American Cancer Society Post-doctoral Fellowship (127531-PF-15-050-01RMC). We thank Tristan Daifuku for computer programming assistance. We thank Daniel Zenklusen and Robert Singer for reagents and training with the single-molecule FISH assay. We thank Jonathan Loeb, Gerald Fink, and Joachim Li for gifts of plasmids. We are grateful to Kristin Patrick, Sarah Ledoux, Megan Mayerle, Michael Marvin, Kelly Nissen, Argenta Price, Christina Homer, Bassem Al-Sady, Jahan-Yar Parsa, Selim Boudoukha, Sandra Catania, Hiten Madhani, and David Tollervey for helpful discussions and comments on the manuscript.
Abbreviations used:
- ChIP
chromatin immunopreciptitation
- ChIPseq
ChIP sequencing
- NET
nuclear envelope transmembrane
- ncRNA
noncoding RNA
- ORF
open reading frame
- RNAseq
RNA sequencing
- RT-qPCR
quantitative reverse transcription PCR
- smFISH
single-molecule fluorescence in situ hybridization
- UTR
untranslated region
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-04-0258) on August 22, 2018.
REFERENCES
- Abruzzi KC, Belostotsky DA, Chekanova JA, Dower K, Rosbash M. (2006). 3'-end formation signals modulate the association of genes with the nuclear periphery as well as mRNP dot formation. EMBO J , 4253–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahverdi A, Yang R, Korolev N, Fan Y, Davey CA, Liu C-F, Nordenskiöld L. (2011). The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res , 1680–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics , 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrulis ED, Neiman AM, Zappulla DC, Sternglanz R. (1998). Perinuclear localization of chromatin facilitates transcriptional silencing. Nature , 592–595. [DOI] [PubMed] [Google Scholar]
- Andrulis ED, Zappulla DC, Ansari A, Perrod S, Laiosa CV, Gartenberg MR, Sternglanz R. (2002). Esc1, a nuclear periphery protein required for Sir4-based plasmid anchoring and partitioning. Mol Cell Biol , 8292–8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR, Pang WL, Ostroff NA, Baumgartner BL, Nayak S, Tsimring LS, Hasty J. (2008). Metabolic gene regulation in a dynamically changing environment. Nature , 1119–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Tong JK, Schreiber SL. (2000). Genomewide studies of histone deacetylase function in yeast. Proc Natl Acad Sci USA , 13708–13713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bone JR, Lavender J, Richman R, Palmer MJ, Turner BM, Kuroda MI. (1994). Acetylated histone H4 on the male X chromosome is associated with dosage compensation in Drosophila. Genes Dev , 96–104. [DOI] [PubMed] [Google Scholar]
- Brickner DG, Sood V, Tutucci E, Coukos R, Viets K, Singer RH, Brickner JH. (2017). Subnuclear positioning and interchromosomal clustering of the GAL1-10 locus are controlled by separable, interdependent mechanisms. Mol Biol Cell , 2980–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner J. (2017). Genetic and epigenetic control of the spatial organization of the genome. Mol Biol Cell , 364–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner JH, Walter P. (2004). Gene recruitment of the activated INO1 locus to the nuclear membrane. PLoS Biol , e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brune C, Munchel SE, Fischer N, Podtelejnikov AV, Weis K. (2005). Yeast poly(A)-binding protein Pab1 shuttles between the nucleus and the cytoplasm and functions in mRNA export. RNA , 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bupp JM, Martin AE, Stensrud ES, Jaspersen SL. (2007). Telomere anchoring at the nuclear periphery requires the budding yeast Sad1-UNC-84 domain protein Mps3. J Cell Biol , 845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabal GG, Genovesio A, Rodriguez-Navarro S, Zimmer C, Gadal O, Lesne A, Buc H, Feuerbach-Fournier F, Olivo-Marin J-C, Hurt EC, Nehrbass U. (2006). SAGA interacting factors confine sub-diffusion of transcribed genes to the nuclear envelope. Nature , 770–773. [DOI] [PubMed] [Google Scholar]
- Czapiewski R, Robson MI, Schirmer EC. (2016). Anchoring a leviathan: how the nuclear membrane tethers the genome. Front Genet , 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruyn Kops A, Guthrie C. (2001). An essential nuclear envelope integral membrane protein, Brr6p, required for nuclear transport. EMBO J , 4183–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo B, Schalch T, Bystricky K, Richmond TJ. (2003). Chromatin fiber folding: requirement for the histone H4 N-terminal tail. J Mol Biol , 85–96. [DOI] [PubMed] [Google Scholar]
- Dultz E, Mancini R, Polles G, Vallotton P, Alber F, Weis K. (2018). Quantitative imaging of chromatin decompaction in living cells. Mol Biol Cell , 1763–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dultz E, Tjong H, Weider E, Herzog M, Young B, Brune C, Müllner D, Loewen C, Alber F, Weis K. (2016). Global reorganization of budding yeast chromosome conformation in different physiological conditions. J Cell Biol , 321–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn EF, Hammell CM, Hodge CA, Cole CN. (2005). Yeast poly(A)-binding protein, Pab1, and PAN, a poly(A) nuclease complex recruited by Pab1, connect mRNA biogenesis to export. Genes Dev , 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekwall K. (2005). Genome-wide analysis of HDAC function. Trends Genet , 608–615. [DOI] [PubMed] [Google Scholar]
- Feuerbach F, Galy V, Trelles-Sticken E, Fromont-Racine M, Jacquier A, Gilson E, Olivo-Marin J-C, Scherthan H, Nehrbass U. (2002). Nuclear architecture and spatial positioning help establish transcriptional states of telomeres in yeast. Nat Cell Biol , 214–221. [DOI] [PubMed] [Google Scholar]
- Fisk DG, Ball CA, Dolinski K, Engel SR, Hong EL, Issel-Tarver L, Schwartz K, Sethuraman A, Botstein D, Michael Cherry J, Project TGD. (2006). Saccharomyces cerevisiae S288C genome annotation: a working hypothesis. Yeast , 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galy V, Olivo-Marin JC, Scherthan H, Doye V, Rascalou N, Nehrbass U. (2000). Nuclear pore complexes in the organization of silent telomeric chromatin. Nature , 108–112. [DOI] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, Seto E. (2005). Acetylation and deacetylation of non-histone proteins. Gene , 15–23. [DOI] [PubMed] [Google Scholar]
- Green EM, Jiang Y, Joyner R, Weis K. (2012). A negative feedback loop at the nuclear periphery regulates GAL gene expression. Mol Biol Cell , 1367–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grund SE, Fischer T, Cabal GG, Antúnez O, Pérez-Ortín JE, Hurt E. (2008). The inner nuclear membrane protein Src1 associates with subtelomeric genes and alters their regulated gene expression. J Cell Biol , 897–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein M, Gasser SM. (2013). Epigenetics in Saccharomyces cerevisiae. Cold Spring Harb Perspect Biol , a017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr JC, Gonzalez-Sandoval A, Gasser SM. (2016). Histones and histone modifications in perinuclear chromatin anchoring: from yeast to man. EMBO Rep , 139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge CA, Choudhary V, Wolyniak MJ, Scarcelli JJ, Schneiter R, Cole CN. (2010). Integral membrane proteins Brr6 and Apq12 link assembly of the nuclear pore complex to lipid homeostasis in the endoplasmic reticulum. J Cell Sci , 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley J, Rubbi L, Grunstein M, Tollervey D, Vogelauer M. (2008). A ncRNA modulates histone modification and mRNA induction in the yeast GAL gene cluster. Mol Cell , 685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. (2003). Global analysis of protein localization in budding yeast. Nat Cell Biol , 686–691. [DOI] [PubMed] [Google Scholar]
- Inada M, Nichols RJ, Parsa J-Y, Homer CM, Benn RA, Hoxie RS, Madhani HD, Shuman S, Schwer B, Pleiss JA. (2016). Phospho-site mutants of the RNA Polymerase II C-terminal domain alter subtelomeric gene expression and chromatin modification state in fission yeast. Nucleic Acids Res , 9180–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin A, Bauer D, Ratti F, Millat G, Méjat A. (2017). Nuclear envelopathies: a complex LINC between nuclear envelope and pathology. Orphanet J Rare Dis , 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kærn M, Elston TC, Blake WJ, Collins JJ. (2005). Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet , 451–464. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol , R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte D, Courtout F, Tollis S, Sagot I. (2016). Quiescent Saccharomyces cerevisiae forms telomere hyperclusters at the nuclear membrane vicinity through a multifaceted mechanism involving Esc1, the Sir complex, and chromatin condensation. Mol Biol Cell , 1875–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AC, Zhu KP, Brouhard EA, Davis MB, Csankovszki G. (2016). An H4K16 histone acetyltransferase mediates decondensation of the X chromosome in C. elegans males. Epigenetics Chromatin , 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP. (2009). The sequence alignment/map format and SAMtools. Bioinformatics , 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Lu C, Yang Y, Fan Y, Yang R, Liu C-F, Korolev N, Nordenskiöld L. (2011). Influence of histone tails and H4 tail acetylations on nucleosome-nucleosome interactions. J Mol Biol , 749–764. [DOI] [PubMed] [Google Scholar]
- Lohr D, Venkov P, Zlatanova J. (1995). Transcriptional regulation in the yeast GAL gene family: a complex genetic network. FASEB J , 777–787. [DOI] [PubMed] [Google Scholar]
- Lone MA, Atkinson AE, Hodge CA, Cottier S, Martínez-Montañés F, Maithel S, Mène-Saffrané L, Cole CN, Schneiter R. (2015). Yeast integral membrane proteins Apq12, Brl1, and Brr6 form a complex important for regulation of membrane homeostasis and nuclear pore complex biogenesis. Eukaryot Cell , 1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol , 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra R, Kerr SC, Harreman MT, Apponi LH, Fasken MB, Ramineni S, Chaurasia S, Valentini SR, Corbett AH. (2007). Actively transcribed GAL genes can be physically linked to the nuclear pore by the SAGA chromatin modifying complex. J Biol Chem , 3042–3049. [DOI] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J , 10-12-13. [Google Scholar]
- Mekhail K, Seebacher J, Gygi SP, Moazed D. (2008). Role for perinuclear chromosome tethering in maintenance of genome stability. Nature , 667–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar CB, Kurdistani SK, Grunstein M. (2004). Acetylation of yeast histone H4 lysine 16: a switch for protein interactions in heterochromatin and euchromatin. Cold Spring Harb Symp Quant Biol , 193–200. [DOI] [PubMed] [Google Scholar]
- Misteli T. (2001). Protein dynamics: implications for nuclear architecture and gene expression. Science , 843–847. [DOI] [PubMed] [Google Scholar]
- Munchel SE, Shultzaberger RK, Takizawa N, Weis K. (2011). Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell , 2787–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neems D, Kosak ST. (2010). Turning down the volume on transcriptional noise. Nat Cell Biol , 929–931. [DOI] [PubMed] [Google Scholar]
- Oppikofer M, Kueng S, Martino F, Soeroes S, Hancock SM, Chin JW, Fischle W, Gasser SM. (2011). A dual role of H4K16 acetylation in the establishment of yeast silent chromatin. EMBO J , 2610–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak C, Aitchison JD, Wozniak RW. (2014). The multifunctional nuclear pore complex: a platform for controlling gene expression. Curr Opin Cell Biol , 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR. (2014). BEDTools: the Swiss-army tool for genome feature analysis. Curr Protoc Bioinform , 11.1211–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Rifkin SA, Andersen E, van Oudenaarden A. (2010). Variability in gene expression underlies incomplete penetrance. Nature , 913–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse I, Groudine M. (2011). On emerging nuclear order. J Cell Biol , 711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randise-Hinchliff C, Coukos R, Sood V, Sumner MC, Zdraljevic S, Meldi Sholl L, Garvey Brickner D, Ahmed S, Watchmaker L, Brickner JH. (2016). Strategies to regulate transcription factor-mediated gene positioning and interchromosomal clustering at the nuclear periphery. J Cell Biol , 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PJJ, An W, Routh A, Martino F, Chapman L, Roeder RG, Rhodes D. (2008). 30 nm chromatin fibre decompaction requires both H4-K16 acetylation and linker histone eviction. J Mol Biol , 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohner S, Gasser SM, Meister P. (2008). Modules for cloning-free chromatin tagging in Saccharomyces cerevisae. Yeast , 235–239. [DOI] [PubMed] [Google Scholar]
- Rohner S, Kalck V, Wang X, Ikegami K, Lieb JD, Gasser SM, Meister P. (2013). Promoter- and RNA polymerase II-dependent hsp-16 gene association with nuclear pores in Caenorhabditis elegans. J Cell Biol , 589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarcelli JJ, Hodge CA, Cole CN. (2007). The yeast integral membrane protein Apq12 potentially links membrane dynamics to assembly of nuclear pore complexes. J Cell Biol , 799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid M, Arib G, Laemmli C, Nishikawa J, Durussel T, Laemmli UK. (2006). Nup-PI: the nucleopore-promoter interaction of genes in yeast. Mol Cell , 379–391. [DOI] [PubMed] [Google Scholar]
- Séron K, Blondel MO, Haguenauer-Tsapis R, Volland C. (1999). Uracil-induced down-regulation of the yeast uracil permease. J Bacteriol , 1793–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shia W-J, Osada S, Florens L, Swanson SK, Washburn MP, Workman JL. (2005). Characterization of the yeast trimeric-SAS acetyltransferase complex. J Biol Chem , 11987–11994. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, Peterson CL. (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science , 844–847. [DOI] [PubMed] [Google Scholar]
- Sood V, Brickner JH. (2014). Nuclear pore interactions with the genome. Curr Opin Genet Dev , 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancheva I, Schirmer EC. (2014). Nuclear envelope: connecting structural genome organization to regulation of gene expression. Adv Exp Med Biol , 209–244. [DOI] [PubMed] [Google Scholar]
- Steglich B, Sazer S, Ekwall K. (2013). Transcriptional regulation at the yeast nuclear envelope. Nucleus , 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Gasser SM. (2012). Structure and function in the budding yeast nucleus. Genetics , 107–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Hediger F, Neumann FR, Bauer C, Gasser SM. (2004). Separation of silencing from perinuclear anchoring functions in yeast Ku80, Sir4 and Esc1 proteins. EMBO J , 1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Van Houwe G, Hediger F, Kalck V, Cubizolles F, Schober H, Gasser SM. (2006). Nuclear pore association confers optimal expression levels for an inducible yeast gene. Nature , 774–778. [DOI] [PubMed] [Google Scholar]
- Tamm T, Grallert A, Grossman EPS, Alvarez-Tabares I, Stevens FE, Hagan IM. (2011). Brr6 drives the Schizosaccharomyces pombe spindle pole body nuclear envelope insertion/extrusion cycle. J Cell Biol , 467–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor GCA, Eskeland R, Hekimoglu-Balkan B, Pradeepa MM, Bickmore WA. (2013). H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res , 2053–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. (2007). Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science , 916–924. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics , 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traven A, Jelicic B, Sopta M. (2006). Yeast Gal4: a transcriptional paradigm revisited. EMBO Rep , 496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Pelechano V, Järvelin AI, Steinmetz LM. (2011). Functional consequences of bidirectional promoters. Trends Genet , 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong X, Luperchio TR, Reddy KL. (2014). NET gains and losses: the role of changing nuclear envelope proteomes in genome regulation. Curr Opin Cell Biol , 105–120. [DOI] [PubMed] [Google Scholar]
- Wu J, Suka N, Carlson M, Grunstein M. (2001). TUP1 utilizes histone H3/H2B-specific HDA1 deacetylase to repress gene activity in yeast. Mol Cell , 117–126. [DOI] [PubMed] [Google Scholar]
- Zenklusen D, Singer RH. (2010). Analyzing mRNA expression using single mRNA resolution fluorescent in situ hybridization. Methods Enzymol , 641–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Erler J, Langowski J. (2017). Histone acetylation regulates chromatin accessibility: role of H4K16 in inter-nucleosome interaction. Biophys J , 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer C, Fabre E. (2011). Principles of chromosomal organization: lessons from yeast. J Cell Biol , 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.