

Abstract
Aqueous in situ one-pot N-Boc-deprotection-cyclization of Nα-Boc-dipeptidyl-tert-butyl and methyl esters under microwave irradiation afforded 2,5-diketopiperazines (DKPs) in excellent yields. This protocol is rapid, safe, environmentally friendly, and highly efficient, and showed that the tert-butoxy moiety is also an excellent leaving group for these cyclizations.
Keywords: 2,5-diketopiperazines; DKPs; cyclic dipeptides; microwave irradiation
1. Introduction
Cyclic peptides are a very important family of bioactive compounds easily available both from natural sources (plants, animals or microorganisms) or by means of synthetic methods. Their chemical properties, like the lack of charges at the amine and carboxylic terminal groups and the lack of zwitterionic character, confer to these molecules high lipophilicity [1], and fast membrane absorption in the digestive tract because of their high permeability [2]. The structural rigidity of the cyclic peptides increases its affinity and selectivity toward protein ligands, making their half-lives in vivo much greater than those of linear peptides [3]. Among them, 2,5-diketopiperazines (DKPs, also known as cyclic dipeptides, 2,5-dioxopiperazines, cyclo(dipeptides), or anhydride dipeptides) are the smallest cyclopeptides. These peptides are most commonly found as natural products [4], showing antimicrobial [5], antitumoral and antiviral [6], cytotoxic [7], and neuroprotective effects [8], among other activities. Some DKPs are stable to proteolysis (enzymatic degradation), an important feature for their high activity. All these properties make DKPs an interesting group of molecules for the development of new therapeutic agents.
The DKP core derives, chemically and biosynthetically, from folding and head-tail cyclization between N and C-terminal amino acids of linear dipeptides [9]. These heterocyclic compounds possess two amide groups (with acceptor-donor properties) with the possibility of including up to four hydrogen bonds [10]. DKPs can be synthesized in solution or in solid phase from commercially available and appropriately protected chiral α-amino acids. Their syntheses can be carried out with the appropriate linear dipeptide [11,12,13], followed by N-deprotection and cyclization using either basic (aminolysis of dipeptide ester in methanolic ammonia, Fischer method) [10,14], neutral (aminolysis of dipeptide ester in methanol, autoaminolysis) [15], or acidic conditions (Suzuki method) [16]. Some of these methods variously result in good reaction yields [17], low reaction yields [18], with or without epimerization [19].
Nowadays, microwave heating can be used to obtain good results from previously unsuccessful or low-yielding reactions. Under microwave heating conditions the reaction times can be reduced from days to hours, from hours to minutes, or from minutes to seconds, and the reaction yields can be greatly increased [20,21]. DKPs syntheses using microwave assistance has barely been investigated. Some examples of the use of this methodology are the syntheses of dimeric structures based on intermolecular DKP formation by activation of C-terminal glycine monomers [22], the solvent-free synthesis of DKPs in one-pot deprotection-cyclization of N-Boc-dipeptidyl ethyl and methyl esters [23], and the DKPs formation using dipeptide methyl ester hydrochlorides in water in three [24] and two steps [25]. In the present study, we report the syntheses of DKPs using Nα-Boc-dipeptidyl methyl and tert-butyl esters in water under microwave irradiation. DKPs were obtained in excellent yields without epimerization employing this general and highly efficient protocol.
2. Results and Discussion
The formation of DKPs occurs through an intramolecular aminolysis depending largely on the nature and the sequence of the amino acids in solution [11]. Under these conditions, when DKP formation is an undesired reaction, tert-butyl ester protection for the carboxy terminus function is used to prevent ring closure of dipeptides under basic conditions, because it provides a poor leaving group and steric hindrance. Nucleophilic removal of this protecting group with ring formation has been observed only in a few cases [26,27]. We studied the cyclization reaction assisted by microwave heating with the purpose of investigating the possible use of Nα-Boc-dipeptidyl tert-butyl esters as DKPs precursors, as part of a synthetic strategy toward the cis-DKP fragment of the natural product cyclo[N-(Lys-Phe)-Orn-Val] (1, Figure 1) [28]. We report our findings herein.
Figure 1.
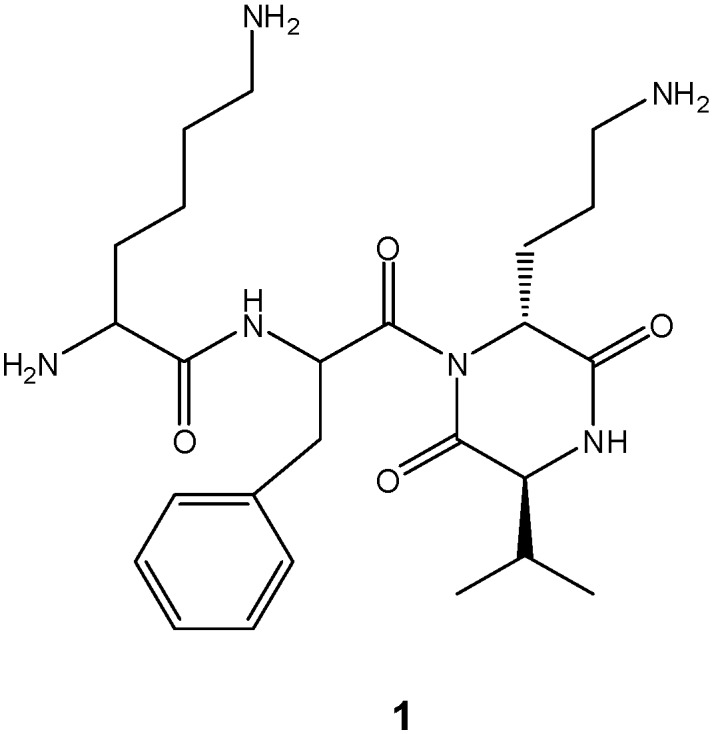
Structure of cyclo[N-(Lys-Phe)-Orn-Val] (1).
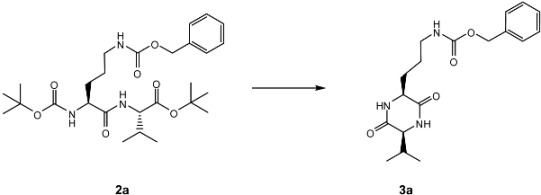
In an effort to explore optimized conditions for the cyclization under microwave irradiation, Boc-Orn(Cbz)-Val-OtBu (2a) was subjected to different reaction variables, such as solvent, temperature (T), irradiation power (W), and exposure time exchange (Table 1). Toluene, toluene-isopropanol (1:1) mixture, and xylene were unsuccessful in the transformation of dipeptide 2a to DKP 3a (entries 1-6). When DMF was used as solvent at 200 °C, 300 W, and 5 min reaction time, DKP 3a was obtained in 61% yield (entry 7). Water resulted a better solvent for the cyclization (entries 8-11). As can be observed in Table 1, with water as solvent, 200 °C, 300 W, and 5 min reaction time resulted in the best conditions, yielding 3a in 89% yield. However, pressure in the sealed reaction tube increased dramatically due to the generation of CO2 gas, increasing the risk of breaking the reaction vessel. Increasing the temperature and the reaction time did not improve the reaction yield any further (entry 11). The optimized conditions were then used in all cyclizations of Nα-Boc-dipeptidyl esters 2a-2l to DKPs 3a-3k.
Table 1.
Cyclization of dipeptide 2a to diketopiperazine 3a under microwave irradiation.a
 | |||||
| Entry | Solvent | T (°C) | Power (W) | Time (min) | 3a (% yield) |
| 1 | toluene | 170 | 160 | 10 | nr |
| 2 | 170 | 180 | 10 | nr | |
| 3 | 200 | 250 | 10 | nr | |
| 4 | 200 | 300 | 10 | nr | |
| 5 | toluene-isopropanol (1:1) | 200 | 150 | 10 | nr |
| 6 | xylene | 200 | 300 | 10 | nr |
| 7 | DMF | 200 | 300 | 5 | 61 |
| 8 | H2O | 200 | 250 | 1 | 22 |
| 9 | 200 | 250 | 2.5 | 85 | |
| 10 | 200 | 300 | 5 | 89 | |
| 11 | 250 | 250 | 10 | 86 | |
nr = no reaction; a Reactions performed in a monomode microwave CEM Discover apparatus.
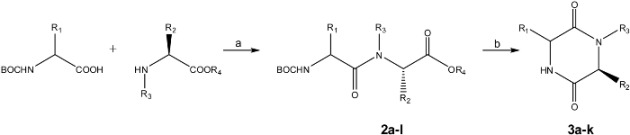
DKPs 3a-3k were synthesized in two steps: dipeptide formation (starting with Nα-Boc-terminal and C-OtBu or C-OMe amino acids, to obtain the corresponding Nα-Boc-dipeptidyl esters 2a-2l) and subsequent cyclization (Table 2).
Table 2.
Coupling of amino acids and ring closure under microwave irradiation.a
 | ||||||||
| (a) EDAC, HOBt, DMAP, TEA/CH2Cl2, 5 °C, then overnight at rt; (b) H2O (1 mL), MW (250 °C, 250 W and 150 psi) for 10 min. | ||||||||
| Entry | R1 | R2 | R3 | R4 | Compound | Yield (%) | Compound | Yield (%) |
| 1 | (S)-Propyl-NH-Cbz | Isopropyl | H | OtBu | 2a | 94 | 3a | 86b |
| 2 | (R)-Propyl-NH-Cbz | Isopropyl | H | OtBu | 2b | 98 | 3b | 99b |
| 3 | Propyl-NH-Cbz | Benzyl | H | OtBu | 2c | 91 | 3c | 95b |
| 4 | Propyl-NH-Cbz | Benzyl | H | OMe | 2d | 77 | 3c | 99b |
| 5 | H | Benzyl | H | OtBu | 2e | 98 | 3e | 99b |
| 6 | Benzyl | Benzyl | H | OtBu | 2f | 98 | 3f | 96b |
| 7 | Isopropyl | Benzyl | H | OtBu | 2g | 99 | 3g | 99b |
| 8 | H | Isopropyl | H | OtBu | 2h | 73 | 3h | 93b |
| 9 | Benzyl | Isopropyl | H | OtBu | 2i | 80 | 3g | 73b |
| 10 | Isopropyl | Isopropyl | H | OtBu | 2j | 83 | 3j | 99b |
| 11 | Benzyl | H | CH3 | OtBu | 2k | 85 | 3k | 84c |
| 12 | Benzyl | H | CH3 | OMe | 2l | 82 | 3k | 99c |
a Reactions performed in a monomode microwave CEM Discover apparatus; b Crude yield (pure by NMR); c Isolated yield after chromatography .
Data in Table 2 clearly shows that the cyclization step assisted by microwave heating yielded DKP compounds 3a-3k in excellent yields. Because N-Boc protecting groups are unstable at temperatures higher than 90 °C, microwave irradiation should deprotect the amines facilitating the spontaneous intramolecular aminolysis [29]. Nature and size of alkyl group (R1) on Cα of Nα-Boc amino acid residue have no effect on the course and reaction yield when the C terminus amino acid is phenylalanine (entries 3-7). However, they do influence the cyclization yield when the amino acid at C terminus is valine (entries 1, 8-10). When valine is present, the amino acid residue sequence is important on the cyclization yield (entries 7 and 9) because β-branched amino acid at C terminus (Cα isopropyl group) exerts a bulky effect on ring closure, diminishing the reaction yield (99 vs 73%). Entries 1 and 3 where a benzyl group replaced an isopropyl one corroborate this steric effect as the reaction yield diminished from 95 to 86%. The leaving groups OtBu and OMe (ester group nature, entries 3,4 and 11,12) have no effect on course and reaction yield, as corroborated by the excellent yields obtained for compounds 3a-3h and 3j-3k. Optical rotation values of compounds 3a-3k and their comparison with literature data showed that cyclization reaction proceeded without Cα chiral center epimerization of the amino acid residues. The cyclization of different Nα-Boc-dipeptidyl esters (even Ot-Bu) in water assisted by microwaves is a rapid, secure, environmentally friendly and highly efficient method to produce cis-DKPs with high optical purity. This reaction conditions are compatible with the presence of Cbz protecting groups. This protocol was used to obtain the trans-DKP fragment 3b of compound 1, which was synthesized in quantitative yield starting of Boc-D-Orn(Cbz)-Val-OtBu (2b, entry 2).
The structures of compounds 2a-2l and 3a-3k were established on the basis of the analysis of their spectroscopic data. For compounds 2a-2l, carbamate, amide, and ester groups showed IR absorptions at 3,364-3,283, 2,979-2,972 and 1,690-1,665, and 1,744-1,712 cm-1, respectively; Carbonyl amide, ester, N-Boc-carbamate, and Nδ-Cbz-carbamate groups showed 13C-NMR resonance signals at δ 172.35-169.07, δ 172.10-167.89, δ 157.09-155.07, and δ 156.98-156.94. Carbamate and amide groups in compounds 3a-3k produce IR absorptions at 3,440-3,426, and 2,977-2,925 and 1,678-1,664 cm-1, respectively. Amide groups showed 13C-NMR resonance signals at δ 167.83-166.00, and Nδ-Cbz-carbamate carbonyls give resonance signals in the δ 156.06-155.79 range.
3. Experimental
3.1. General procedures
Reactions were performed in sealed vessels in a monomode microwave CEM Discover apparatus and the temperature was evaluated by infrared. Melting points were obtained in a Fisher Johns melting point apparatus and are uncorrected. Infrared (IR) spectra were obtained in KBr on a Bruker Vector 22 IR spectrometer. Optical rotations were measured with sodium light (unless otherwise specified) on a Perkin-Elmer 341 MC polarimeter. 1H- and 13C-NMR spectra were recorded on a Varian Unity 400 spectrometer at 400 MHz for 1H-NMR, 1H-1H COSY, HSQC, and HMBC, and at 100 MHz for 13C- NMR, using CDCl3 or DMSO as solvents, as indicated. Chemical shifts are reported in ppm (δ) relative to the TMS signal. FAB+MS, and HRFAB+MS were recorded on a JEOL JMStation-JM 700 mass spectrometer at 70 eV in a matrix of glycerol. Flash column chromatography (FCC) and analytical thin-layer chromatography (TLC) were performed using silica gel 230-400 mesh and pre-coated silica gel 60 F254 Merck plates, respectively. Boc-Phe-OH, Boc-Val-OH, Boc-Orn(Cbz)-OH, Boc-Gly-OH, Sar-OMe, Sar-OtBu, Phe-OMe, Phe-OtBu, Val-OtBu, EDAC, TEA, and DMAP were obtained from Aldrich, HOBt was obtained from ANASPEC, and all chemicals were used without further purification.
3.2. General procedure for the syntheses of dipeptides 2a-2j
A mixture of Boc-amino acid (1 mmol), amino ester hydrochloride (1 mmol), EDAC (1.5 mmol), HOBt (1 mmol) and DMAP (0.1 mmol) were dissolved in dry CH2Cl2 (10 mL). Mixture was cooled to 5 °C and then TEA (1 mmol) was added. Reaction was stirred at 5 °C for further 30 min, then allowed to warm up to room temperature and stirred overnight. Reaction mixture was treated with sat. NH4Cl soln. (20 mL). The organic phase was separated and the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). Combined organic layers were washed with brine (2 × 15 mL) and with water (2 ×15 mL) and dried over Na2SO4. Solvent was removed under vacuum. The residue was purified by FCC.
3.3. General procedure for the syntheses of dipeptides 2k-2l
A mixture of Boc-amino acid (2.4 mmol), amino ester hydrochloride (2 mmol), EDAC (2.4 mmol), HOBt (2.4 mmol) and DMAP (0.1 mmol) were dissolved in dry CH2Cl2 (5 mL). Mixture was cooled to 5 °C and then TEA (2.4 mmol) was added. Reaction was stirred at 5 °C for further 30 min, then allowed to warm up to room temperature and stirred for two days. Reaction mixture was treated with sat. NH4Cl soln. (20 mL). The organic phase was separated and the aqueous layer was extracted with CH2Cl2 (3 × 15 mL). Combined organic layers were washed with brine (2 × 15 mL) and with water (2 × 15 mL) and dried over Na2SO4. Solvent was removed under vacuum. The residue was purified by FCC.
Boc-Orn(Cbz)-Val-OtBu (2a): Colorless syrup; [α] + 8.5 (c 1.09, CHCl3); IR: 3,336, 2,973, 2,935, 2,878, 1,712, 1,666, 1,532, 1,454, 1,368, 1,254, 1,162, 1,018 cm-1; 1H-NMR (CDCl3) δ 7.28-7.20 (5H, m, Ar), 6.85 (1H, bs, NH-Val), 5.29 (1H, bs, NH-Orn), 5.18 (1H, bs, NHδ-Orn), 5.04 (1H, d, J = 12.4 Hz, CH2-Cbz), 5.01 (1H, d, J = 12.4 Hz, CH2-Cbz), 4.33 (1H, dd, J = 9.2, 4.8 Hz, Hα-Val), 4.24 (1H, bs, Hα-Orn), 3.29 (1H, bs, Hδ-Orn), 3.08 (1H, bd, J = 13.2 Hz, Hδ’-Orn), 2.09 (1H, dh, J = 9.2, 7.2 Hz, Hβ-Val), 1.78 (1H, m, Hβ-Orn), 1.51 (3H, m, Hβ’-Orn, Hγ-Orn), 1.37 (9H, s, CH3Boc), 1.35 (9H, s, CH3tBu), 0.86 (3H, d, J = 7.2 Hz, Hγ-Val), 0.84 (3H, d, J = 7.2 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 172.26 (s, CO-Orn), 170.84 (s, CO-Val), 156.97 (s, CO-Cbz), 155.79 (s, CO-Boc), 136.63 (s, Ar), 128.49 (d, Ar), 128.11 (d, Ar), 128.07 (d, Ar), 81.87 (s, C-Boc), 79.89 (s, C-tBu), 66.80 (t, CH2-Cbz), 57.65 (d, Cα-Val), 53.41 (d, Cα-Orn), 40.02 (t, Cδ-Orn), 31.28 (d, Cβ-Val), 30.10 (t, Cβ-Orn), 28.50 and 28.20 (q, CH3-Boc, CH3-tBu), 26.35 (t, Cγ-Orn), 19.17 (q, Cγ-Val), 17.25 (q, Cγ’-Val); FAB+MS m/z: 522 (53) [M + H]+, 466 (11) [M + H - C4H8]+, 422 (13) [M + H - C5H8O2]+, 414 (15) [M - C7H7O]+, 366 (100) [M + H - C5H8O2 - C4H8]+, 258 (13), 213 (20), 91 (100) [C7H7]+, 57 (44) [C4H9]+; HRFAB+MS: observed 522.3176 [M + H]+, (calcd. for C27H44N3O7, 522.3179).
Boc-D-Orn(Cbz)-Val-OtBu (2b): Colorless syrup; [α] + 15.7 (c 1.04, CHCl3); IR: 3,339, 2,973, 2,935, 2,877, 1,712, 1,666, 1,525, 1,456, 1,391, 1,368, 1,254, 1,165, 1,022, 738, 699 cm-1; 1H-NMR (CDCl3) δ 7.36-7.25 (5H, m, Ar), 6.89 (1H, bs, NH-Val), 5.36 (1H, bd, J = 6.6 Hz, NH-Orn), 5.25 (1H, bs, NHδ-Orn), 5.08 (2H, s, CH2-Cbz), 4.39 (1H, dd, J = 8.8, 4.4 Hz, Hα-Val), 4.23 (1H, bs, Hα-Orn), 3.21 (2H, m, Hδ-Orn), 2.10 (1H, dh, J = 9.2, 7.2 Hz, Hβ-Val), 1.85 (1H, m, Hβ-Orn), 1.60 (3H, m, Hβ’-Orn, Hγ-Orn), 1.45 (9H, s, CH3Boc), 1.43 (9H, s, CH3tBu), 0.92 (3H, d, J = 7.0 Hz, Hγ-Val), 0.89 (3H, d, J = 7.0 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 172.06 (s, CO-Orn), 170.78 (s, CO-Val), 156.74 (s, CO-Cbz), 155.71 (s, CO-Boc), 136.69 (s, Ar), 128.50 (d, Ar), 128.06 (d, Ar), 82.00 (s, C-Boc), 80.06 (s, C-tBu), 66.68 (t, CH2-Cbz), 57.60 (d, Cα-Val), 54.09 (d, Cα-Orn), 40.37 (t, Cδ-Orn), 31.46 (d, Cβ-Val), 30.07 (t, Cβ-Orn), 28.47 and 28.19 (q, CH3-tBu, CH3-tBu), 26.39 (t, Cγ-Orn), 19.11 (q, Cγ-Val), 17.77 (q, Cγ’-Val); FAB+MS m/z: 522 (9) [M + H]+, 466 (3) [M + H - C4H8]+, 422 (4) [M + H - C5H8O2]+, 410 (9), 366 (39) [M + H - C9H17O2]+, 258 (8), 213 (15), 91 (100) [C7H7]+, 72 (28) [C4H8O] +, 57 (38) [C4H9]+; HRFAB+MS: observed 522.3176 [M + H]+, (calcd. for C27H44N3O7, 522.3179).
Boc-Orn(Cbz)-Phe-OtBu (2c): White solid; Mp 104-105 °C {lit [30] 102 °C}; [α] + 25.8 (c 1.01, CHCl3); IR: 3,364, 2,977, 2,880, 1,732, 1,690, 1,668, 1,524, 1,452, 1,368, 1,280, 1,240, 1,164, 1,027 cm-1; 1H-NMR (CDCl3): δ 7.35-7.15 (10H, m, Ar), 6.82 (1H, d, J = 6.8 Hz, NH-Phe), 5.18 (1H, d, J = 6.8 Hz, NH-Orn), 5.06 (2H, d, J = 12.8 Hz, CH2-Cbz, NHδ-Orn), 5.02 (1H, d, J = 12.8 Hz, CH2-Cbz), 4.70 (1H, dd, J = 14.0, 6.0 Hz, Hα-Phe), 4.24 (1H, bs, Hα-Orn), 3.33 (1H, bs, Hδ-Orn), 3.13 (1H, m, Hδ’-Orn), 3.08 (1H, dd, J = 13.6, 14.0 Hz, Hβ-Phe), 3.04 (1H, dd, J = 13.6, 6.0 Hz, Hβ’-Phe), 2.14 (1H, m, Hγ-Orn), 1.81 (1H, m, Hγ’-Orn), 1.53 (2H, m, Hβ, Hβ’-Orn), 1.43 (9H, s, CH3tBu), 1.37 (9H, s, CH3Boc); 13C-NMR (CDCl3): δ 171.80 (s, CO-Orn), 170.48 (s, CO-Phe), 156.94 (s, CO-Cbz), 155.66 (s, CO-Boc), 136.61 (s, Ar-Cbz), 136.23 (s, Ar), 129.58 (d, Ar), 128.58 (d, Ar), 128.49 (d, Ar), 128.16 (d, Ar), 127.03 (d, Ar), 82.41 (s, C-Boc), 80.07 (s, C-tBu), 66.88 (t, CH2-Cbz), 53.81 (d, Cα-Phe), 53.35 (d, Cα-Orn), 39.97 (t, Cδ-Orn), 38.16 (t, Cβ-Phe), 30.25 (t, Cγ-Orn), 28.44 and 28.04 (q, CH3-tBu, CH3-tBu), 26.27 (t, Cβ-Orn); FAB+MS m/z: 570 (56) [M + H]+, 556 (9) [M + H - CH2]+, 514 (8) [M + H - C4H8]+, 470 (56) [M + H - C5H8O2]+, 414 (90) [M + H - C5H8O2 - C4H8]+, 306 (12) [M + H - C14H19NO3 - CH3]+, 261 (17), 204 (14), 154 (27), 120 (27), 91 (100) [C7H7]+, 57 (44) [C4H9]+; HRFAB+MS: observed 570.3152 [M + H]+, (calcd. for C31H44N3O7, 570.3179).
Boc-Orn(Cbz)-Phe-OMe (2d): White solid; Mp 118-120 °C {lit [31] 106-118 °C}; [α] - 7.5 (c 1.01, MeOH) {lit [31] - 7.6 (c 1.1, MeOH)}; IR: 3,339, 2,974, 2,939, 2,876, 1,743, 1,677, 1,531, 1,449, 1,369, 1,278, 1,249, 1,172, 1,032 cm-1; 1H-NMR (CDCl3) δ 7.36-7.10 (10H, m, Ar), 7.00 (1H, d, J = 7.6 Hz, NH-Phe), 5.24 (1H, d, J = 8.0 Hz, NH-Orn), 5.12 (1H, t, J = 6.0 Hz, NHδ-Orn), 5.03 (1H, d, J = 12.8 Hz, CH2-Cbz), 4.99 (1H, d, J = 12.8 Hz, CH2-Cbz), 4.83 (1H, dd, J = 13.2, 6.4 Hz, Hα-Phe), 4.25 (1H, bs, Hα-Orn), 3.67 (3H, s, OCH3), 3.33 (1H, m, Hδ-Orn), 3.12 (1H, m, Hδ’-Orn), 3.11 (1H, dd, J = 13.6, 5.6 Hz, Hβ-Phe), 3.05 (1H, dd, J = 13.6, 6.8 Hz, Hβ’-Phe), 1.78 (1H, m, Hγ-Orn), 1.52 (3H, m, Hβ, Hβ’, Hγ-Orn), 1.42 (9H, s, CH3Boc); 13C-NMR (CDCl3) δ 172.10 (s, CO-Phe), 171.91 (s, CO-Orn), 156.98 (s, CO-Cbz), 155.68 (s, CO-Boc), 136.58 (s, Ar-Cbz), 135.95 (s, Ar), 129.31 (d, Ar), 128.64 (d, Ar), 128.55 (d, Ar), 128.14 (d, Ar), 127.14 (d, Ar), 80.04 (s, C-Boc), 66.82 (t, CH2-Cbz), 53.48 (d, Cα-Phe), 53.28 (d, Cα-Orn), 52.47 (q, OCH3), 39.95 (t, Cδ-Orn), 38.05 (t, Cβ-Phe), 30.26 (t, Cγ-Orn), 28.52 (q, CH3Boc), 26.24 (t, Cβ-Orn); FAB+MS m/z: 528 (11) [M + H]+, 472 (7) [M + H - C4H9]+, 428 (62) [M + H - C5H8O2]+, 320 (9) [C17H24N2O4]+, 275(10), 180 (19) [C10H14NO2]+, 120 (31) [C8H10N]+, 91 (100) [C7H7]+, 57 (44) [C4H9]+; HRFAB+MS: observed 528.2686 [M + H]+, (calcd. for C28H38N3O7, 528.2710).
Boc-Gly-Phe-OtBu (2e): Colorless syrup; [α] + 47.2 (c 1.1, CHCl3); IR: 3,414, 3,336, 2,979, 2,934, 1,727, 1,671, 1,521, 1,452, 1,369, 1,220, 1,160, 1,044, 942, 850, 743, 701 cm-1; 1H-NMR (CDCl3): δ 7.30-7.13 (5H, m, Ar), 6.72 (1H, d, J = 8.0 Hz, NH-Phe), 5.32 (1H, dd, J = 4.8, 4.8 Hz, NH-Gly), 4.75 (1H, dd, J = 14.0, 6.0 Hz, Hα-Phe), 3.83 (1H, dd, J = 16.4, 5.2 Hz, Hα-Gly), 3.74 (1H, dd, J = 16.4, 5.2 Hz, Hα’-Gly), 3.08 (2H, d, J = 6.0 Hz, Hβ-Phe), 1.44 (9H, s, CH3-tBu), 1.39 (9H, s, CH3-Boc); 13C-NMR (CDCl3) δ 170.39 (s, CO-Phe), 169.07 (s, CO-Gly), 155.97 (s, CO-tBu), 136.63 (s, Ar), 129.54 (d, Ar), 128.43 (d, Ar), 127.00 (d, Ar), 82.54 (s, C-Boc), 80.21 (s, C-tBu), 53.66 (d, Cα-Phe), 44.32 (t, Cα-Gly), 38.22 (t, Cβ-Phe), 28.47 and 28.09 (q, CH3-tBu, CH3-tBu); FAB+MS m/z: 379 (41) [M + H]+, 323 (16) [M + H - C4H8]+, 267 (100) [M + H - C4H8 - C4H8]+, 223 (25) [M + H - C7H10NO3]+, 166 (13), 154 (57), 120 (28), 57 (38) [C4H9]+; HRFAB+MS: observed 379.2267 [M + H]+, (calcd. for C20H31N2O5, 379.2233).
Boc-Phe-Phe-OtBu (2f): Colorless crystals; Mp 125-127 °C; [α] + 34.1 (c 1.0, CHCl3); IR, 1H-NMR and 13C-NMR (CDCl3) are in agreement with previously reported data [32]; FAB+MS m/z: 469 (27) [M + H]+, 413 (10) [M + H - C4H8]+, 357 (58) [M + H - C4H8 - C4H8]+, 313 (78) [M + H - C4H8 - C5H8O2]+, 166 (15), 120 (100), 57 (45) [C4H9]+; HRFAB+MS: observed 469.2724 [M + H]+, (calcd. for C27H37N2O5, 469.2702).
Boc-Val-Phe-OtBu (2g): Colorless crystals; Mp 112-115 °C; [α] + 29.0 (c 1.01, CHCl3); IR: 3,337, 3,282, 2,972, 2,936, 2,874, 1,733, 1,689, 1,659, 1,525, 1,457, 1,371, 1,248, 1,162, 1,020, 848, 752, 696 cm-1; 1H-NMR (CDCl3): δ 7.30-7.15 (5H, m, Ar), 6.50 (1H, d, J = 7.2 Hz, NH-Phe), 5.16 (1H, d, J = 8.8 Hz, NH-Val), 4.74 (1H, dd, J = 14.0, 6.4 Hz, Hα-Phe), 3.94 (1H, dd, J = 8.4, 6.8Hz, Hα-Val), 3.08 (2H, dd, J = 6.4, 4.4 Hz, Hβ-Phe), 2.09 (1H, m, Hβ-Val), 1.45 (9H, s, CH3-tBu), 1.38 (9H, s, CH3-Boc), 0.93 (3H, d, J = 6.8 Hz, Hγ-Val), 0.88 (3H, d, J = 6.4 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 171.16 (s, CO-Val), 170.46 (s, CO-Phe), 155.79 (s, CO-Boc), 136.10 (s, Ar), 129.57 (d, Ar), 128.45 (d, Ar), 127.02 (d, C4), 82.42 (s, C-Boc), 79.89 (s, C-tBu), 60.03 (d, Cα-Val), 53.78 (d, Cα-Phe), 38.34 (t, Cβ-Phe), 31.16 (d, Cβ-Val), 28.52 and 28.09 (q, CH3-tBu, CH3-Boc), 19.44 (q, Cγ-Val), 17.97 (q, Cγ’-Val); FAB+MS m/z: 421 (59) [M + H]+, 365 (17) [M + H - C4H8]+, 309 (100) [M + H - C4H8 - C4H8]+, 265 (95) [M + H - C4H8 - C5H8O2]+, 166 (30), 120 (60), 72 (53), 57 (48) [C4H9]+; HRFAB+MS: observed 421.2666 [M + H]+, (calcd. for C23H37N2O5, 421.2702).
Boc-Gly-Val-OtBu (2h): Colorless oil; [α] + 23.8 (c 1.02, CHCl3); IR: 3,333, 2,975, 2,935, 2,878, 1,726, 1,671, 1,524, 1,458, 1,391, 1,369, 1,281, 1,251, 1,166, 1,052, 943, 848, 786 cm-1; 1H-NMR (CDCl3) δ 6.80 (1H, bs, NH-Val), 5.48 (1H, bd, J = 5.6 Hz, NH-Gly), 4.46 (1H, dd, J = 9.2, 4.4 Hz, Hα-Val), 3.87 (1H, dd, J = 16.4, 5.6 Hz, Hα-Gly), 3.80 (1H, dd, J = 16.4, 5.6 Hz, Hα’-Gly), 2.17 (1H, hd, J = 7.2, 4.4 Hβ-Val), 1.47 (9H, s, CH3Boc), 1.46 (9H, s, CH3tBu), 0.94 (3H, d, J = 7.2 Hz, Hγ-Val), 0.89 (3H, d, J = 7.2 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 170.95 (s, CO-Val), 169.52 (s, CO-Gly), 156.12 (s, CO-Boc), 82.16 (s, C-Boc), 80.22 (s, C-tBu), 57.42 (d, Cα-Val), 44.50 (t, Cα-Gly), 31.58 (d, Cβ-Val), 28.46 and 28.20 (q, CH3-Boc, CH3-tBu), 19.08 (q, Cγ-Val), 17.68 (q, Cγ’-Val); FAB+MS m/z: 331 (28) [M + H]+, 275 (18) [M + H - C4H8]+, 219 (100) [M + H - C4H8 - C4H8]+, 175 (20) [M + H - C4H8 - C5H8O2]+, 72 (17) [C4H8O]+, 57 (20) [C4H9]+; HRFAB+MS: observed 331.2251 [M + H]+, (calcd. for C16H31N2O5, 331.2233).
Boc-Phe-Val-OtBu (2i): White solid; Mp 119-121 °C; [α]DHg (365 nm) + 9.0 (c 0.5, CHCl3); IR: 3,327, 2,975, 2,933, 1,735, 1,687, 1,651, 1,538, 1,367, 1,252, 1,165, 1,025, 855 cm-1; 1H-NMR (CDCl3) δ 7.32 - 7.10 (5H, Ar), 6.56 (1H, d, J = 8.4 Hz, NH-Phe), 5.20 (1H, d, J = 8.0 Hz, NH-Val), 4.41 (1H, m, Hα-Phe), 4.36 (1H, dd, J = 8.0, 4.8 Hz, Hα-Val), 3.10 (2H, dd, J = 13.6, 6.4 Hz, Hβ-Phe), 3.04 (1H, dd, J = 13.6, 6.8 Hz, Hβ’-Phe), 2.17 (1H, hd, J = 6.8, 4.8 Hβ-Val), 1.45 (9H, s, CH3Boc), 1.41 (9H, s, CH3tBu), 0.88 (3H, d, J = 6.8 Hz, Hγ-Val), 0.89 (3H, d, J = 6.8 Hz, Hγ’-Val); 13C-NMR (CDCl3) δ 171.07 (s, CO-Phe), 170.45 (s, CO-Val), 155.39 (s, CO-Boc), 136.69 (s, C1), 129.36 (d, C2 and C6), 128.58 (d, C3 and C5), 126.85 (d, C4), 81.98 (s, C-Boc), 80.10 (s, C-tBu), 57.65 (d, Cα-Val), 55.95 (d, Cα-Phe), 38.23 (t, Cβ-Phe), 31.62 (d, Cβ-Val), 28.43 and 28.26 (q, CH3-Boc, CH3-tBu), 18.92 (q, Cγ-Val), 17.91 (q, Cγ’-Val); FAB+MS m/z: 421 (46) [M + H]+, 365 (15) [M + H - C4H8]+, 309 (100) [M + H - C4H8 - C4H8]+, 265 (96) [M + H - C4H8 - C5H8O2]+, 120 (40), 72 (32) [C4H8O]+, 57 (44) [C4H9]+; HRFAB+MS: observed 421.2688 [M + H]+, (calcd. for C23H37N2O5, 421.2702).
Boc-Val-Val-OtBu (2j): White solid; Mp 132-134 °C; [α] - 6.8 (c 1.1, CHCl3); IR: 3,309, 2,972, 2,934, 2,888, 1,744, 1,685, 1,651, 1,536, 1,464, 1,372, 1,301, 1,254, 1,219, 1,157, 1,017, 855 cm-1; 1H-NMR (CDCl3) δ 6.35 (1H, d, J = 7.6 Hz, NH-Val), 5.09 (1H, d, J = 8.8 Hz, NH-Val), 4.36 (1H, dd, J = 8.4, 4.4 Hz, Hα-Val), 3.86 (1H, dd, J = 7.6, 7.6 Hz, Hα-Val), 2.08 (2H, m, Hβ-Val, Hβ-Val), 1.40 (9H, s, CH3Boc), 1.38 (9H, s, CH3tBu), 0.90, 0.87, 0.86, 0.84 (3H each, d, J = 6.8 Hz, Hγ-Val, Hγ’-Val, Hγ-Val, Hγ’-Val); 13C-NMR (CDCl3) δ 171.58 (s, CO-Val), 170.81 (s, CO-Val), 155.93 (s, CO-Boc), 81.16 (s, C-Boc), 80.00 (s, C-tBu), 60.38 (d, Cα-Val), 57.70 (d, Cα-Val), 31.64 (d, Cβ-Val), 31.06 (d, Cβ-Val), 28.56 and 28.29 (q, CH3-Boc, CH3-tBu), 19.57, 19.16, 18.21, 17.96 (q, Cγ-Val, Cγ’-Val, Cγ-Val, Cγ’-Val); FAB+MS m/z: 373 (46) [M + H]+, 317 (28) [M + H - C4H8]+, 261 (89) [M + H - C4H8 - C4H8]+, 217 (94) [M + H - C4H8 - C5H8O2]+, 116 (26) [C5H10NO2]+, 72 (100) [C4H8O]+, 57 (47) [C4H9]+; HRFAB+MS: observed 373.2711 [M + H]+, (calcd. for C19H37N2O5, 373.2702).
Boc-Phe-Sar-OtBu (2k): Colorless syrup (73:27 rotamer mixture); [α] - 22.5 (c 1.58, CHCl3); IR: 3,427, 3,322, 2,978, 2,933, 1,741, 1,710, 1,652, 1,491, 1,367, 1,236, 1,164, 1,049, 1,020, 952, 850, 759 and 701 cm-1; 1H-NMR (CDCl3) δ 7.30-7.00 (5H, m, Ar), 5.31 (1H, d, J = 8.8 Hz, NH-Phe), 4.80 (1H, dd, J = 15.4, 6.6 Hz, Hα-Phe), 3.92 (1H, d, J = 17.2 Hz, Hα-Sar), 3.84 (1H, d, J = 17.2 Hz, Hα’-Sar), 2.83 (3H, s, NCH3-Sar), 2.97 (1H, dd, J = 13.6, 7.2 Hz, Hβ-Phe), 2.90-2.86 (1H, m, Hβ’-Phe), 1.38 (9H, s, CH3-Boc), 1.31 (9H, s, CH3-tBu); 13C-NMR (CDCl3) δ 172.11 (s, CO-Phe), 167.89 (s, CO-Sar), 155.09 (s, CO-Boc), 136.42 (s, C1), 129.65 (d, C2,C6), 128.38 (d, C3,C5), 126.85 (d, C4), 82.03 (s, C-tBu), 79.67 (s, C-Boc), 51.52 (d, Cα-Phe), 50.45 (t, Cα-Sar), 39.57 (t, Cβ-Phe), 36.34 (q, NCH3-Sar), 28.49 and 28.24 (q, CH3-Boc, CH3-tBu); FAB+MS m/z: 393 (27) [M + H]+, 337 (15) [M + H - C4H8]+, 281 (76) [M + H - 2 C4H8]+, 263 (15) [M + H - C4H8 - C4H8O]+, 237 (100) [M + H - C4H8 - C5H8O2]+, 164 (18), 120 (81), 90 (43) [C7H6]+, 57 (78) [C4H9]+; HRFAB+MS: observed 393.2409 [M + H]+, (calcd. for C21H33N2O5, 393.2389).
Boc-Phe-Sar-OMe (2l): Colorless syrup (77:23 rotamer mixture); [α] + 19.1 (c 0.54, CHCl3); IR: 3,427, 3,322, 2,977, 2,943, 1,751, 1,708, 1,652, 1,490, 1,407, 1,365, 1,250, 1,212, 1,171, 1,047, 1,021, 751 and 702 cm-1; 1H-NMR (CDCl3) δ 7.32-7.18 (5H, m, Ar), 5.34 (1H, d, J = 8.8 Hz, NH-Phe), 4.88 (1H, dd, J = 14.8, 6.8 Hz, Hα-Phe), 4.14 (1H, d, J = 17.2 Hz, Hα-Sar), 3.99 (1H, d, J = 17.2 Hz, Hα'-Sar), 3.73 (3H, s, OCH3), 3.04 (1H, dd, J = 13.6, 7.2 Hz, Hβ-Phe), 2.95 (1H, dd, J = 13.6, 6.4 Hz, Hβ’-Phe), 2.87 (3H, s, NCH3-Sar), 1.40 (9H, s, CH3-Boc); 13C-NMR (CDCl3) δ 172.35 (s, CO-Phe), 169.28 (s, CO-Sar), 155.07 (s, CO-Boc), 136.33 (s, Ar), 129.65 (d, Ar), 128.45 (d, Ar), 126.93 (d, C4), 79.81 (s, C-Boc), 52.38 (q, OCH3), 51.59 (d, Cα-Phe), 49.66 (t, Cα-Sar), 39.80 (t, Cβ-Phe), 36.43 (q, NCH3-Sar), 28.55 (q, CH3-Boc); FAB+MS m/z: 351 (33) [M + H]+, 295 (55) [M + H - C4H8]+, 251 (100) [M + H - C5H9O2]+, 164 (16), 120 (67), 104 (70) [C4H10NO2]+, 57 (44) [C4H9]+, 44 (18); HRFAB+MS: observed 351.1907 [M + H]+, (calcd. for C18H27N2O5, 351.1920).
3.4. General procedure for the syntheses of 2,5-diketopiperazines 3a-3k
Each Nα-Boc-dipeptidyl ester (0.25 mmol) was dissolved or suspended in water (1 mL) and heated during 10 minutes at 250 °C and 150 psi, using a monomode CEM Discover microwave apparatus at 250 W. The resulting suspension was filtered through a Hirsch funnel and washed with water (5 mL), the solid was dried under high vacuum and analyzed without further purification by NMR. Compounds 3h and 3k were water soluble, and in these cases, resulting solutions were lyophilized and the solids purified as indicated in Table 2 and analyzed by NMR.
Cyclo[Val-Orn(Cbz)] (3a): White solid; Mp 202-204 °C {lit [33] 206-208 °C}; [α] - 26.0 (c 0.27, DMSO) {lit [33] - 47.4 (c 1 %)}; IR: 3,434, 3,333, 3,201, 3,094, 3,052, 2,965, 2,878, 1,678, 1,531, 1,444, 1,258, 1,142, 1,025, 774, 696, 629 cm-1; 1H-NMR (DMSO): δ 8.14 (1H, s, NH-Orn), 8.04 (1H, s, NH-Val), 7.39-7.26 (6H, m, Ar, NHδ-Orn), 5.00 (2H, s, CH2-Cbz), 3.81 (1H, t, J = 4.8 Hz, Hα-Orn), 3.66 (1H, bs, Hα-Val), 2.97 (1H, dd, J = 12.8, 6.4 Hz, Hδ-Orn), 2.14 (1H, m, Hβ-Val), 1.69 (1H, m, Hβ-Orn), 1.62 (1H, m, Hβ’-Orn), 1.46 (2H, m, Hγ-Orn), 0.93 (3H, d, J = 7.2 Hz, Hγ-Val), 0.82 (3H, m, d, J = 7.2 Hz, Hγ’-Val); 13C-NMR (DMSO): δ 167.83 (s, CO-Orn), 166.88 (s, CO-Val), 156.06 (s, CO-Cbz), 137.21 (s, Ar), 128.33 (d, Ar), 127.64 (d, Ar), 65.18 (t, CH2-Cbz), 59.43 (d, Cα-Val), 53.75 (d, Cα-Orn), 40.18 (t, Cδ-Orn), 31.33 (d, Cβ-Val), 31.16 (t, Cβ-Orn), 25.35 (t, Cγ-Orn), 18.74 (q, Cγ-Val), 17.29 (q, Cγ’-Val); FAB+MS m/z: 348 (87) [M + H]+, 307 (100) [M + H - C3H6]+, 289 (61), 240 (22) [M + H - C7H8O]+, 219 (25) [C12H15N2O2]+, 214 (12), 195 (12), 165 (12); HRFAB+MS: observed 348.1887 [M + H]+, (calcd. for C18H26N3O4, 348.1923).
Cyclo[Val-D-Orn(Cbz)] (3b): White solid; Mp 212-214 °C;[α] + 11.7 (c 0.5, MeOH);IR: 3,347, 3,192, 3,054, 2,962, 1,675, 1,540, 1,460, 1,265, 1,143, 1,031, 852, 696 cm-1; 1H-NMR (DMSO): δ 8.11 (1H, s, NH-Orn), 7.32 (6H, m, Ar, NH-Val), 4.99 (2H, s, CH2-Cbz), 4.03 (1H, bs, Hα-Val), 3.88 (1H, bt, Hα-Orn), 2.96 (1H, d, J = 5.6 Hz, Hδ-Orn), 2.09 (1H, m, Hβ-Val), 1.65 (1H, m, Hβ-Orn), 1.62 (1H, m, Hβ’-Orn), 1.41 (2H, m, Hγ-Orn), 0.92 (3H, d, J = 6.8 Hz, Hγ-Val), 0.83 (3H, m, d, J = 7.0 Hz, Hγ’-Val); 13C-NMR (DMSO): δ 168.13 (s, CO-Orn), 167.67 (s, CO-Val), 156.17 (s, CO-Cbz), 137.25 (s, Ar), 128.44 (d, Ar), 127.77 (d, Ar), 65.30 (t, CH2-Cbz), 59.79 (d, Cα-Val), 53.27 (d, Cα-Orn), 40.18 (t, Cδ-Orn), 32.19 (d, Cβ-Val), 29.64 (t, Cβ-Orn), 24.41 (t, Cγ-Orn), 18.54 (q, Cγ-Val), 17.08 (q, Cγ’-Val); FAB+MS m/z: 348 (3) [M + H]+, 219 (5), 154 (12), 130 (30), 107 (8), 91 (100), 85 (28), 72 (25); HRFAB+MS: observed 348.1949 [M + H]+, (calcd. for C18H26N3O4, 348.1923).
Cyclo[Phe-Orn(Cbz)] (3c): White solid; Mp 200-202 °C {lit [33] 210-212 °C}; [α] - 24.8 (c 0.24, DMSO) {lit [33] - 12.9 (c 1 %)}; IR: 3,322, 3,189, 3,039, 2,965, 2,894, 1,669, 1,532, 1,458, 1,338, 1,246, 1,134, 1,101, 1,016, 852, 753, 669 cm-1; 1H-NMR (DMSO): δ 8.16 (1H, s, NH-Phe), 8.05 (1H, s, NH-Orn), 7.40 - 7.05 (10H, m, Ar), 5.00 (2H, s, CH2-Cbz), 4.17 (1H, s, Hα-Phe), 3.57 (1H, bs, Hα- Orn), 3.13 (1H, dd, J = 13.6, 4.0 Hz, Hβ-Phe), 2.83 (1H, dd, J = 13.6, 5.2 Hz, Hβ’-Phe), 2.68 (1H, dd, J = 12.8, 6.4 Hz, Hδ-Orn), 1.04 (1H, m, Hβ-Orn), 0.85 (2H, m, Hγ-Orn), 0.65 (2H, m, Hβ’-Orn); 13C-NMR (DMSO): δ 166.70 (s, CO-Orn), 166.00 (s, CO-Phe), 155.79 (s, CO-Cbz), 137.16 (s, Ar-Cbz), 135.92 (s, Ar), 130.16 (d, Ar), 128.27 (d, Ar), 127.92 (d, Ar), 127.69 (d, Ar), 126.57 (d, Ar), 65.10 (t, CH2-Cbz), 55.30 (d, Cα-Phe), 53.61 (d, Cα-Orn), 39.95 (t, Cδ-Orn), 38.16 (t, Cβ-Phe), 30.66 (t, Cβ-Orn), 24.33 (t, Cγ-Orn); FAB+MS m/z: 396 (100) [M + H]+, 352 (32) [M + H - H2NCO]+, 335 (9), 307 (53) [M + H - C7H6]+, 289 (32) [M + H - C7H7O]+, 243 (16) [M + H - C14H16NO2]+, 219 (18) [M + H - C10H12NO2]+, 165 (14); HRFAB+MS: observed 396.1949 [M + H]+, (calcd. for C22H26N3O4, 396.1923).
Cyclo(Phe-Gly) (3e): White solid; Mp 266-268 °C {lit [34] 271-273 °C }; [α] + 26.6 (c 0.95, DMSO) {lit [34] + 7.3 (c 0.95, DMSO)}; IR: 3,426, 3,190, 3,057, 2,977, 2,921, 2,878, 1,676, 1,462, 1,332, 1,086, 1,004, 847, 794, 758, 702 cm-1; 1H- and 13CNMR (DMSO) are in agreement with previously reported data [35]; FAB+MS m/z: 205 (13) [M + H]+, 169 (15), 154 (24), 130 (43) [M + H - C4H4]+, 85 (100) [C3H3NO2]+; HRFAB+MS: observed 205.1058 [M + H]+, (calcd. for C11H13N2O2, 205.0977).
Cyclo(Phe-Phe) (3f): White solid; [α] - 42.8 (c 0.2, AcOH); 1H- and 13C-NMR (DMSO), and HRFAB+MS are in agreement with previously reported data [36]; IR: 3,440, 3,318, 3,198, 3,056, 2,970, 2,929, 1,669, 1,458, 1,338, 1,197, 1,088, 1,013, 759 and 700 cm-1.
Cyclo(Val-Phe) (3g): White solid; Mp 264-266 °C {lit [36] 263-265 °C}; [α] - 66.0 (c 0.28, DMSO) {lit [37] - 64 (c 0.2 AcOH); [34] - 43.3 (c 0.27 DMSO)}; IR: 3,440, 3,316, 3,192, 3,056, 2,967, 2,885, 1,668, 1,454, 1,341, 1,090, 859, 758, 699, cm-1; 1H- and 13C-NMR (DMSO) are in agreement with previously reported data [34,37]; FAB+MS m/z: 247 (100) [M + H]+, 219 (13) [M + H - CO]+, 217 (10), 203 (7), 165 (5); HRFAB+MS: observed 247.1446 [M + H]+, (calcd. for C14H19N2O2, 247.1447).
Cyclo(Val-Gly) (3h): White solid; Mp 210-212 °C {lit [38] 256 °C}; [α] + 32.9 (c 0.46, H2O) {lit [38] + 23.7 (c 1.0, H2O)}; IR: 3,431, 3,199, 3,055, 2,925, 2,859, 1,670, 1,461, 1,381, 1,346, 1,109, 1,047, 808, 621 cm-1; 1H- and 13C-NMR (DMSO) are in agreement with previously reported data [38,39]; FAB+MS m/z: 157 (94) [M + H]+, 154 (100) [M - H2], 136 (78), 120 (12), 107 (25), 85 (58) [C5H9O]+, 77 (25), 55 (32), 43 (27) [C3H7]+, 41 (25); HRFAB+MS: observed 157.0957 [M + H]+, (calcd. for C7H13N2O2, 157.0957).
Cyclo(Val-Val) (3j): Colorless needless; Mp 268 °C with sublimation {lit [40] 268 °C}; [α] - 54.8 (c 0.5, AcOH) {lit [15] - 62 (c 0.5, AcOH); IR: 3,434, 3,325, 3,193, 3,100, 3,057, 2,966, 2,880, 1,664, 1,449, 1,346, 1,293, 847 cm-1; 1H-NMR (DMSO): δ 7.96 (1H, s, NH-Val), 3.69 (1H, s, Hα-Val), 2.18 (1H, m, Hβ-Val), 0.95 (3H, d, J = 7.6 Hz, Hγ-Val), 0.83 (3H, d, J = 7.6 Hz, Hγ’-Val); 13C-NMR (DMSO): δ 167.26 (s, CO), 59.09 (d, Cα), 31.06 (d, Cβ), 18.73 (q, Cγ), 17.33 (q, Cγ’); FAB+MS m/z: 199 (100) [M + H]+, 197 (19), 169 (8); HRFAB+MS: observed 199.1463 [M + H]+, (calcd. for C10H19N2O2, 199.1447).
Cyclo(Sar-Phe) (3k): Colorless needles; Mp 180-183 °C {lit [41] 184-185 °C}; [α] + 56.0 (c 1.01, MeOH) {lit [41] + 47.5 (c 2.3 MeOH)}; IR: 3,440, 3,253, 2,963, 2,929, 2,895, 1,656, 1,470, 1,441, 1,322, 1,102, 1,033, 870, 754, 699, cm-1; 1H-NMR (CDCl3): δ 7.60 (1H, bs, NH-Phe), 7.35-7.16 (5H, m, Ar), 4.31 (1H, bs, Hα-Phe), 3.48 (1H, d, J = 17.6 Hz, Hα-Sar), 3.24 (1H, dd, J = 13.6, 5.2 Hz, Hβ-Phe), 3.07 (1H, dd, J = 13.6, 4.4 Hz, Hβ’-Phe), 2.81 (3H, s, NCH3-Sar), 2.79 (1H, d, J = 17.6 Hz, Hα’-Sar); 13C-NMR (CDCl3): δ 166.32 (s, CO-Sar), 165.52 (s, CO-Phe), 134.99 (s, Ar), 130.07 (d, Ar), 128.63 (d, Ar), 127.58 (d, C4), 56.53 (d, Cα-Phe), 50.92 (t, Cα-Sar), 40.94 (t, Cβ-Phe), 33.66 (s, NCH3-Sar); FAB+MS m/z: 219 (100) [M + H]+, 154 (50) [M + H - CO]+, 136 (56), 91 (35) [C7H7]+, 73 (58); HRFAB+MS: observed 219.1134 [M + H]+, (calcd. for C12H15N2O2, 219.1134).
4. Conclusions
Optically pure cis-DKPs could be synthesized in one-pot from the corresponding Nα-Boc-dipeptidyl-tert-butyl esters in water under microwave irradiation for ten minutes. Employing these conditions, the tert-butoxy group is efficiently removed, leading to cyclization in excellent yields. This is the first protocol for ring closures using Nα-Boc-dipeptidyl-tert-butyl esters. The trans-DKP fragment present in the natural product 1 was synthesized in quantitative yield. The reaction is rapid, secure, environmentally friendly and highly efficient.
Acknowledgements
This work was financially supported by CONACyT (Grants number 79584-Q and 2006-C01-LN-56431). Lemuel Pérez-Picaso thanks CONACyT for a doctoral fellowship (number 181754). We are grateful to Dr. A. Berenice Aguilar-Guadarrama, Dr. Blanca Domínguez Mendoza, Dr. Diana Gabriela Vargas Pineda, Ing. Victoria Labastida, and T. C. María Medina Pastor for technical assistance.
Footnotes
Sample Availability: Samples of the compounds 2a-2l and 3a-3k are available from the authors.
References and Notes
- 1.Wipf P. Synthetic studies of biologically active marine cyclopeptides. Chem. Rev. 1995;95:2115–2134. doi: 10.1021/cr00038a013. [DOI] [Google Scholar]
- 2.Amidon G.L., Lee H.J. Absorption of peptide and peptidomimetic drugs. Annu. Rev. Pharmacol. Toxicol. 1994;34:321–341. doi: 10.1146/annurev.pa.34.040194.001541. [DOI] [PubMed] [Google Scholar]
- 3.Liu S., Gu W., Lo D., Ding X., Ujiki M., Adrian T.E., Soff G.A., Silverman R.B. N-Methylsansalvamide A peptide analogues. Potent new antitumor agents. J. Med. Chem. 2005;48:3630–3638. doi: 10.1021/jm048952t. [DOI] [PubMed] [Google Scholar]
- 4.Prasad C. Bioctive cyclic dipeptides. Peptides. 1995;16:151–164. doi: 10.1016/0196-9781(94)00017-Z. [DOI] [PubMed] [Google Scholar]
- 5.Houston D.R., Synstad B., Eijsink V.G.H., Stark M.J.R., Eggleston I.M., van Aalten D.M.F. Structure-based exploration of cyclic dipeptide chitinase inhibitors. J. Med. Chem. 2004;47:5713–5720. doi: 10.1021/jm049940a. [DOI] [PubMed] [Google Scholar]
- 6.Martins M.B., Carvalho I. Diketopiperazines: biological activity and synthesis. Tetrahedron. 2007;63:9923–9932. doi: 10.1016/j.tet.2007.04.105. [DOI] [Google Scholar]
- 7.Graz C.J.M., Grant G.D., Brauns S.C., Hunt A., Jamie H., Milne P.J. Cyclic dipeptides in the induction of maturation for cancer therapy. J. Pharm. Pharmacol. 2000;52:75–82. doi: 10.1211/0022357001773535. [DOI] [PubMed] [Google Scholar]
- 8.Prakash K.R.C., Tang Y., Kozikowski A.P., Flippen-Anderson J.L., Knoblach S.M., Faden A.I. Synthesis and biological activity of novel neuroprotective diketopiperazines. Bioorg. Med. Chem. 2002;10:3043–3048. doi: 10.1016/S0968-0896(02)00132-3. [DOI] [PubMed] [Google Scholar]
- 9.Lambert J.N., Mitchell J.P., Roberts K.D. The synthesis of cyclic peptides. J. Chem. Soc. Perkin Trans. I. 2001:471–484. [Google Scholar]
- 10.Rajappa S., Natekar M.V. Piperazine-2,5-diones and related lactam ethers. Adv. Het. Chem. 1993;57:187–289. [Google Scholar]
- 11.Fischer P.M.J. Diketopiperazines in peptide and combinatorial chemistry. J. Pept. Sci. 2003;9:9–35. doi: 10.1002/psc.446. [DOI] [PubMed] [Google Scholar]
- 12.Dinsmore C.J., Beshore D.C. Recent advances in the synthesis of diketopiperazines. Tetrahedron. 2002;58:3297–3312. doi: 10.1016/S0040-4020(02)00239-9. [DOI] [Google Scholar]
- 13.Rodionov I.L., Rodionova L.N., Baidakova L.K., Romashko A.M., Balashova T.A., Ivanov V.T. Cyclic dipeptides as building blocks for combinatorial libraries. Part 2: Synthesis of bifunctional diketopiperazines. Tetrahedron. 2002;58:8515–8523. doi: 10.1016/S0040-4020(02)01021-9. [DOI] [Google Scholar]
- 14.Fischer E. Synthese von polypeptiden. Chem. Ber. 1906;39:2893–2931. doi: 10.1002/cber.190603903103. [DOI] [Google Scholar]
- 15.Ueda T., Saito M., Kato T., Izumiya N. Facile Synthesis of Cyclic Dipeptides and Detection of Racemization. Bull. Chem. Soc. Jpn. 1983;56:568–572. doi: 10.1246/bcsj.56.568. [DOI] [Google Scholar]
- 16.Suzuki K., Sasaki Y., Endo N., Mihara Y. Acetic acid-catalized diketopiperazine synthesis. Chem. Pharm. Bull. 1981;29:233–237. [Google Scholar]
- 17.Nitecki D.E., Halpern B., Westley J.W. Simple route to sterically pure dioxopiperazines. J. Org. Chem. 1968;33:864–866. doi: 10.1021/jo01266a091. [DOI] [PubMed] [Google Scholar]
- 18.Eriksson J., Arvidsson P.I., Davidsson O. Solution structure of a dilithiumamide/diethylzinc heterocomplex that catalyzes asymmetric alkylation reactions. Chem. Eur. J. 1999;5:2356–2361. doi: 10.1002/(SICI)1521-3765(19990802)5:8<2356::AID-CHEM2356>3.0.CO;2-K. [DOI] [Google Scholar]
- 19.Lee S., Kanmera T., Aoyagi H., Izumiya N. Cyclic peptides. VI. Asymmetric hydrogenation of dehydroalanine or dehydroaminobutanoic acid residue in cyclodipeptides. Int. J. Pept. Protein Res. 1979;13:207–217. [PubMed] [Google Scholar]
- 20.Kappe C.O., Dallinger D. Controlled microwave heating in modern organic synthesis: highlights from the 2004-2008 literature. Mol. Divers. 2009;13:71–193. doi: 10.1007/s11030-009-9138-8. [DOI] [PubMed] [Google Scholar]
- 21.Kappe C.O., Dallinger D., Murphree S.S. Practical microwave synthesis for organic chemists: Strategies, Instruments, and Protocols. 1st. Wiley-VCH; Darmstadt, Germany: 2009. [Google Scholar]
- 22.Santagada V., Fiorino F., Perissutti E., Severino B., Terracciano S., Cirino G., Caliendo G. A convenient strategy of dimerization by microwave heating and using 2,5-diketopiperazine as scaffold. Tetrahedron Lett. 2003;44:1145–1148. [Google Scholar]
- 23.López-Cobeñas A., Cledera P., Sánchez J.D., Pérez-Contreras R., López-Alvarado P., Ramos M.T., Avendaño C., Menéndez J.C. Solvent-Free, Efficient Synthesis of 2,5-Piperazinediones from Boc-protected dipeptide esters under microwave irradiation. Synlett. 2005;7:1158–1160. [Google Scholar]
- 24.Tullberg M., Grøtli M., Luthman K. Efficient synthesis of 2,5-diketopiperazines using microwave assisted heating. Tetrahedron. 2006;62:7484–7491. [Google Scholar]
- 25.Jam F., Tullberg M., Luthman K., Grøtli M. Microwave assisted synthesis of spiro-2,5-diketopiperazines. Tetrahedron. 2007;63:9881–9889. [Google Scholar]
- 26.Fischer P.M., Solbakken M., Undheim K. Solution synthesis of a dimeric pentapeptide: diketopiperazine cyclisation of Glu-Asp dipeptide esters and Asp-racemisation during segment condensation. Tetrahedron. 1994;50:2277–2288. [Google Scholar]
- 27.Besser D., Greiner G., Reissmann S. Side reaction with N-carboxymethyl amino acids in the synthesis of lactone cyclized peptides. Lett. Pept. Sci. 1998;5:299–303. [Google Scholar]
- 28.Pérez-Picaso L., Rios M.Y., Hernández A.N., Martínez J. 1H and 13C assignments of cyclo [N-(Lys-Phe)-Orn-Val], a semicyclic imide tetrapeptide from Burkholderia cepacia. Magn. Reson. Chem. 2006;44:959–961. doi: 10.1002/mrc.1864. [DOI] [PubMed] [Google Scholar]
- 29.Pandey S.K., Awasthi K.K., Saxena A.K. Microwave assisted stereospecific synthesis of (S)-3-substituted2,3,6,7,12,12a-hexahydropyrazino[1′,2′:1,6]pyrido[3,4-b]indole-1,4-diones. Tetrahedron. 2001;57:4437–4432. [Google Scholar]
- 30.Kobayashi N., Higuchi T., Urano Y., Kikuchi K., Hirobe M., Nagano T. Dipeptides Containing L-arginine analogs: New isozyme-selective inhibitors of nitric oxide synthase. Biol. Pharm. Bull. 1999;22:936–940. doi: 10.1248/bpb.22.936. [DOI] [PubMed] [Google Scholar]
- 31.Shen H.Y., Tian G.L., Ye Y.H., Wang J. Non-coded amino acids as acyl donor substrates for peptide bond formation catalyzed by thermoase in toluene. J. Mol. Catal. B-Enzym. 2005;37:26–29. doi: 10.1016/j.molcatb.2005.09.002. [DOI] [Google Scholar]
- 32.Dineen T.A., Zajac M.A., Myers A.G. Efficient transamidation of primary carboxamides by in situ activation with N,N-dialkylformamide dimethyl acetals. J. Am. Chem. Soc. 2006;128:16406–16409. doi: 10.1021/ja066728i. [DOI] [PubMed] [Google Scholar]
- 33.Izumiya N., Kato T., Fujita Y., Ohno M., Kondo M. Studies of peptide antibiotics, I. Dipeptide anhydrides as models of cyclic peptide antibiotics. Bull. Chem. Soc. Jpn. 1964;37:1809–1816. doi: 10.1246/bcsj.37.1809. [DOI] [Google Scholar]
- 34.López-Cobeñas A., Cledera P., Sánchez J.D., López-Alvarado P., Ramos M.T., Avendaño C., Menéndez J.C. Microwave-assisted synthesis of 2,5-piperazinediones under solvent-free conditions. Synthesis. 2005;19:3412–3422. [Google Scholar]
- 35.Huang H., She Z., Lin Y., Vrijmoed L.L.P., Lin W. Cyclic Peptides from an Endophytic Fungus Obtained from a Mangrove Leaf (Kandelia candel) J. Nat. Prod. 2007;70:1696–1699. doi: 10.1021/np0605891. [DOI] [PubMed] [Google Scholar]
- 36.Joshi K.B., Verma S. Participation of aromatic side chains in diketopiperazine ensembles. Tetrahedron Lett. 2008;49:4231–4234. doi: 10.1016/j.tetlet.2008.04.156. [DOI] [Google Scholar]
- 37.Tullberg M., Luthman K., Grøtli M. Microwave-assisted solid-phase synthesis of 2,5-diketopiperazines: Solvent and resin dependence. J. Comb. Chem. 2006;8:915–922. doi: 10.1021/cc0600876. [DOI] [PubMed] [Google Scholar]
- 38.Bull S.D., Davies S.G., Moss W.O. Practical synthesis of Schöllkopf's bis-lactim ether chiral auxiliary: (3S)-3,6-dihydro-2,5-dimethoxy-3-isopropyl-pyrazine. Tetrahedron: Asym. 1998;9:321–327. doi: 10.1016/S0957-4166(97)00623-X. [DOI] [Google Scholar]
- 39.Cledera P., Avendaño C., Menéndez J.C. Comparative study of synthetic approaches to 1-arylmethylenepyrazino[2,1-b]quinazoline-3,6-diones. Tetrahedron. 1998;54:12349–12360. doi: 10.1016/S0040-4020(98)00744-3. [DOI] [Google Scholar]
- 40.Tanihara M., Hiza T., Imanishi Y., Higashimura T. Solution conformation of cyclic dipeptides having aliphatic side chains. Bull. Chem. Soc. Jpn. 1983;56:1155–1160. [Google Scholar]
- 41.Lucente G., Pinnen F., Zanotti G. Cyclization of activated N-benzyloxycarbonyl-tripeptides. Tetrahedron Lett. 1978;11:1009–1012. [Google Scholar]