

Abstract
A versatile method for the synthesis of chiral 1,4-disubstituted-1,2,3-triazole derivatives starting from easily accessible naturally occurring D-or L-amino acids as chiral synthons is described. The amino acids were converted into azido alcohols, followed by copper catalyzed [3+2] cycloaddition reactions between the azido alcohols and methyl propiolate and subsequent ester aminolysis with primary and secondary amines furnished the target compounds, which were obtained in excellent yields with no racemization. Docking of selected target compounds shows that the chiral 1,4-disubstituted-1,2,3-triazoles derivatives has the potential of mimicking the binding mode of known purine analogues.
Keywords: [3+2] cycloaddition; azido alcohols; 1,4-disubstituted-1,2,3-triazoles
1. Introduction
1,2,3-Triazoles are an important class of heterocycles due to their wide range of applications as synthetic intermediates and pharmaceuticals [1,2]. Several therapeutically interesting 1,2,3-triazoles have been reported, including anti-HIV agents [3,4,5,6], antimicrobial compounds [7], β3-selective adrenergic receptor agonists [8], kinase inhibitors [9,10] and other enzyme inhibitors [11,12]. The 1,2,3-triazole moiety is also present in a number of drugs, for example, the β-lactam antibiotic tazobactam [13] and the cephalosporin cefatrizine [14].
In our search for synthetic non-natural frameworks that could mimic nucleosides, we wanted to investigate the possibility of using 1,2,3-triazole moiety as a mimic of the imidazole part of the adenine system (Figure 1). Imidazole and triazole based nucleoside derivatives have been reported as potent enzyme inhibitors. Ribavarin [15], a 1,2,4-triazole based purine analogue, has high potency as an antiviral agent and is currently used in the clinic to inhibit RNA virus infections. Furthermore, imidazole based derivatives have been reported as potent adenosine deaminase (ADA) inhibitors (Figure 1) [16,17,18,19]. Therefore, we wanted to investigate the possibility of the 1,2,3-triazole scaffold to be docked into proteins that are inhibited of these nucleoside analogues and to develop efficient synthesis protocols for the preparation of 1,4-disubstituted-1,2,3-triazoles.
Figure 1.

(a) Docked binding mode of (R)-3 (yellow) compared to X-ray structure (1R6A) of ribavirin-5’-triphosphate (blue) 1 in the active site of the NS5MTaseDV in the dengue virus; (b) The docking shows that the NH2-groups of compound (R)-3 and ribavirin-5’-monophosphate are almost superimposed and makes the same important interaction with the carbonyl functions of Leu17 and Leu20 in the protein; (c) Docked binding mode of (R)-3 (yellow) compared to x-ray structure (1v7a) of inhibitor 2 in the active site of the ADA enzyme; (d) The imidazole part of compound (R)-3 is almost superimposed over compound 2 and the phenyl ring points in (R)-3 in the same direction as the naphtyl ring in 2.
2. Results and Discussion
In order to explore the potential of the 1,4-disubstituted-1,2,3-triazoles as a possible scaffold for nucleoside inhibitors, compound (R)-3 derived from D-phenylalanine, was docked into the nucleoside binding pocket of a viral enzyme of the Dengue virus (NS5MTaseDV) [20] and adenosine deaminase [16,17,18,19]. Initially, the triazole scaffold was compared with the ribavirin nucleotide that inhibits the NS5MTaseDV enzyme. Upon entry into the cell, ribavirin gets converted to the 5’-triphosphate derivative before it binds to the enzyme (Figure 1A). However, due to poor electron density in the X-ray structure (1R6A) only the monophosphate has been modelled. Using Schrödinger’s Glide (XP docking mode), compound (R)-3 docks into the same pose (yellow ligand) as the ribavirin 5’-monophosphate in the NS5MTaseDV enzyme suggesting that compound (R)-3 mimics the binding mode of ribavirin (dark blue) (Figure 1B). The second example comes from the inhibition of the ADA enzyme. In this case, several crystal structures have been reported with imidazole-based nano-molar inhibitors [16,17,18,19].
Therefore, (R)-3 was also docked into the ADA system in order to investigate the similarity of the 1,4-disubstituted-1,2,3-triazoles to the imidazole-based nucleoside analogue 2 (Figure 1C). The docking results from Glide (XP docking mode) show that our model compounds (yellow) superimposes the imidazole of the inhibitor in the binding pocket of the X-ray structure (Figure 1D).
The docking results from NS5MTaseDV and adenosine deaminase (ADA) suggest that chiral 1,4-disubstituted-1,2,3-triazoles derivatives has the potential of mimicking the binding mode of known purine analogues.
The 1 and 4 position of the triazole scaffold allow diversification and the 1,4-disubstituted-1,2,3-triazoles should be easy accessible from [3+2] cycloaddition reactions between azides and alkynes using the copper catalyzed procedure reported by Meldal [21] and Sharpless [22] (Figure 2). We anticipated that by using azido alcohol derivatives, prepared from amino acids, a variety of different R-groups could be introduced resulting in chiral 1,4-disubstituted-1,2,3-triazoles. Furthermore, by using propiolate in the [3+2] cycloaddition reactions an ester functionality would be present in 4 position of the disubstituted-1,2,3-triazoles and allow further derivatisation, for example by reactions with primary or secondary amines.
Figure 2.

Retrosynthetic strategy for the generation of chiral 1,4-disubstituted-1,2,3-triazole derivatives.
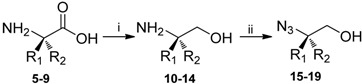
Here, we report an efficient synthesis of compounds represented by structure 4 (Figure 2) by performing [3+2] cycloaddition reactions followed by ester aminolysis. According to the general strategy outlined in Figure 2, our synthesis of the 1,4-disubstituted-1,2,3-triazoles commenced with the preparation of amino alcohols 10-14 by reduction of the corresponding amino acids 5-9 (Table 1).
Table 1.
Synthesis of azido alcohols.
| Compound | R1 | R2 | Yield (%) |
|---|---|---|---|
| 5 | H | CH2Ph | - |
| 6 | CH2Ph | H | - |
| 7 | H | CH2CH2Ph | - |
| 8 | H | CH(CH3)2 | - |
| 9 | H | CH2OCH2Ph | - |
| 10 | H | CH2Ph | 79 |
| 11 | CH2Ph | H | 91 |
| 12 | H | CH2CH2Ph | 92 |
| 13 | H | CH(CH3)2 | 96 |
| 14 | H | CH2OCH2Ph | 97 |
| 15 | H | CH2Ph | 76 |
| 16 | CH2Ph | H | 72 |
| 17 | H | CH2CH2Ph | 74 |
| 18 | H | CH(CH3)2 | 70 |
| 19 | H | CH2OCH2Ph | 73 |
(i) LiAlH4, THF, 16 h, reflux. (ii) TfN3, DMAP, DCM, 15 h, RT.
Both L-Phe and D-Phe were selected in order to obtain the two different stereoisomers of 1,4-disubstituted-1,2,3-triazoles containing an aromatic R-group. L-Homo-Phe was selected in order to obtain 1,4-disubstituted-1,2,3-triazoles with the phenyl group further away from the triazole ring. These aromatic groups could potentially make interactions with hydrophobic pockets adjacent to the nucleoside binding site in target proteins [23]. We also wanted to explore the use of amino acids with alkyl side chains and selected L-Val and L-Ser. Although the copper(I)-catalyzed 1,3-dipolar cycloaddition is insensitive to functional group interference, a benzyl ether derivative of L-Ser was used in order to facilitate column chromatography of the target compound. The amino acids were reduced using LiAlH4 and the resulting amino alcohols converted to the corresponding azides via treatment with TfN3. In this way, a variety of azides derived from various amino alcohols were obtained in good yields (70-76% yields) [24,25,26,27,28,29].
In general, the copper(I)-catalyzed 1,3-dipolar cycloaddition is known as a mild reaction proceeding well in aqueous solutions without protection from oxygen. A range of different reaction conditions have been reported, particularly with respect to generation of the active CuI species. Sources of CuI include CuI salts, most commonly copper iodide [21], in situ reduction of CuII salts, particularly CuII sulfate [22], and comproportionation of Cu0 and CuII [30,31]. Recent reports suggest that nitrogen-based ligands can stabilize the CuI oxidation state under aerobic, aqueous conditions and promote the desired transformation [32].
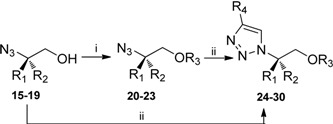
We got very good results using 5 mol % of sodium ascorbate and 1 mol % of copper(II) sulphate in a 1:1 mixture of water and tert-butyl alcohol (Table 2). The reaction between methylene propiolate and 15 or 18 using these reaction conditions furnished the two 1,4-disubstituted triazole products 24 and 25 in 71% and 74% isolated yields respectively, after stirring at RT in a capped vial. The reactions were monitored by TLC and showed complete conversion after 15 h. The product yields in the reactions therefore reflect the efficiency of product purification rather than the coupling efficiency.
Table 2.
Synthesis of 1,4-disubstituted-1,2,3-triazoles.
| Compound | R1 | R2 | R3 | R4 | Yield (%) |
|---|---|---|---|---|---|
| 20 | H | CH2Ph | CH2Ph | - | 87 |
| 21 | CH2Ph | H | CH2Ph | - | 91 |
| 22 | H | CH2CH2Ph | CH2Ph | - | 92 |
| 23 | H | CH2OCH2Ph | Si(CH3)2C(CH3)3 | - | 67 |
| 24 | H | CH2Ph | H | CO2CH3 | 71 |
| 25 | H | CH(CH3)2 | H | CO2CH3 | 74 |
| 26 | H | CH2Ph | H | Ph | 97 |
| 27 | H | CH2Ph | CH2Ph | CO2CH3 | 83 |
| 28 | CH2Ph | H | CH2Ph | CO2CH3 | 90 |
| 29 | H | CH2CH2Ph | CH2Ph | CO2CH3 | 82 |
(i) BnCl, NaH, TBAI, THF, 15 h, RT; or TBDMSiCl, imidazole, DMF, 15 h, RT; (ii) sodium ascorbate, CuSO4 in H2O:t-BuOH (1:1 v/v), 15h, RT.
In order to improve the efficiency of purification of the products by column chromatography we decided to block the hydroxyl function of the azido alcohols either as benzyl ethers (as in compounds 20-22) or as TBDMS ethers (as in compound 23). In addition, the benzyl ethers could make potential interactions in the phosphate-and sugar binding area of the ATP binding site of target kinases [23].
The protected azido derivatives 20-22 were only partially soluble in water: t-BuOH (1:1 v/v) and the cycloaddition reactions with methylene propiolate were attempted in water: THF (1:2 v/v) instead. The corresponding 1,4-disubstituted triazole products 26-28 were obtained high yields (83–97% yields). However, the reaction between methylene propiolate and the TBDMS-protected azido alcohol did not proceed to completion, and the corresponding 1,4-disubstituted triazole 30 was only obtained in 66% yield. Prolonged reaction times (30 h) or running the reaction at elevated temperature (50 oC for 15 h) did not improve the yield (data not shown). Steric interference of the bulky TBDMS ether in the cycloaddition reaction is a possible explanation for this effect.
As well as high yields and prevention of by-product formation, it is essential to avoid racemization when functionalizing enantiomerically pure compounds. Hence, a chiral HPLC system was used to analyze the [3+2] cycloaddition products 27 and 28. The two enantiomers separated well on the chiral HPLC column with retention times of 16.6 min (27) and 13.4 min (28) respectively. No racemization could be detected. (Scheme 1)
Scheme 1.

Derivatisation of ester functionality with different amides. i. Amines, NaOMe, MeOH, 15 h, RT.
The ester functionality of the disubstituted-1,2,3-triazoles was further derivatised by reaction with primary and secondary amines. Compound 24 and 25 were reacted with 7N NH3 in MeOH for 15 h at RT and the corresponding target compounds were obtained in 97% and 93% isolated yields respectively. However, reacting 24 with other amines (3 eq) such as butylamine, benzylamine, diethylamine, morpholine or ethanolamine in MeOH, resulted in sluggish reactions. Prolonged reaction times (30 h), larger excess of amines (10 eq) or elevated temperature, either by classical thermal heating (50 oC, 10 h) or by microwave assisted heating (100 oC, 10-60 min in a sealed vessel) had only a marginal effect on the yield.
The reactions were repeated in the presence of 2 eq of sodium methoxide and TLC showed complete conversion after 15 h at RT. Nevertheless, several products were formed and the target compounds 31-35 were only isolated in low to moderate yields (4-58%). In general, the primary amines gave the best result while the secondary amines proceeded more slowly (Table 3).
Table 3.
Synthesis of 4-carbamoyl-1,2,3-triazoles.
| Compound | R1 | R2 | R3 | R4 | Yield (%) |
|---|---|---|---|---|---|
| (S)-3 | H | CH2Ph | H | NH2 | 97 |
| 31 | H | CH2Ph | H | NH(CH2)3CH3 | 58 |
| 32 | H | CH2Ph | H | NH(CH2)2OH | 4 |
| 33 | H | CH2Ph | H | NHCH2Ph | 35 |
| 34 | H | CH2Ph | H | N(CH2CH3)2 | 12 |
| 35 | H | CH2Ph | H | N(CH2CH2)2O | 30 |
| 36 | H | CH(CH3)2 | H | NH2 | 35 |
| 37 | H | CH2Ph | CH2Ph | NH2 | 92 |
| 38 | H | CH2Ph | CH2Ph | NH(CH2)3CH3 | 93 |
| 39 | H | CH2Ph | CH2Ph | NH(CH2)2OH | 81 |
| 40 | H | CH2Ph | CH2Ph | NHCH2Ph | 87 |
| 41 | H | CH2Ph | CH2Ph | N(CH2CH3)2 | 8 |
| 42 | H | CH2Ph | CH2Ph | N(CH2CH2)2O | 68 |
| 43 | CH2Ph | H | CH2Ph | NH(CH2)3CH3 | 98 |
| 44 | H | CH2OCH2Ph | CH2Ph | NH2 | 88 |
| 45 | H | CH2OCH2Ph | CH2Ph | NH(CH2)3CH3 | 93 |
| 46 | H | CH2OCH2Ph | Si(CH3)2C(CH3)3 | NH2 | 92 |
On the other hand, when the 4-carboxy methyl ester-triazole 27 was reacted with 7N NH3 in MeOH or amines in the presence of sodium methoxide the 4-carbamoyltriazoles 37-42 were obtained in high yields. Ammonia and primary amines gave excellent yields (81–92%) while diethyl amine and morpholine did not go to completion and gave the corresponding 4-carbamoyltriazoles in 8 and 68% yields respectively. The 4-carbamoyltriazoles 43-46 were also obtained in excellent yields (88–98%). In all cases TLC showed clean reactions with only formation of target compounds. The results clearly show that it is essential to block the primary alcohol in order to prevent side-reaction and to secure high yields of the 4-carbamoyltriazoles.
The two enantiomers 38 and 43 were analyzed by chiral HPLC to determine if any racemization occurred in the amination step. The two enantiomers separated well on the chiral HPLC column with retention times of 21.2 min (38) and 14.9 min (43) respectively. No racemization could be detected.
3. Experimental
3.1. General
(S)-2-Amino-3-phenylpropionic acid, (S)-2-amino-3-methylbutyric acid, (R)-2-amino-3-phenyl-propionic acid, (S)-2-amino-4-phenylbutyric acid and (S)-2-amino-3-benzyloxypropionic acid (40) were obtained from Sigma Aldrich and Bachem. Trifluoromethanesulfonic anhydride was obtained from Acros. All other chemicals were reagent grade or better obtained from reputable suppliers. 1H- and 13C-NMR spectra were recorded on a JEOL JNM-EX 400 spectrometer at 400 and 100 MHz, respectively. Optical rotation was measured with a Perkin Elmer Polarimeter 341 LC. The reactions were monitored by TLC, on silica plated aluminium sheets (Silica gel 60F254, E. Merck) in case of flash chromatography, Merck silica gel CC (230-400 mesh) was used. Elemental analyses were performed at H Kolbe Mikroanalytisches Laborium, Mühlheim an der Ruhr, Germany.
Chiral HPLC analysis was performed on a Varian system (9012 pump, flow 1 mL min−1; 9050 detector at 254 nm) using a Chiralpac AD column (250 × 4.6 mm) and isocratic elution with 10% i-PrOH/n-hexane (method A) or 5% i-PrOH/n-hexane (method B).
3.2. General procedure for preparation of amino alcohols
A solution of lithium aluminium hydride (1.4 eq) in dry THF (2 mL/mmol amino acid) was stirred and cooled in an ice bath. The amino acid (1 eq) was added and the mixture was stirred at 0 oC for 1 h, warmed to room temperature, and stirred for an additional 1 h. The reaction mixture was then heated at reflux for 16 h. The solution was cooled in an ice bath and diluted slowly with ether (30 mL). Water (7.5 mL) and 15% NaOH (2.5 mL) were added dropwise to the solution in succession. The mixture was filtered through Celite to remove the aluminium salts. The Celite was washed with ether (2 × 15 mL). The filtrate was collected and the solvent was removed under reduced pressure affording the amino alcohol. The amino alcohols were used in the following steps without further purification.
(S)-2-Amino-3-phenylpropan-1-ol (10): The target compound was prepared from 5 (24.2 mmol, 4 g) using the general procedure described above and obtained as a yellow solid (2.9 g, 79% yield). Analytical data was consistent with the literature [33].
(R)-2-Amino-3-phenylpropan-1-ol (11): The target compound was prepared from 6 (18.2 mmol, 3.00 g) using the general procedure described above and obtained as a yellow oil (2.49 g, 91% yield). The amino alcohol was used for the following steps without further purification [34].
(S)-2-Amino-4-phenylbutan-1-ol (12): The target compound was prepared from 7 (5.5 mmol, 1.00 g) using the general procedure described above and obtained as a yellow oil (50 mg, 92% yield). Analytical data was consistent with the literature [34].
(S)-2-Amino-3-methylbutan-1-ol (13): The target compound was prepared from 8 (8.23 mmol, 965 mg) using the general procedure described above and obtained as a yellow oil (805 mg, 96% yield). Analytical data was consistent with the literature [35].
(R)-2-Amino-3-benzyloxypropan-1-ol (14): The target compound was prepared from 9 (10.24 mmol, 2.00 g) using the general procedure described above and obtained as an oil (1.80 g, 97% yield). The amino alcohol was used for the following steps without further purification. 1H-NMR (CDCl3) δ 3.03–3.10 (1H, m), 3.37–3.62 (2H, m), 3.43–3.50 (2H, m), 4.49 (2H, s), 7.24–7.37(5H, m). Analytical data was consistent with the literature [35].
3.3. General procedure for preparation of azido alcohols by diazo transfer
To a solution of amino alcohol (1 eq) and DMAP (2.5 eq) in CH2Cl2 (1mL/mmol amino alcohol) triflyl azide solution (4.4 eq) was added dropwise with stirring and exclusion of moisture. The reaction mixture was stirred at rt for 15 h. The reaction mixture was concentrated under reduced pressure and purified by flash chromatography.
(S)-2-Azido-3-phenylpropan-1-ol (15): The target compound was prepared from 10 (19.27 mmol, 2.91 g) using the general procedure described above and purified by flash chromatography on silica gel using EtOAc/hexane(1:2 v/v) as eluent. The target compound was obtained as a yellow oil (2.45 g, 66% yield). Analytical data was consistent with the literature [28].
(R)-2-Azido-3-phenylpropan-1-ol (16): The target compound was prepared from 11 (16.4 mmol, 2.49 g) using the general procedure described above and purified by flash chromatography on silica gel using EtOAc/hexane (1:2 v/v) as eluent. The target compound was obtained as a yellow oil (2.10 g, 72% yield). 1H-NMR (CDCl3) δ 2.76–3.14 (2H, m), 3.55–3.69 (2H, m), 3.77–3.85 (1H, m), 7.20–7.35 (5H, m); 13C-NMR (CDCl3) δ 36.79 (-), 62.9 (+), 70.51 (-), 127.43 (+), 128.59 (+), 128.91 (+), 141.05 (q); Anal. Calc. for C8H11N3O: C, 64.39; H, 7.45; N, 28.17. Found: C, 64.44; H, 7.46; N, 28.28.
(S)-2-Azido-4-phenylbutan-1-ol (17): The target compound was prepared from 12 (5.03 mmol, 830 mg) using the general procedure described above and purified by flash chromatography on silica gel using EtOAc/hexane (1:2 v/v) as eluent. The target compound was obtained as a clear oil (710 mg, 74% yield). 1H-NMR (CDCl3) δ 1.78–1.85 (2H, m), 2.67–2.85 (2H, m), 3.42–3.49 (1H, m), 3.55–3.73 (2H, m), 3.68–3.72 (2H, m), 7.18–7.34 (5H, m); 13C-NMR (CDCl3) δ 32.3 (-), 32.4 (-), 63.5 (+), 65.5 (-), 126.4 (+), 128.6 (+), 128.8 (+), 141.0 (q); Anal. Calc. for C10H13N3O: C, 62.81; H, 6.85; N, 21.97. Found: C, 62.69; H, 6.79; N, 21.88.
(S)-2-Azido-3-methylbutan-1-ol (18): The target compound was prepared from 13 (7.8 mmol, 805 mg) using the general procedure described above and purified by flash chromatography on silica gel using EtOAc/hexane (1:3 v/v) as eluent, affording the target compound as a yellow oil (698 mg, 70% yield). Analytical data was consistent with the literature [35].
(R)-2-Azido-3-benzyloxypropan-1-ol (19): The target compound was prepared from 14 (10.7 mmol, 1.8 g) using the general procedure described above and purified by flash chromatography on silica gel using EtOAc/hexane (1:2 v/v) as eluent. The target compound was obtained as an oil (1.51 g, 73% yield). Analytical data was consistent with the literature [36].
3.4. General procedure for benzylation of the hydroxyl functionality
The azido alcohol (1 eq) was dissolved in dry THF (10 mL), NaH (suspension in mineral oil 60%, 2 eq) was added and the suspension was stirred 5 min at rt. Benzyl bromide (2 eq) and TBAI (0.05 eq) were added and the reaction mixture stirred for 15 h. The reaction was quenched with sat. NH4Cl aq (15 mL) at which the reaction mixture became clear, and was then extracted with EtOAc (2 × 30 mL). The organic phases were pooled and washed once with brine, dried with MgSO4, filtered and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using hexane and hexane/EtOAc (95:5 v/v) as eluents.
(S)-2-Azido-1-benzyloxy-3-phenylpropane (20): The target compound was prepared from 15 (12.18 mmol, 2.16 g) using the general procedure described above and obtained as a yellow oil (2.8 g, 87% yield). 1H-NMR (CDCl3) δ 2.83–2.99 (2 H, m), 3.54–3.59 (2H, m), 3.63–3.68 (1H, m), 4.63 (2H, m), 7.24–7.45 (10 H, m); 13C-NMR (CDCl3) δ 37.30 (-), 62.81 (+), 71.91 (-), 73.46 (-), 126.90 (+), 127.73 (+), 127.89 (+), 128.52 (+), 128.68 (+), 129.38 (+), 137.43 (q), 137.91 (q); Anal. Calc. for C16H17N3O: C,71.87; H, 6.42; N, 15.72. Found: C, 71.90; H, 6.44; N, 15.75.
(R)-2-Azido-1-benzyloxy-3-phenylpropane (21): The target compound was prepared from 16 (5.64 mmol, 1.00 mg) using the general procedure described above and was obtained as a yellow oil (1.37 g, 91% yield). 1H-NMR (CDCl3) δ 2.82–3.01 (2 H, m), 3.54–3.62 (2H, m), 3.79–3.88 (1H, m), 4.65–4.72 (2H, m), 7.29–7.48 (10 H, m); 13C-NMR (CDCl3) δ 37.24 (-), 62.75 (+), 71.86 (-), 73.39 (-), 126.84 (+), 127.68 (+), 127.84 (+), 128.53 (+), 128.63 (+), 129.34 (+), 137.39 (q), 137.88 (q); Anal. Calc. For C16H19N3O: C, 72.57; H, 6.81; N, 14.93. Found: C, 71.67; H, 6.72; N, 15.04.
(S)-2-Azido-1-benzyloxy-4-phenylbutane (22): The target compound was prepared from 17 (3.32 mmol, 635 mg) using the general procedure described above and was obtained as a yellow oil (861 mg, 92% yield). 1H-NMR (CDCl3) δ 1.82–1.92 (2H, m), 2.68–2.91 (2H, m), 3.53–3.68 (3H, m), 4.62 (2H, s), 7.23–7.48 (10 H, m); 13C-NMR (CDCl3) δ 32.27 (-), 32.63 (-), 61.08 (+), 72.96 (-), 73.44 (-), 126.25 (+), 127.6 (+), 127.8 (+), 128.5 (+), 128.6 (+), 128.8 (+), 137.9 (q), 141.0 (q), Anal. Calc. for C17H19N3O: C, 72.57; H, 6.81; N, 14.93. Found: C, 72.59; H, 6.77; N, 15.00.
((S)-2-Azido-3-benzyloxypropoxy)-tert-butyl-dimethylsilane (23): Compound 19 (2.59 mmol, 538 mg) was dissolved in DMF (5 mL), TBDMS-Cl (3.38 mmol, 506 mg) and imidazole (6.5 mmol, 441 mg) were added and the mixture stirred 15 h at rt. The reaction mixture was concentrated under reduced pressure, extracted with EtOAc and NH4Claq and purified by column chromatography on silica gel using hexane/EtOAc (99:1 v/v) as eluent. The target compound was obtained as an oil (775 mg, 93% yield). 1H-NMR (CDCl3) δ 0.13 (6H, s), 0.95 (9H, s), 3.54–3.85 (5H, m), 4.60 (2H, s), 7.29–7.43 (5H, m); 13C-NMR (CDCl3) δ -5.39 (+), 18.35 (q), 25.93 (+), 62.50 (N3CHCH2), 63.40 (+), 69.37 (-), 73.59 (-), 127.80 (+), 127.90 (+), 128.59 (+), 137.93 (q); Anal. Calc. for C16H27N3O2Si: C, 59.78; H, 8.47; N, 13.07. Found: C, 59.74; H, 8.42; N, 13.02.
3.5. General procedure for copper catalyzed cycloaddition reactions
The unprotected azido alcohol (1eq) and the alkyne (1.5 eq) were suspended in a 1:1 mixture of water and tert-butyl alcohol (5 mL/mmol azido alcohol). Benzyl protected azido alcohol (1 eq) and the alkyne (1.5 eq) were suspended in H2O/THF (1:2, v/v). Sodium ascorbate (0.1 eq as a 1 M solution in water) was added, followed by copper(II) sulfate (0.01 eq as a 0.1 M solution in water). The reaction mixture was stirred at rt for 15 h. The solvents were removed under reduced pressure and the residue adsorbed on silica gel and purified by column chromatography.
1-((S)-1-Hydroxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid methyl ester (24): The target compound was prepared from 15 (14.9 mmol, 2.64 g) and methyl propiolate (22.35 mmol, 1.87 g) using the general procedure described above. The product was purified by column chromatography on silica gel (2:1 EtOAc/hexane) affording the target compound as a pale oil (3.15 g, 81% yield). 1H- NMR (CDCl3) δ 3.28 (2H, d, J = 7.7 Hz), 3.89 (3H, s), 4.04–4.17 (2H, m), 4.75–4.81 (1H, m), 7.05–7.27 (5H m), 7.95 (1H, s); 13C-NMR (CDCl3) δ 37.35 (-), 52.15 (+), 63.50 (+), 65.56 (+), 127.25 (+), 128.46 (q), 129.10 (2x+), 135.89 (q), 139.06 (q), 161.03 (q); Anal. Calc. for C13H15N3O3: C, 59.76; H, 5.79; N, 16.08. Found: C, 59.87; H, 5.74; N, 15.93.
1-((S)-1-Hydroxymethyl-2-methylpropyl)-1H-1,2,3-triazole-4-carboxylic acid methyl ester (25): The target compound was prepared from 18 (5.43 mmol, 700 mg) and methyl propiolate (8.14 mmol, 684 mg) using the general procedure described above. The product was purified by column chromatography on silica gel (2:1 EtOAc/hexane) affording the target compound as a pale oil (984 mg, 85% yield). 1H-NMR (CDCl3) δ 0.76 (3H, d, 6.6 Hz), 1.08 (3H, d, 6.6 Hz), 2.30–2.43 (1H, m), 3.92 (3H, s), 4.00–4.07 (1H, m), 4.17–4.25 (1H, m), 8.17 (1H, s); 13C-NMR (CDCl3) δ 19.33 (+), 19.61 (+), 29.73 (+), 52.31 (+), 62.35 (-), 70.24 (+), 128.15 (+), 139.73 (q), 161.31 (q); Anal. Calc. for C9H15N3O3: C, 50.69; H, 7.09; N, 19.71; Found: C, 50.90; H, 6.92; N, 19.84.
1-((S)-(1-Hydroxymethyl-2-phenethyl)-4-phenyl-1H,1,2,3-triazole (26): The target compound was prepared from 15 (2 mmol, 365 mg) and phenylacetylene (2.5 mmol, 275 μL) using the general procedure described above. The crude product was purified by column chromatography on silica gel (2:3 EtOAc/hexane) affording the desired product as a colourless solid (512 mg, 98% yield). 1H-NMR (CDCl3) δ 3.22–3.35 (2H, m), 4.05–4.25 (2H, m), 4.67–4.73 (1H, m), 7.07–7.12 (2H, m), 7.19–7.39 (6H, m), 7.50 (1H, s), 7.61–7.67 (2H, m); 13C-NMR (CDCl3) δ 37.97(+), 64.19 (-), 65.55 (+), 120.70 (+), 125.75 (+), 127.30 (+), 128.30 (+), 128.96 (+), 129.00 (+), 129.21 (+), 130.46 (q), 135.76 (q), 136.77 (q); Anal. Calc. for C17H17N3O: C, 73.10; H, 6.13; N, 15.04. Found: C, 72.94; H, 6.09; N, 14.98.
1-((S)-1-Benzyloxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid methyl ester (27): The target compound was prepared from 20 (5.6 mmol, 1.50 g) and methyl propiolate (8.4 mmol, 711 mg) using the general procedure described above. The crude product was obtained as a pale oil (1.63 g, 83% yield). 1H-NMR (CDCl3) δ 3.21–3.32 (2H, m), 3.75–3.84 (2H, m), 3.92 (3H, s), 4.41–4.51 (2H, m), 4.84–4.98 (1H, m), 7.01–7.05 (2H, m), 7.16–7.36 (8H, m), 8.08 (1H, s); 13C-NMR (CDCl3) δ 37.81 (-), 52.30 (+), 63.59 (+), 70.13 (-), 73.66 (-), 127.38 (+), 127.96 (+), 128.23 (+), 128.71 (+), 128.96 (+), 129.04 (+), 135.94 (q), 137.19 (q), 139.51 (+), 161.31 (q); Anal. Calc. for: C20H21N3O3: C, 68.36; H, 6.02; N, 13.66. Found: C, 68.29; H, 5.98; N, 13.55; Optical rotation: [α]20D= +53,33 (c 1,06 in CHCl3); tR (method A): 16.3.
1-((R)-1-Benzyloxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid methyl ester (28): The target compound was prepared from 21 (3.7 mmol, 1.00 g) and methyl propiolate (5.6 mmol, 474 mg) using the general procedure described above. The crude product was obtained as a yellow oil (1.18 mg, 90% yield). 1H-NMR (CDCl3) δ 3.21–3.33 (2H, m), 3.75–3.84 (2H, m), 3.93 (3H, s), 4.41–4.52 (2H, m), 4.89–4.97 (1H, m), 7.01–7.06 (2H, m), 7.17–7.37 (8H, m), 8.08 (1H, s); 13C-NMR (CDCl3) δ 37.85 (-), 52.30 (+), 63.56 (+), 70.16 (-), 73.68 (-), 127.39 (+), 127.98 (+), 128.24 (Ph), 128.73 (Ph), 128.98 (Ph), 129.06 (Ph), 135.98 (q), 137.21 (q), 139.47 (+), 161.40 (q); Anal. Calc. for C20H21N3O3: C, 68.36; H, 6.02; N, 11.96. Found: C, 68.30; H, 5.99; N, 11.87; Optical rotation: [α]20D= -50,94 (c 1,02 in CHCl3); tR (methode A): 13.4.
1-((S)-1-Benzyloxymethyl-3-phenylpropyl)-1H-1,2,3-triazole-4-carboxylic acid methyl ester (29): The target compound was prepared from 22 (1.49 mmol, 400 mg) and methyl propiolate (2.14 mmol, 180 mg) using the general procedure described above. The crude product was obtained as a yellow oil (473 mg, 88% yield). 1H-NMR (CDCl3) δ 1.85–2.02 (2H, m), 2.57–2.71 (2H, m), 3.44–3.55 (2H, m), 3.84 (3H, s), 4.18–4.28 (2H, m), 4.46–4.59 (1H, m), 7.13–7.19 (2H, m), 7.23–7.37 (8H, m), 7.82 (1H, s); 13C-NMR (CDCl3) δ 32.62 (-), 33.98 (-), 49.67 (+), 52.54 (+), 71.75 (-), 73.38 (-), 126.06 (+), 127.86 (+), 128.58 (+), 138.36 (q), 139,81 (q), 141.84 (q), 161.38 (q); Anal. Calc. for C21H23N3O3: C, 68.82; H, 6.34; N, 11.74. Found: C, 69.03; H, 6.39; N, 11.85.
1-[(S)-1-Benzyloxymethyl-2-(tert-butyl-dimethylsilanyloxy)-ethyl]-1H-1,2,3-triazole-4-carboxylic acid methyl ester (30): The target compound was prepared from 23 (1.21 mmol, 390 mg) and methyl propiolate (1.82 mmol, 153 mg) using the general procedure described above. The crude product was purified by column chromatography on silica gel (1:3 EtOAc/hexane) affording the product as an oil (382 mg, 82% yield). 1H-NMR (CDCl3) δ -0.11(3H, s), -0.10 (3H, s), 0.74 (9H, s), 3.77–3.88 (5H, m), 3.89–4.04 (2H, m), 4.37–4.46 (2H, m), 4.79–4.87 (1H, m), 7.11–7.25 (5H, m), 8.16 (1H, s); 13C-NMR (CDCl3) δ -5.88 (+), -5.85 (+), 17.87 (Q), 25.50 (+), 51.80 (+), 61.99 (-), 62.64 (+), 67.74 (-), 73.24 (-), 125.56 (+), 127.79 (+), 128.31 (+), 137.01 (q), 139.15 (q), 161.03 (q); Anal. Calc. for C20H31N3O4Si: C, 59.23; H, 7.70; N, 10.36. Found: C, 58.99; H, 7.79; N, 10.28.
3.6. General procedure for ester-amide conversion
Reaction with ammonia: The ester was dissolved in 7N NH3 in MeOH (1.5 mL) and stirred at rt overnight. The solvent was removed under reduced pressure and the desired product was purified by recrystallization or by column chromatography on silica gel.
Reaction with all other amines: Sodium (2 eq) was dissolved in MeOH (3 mL) under nitrogen followed by addition of amine (3 eq). A solution of the ester (1 eq) dissolved in MeOH (0.5 mL) was added to the reaction mixture. The reaction was stirred under nitrogen at rt for 15 h. The reaction was quenched with 1M HCl aq (1 mL) and extracted with CHCl3 (2 x 20 mL). The organic layer was washed with brine, separated, dried with MgSO4. The solvent was removed under reduced pressure and the crude product purified by column chromatography on silica gel or by recrystallization.
1-((S)-1-Benzyl-2-hydroxyethyl)-1H-1,2,3-triazole-4-carboxylic acid amide ((S)-3): The target compound was obtained by reacting 24 (0.89 mmol, 233 mg) with NH3 following the procedure above. The desired product was recrystallized from EtOH affording 3 as a white solid (184 mg, 97% yield). 1H- NMR (DMSO) δ 3.13–3.26 (2H, m), 3.75–3.84 (2H, m), 4.86–4.93 (1H, m), 5.16 (1H, t), 7.09–7.23 (5H, m), 7.40 (1H, s), 7.77 (1H, s), 8.53 (1H, s); 13C-NMR (DMSO) δ 36.53 (-), 63.10 (-), 64.62 (+), 125.96 (+), 126.49 (+), 128.43 (+), 128.80 (+), 137.12 (q), 142.46 (q), 161.65 (q); Anal. Calc. for: C12H14N4O2: C, 58.53; H, 5.73; N, 22.75. Found: C, 58.39; H, 5.64; N, 22.78.
1-((S)-1-Benzyl-2-hydroxyethyl)-1H-1,2,3-triazole-4-carboxylic acid butylamide (31): The target compound was obtained by reacting 24 (0.76 mmol, 200 mg) with butyl amine (2.38 mmol, 230 μL) following the procedure described above. The crude product was purified by column chromatography on silica gel (2:3 EtOAc/hexane, then 2:1 EtOAc/hexane + 10% MeOH) affording compound 31 as a white solid (100 mg, 58% yield). 1H-NMR (CDCl3) δ 0.87–0.95 (3H, m), 1.30–1.61 (4H, m), 3.19–3.28 (2H, m), 3.37–3.42 (2H, m), 3.98–4.05 (2H, m), 4.77–4.84 (1H, m), 7.06–7.10 (2H, m), 7,14 (1H, t), 7.18–7.28 (3H, m), 8.07 (1H, s); 13C-NMR (CDCl3) δ 13.95 (+), 20.29 (-), 31.71 (-), 37.73 (-), 39.12 (-), 63.61(-), 65.46 (+), 126.00 (+), 127.43 (+), 129.05 (+), 129.13 (+), 136.27 (q), 142.19 (q), 160.48 (q); Anal. Calc. for: C16H22N4O2: C, 63.56; H, 7.33; N, 18.53. Found: C, 63.51; H, 7.23; N, 18.41.
1-((S)-1-Benzyl-2-hydroxyethyl)-1H-1,2,3-triazole-4-carboxylic acid (2-hydroxy-ethyl)-amide (32): The target compound was obtained by reacting 24 (0.55 mmol, 145 mg) with ethanol amine (1.6 mmol, 100 μL) following the procedure described above. The crude product was purified by column chromatography on silica gel using CHCl3/MeOH (9:1 v/v) as eluent. The title compound was obtained as an oil (7 mg, 4% yield). 1H-NMR (CD3OD) δ 3.18–3.36 (2H, m), 3.46–3.52 (2H, m), 3.66–3.71 (2H, m), 3.91–4.04 (2H, m), 4.64–4.69 (1H, m), 7.07–7.29 (5H, m), 8.27 (1H, s); 13C-NMR (CDCl3) δ 38.14 (-), 42.46 (-), 61.37 (-), 64.40 (-), 66.84 (+), 126.99 (+), 127.73 (+), 129.43 (+), 129.81 (+), 136.31 (q), 142.67 (q), 160.52 (q); Anal. Calc. for C14H18N4O3: C, 57.92; H, 6.25; N, 19.30. Found: C, 58.18; H, 6.24; N, 19.32.
1-((S)-1-Hydroxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid benzylamide (33): The target compound was obtained by reacting 24 (0.76 mmol, 200 mg) with benzylamine (2.3 mmol, 250 μL) following the procedure described above. The crude product was purified by column chromatography on silica gel using EtOAc/hexane (2:3 v/v) and EtOAc/hexane + 10% MeOH) as eluents to afford 33 as an oil (80 mg, 35% yield). 1H-NMR (CDCl3) δ 3.22–3.34 (2H, m), 3.99–4.10 (2H, m), 4.56–4.67 (2H, m), 4.76–4.85 (2H, m), 7.08–7.12 (2H, m), 7.21–7.38 (8H, m), 7.46 (1H, t), 8.06 (1H, s); 13C-NMR (CDCl3) δ 37.75(-), 43.46 (-), 63.71 (-), 65.46 (+), 126.23 (+), 127.51 (+), 127.85 (+), 128.08 (+), 128.97 (+), 129.11 (+), 136.15 (q), 137.93 (q), 142.66 (q), 160.27 (q); Anal. Calc. for: C19H20N4O2: C, 67.84; H, 5.99; N, 16.65. Found: C, 67.68; H, 5.78; N, 16.47.
1-((R)-1-Hydroxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid diethylamide (34): The target compound was obtained by reacting 24 (0.69 mmol, 180 mg) with diethyl amine (2.1 mmol, 220 μL) following the procedure described above. The crude product was purified by column chromatography on silica gel using CHCl3/EtOAc (2:1 v/v) and, DCM/MeOH (9:1 v/v) as eluents to afford 34 as an oil (25 mg, 12% yield). 1H-NMR (CDCl3) δ 0.98–1.48 (6H, m), 3.22–3.35 (2H, m), 3.88–3.92 (2H, m), 3.94–4.15 (2H, m), 4.78–4.85 (1H, m), 7.00–7.41 (5H, m), 8.05 (1H, s); 13C-NMR (CDCl3) δ 12.92 (+), 14.76 (+), 37.61 (-), 41.56 (-), 43.34 (-), 63.61 (-), 65.81 (-), 127.31 (+), 128.12 (+), 128.25 (+), 128.79 (+), 128.91 (+), 129.1 (+), 136.38 (q), 144.51 (q), 160.90 (q); Anal. Calc. for C16H22N4O2: C, 63.56; H, 7.33; N, 18.53; Found: C, 63.70; H, 7.36; N, 18.57.
[1-((S)-1-Benzyl-2-hydroxyethyl)-1H-1,2,3-triazol-4-yl]-morpholin-4-yl-methanone (35): The target compound was obtained by reacting 24 (0.47 mmol, 124 mg) with morpholine (1.4 mmol, 125 μL) following the procedure above. The crude product was purified by column chromatography on silica gel using EtOAc/hexane (3:1v/v) and DCM/MeOH (9;1 v/v) as eluents affording the target compound as an oil (52 mg, 30% yield). 1H-NMR (CDCl3) δ 3.22–3.33 (2H, m), 3.60–3.85 (4H, d), 3.91–4.04 (2H, m), 4.23–4.31 (4H, d), 4.72–4.83 (1H, m), 7.08–7.09 (2H, m), 7.15–7.26 (3H, m); 13C-NMR (CDCl3) δ 37.61 (-), 43.22 (-), 47.48 (-), 63.51 (-), 65.34 (+), 66.97 (-), 67.29 (-), 127.34 (+), 128.95 (+), 129.08 (+), 136.28 (q), 143.24 (q), 160.15 (q); Anal. Calc. for: C16H20N4O3: C, 60.75; H, 6.37; N, 17.71. Found: C, 60.80; H, 6.39; N, 17.75.
1-((S)-1-Hydroxymethyl-2-methylpropyl)-1H-1,2,3-triazole-4-carboxylic acid amide (36): The target compound was obtained by reacting 25 (0.49 mmol, 106 mg) with NH3 following the procedure above. The crude product was obtained as an oil (32 mg, 35% yield). 1H-NMR (CD3OD) δ 0.75 (3H, d, J = 6.6 Hz), 1.07 (3H, d, J = 6.6 Hz ), 2.24–2.37 (1H, m), 3.91–4.12 (2H, m), 4.32–4.44 (1H, m), 8.42 (1H, s); 13C-NMR (CD3OD) δ 19.31 (+), 19.90 (+), 31.08 (+), 62.83 (-), 71.49(+), 127.51 (+), 133.36 (q), 164.94 (q); Anal. Calc. for C8H14N4O2: C, 48.46; H, 7.13; N, 28.27. Found: C, 48.68; H, 7.16; N, 28.35.
1-((S)-1-Benzyloxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid amide (37): The target compound was obtained by reacting 27 (0.51 mmol, 180 mg) with NH3 following the procedure above. The product was recrystallized from EtOH affording the target compound as a white solid (158 mg, 92% yield). 1H-NMR (CDCl3) δ 3.20–3.30 (2H, m,, 3.73–3.82 (2H, m), 4.42–4.52 (2H, m), 4.89–4.98 (1H, m), 5.79 (2H, s), 6.99–7.10 (2H, m), 7.16–7.36 (8H, m), 8.11 (1H, s); 13C-NMR (CDCl3) δ 37.98 (-), 63.54 (+), 70.22 (-), 73.72 (-), 126.24 (+), 127.44 (+), 128.01 (+), 128.27(+), 128.77 (+), 129.00 (+), 129.09 (+), 136.0 (q), 137.2 4(q), 142.34 (q), 162.31 (q); Anal. Calc. for: C19H20N4O2: C, 67.84; H, 5.99; N, 16.55. Found: C, 67.73; H, 5.93; N,16.49.
1-((S)-1-Benzyloxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid butylamide (38): The target compound was obtained by reacting 27 (0.56 mmol 200 mg) with butyl amine (1.7 mmol, 124 mg) following the procedure above. The target compound was obtained as an oil (207 mg, 93% yield) without any purification. 1H-NMR (CDCl3) δ 0.92–0.98 (3H, m), 1.37–1.45 (2H, m), 1.55–1.65 (2H, m), 3.19–3.33 (2H, m), 3.40–3.48 (2H, m), 3.72–3.83 (2H, m), 4.41–4.53 (2H, dd), 4.89–4.99 (1H, m), 7.01–7.07 (2H, m), 7.17–7.37 (8H, m), 8.12 (1H, s); 13C-NMR (CDCl3) δ 13.87 (+), 20.19 (-), 31.78 (-), 37.87 (-), 38.94 (-), 63.33 (+), 70.22 (-), 73.56 (-), 125.51 (+), 127.28 (+), 127.89 (+), 128.12 (+), 128.64 (+), 128.87 (+), 128.99 (+), 136.05 (q), 137.21 (q), 143.10 (q), 160.24 (q); Anal. Calc. for: C23H28N4O2: C, 70.38; H, 7.19; N, 14.27; Found: C, 70.34; H, 7.24; N, 14.36; tR (Method B): 21.2.
1-((S)-1-Benzyloxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid (2-hydroxyethyl)-amide (39): The target compound was obtained by reacting 27 (0.56 mmol, 200 mg) with ethanol amine (1.7 mmol, 104 mg) following the procedure above. The target compound was obtained as an oil (170 mg, 81% yield) without any purification. 1H-NMR (CDCl3) δ 3.16–3.30 (2H, m), 3.55–3.61 (2H, m), 3.71–3.80 (4H, m), 4.39–4.50 (2H, m), 4.88–4.97 (1H, m), 7.00–7.04 (2H, m), 7.14–7.34 (8H, m), 7.87 (1H, m), 8.18 (1H, s); 13C-NMR (CDCl3) δ 37.77 (-) , 42.18 (-) , 61.88 (-), 63.31 (+), 70.19 (-), 73.47 (-), 125.86 (+), 127.23 (+), 127.84 (+), 128.08 (+), 128.59 (+), 128.82 (+), 128.94 (+), 135.97 (q), 137.17 (q), 142.72 (q), 161.1 (q); Anal. Calc. for: C21H24N4O3: C, 65.38; H, 6.31; N, 15.25; Found: C, 65.45; H, 6.29; N, 15.23.
1-((S)-1-Benzyloxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid benzylamide (40): The target compound was obtained by reacting 27 (0.42 mmol, 150 mg) with benzyl amine (1.2 mmol, 137 mg) following the procedure above. The target compound was obtained as an oil (161 mg, 87% yield) without any purification. 1H-NMR (CDCl3) δ 3.19–3.33 (2H, m), 3.73–3.82 (2H, m), 4.42–4.53 (2H, m), 4.56 (2H, d, J = 5.9 Hz), 4.89–4.98 (1H, m), 7.02–7.07 (2H, m), 7.18–7.38 (13H, m), 7.49 (1h, t, J = 5.9Hz), 8.12 (1H, s); 13C-NMR (CDCl3) δ 37.96 (-), 43.31 (-), 63.47 (+), 70.26 (-), 73.70 (+), 125.80 (+), 127.40 (+), 127.73 (+), 128.00 (+), 128.06 (+), 128.24 (+), 128.75 (+), 128.90 (+), 128.98 (+), 129.08 (+), 136.09 (q), 137.26 (q), 138.12 (q), 142.90 (q), 160.24 (q); Anal. Calc. for C26H25N4O2: C: 73.22; H, 6.14; N, 13.14. Found: C, 73.28; H, 6.12; N, 13.19.
1-((S)-1-Benzyloxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid diethylamide (41): The target compound was obtained by reacting 27 (0.56 mmol, 200 mg) with diethylamine (1.7 mmol, 125 mg) following the procedure above. The crude product was purified by column chromatography on silica gel (hexane, then 1:1 EtOAc/hexane) to afford the desired amide as an oil (17 mg, 8% yield). 1H- NMR (CDCl3) δ 1.18–1.35 (6H, m), 3.20–3.36 (2H, m), 3.46–3.58 (2H, m), 3.70–3.98 (4H, m), 4.42–4.55 (2H, m), 4.87–4.97 (1H, m), 7.00–7.39 (10H, m, Ph), 8.09 (1H, s, CCHN); 13C-NMR (CDCl3) δ 13.04 (+), 14.81 (+), 38.08 (-), 41.35 (-), 43.27 (-), 63.14 (+), 70.34 (-), 73.69 (-), 127.33 (+), 127.9 (+), 128.1 (+), 128.2 (+), 128.7 (+), 129.1 (+), 136.3 (q), 137.18 (q), 144.5(q), 160.9 (q); Anal. Calc. for C23H28N4O2: C, 70.38; H, 7.19; N, 14.27. Found: C, 70.40; H, 7.16; N, 14.28.
[1-((S)-1-Benzyloxymethyl-2-phenyethyl)-1H-1,2,3-triazol-4-yl]-morpholin- 4-yl-methanone (42): The target compound was obtained by reacting 27 (0.56 mmol, 200 mg) with morpholine (1.7 mmol, 148 mg) following the procedure above. The target compound was obtained as an oil (182 mg, 82% yield) without any purification. 1H-NMR (CDCl3) δ 3.18–3.32 (2H, m), 3.71–3.78 (8H, m), 4.27–4.33 (2H, m), 4.36–4.49 (2H, m), 4.88–4.96 (1H, m), 6.97–7.32 (10H, m), 8.13 (1H, s); 13C-NMR (CDCl3) δ 37.84 (-), 43.07 (-), 47.35 (-), 63.14 (+), 66.95 (-), 67.28 (-), 70.16 (-), 73.52 (-), 127.25 (+), 127.83 (+), 128.08 (+), 128.61 (+), 128.67 (+),128.82 (+), 128.99 (+), 136.07 (q), 137.23 (q), 143.65 (q), 160.01 (q); Anal. Calc. for C22H23N4O3: C, 67.96; H, 6.45; N, 13.78. Found: C, 68.04; H, 6.41; N, 13.72.
1-((R)-1-Benzyloxymethyl-2-phenethyl)-1H-1,2,3-triazole-4-carboxylic acid butylamide (43): The target compound was obtained by reacting 28 (0.56 mmol, 200 mg) with butyl amine (1.7 mmol, 124 mg) following the procedure above. The target compound was obtained as an oil (220 mg, 98% yield) without any purification. 1H-NMR (CDCl3) δ, 0.96 (3H, t, J = 7.3Hz), 1.37–1.47 (2H, m), 1.56–1.65 (2H, m), 3.19–3.34 (2H, m), 3.41–3.47 (2H, m), 3.72–3.83 (2H, m), 4.42–4.57 (2H, dd, J = 11.9 Hz, J = 25.5Hz), 4.90–4.98 (1H, m), 7.03–7.07 (2 H, m), 7.17–7.38 (8H, m), 8.13 (1H, s); 13C-NMR (CDCl3) δ 13.88 (+), 20.21 (-), 31.79 (-), 37.88 (-), 38.96 (-), 63.34 (+), 70.25 (-), 73.59 (-), 125.53 (+), 127.30 (+), 127.90 (+), 128.12 (+), 128.66 (+), 128.89 (+), 129.01 (+), 136.06 (q), 137.22 (q), 143.10 (q), 160.25 (q); Anal. Calc. for C23H28N4O2: C, 70.38; H, 7.19; N, 14.27. Found: C, 70.46; H, 7.16; N, 14.21; tR (Method B): 14.9.
1-((S)-1-Benzyloxymethyl-3-phenylpropyl)-1H-1,2,3-triazole-4-carboxylic acid amide (44): The target compound was obtained by reacting 29 (0.15 mmol, 57 mg) with NH3 following the procedure above. The crude product was purified by column chromatography using EtOH/hexane (1:1 v/v) as eluent. The target compound was obtained as a white solid (44 mg, 88% yield). 1H-NMR (CDCl3) δ 2.15–2.28 (1H, m), 2.33–2.57 (3H, m), 3.68–3.79 (2H, m), 4.38–4.52 (2H, dd, J = 12.1 Hz, J = 30.0 Hz), 4.67–4.75 (1H, m), 7.05–7.34 (10H, m), 8.22 (1H, s); 13C-NMR (CDCl3) δ 31.90 (-), 33.08 (-), 61.41 (+), 71.39 (-), 73.63 (-), 126.04 (+), 126.64 (+), 127.93 (+), 128.24 (+), 128.59 (+), 128.75 (+), 128.85 (+), 137.27 (q), 140.01 (q), 142.65 (q), 162.47 (q); Anal. Calc. for C20H22N4O2: C, 68.55; H, 6.33; N, 15.99. Found: C, 68.48; H, 6.26; N, 15.88.
1-((S)-1-Benzyloxymethyl-3-phenylpropyl)-1H-1,2,3-triazole-4-carboxylic acid butylamide (45): The target compound was obtained by reacting 29 (0.3 mmol, 113 mg) with butylamine (0.9 mmol, 67 mg) following the procedure above. The target compound was obtained as an oil (113 mg, 93% yield) without any purification. 1H-NMR (CDCl3) δ 0.95 (3H, t, J = 7.3 Hz), 1.37–1.47 (2H, m), 1.56–1.67 (2H, m), 2.15–2.56 (4H, m), 3.44–3.49 (2H, m), 3.67–3.77 (2H, m), 4.43 (2H, dd, J = 12.1 Hz, J = 29.3 Hz), 4.65–4.73 (1H, m), 7.04–7.35 (10H, m), 8.18 (1H, s); 13C-NMR (CDCl3) δ 13.89 (+), 20.22 (-), 31.82 (-), 31.82 (-), 33.03 (-), 38.98 (-), 61.24 (-), 71.39 (-), 73.52 (-),125.30 (+), 126.52 (+), 127.82 (+),128.11 (+), 128.51 (+), 128.64 (+), 128.75 (+), 137.24 (q), 140.01 (q), 143.33 (q), 160.30 (q); Anal. Calc. for C24H30N4O2: C, 70.91; H, 7.44; N, 13.78. Found: C, 71.05; H, 7.39; N, 13.86.
1-[(S)-1-Benzyloxymethyl-2-(tert-butyl-dimethylsilanyloxy)-ethyl]-1H-1,2,3-triazole-4-carboxylic acid amide (46): The target compound was obtained by reacting 30 (0.23 mmol, 95 mg) with NH3 following the procedure above. The product was recrystallized from EtOH affording 46 as a white solid (88 mg, 92% yields). 1H-NMR (CDCl3) δ -0.09(3H, s), -0.01 (3H, s), 0.82 (9H, s), 3.81–4.03 (5H, m), 4.41–4.52 (2H, m), 4.81–4.87 (1H, m), 7.11–7.25 (5H, m), 8.16 (1H, s); 13C-NMR (CDCl3) δ -5.48 (+), -5.42 (+), 18.31 (q), 62.10 (-), 62.64 (+), 67.74 (-), 73.24 (-), 125.56 (+), 127.79 (+), 128.31 (+), 128.73 (+), 137.01 (q), 142.23 (q), 161.03 (q); Anal. Calc. for C19H30N4O3Si: C, 58.43; H, 7.74; N, 14.35. Found: C, 58.58; H, 7.79; N, 14.42.
3.7. Docking
X-ray structures with inhibitors were used as starting point for all dockings. The enzymes were prepared according to the standard procedure in the Schrödinger package. Docking was performed by using Glide (Schrödinger) [37,38,39] with extra precision (XP) settings and standard parameters for ligand docking.
4. Conclusions
An efficient synthetic route was developed for the synthesis of chiral 1,4-disubstituted-1,2,3-triazole derivatives starting from D-or L-amino acids. The target compounds were obtained in excellent yields with no racemization. This straight forward and flexible strategy allow the synthesis of novel, 4-disubstituted-1,2,3-triazole derivatives with desired properties and is currently being explored in our laboratory for the development of kinase inhibitors.
Acknowledgements
Financial support was obtained from the European Commission (the QUASI project, contract LSHG-CT2003-503230 and the CELLCOMPUTE project, contract 040451).
Footnotes
Sample Availability: Samples of the compounds are available from the authors.
References and Notes
- 1.Fan W.Q., Katritzky A.R. 1,2,3 Triazoles. In: Katritzky A.R., Rees C.W., Scriven E.F.V., editors. Comprehensive Heterocycle Chemistry II. Vol. 4. Pergamon Press; New York, NY, USA: 1996. pp. 1–126. [Google Scholar]
- 2.Katritzky A.R., Zhang Y., Singh S.K. 1,2,3-Triazole formation under mild conditions via 1,3-dipolar cycloaddition of acetylenes with azides. Heterocycles. 2003;60:1225–1239. doi: 10.3987/REV-02-562. [DOI] [Google Scholar]
- 3.Alvarez R., Velazquez S., San R., Aquaro S., De C.E., Perno C.F., Karlsson A., Balzarini J., Camarasa M.J. 1,2,3-Triazole-[2',5'-bis-O-(tert-butyldimethylsilyl)-beta-D- ribofuranosyl]-3'-spiro-5"-(4"-amino-1",2"-oxathiole 2",2"-dioxide) (TSAO) analogues: Synthesis and anti-HIV-1 activity. J. Med. Chem. 1994;37:4185–4194. doi: 10.1021/jm00050a015. [DOI] [PubMed] [Google Scholar]
- 4.Velazquez S., Alvarez R., Perez C., Gago F., De C.E., Balzarini J., Camarasa J.M. Regiospecific synthesis and anti-human immunodeficiency virus activity of novel 5-substituted N-alkylcarbamoyl and N,N-dialkylcarbamoyl 1,2,3-triazole-TSAO analogues. Antiviral Chem. Chemother. 1998;9:481–489. doi: 10.1177/095632029800900604. [DOI] [PubMed] [Google Scholar]
- 5.Whiting M., Muldoon J., Lin Y.C., Silverman S.M., Lindstrom W., Olson A.J., Kolb H.C., Finn M.G., Sharpless B.K., Elder J.H., Fokin V.V. Inhibitors of HIV-1 protease by using in situ click chemistry. Angew. Chem. Int. Ed. 2006;45:1435–1439. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]
- 6.Brik A., Muldoon J., Lin Y.C., Elder J.C., Goodsell D.S., Olson A.J., Fokin V.V., Sharpless B.K., Wong C.H. Rapid diversity-oriented synthesis in microtiter plates for in situ screening of HIV protease inhibitors. Chem. Bio. Chem. 2003;4:1246–1248. doi: 10.1002/cbic.200300724. [DOI] [PubMed] [Google Scholar]
- 7.Genin M.J., Allwine D.A., Anderson D.J., Barbachyn M.R., Emmert D.M., Garmon S.A., Graber D.R., Grega K.C., Hester J.B., Hutchinson D.K., Morris J., Reischer R.J., Ford C.W., Zurenko G.E., Hamel J.C., Schaadt R.D., Stapert D., Yagi B.H. Substituent effects on the antibacterial activity of nitrogen-carbon-linked (azolylphenyl)oxazolidinones with expanded activity against the fastidious gram-negative organisms Haemophilus influenzae and Moraxella catarrhalis. J. Med. Chem. 2000;43:953–970. doi: 10.1021/jm990373e. [DOI] [PubMed] [Google Scholar]
- 8.Brockunier L.L., Parmee E.R., Ok H.O., Candelore M.R., Cascieri M.A., Colwell L.F., Deng L., Feeney W.P., Forrest M.J., Hom G.J., MacIntyre D.E., Tota L., Wyvratt M.J., Fisher M. H., Weber A.E. Human beta3-adrenergic receptor agonists containing 1,2,3-triazole-substituted benzenesulfonamides. Bioorg. Med. Chem. Lett. 2000;10:2111–2114. doi: 10.1016/S0960-894X(00)00422-4. [DOI] [PubMed] [Google Scholar]
- 9.Pande V., Ramos M.J. Structural basis for the GSK-3beta binding affinity and selectivity against CDK-2 of 1-(4-aminofurazan-3yl)-5-dialkylaminomethyl-1H-[1,2,3]triazole-4-carboxylic acid derivatives. Bioorg. Med. Chem. Lett. 2005;15:5129–5135. doi: 10.1016/j.bmcl.2005.08.077. [DOI] [PubMed] [Google Scholar]
- 10.Olesen P.H., Sørensen A.R., Ursö B., Kurtzhals P., Bowler A.N., Ehrbar U., Hansen B.F. Synthesis and in vitro characterization of 1-(4-Aminofurazan-3-yl)-5-dialkylaminomethyl-1H-[1,2,3]triazole-4-carboxylic acid derivatives. A new class of selective GSK-3 Inhibitors. J. Med. Chem. 2003;46:3333–3341. doi: 10.1021/jm021095d. [DOI] [PubMed] [Google Scholar]
- 11.Krasinski A., Radic Z., Manetsch R., Raushel J., Taylor P., Sharpless B.K., Kolb H.C. In situ selection of lead compounds by click chemistry: Target-guided optimization of aceylcholinesterase inhibitors. J. Am. Chem. Soc. 2005;127:6686–6692. doi: 10.1021/ja043031t. [DOI] [PubMed] [Google Scholar]
- 12.Mocharla V.P., Colasson B., Lee L.V., Roeper S., Sharpless B.K., Wong C.H., Kolb H.C. In situ click chemistry: Enzyme-generated inhibitors of carbonic anhydrase II. Angew. Chem. Int. Ed. 2005;44:116–120. doi: 10.1002/anie.200461580. [DOI] [PubMed] [Google Scholar]
- 13.Micetich R.G., Maiti S.N., Spevak P., Hall T.W., Yamabe S., Ishida N., Tanaka M., Yamazaki T., Nakai A., Ogawa K. Synthesis and .beta.-lactamase inhibitory properties of 2.beta.-[(1,2,3-triazol-1-yl)methyl]-2.alpha.-methylpenam-3.alpha.-carboxylic acid 1,1-dioxide and related triazolyl derivatives. J. Med. Chem. 1987;30:594–601. doi: 10.1021/jm00391a032. [DOI] [PubMed] [Google Scholar]
- 14.Actor P., Uri J.V., Phillips L., Sachs C.S., Zajac J.R.G., Berges D.A., Dunn G.L., Hoover J.R.E., Weisbach J.A. Laboratory studies with cefatrizine (SK + F 60771), a new broad-spectrum orally-active cephalosporin. J. Antibiot. 1975;28:594–601. doi: 10.7164/antibiotics.28.594. [DOI] [PubMed] [Google Scholar]
- 15.Sidwell R.W., Huffman J.H., Khare G.P., Allen L.B., Witkowski J.T., Robins R.K. Broad-spectrum antiviral activity of Virazole: 1-Beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science. 1972;177:705–706. doi: 10.1126/science.177.4050.705. [DOI] [PubMed] [Google Scholar]
- 16.Terasaka T., Kinoshita T., Kuno M., Nakanishi I. A highly potent non-nucleoside adenosine deaminase inhibitor: Efficient drug discovery by intentional lead hybridization. J. Am. Chem. Soc. 2004;126:34–35. doi: 10.1021/ja038606l. [DOI] [PubMed] [Google Scholar]
- 17.Terasaka T., Kinoshita T., Kuno M., Seki N., Tanaka K., Nakanishi I. Structure-based design, synthesis, and structure-activity relationship studies of novel non-nucleoside adenosine deaminase inhibitors. J. Med. Chem. 2004;47:3730–3743. doi: 10.1021/jm0306374. [DOI] [PubMed] [Google Scholar]
- 18.Terasaka T., Okumura H., Tsuji K., Kato T., Nakanishi I., Kinoshita T., Kato Y., Kuno M., Seki N., Naoe Y., Inoue T., Tanaka K., Nakamura K. Structure-based design and synthesis of non-nucleoside, potent, and orally bioavailable adenosine deaminase inhibitors. J. Med. Chem. 2004;47:2728–2731. doi: 10.1021/jm0499559. [DOI] [PubMed] [Google Scholar]
- 19.Terasaka T., Tsuji K., Kato T., Nakanishi I., Kinoshita T., Kato Y., Kuno M., Inoue T., Tanaka K., Nakamura K. Rational design of non-nucleoside, potent, and orally bioavailable adenosine deaminase inhibitors: predicting enzyme conformational change and metabolism. J. Med. Chem. 2005;48:4750–4753. doi: 10.1021/jm050413g. [DOI] [PubMed] [Google Scholar]
- 20.Benarroch D., Egloff M.P., Mulard L., Guerreiro C., Romette J.L., Canard B. A structural basis for the inhibition of the NS5 dengue virus mRNA 2′-O-methyltransferase domain by ribavirin 5′-triphosphate. J. Biol. Chem. 2004;279:35638–35643. doi: 10.1074/jbc.M400460200. [DOI] [PubMed] [Google Scholar]
- 21.Tornøe C., Christensen C., Meldal M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002;67:3057–3064. doi: 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- 22.Rostovtsev V.V., Green L.G., Fokin V.V., Sharpless B.K. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective ligation of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 23.Laufer S.A., Domeyer D.M., Scior T.R.F., Albrecht W., Hauser D.R.J. Synthesis and biological testing of purine derivatives as potential ATP-competitive kinase inhibitors. J. Med. Chem. 2005;48:710–722. doi: 10.1021/jm0408767. [DOI] [PubMed] [Google Scholar]
- 24.Zaloom J., Roberts D.C. Preparation of azido derivatives from amino acids and peptides by diazo transfer. J. Org. Chem. 1981;46:5173–5176. doi: 10.1021/jo00338a022. [DOI] [Google Scholar]
- 25.Vasella A., Witzig C., Chiara J.L., Lomas M.M. Convenient synthesis of 2-Azido-2-deoxy-aldoses by diazo transfer. Helv. Chim. Acta. 1991;74:2073–2077. doi: 10.1002/hlca.19910740842. [DOI] [Google Scholar]
- 26.Hedenström M., Holm L., Yuan Z.Q., Emtenas H., Kihlberg J. Stereoselective Synthesis of Ψ[CH2O] Pseudodipeptides and Conformational Analysis of a PheΨ[CH2O]Ala Containing Analogue of the Drug Desmopressin. Bioorg. Med. Chem. Lett. 2002;12:841–844. doi: 10.1016/S0960-894X(02)00016-1. [DOI] [PubMed] [Google Scholar]
- 27.Hedenström M., Yuan Z., Brickmann K., Carlsson J., Ekholm K., Johansson B., Kreutz E., Nilsson A., Sethson I., Kihlberg J. Conformations and receptor activity of desmopressin analogues, which contain gamma-turn mimetics or a psi[CH(2)O] isostere. J. Med. Chem. 2002;45:2501–2511. doi: 10.1021/jm011073b. [DOI] [PubMed] [Google Scholar]
- 28.Ramanathan S.K., Keeler J., Lee H.L., Reddy D.S., Lushington G., Aube J. Modular synthesis of cyclic peptidomimetics inspired by γ-turns. Org. Lett. 2005;7:1059–1062. doi: 10.1021/ol047323a. [DOI] [PubMed] [Google Scholar]
- 29.Gerard B., Ryan J., Beeler A.B., Porco J.A., Jr. Synthesis of 1,4,5-trisubstituted-1,2,3-triazoles by copper-catalyzed cycloaddition-coupling of azides and terminal. Tetrahedron. 2006;62:6405–6411. doi: 10.1016/j.tet.2006.04.025. [DOI] [Google Scholar]
- 30.Wang Q., Chan T.R., Hilgraf R., Fokin V.V., Sharpless B.K., Finn M.G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 2003;125:3192–3193. doi: 10.1021/ja021381e. [DOI] [PubMed] [Google Scholar]
- 31.Himo F., Lovell T., Hilgraf R., Rostovtsev V.V., Noodleman L., Sharpless B.K., Fokin V.V. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 2005;127:210–216. doi: 10.1021/ja0471525. [DOI] [PubMed] [Google Scholar]
- 32.Chan T.R., Hilgraf R., Sharpless B.K., Fokin V.V. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004;6:2853–2855. doi: 10.1021/ol0493094. [DOI] [PubMed] [Google Scholar]
- 33.Merlo A.A., Fernandes M.S. An efficient strategy for the synthesis of chiral liquid crystals using Evans' methodology. Synth. Commun. 2003;33:1167–1178. doi: 10.1081/SCC-120017193. [DOI] [Google Scholar]
- 34.Park C.S., Kim M.S., Sim T.B., Pyun D.K., Lee C.H., Choi D., Lee W.K., Chang J.W., Ha H.J. Novel stereoselective synthesis of functionalized oxazolidinones from chiral aziridines. J. Org. Chem. 2003;68:43–49. doi: 10.1021/jo025545l. [DOI] [PubMed] [Google Scholar]
- 35.Welch J.T., Seper K.W. Synthesis, regioselective deprotonation, and stereoselective alkylation of fluoro ketimines. J. Org. Chem. 1988;53:2991–2999. doi: 10.1021/jo00248a017. [DOI] [Google Scholar]
- 36.Danklmaier J., Hoenig H. Synthese diastereomerenreiner 2,5-disubstituierter 3-Oxoperhydro-1,4-oxazine. Liebigs Ann. Chem. 1988:851–854. doi: 10.1002/jlac.198819880906. [DOI] [Google Scholar]
- 37.Macromodel [9.1] Schrödinger, LLC; New York, NY, USA: 2007. [Google Scholar]
- 38.Glide [4.0] Schrödinger, LLC; New York, NY, USA: 2005. [Google Scholar]
- 39.Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006;49:6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]