

Abstract
The establishment of Leishmania infection in mammalian hosts and the subsequent manifestation of clinical symptoms require internalization into macrophages, immune evasion and parasite survival and replication. Although many of the genes involved in these processes have been described, the genetic and genomic variability associated to differences in virulence is largely unknown. Here we present the genomic variation of four Leishmania (Viannia) panamensis strains exhibiting different levels of virulence in BALB/c mice and its application to predict novel genes related to virulence. De novo DNA sequencing and assembly of the most virulent strain allowed comparative genomics analysis with sequenced L. (Viannia) panamensis and L. (Viannia) braziliensis strains, and showed important variations at intra and interspecific levels. Moreover, the mutation detection and a CNV search revealed both base and structural genomic variation within the species. Interestingly, we found differences in the copy number and protein diversity of some genes previously related to virulence. Several machine-learning approaches were applied to combine previous knowledge with features derived from genomic variation and predict a curated set of 66 novel genes related to virulence. These genes can be prioritized for validation experiments and could potentially become promising drug and immune targets for the development of novel prophylactic and therapeutic interventions.
Introduction
Leishmaniasis is a group of neglected tropical diseases caused by parasites belonging to the Leishmania genus, affecting around 14 million people worldwide and with 350 million people at risk of infection. There are three main forms of the disease: cutaneous leishmaniasis (CL), mucocutaneous leishmaniasis and visceral leishmaniasis1–3. Whether one of those clinical forms or the asymptomatic infection is developed, depends on a complex interaction between host- and parasite-derived factors. A better understanding the mechanisms of pathogenesis and immunity in Leishmania infections is crucial for the study of parasite-host interaction and the development of new therapies and vaccines for leishmaniasis.
Comparative genomic analysis is a powerful tool to discover genetic features that might underlie the variability in pathogenesis and clinical manifestations among different forms of leishmaniasis. Genome comparison between Leishmania donovani and L. major allowed to identify genes involved in virulence and tissue tropism after infections in animal models4,5. Genome sequencing has also allowed the identification of genes associated with the intracellular amastigote stage in the pathogenic Leishmania species6. Similarly, whole genome sequencing (WGS) confirmed the deletion of virulence genes in genetically modified strains of L. donovani with attenuated virulence7. Moreover, genomic variations such as aneuploidies, single nucleotide polymorphisms (SNPs) and structural variants (SVs) such as copy number variation (CNV) can affect the presence, dosage, and consequently the expression of alleles of genes related to virulence. SNPs, CNVs and aneuploidies were suggested as drivers of tropism towards cutaneous or visceral tissue and virulence in L. donovani8. In addition, WGS also showed differences in SNPs between strains of Trypanosoma brucei that generate chronic or acute infections9.
Recent years, the importance of parasite virulence factors has become evident. Supplementary Table S1 shows a list of 94 genes reported to be involved in virulence or up-regulated in the amastigote stage. Although these genes have been experimentally associated to processes necessary to establish infection and response to different pressures or stress, the variability of these genes among and within Leishmania species is largely unknown.
Leishmania species belonging to the Viannia subgenus, such as Leishmania (Viannia) panamensis, are major causal agents of American cutaneous leishmaniasis (ACL). A wide spectrum of clinical manifestations caused by this group of parasites has been reported in humans1,10 and animal models11. This variation is attributed to variability not only in the host immune response but also in parasite virulence12–16. To investigate the mechanisms that mediate virulence in the ACL caused by L. panamensis, we report here the genome of the virulent UA946 strain, and the genomic variability between this and another three L. panamensis strains exhibiting different levels of virulence in BALB/c mice. Moreover, we compare the assembled genome with the previously reported draft genome for the species and the Viannia reference genome L. braziliensis. Following machine learning approaches we predict new possible genes involved in virulence. Our results suggest that differences in dosage of some genes involved in virulence and allelic diversity through single nucleotide mutations may be determinant in the level of virulence of these strains in the murine model.
Results
L. panamensis strains exhibit different levels of virulence in BALB/c mice
The availability of a collection of L. panamensis strains with a range of virulence levels in mice, motivated us to perform the genomic comparative study reported here. We began by comparing four strains together in the same experiment. All BALB/c mice infected with UA946 develop lesions that progressed to large ulcers within 8 weeks, whiles mice infected with UA140 develop no lesion or a mild disease (Fig. 1A–E). Interestingly, the disease induced by the strains UA1114 and UA1511 was intermediate between UA946 and UA140, as determined by the size of the lesion and the severity score (Fig. 1A–E). When the parasitic loads at the site of infection (a parameter that reflects in vivo parasite multiplication) were determined, a striking correlation with the clinical behaviour was observed (Fig. 1F). Thus, the integration of the three parameters, namely lesion size, score and parasitic load, permitted to confirm that the four strains presented one of the following three patterns of virulence: high (UA946), moderate (UA1114 and UA1511) or low (UA140) (Table 1). An independent in vivo second experiment performed under similar conditions, revealed very similar results (Supplementary Fig. S1). Kruskal-Wallis test showed significant differences (p < 0,005) between strains behavior in size of the lesion and severity score data. Mann-Whitney test with Bonferroni corrections of p value to do pairwise comparisons showed significant differences between UA946 strain and the other three strains tested. Also, non-significant differences between the strains UA1114 and UA1511 for the two variables were observed. The parasite load was different between UA946 and UA140 (Supplementary Table 1).
Figure 1.

Virulence of four L. panamensis strains in BALB/c mice. BALB/c mice were infected as described in material and methods and the size of the lesion was determined weekly (A) or at the 8th week post-infection and reported in individually (B). The severity score was also monitored weekly (C) or 8 weeks post-infection in individual mice (D). Representative photographs of the infected ears at the end of the experiment (8th week post-infection) are presented for each experimental group (E). Parasitic loads records (F). Graphs show the mean+/−SEM (A,B,C), the median (D) or the geometric mean +/− 95% CI (F).
Table 1.
Virulence of the four strains of L. panamensis used in the present study according to the Mean (Median) ± SD values from the experimental variables.
| Strain | Lesion intensity scale | Lesion area (mm2) | Parasitic load (parasites/ear) | Virulence (Inferred) |
|---|---|---|---|---|
| UA140 | 1, 0 (1) ± 0, 8 | 3, 2 (1, 8) ± 3, 6 | 4 × 1010 (4 × 105) ± 8 × 1010 | Low |
| UA1114 | 1, 8 (2) ± 0, 8 | 9, 5 (6, 9) ± 9, 8 | 9 × 1010 (7 × 1010) ± 8 × 1010 | Moderate |
| UA1511 | 1, 9 (2) ± 0, 9 | 8, 7 (7, 7) ± 7, 5 | 8 × 1010 (7 × 1010) ±9 × 1010 | Moderate |
| UA946 | 2, 6 (3) ± 1 | 15, 4 (12, 5) ± 12 | 2 × 1011 (2 × 1011) ±6 × 1010 | High |
This confirmed not only the value of the UA946 strain for genome sequencing, but also the sequencing of the other three strains for comparative genomics analysis.
The genome of the virulent UA946 strain of L. panamensis
Based on the evaluation shown above, the virulent L. panamensis UA946 strain was selected for genome sequencing and de-novo assembly, aiming to use it as reference for comparison to less virulent strains. Using a hybrid sequencing strategy we achieved a chromosome-level assembly for UA946 spanning 31,312,330 bp of genome size, distributed in 35 chromosomes. This is 2% larger than the genome of strain PSC-1 previously reported for the species (30,688,794 bp)17. Gene annotation revealed 8,094 gene models from which 8,034 showed high conservation relative to the annotated genes of L. braziliensis (M2904) and L. panamensis (PSC-1). The 60 new gene models have an average length of 642 bp, Codon Adaptation Index (CAI)18 of 0.68 (Supplementary Table S2), usage codon according to L. braziliensis genes and transcriptome mapping of amastigote and promastigote stage. One of the new gene models was located in a gap in the genome of strain PSC-1 and another two genes overlap a predicted deletion in the PSC-1 genome. However, these genes were not completely missing as indicated by the alignment of raw Illumina reads from PSC-1 to the gene models annotated in the UA946 assembly. One UA946 gene was partially deleted in PSC-1. Seven UA946 genes have sequences in PSC-1 without open reading frames (ORFs) and finally 49 genes have ORFs in PSC-1 without any annotation. 56% of the 8,094 genes correspond to “hypothetical protein, conserved” genes, 6% to “hypothetical protein, unknown function” genes, 1% to undefined genes and 37% to known genes (Fig. 2A). We also performed a reciprocal blast of the gene annotations to identify paralogous genes (Supplementary Table S3).
Figure 2.

Functional annotation and synteny with L. braziliensis and L. panamensis genomes. (A) Distribution of ontologies of genes annotated in the UA946 assembly. (B) Structural comparison between the assemblies of the L. panamensis strains PSC-1 and UA946. (C) Structural comparison between the assembly of the L. braziliensis strain M2904 and the L. panamensis strain UA946.
We performed pairwise synteny comparisons between the L. panamensis UA946 strain assembly and the assemblies of L. braziliensis M2904 and L. panamensis PSC-1 strains. Consistent with previous comparisons17, the three genomes were in general highly co-linear (Fig. 2B,C). As expected, the comparison between the assemblies of M2904 and UA946 strains revealed 365 potential structural variation events (SVs), whereas the comparison between the assemblies of L. panamensis strains only showed 111 potential SVs. However, half of the predicted SVs between M2904 and UA946 correspond to DNA that could not be placed in the correct contig segment of the M2904 assembly (First horizontal sequence in Fig. 2C).
We also aligned raw Illumina reads taken from the strains PSC-1 and M2904 to the UA946 assembly to look for confirmation signals of the structural events identified by the synteny analysis (See methods for details). As expected by the genetic distance between the strains, 80% of the PSC-1 reads and 71% of the L. braziliensis M2904 reads aligned to the UA946 genome. As a first attempt to find genes that could be related to virulence, we investigated insertions that could possibly include private UA946 genes and could be functionally related to virulence. Relative to the previously assembled genomes, we found 22 large (>200 bp) insertions in UA946 relative to PSC-1 confirmed by mapping (Supplementary Table S4). The two largest insertions (6,875 bp and 5,169 bp) located on chromosomes 19 and 29, respectively, completely cover two annotated genes: a COA ligase like protein and a tuzin. Most of the other insertions are shorter (less than 500 bp) and only one insertion partially spanned an annotated hypothetical protein. The same comparison procedure applied against the L. braziliensis strain M2904 revealed a larger number of 178 insertions (Supplementary Table S5, Supplementary File 1, Fig. 2). However, only 18 insertions completely covered annotated genes, 9 of them annotated as hypothetical proteins. Validation of the predicted insertions using alignments of short reads showed that none of the predicted insertions covering annotated genes shows the signatures of a novel DNA element. Hence, the insertions of complete genes identified by whole genome alignments were caused by copy number expansion of genes already present in the genome of M2904 and PSC-1 strains.
Sequencing of L. panamensis strains with different levels of virulence
To compare the genomic variation among the different L. panamensis strains that are less virulent than UA946, Illumina sequencing was also performed on the strains UA140, UA1114 and UA1511. Raw reads obtained from each sample were independently aligned to the UA946 assembly. The alignment rates for these samples were in a range between 88 and 94% (Supplementary File 1, Fig. 3).
Based on Illumina read alignments to the UA946 assembly, we identified 656,782 variants, including copy number variants (CNVs, Supplementary File 2) and single nucleotide polymorphisms (SNPs, Supplementary File 3) in six strains including L. panamensis PSC-1 and L. braziliensis M2904 alignments for comparison in downstream analysis. These variants were distributed in 593,361 (90.34%) biallelic SNPs, 42,376 (6.45%) biallelic indels and 21,045 (3.21%) multiallelic variants. The high average read depth (>80x) of these 6 samples allowed to obtain accurate individual genotype calls for each variant. As expected, 88.6% of the SNPs and 79.4% of the indels correspond to differences interspecific between L. braziliensis and L. panamensis (Supplementary Table S6). Removing the L. braziliensis strain to identify polymorphisms within L. panamensis the total number of variants becomes 68,992 (10.5% of the total) distributed in 51,267 (74.31%) SNPs, 5,358 (7.77%) biallelic indels and 12,367 (17.92%) multiallelic variants. The percentage of biallelic SNPs located within protein coding regions (30% to 35%) is larger than the same percentage for other types of variants (4% to 5%). Including L. braziliensis, the total number of variants producing stop codons was 783 nonsense SNPs and 724 frameshift indels. Within L. panamensis, these types of mutations were reduced to 140 nonsense SNPs and 118 frameshift indels.
We estimated ploidy levels and compared them with the previously reported aneuploidies for the L. braziliensis19 and L. panamensis17 sequenced strains. Consistent with previous reports, this analysis shows that the five sequenced L. panamensis strains are predominantly diploid but the L. braziliensis strain is predominantly triploid (Fig. 3A). The overall distribution of relative allele frequencies (estimated from read counts) across the genome is clearly centred at 0.33 for the L. braziliensis strain M2904 (Fig. 3B). This indicates that the allele frequencies of heterozygous mutations should be predominantly in 2:1 proportions. For the L. panamensis strains, their allele distributions are flat with a small increase towards 0.5. This distribution combined with the low number of heterozygous SNPs identified for these strains indicates an overall ploidy of 2 with low levels of heterozygous. The main exception is chromosome 31, which shows a copy number of 4 for the 5 L. panamensis strains. Normalized average read depths per chromosome also show an increased ploidy in chromosomes of individual L. panamensis strains, including chromosome 23 of PSC-1, chromosome 29 of UA140, and chromosome 30 of UA946 (Fig. 3A).
Figure 3.
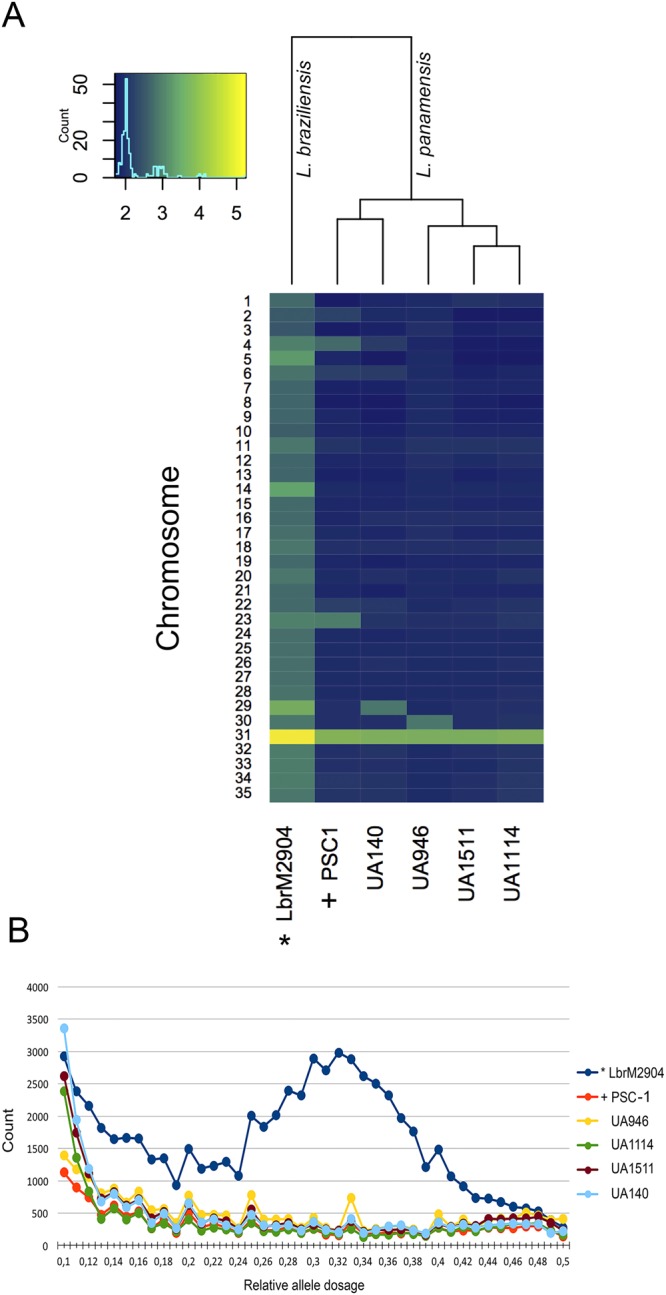
Comparison of ploidy between the four strains of L. panamensis with different virulence and reference genomes previously reported for the subgenus Viannia. (A) Normalized median read depth coverage using UA946 genome as reference. (B) Distribution of relative allele dosage among the six strains predicted from the relative allele counts at each site in the reference genome having base calls for at least two observed base pairs. *Reference genome of L. braziliensis reported19. +Reference genome of PSC-1 reported17.
Analysis of Copy Number Variation (CNV)
We performed a read depth analysis of the aligned data to identify regions of potential copy number variation (CNV) and predict the copy number (CN) for each gene within each L. panamensis sequenced strain (See methods for details). Considering that all L. panamensis strains look diploid, in this analysis the average read depth of each sample after correction for GC-content biases is related to the “normal” copy number (CN = 2) and significant local alterations of the average read depth are related to “abnormal” copy number. Combining the predictions for the five samples, we identified a set of 3,887 predicted CNVs spanning 12.6 Mbp of the genome (Supplementary Table S7). Consistent with the aneuploidies described above, predicted CNVs span more than 80% of the chromosomes 23, 29, 30 and 31, mainly because the span of duplications predicted separately for each strain reflects the copy number predicted from the relative read counts (Fig. 4A). In general, it is well known that the main source of false positive calls in any read depth analysis is the confounding effect of misalignments in repetitive regions of the genome. However, in this case, the assembly contains a very small portion of repetitive content for alignment purposes and hence 3,333 CNVs (85.8%) show less than 10% of intersection with repetitive elements.
Figure 4.

Analyses of duplications and copy number variation within L. panamensis genomes. (A) Percentage of each chromosome within each strain covered by predicted CNVs in which the copy number value (CN) is greater than 2 (e.g. duplications). (B) Distribution of CNVs by number of different observed CN values over the 5 strains for the complete CNV dataset and for the CNVs in non repetitive (NR) regions. (C) Distribution of predicted CN values on each strain for the complete CNVs dataset and of CNVs in non-repetitive (NR) regions. CNVs are classified by number of different CN values as fixed (only one value observed across the 5 samples) or non-fixed. (D) Differences in CN values among the 4 strains of L. panamensis evaluated with different virulence. *Genes involved in virulence or up-regulated in the amastigote state reported by different authors (Supplementary Table S1). The colours of the heatmap and the dendrogram were included with the purpose of highlighting the differences and similarity in CNVs among the strains analyzed. Reads of PSC-1 strain were included for comparison purposes17.
Comparing the predictions of copy number within each CNV and within each strain, we identified 2,012 CNVs (51.8% of the total) showing a “fixed” abnormal copy number (CN ≠ 2) over the five samples. A total of 1,668 CNVs (42.9%) showed two different CN values across the five samples. The 207 (5.3%) remaining CNVs showed between three and five different CN values (Fig. 4B). The latter two categories are called “non-fixed” hereafter. The distribution of CN values predicted over the five strains and across all CNVs shows that CN = 1 (heterozygous deletion) was most common, followed by normal copy number calls (CN = 2) (Fig. 4C).
From the 3,333 CNVs in non-repetitive regions, 1,871 (54.7%) span annotated genes and furthermore 888 CNVs (26%) completely cover at least one gene. Considering only the 1,471 non-fixated CNVs in non-repetitive regions, 1,047 CNVs (71.2%) span annotated genes and 654 CNVs (44,5%) completely overlap at least one gene. This is encouraging to further investigate relationships between gene copy number variation and virulence. As expected, 403 of the latter cases are annotated as hypothetical proteins. Eight of these genes show differences ≥3 CN between UA946 and UA140. Considering the 94 genes previously related to virulence (Supplementary Table S1), whereas in general 548 (6.78%) genes seem to be duplicated within UA946 (either by aneuploidies or by copy number variation) according to the read depth analysis, 36 (37.5%) of the 94 virulence genes seem to be duplicated in UA946. Moreover, eleven of these genes showed differences ≥3 CN between the virulent UA946 strain and the less virulent UA140 strain. Functional annotations of these genes include heat shock protein, beta-tubulin, pteridine transporter, histones, peptidases, ATG8, phosphatidic acid phosphatase, tuzin protein and GP63 or leishmanolysin. Predictions of copy number for other genes possibly involved in virulence according to their functional annotations and having differences ≥3 CN between UA946 and UA140 are shown in Fig. 4D.
Finally, the analysis was also useful to test the presence of minichromosomes or circular episomes as reported in the previous assemblies17,20. Duplications were consistently reported for all L. panamensis strains on a segment of 37 kbp within chromosome 34 (1,351,000–1,388,000), which corresponds to about 70% of a previously reported minichromosome17 aligned by homology search with BLAST between 1,340,652 to 1,388,378. This region is interesting because it harbours approximately 13 genes involved in biological regulation, localization, response to stimulus, signalling and single-organism process according to gene ontologies. A larger minichromosome of close to 100 Kbp at the end of the same chromosome previously reported by Llanes et al.17 was consistently supported by two long duplications in PSC-1 spanning 80% of the region between 1,884,400 and 1,985,800, where the minichromosome is identified in the sequence. However, in contrast with the previous case, copy number estimation of the four strains UA sequenced in this study supports reference CN = 2 alleles for this region. Sampaio et al., showed that the presence of a similar mini-chromosome found in L. braziliensis favours the survival and infectivity21. Nevertheless, the copy number estimation for this region in the virulent strain UA946 suggests that the number of copies of this minichromosome is not related to the virulence level observed in this study.
Gene diversity within L. panamensis and between L. panamensis and L. braziliensis
Using a filtered dataset of 14,793 SNPs genotyped on the five L. panamensis strains after removing repetitive regions or CNV regions for at least one strain, we built a neighbour-joining dendrogram to compare SNP based predicted genetic distances between the strains (Fig. 5A). The strain PSC-1 is clearly separated from the UA strains sequenced in this study. UA946 clusters together with strain UA1114 whereas the less virulent strains UA140 and UA1511 appear more separated. In contrast to the number of heterozygous variants, ranging from 2,000 to 3,000 for the five strains, the number of homozygous differences with UA946 is more than 2,500 for UA140 and UA1511 and grows to 7,653 for PSC-1.
Figure 5.
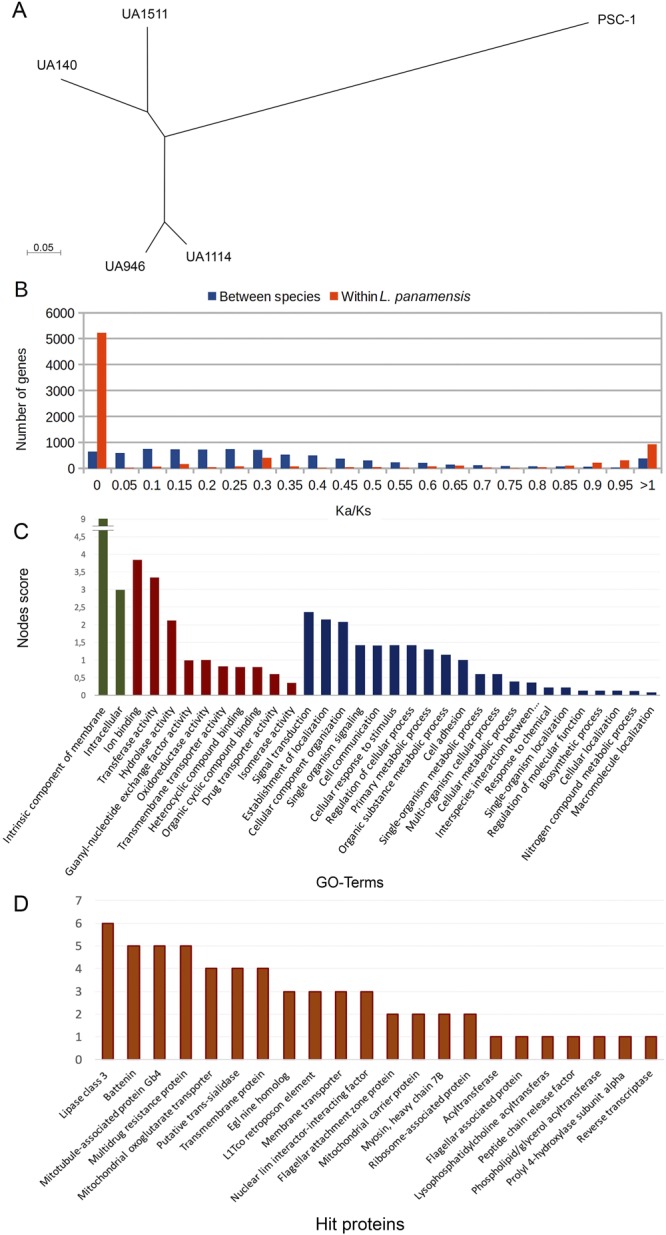
(A) Neighbor Joining dendrogram of L. panamensis strains based in Kimura 2-parameter distances (scale at bottom) expressed in units of number of base substitutions per site. The tree is drawn to scale with branch lengths, which is in the same units as those of the evolutionary distances inferred. (B) Distribution of Ka/Ks values estimated from SNPs in protein coding regions. Blue bars represent Ka/Ks values predicted from substitutions between L. braziliensis and L. panamensis. Red bars are Ka/Ks values predicted from polymorphisms within L. panamensis. (C) Gene ontology analysis at level 3 of the 81 hypothetical proteins possibly implicated in virulence according to machine learning techniques. Green: cellular component; red: molecular function; and blue, biological process. (D) Proteins identified in the annotation of the 81 hypothetical proteins that are part of 230 possible new genes associated to virulence (Blast2GO program).
Because the number of samples to assess variation in virulence is limited, it is generally difficult to identify associations between genetic variants and virulence levels using conventional techniques such as Genome-Wide Association Studies (GWAS) or Bulk Segregant Analysis (BSA). Hence, we investigated the patterns of protein allelic diversity between and within species that could be inferred from SNPs using the Ka/Ks ratio (also known as dN/dS) as a standardized measure22,23 and investigated if these patterns could provide information about genes related to virulence.
We estimated Ka/Ks values separately from SNPs differentiating L. braziliensis and L. panamensis (between species, also called substitutions) and from the SNPs identified only within L. panamensis (within species, also called polymorphisms). As expected based on the overall number of SNPs, Ka/Ks values between species showed an average of 0.36 calculated over the 7,854 genes with at least one synonymous mutation whilst within L. panamensis was 0.27, calculated over only 2,779 genes with at least one synonymous mutation. Figure 5B shows the distribution of Ka/Ks values in both datasets. Consistent with the distribution of SNPs, Ka/Ks values indicate that the observed variability between species is much larger than the variability within L. panamensis. Interestingly, the last four bars of the histogram seem to show an unexpectedly large number of genes with high Ka/Ks values within L. panamensis. A closer look to these categories revealed that they were mostly composed by 1,417 genes having zero synonymous mutations and a moderate number (1.43 on average) of non-synonymous mutations. The same pattern of variation was observed between species, but only in 118 genes, and with an average of 2.63 non-synonymous mutations.
Ka/Ks has been proposed as a statistic to test selection in protein evolution. Informally, under a Wright-Fisher model without selection, mutations in synonymous and non-synonymous sites should appear at a similar rate and hence the Ka/Ks ratio should be close to 123. Whereas values significantly smaller than 1 could indicate purifying selection, values larger than 1 could indicate positive selection. However, recent simulation studies show that this test should only be applied to substitutions between species because the assumption of sampling from divergent lineages is violated for polymorphisms within species24. Because other tests of intraspecies selection require large sample sizes, we only looked for signatures of positive selection from the SNPs differentiating L. braziliensis and L. panamensis. Only 7 genes showed Ka/Ks ratios significantly larger than 1, none of them previously related to virulence. Three of these genes were annotated respectively as a viscerotropic leishmaniasis antigen, an argininosuccinate synthase and a cysteine peptidase.
Taking into account the large overall amount of observed variability between species compared to that within species, we also investigated genes in which higher Ka/Ks values are observed within L. panamensis than between species. Whereas in general, these were only 2,188 (26.9%) genes, within the genes related to virulence, 38 (39.6%) present the same pattern of variability. This suggests that non-synonymous point mutations within virulence genes can be drivers of the observed variability in virulence within the sequenced strains. High Ka/Ks values were observed in virulence genes involved in intracellular transport, autophagy and cell remodelling, pteridine transport and gene expression. Whereas 36 virulence factors only showed variability between species, six surface protein genes were variable only within L. panamensis (Supplementary Table S8).
Machine learning to predict virulence genes from genomic variation of limited samples
Although Ka/Ks values within L. panamensis and between species, as well as predicted values of copy number showed interesting patterns within the genes related to virulence, none of the Ka/Ks ratios or estimations or copy number can be used independently as good predictors to completely distinguish virulence genes. Other annotated genes may be involved in virulence or may be important in the amastigote state. Moreover, more than 60% of the L. panamensis genes products are annotated as “hypothetical proteins”. However, the observed partial associations are encouraging to try to identify novel candidate genes for virulence using a supervised learning approach able to combine the partial information that seems to be provided by the different analyses of genomic variability. Hence, we tried several machine learning approaches to perform automated identification of virulence genes using as features information of gene diversity between and within species (non-synonymous mutations and Ka/Ks values), as well as predictions of copy number and heterozygous within individual strains (see Methods for details). Genes not previously related to virulence were tested against 200 models built running commonly used machine learning techniques (Support Vector Machines, Naive Bayes and Random forests) using different random subsets of genes as negative cases. As expected, each experiment predicted association with virulence for a different set of genes. Whereas the random forest approach consistently predicted relation to virulence for the largest number of genes (1113 on average), the support vector machine predicted virulence for the lowest number of genes (97 on average). Within each prediction model, almost all genes predicted by the support vector machine and over 80% of the genes predicted by naive Bayesian approaches were predicted by at least one additional method (Supplementary File 1, Fig. 4, Supplementary Table S9). Taking the union of all models, 230 genes (2.88%) were constantly associated to virulence in 100 or more models. 59 of these were homologous to previous list of 94 genes reported to be involved in virulence or up-regulated in amastigote stage and 80 genes were annotated as hypothetical proteins are predicted as new genes involved in virulence and represent new potential targets for antileishmania drug design. However, a homology search with BLAST itself found 14 genes duplicated which may not be good targets for drug design. An ontology analysis of 66 remaining genes using the Blast2GO tool25, showed the biological processes and molecular function in which these genes are involved. Most of these processes occur at the membrane level (Fig. 5C). Similarly, the annotation revealed by Blast2GO showed 18 proteins with diverse functions (Fig. 5D). Additionally, the use of the TargetP, SignalP and TMHMM programs showed that within the 66 possible new proteins involved in virulence, 14 are possibly located in the kinetoplast, 8 contain signal peptide, 4 have transmembrane helices, and two of them more than 10 helices (Supplementary Table S10).
Discussion
The virulence in leishmaniasis is not only the result of the genotypic characteristics of the strains, but it is also the result of the response of vertebrate host14,26,27. In the present study, all the parameters of experimental inoculation in mice were controlled. BALB/c mice were used, which are considered a susceptible strain to typical lesions of CL28, and a constant amount of inoculum of 105 promastigotes was used for all the experiments. A previous study in L. major showed that inoculations of 105–107 parasites produce large lesions in BALB/c mice unlike inoculations with fewer parasites29. Ear (intradermal) was also chosen as the site of inoculum. This site has been previously reported as capable to generate Th2 immune response, leading to the development of persistent lesions in infections with L. major30. However, the mosaicism31, constitutes a major methodological problem for the analysis of structural variation genomics in Leishmania. Prieto Barja et al.32, found that L. donovani is more aneuploid in vitro culture than in vertebrate host, establishing trisomy in chromosomes 5, 9, 23 and 26 in vitro passages. It is possible that the number of subpopulations of karyotypically different parasites in vitro culture increases. Likewise, there are fluctuations in allele frequencies during the in vitro culture, making it difficult to interpret the chromosome somy based on allele frequency. Therefore, it is important to perform DNA extraction in each passage. In this work all strains, before being subjected to whole genome sequencing, were isolated from the ear of the infected mice and grown in vitro by the same short period of time in order to minimize the effect of aneuploid variation on the analysis. In this regard, the experimental variables that could affect the results, different from the characteristics of the strains used, were controlled to the maximum. Previous studies have shown a relationship between the genotypic variation in strains of L. major and heterogeneity in size of the lesion and immune response generated in BALB/c mice under controlled conditions33–35.
This work is the first attempt to assess the role of genomics variation in virulence within the subgenus Viannia. Analysis of CNVs between the strain with increased virulence (UA946) and the strain with low virulence (UA140) showed 22 genes with variation in CN involved in survival and replication of amastigotes (Supplementary Table 1, Fig. 4D). Several studies had shown how the overexpression or depletion of some of them directly affect the virulence in BALB/c mice and cellular remodelling and differentiation, such as GP6336–40, glycoconjugates and GPI-anchored proteins secreted and/or expressed on the surface41,42, Beta 1,3 galactosyltransferase43–45, Phosphatidic acid phosphatase proteins46–48, amastin surface proteins49, biopterin transporter50–54, ATG855–60, and histone proteins61–63.
Whole-genome sequencing (WGS) studies in L. donovani strains from two regions in Ethiopia also found CNVs in genes involved in virulence or up-regulated in amastigote stage (Folate/biopterin transporter, Hydrophilic acylated surface protein, Mannosyltransferase, Amastin-like protein)64. Dumetz et al., demonstrated that Leishmania has the ability to pre-adapt to different stress conditions65. Strains of L. donovani studied have an intrachromosomal amplification of genes involved in resistance to pentavalent antimonials (Sb) that allow them to survive to the direct exposure to the maximum concentration of the drug. In this amplified fragment, in addition to genes involved in redox pathways and drug resistance, genes associated with virulence were also found (Hydrophilic acylated surface and Membrane-bound acid phosphatase proteins). The L. panamensis UA946 strain, sequenced in the present work, was isolated more than 15 years ago from a patient with cutaneous leishmaniasis and maintained in vivo passages in BALB/c mice. Hence, UA946 strain could have undergone a process of selection that favors infection in BALB/c. This would be a desired scenario for future studies of pathogenesis and could explain the results shown in this work.
One of the main factors thought to affect gene dosage is the chromosome copy number variation, which makes it possible to find copies of extra chromosomes due to the extensive aneuploidy confirmed at the population level in the genus Leishmania19,20. In this study, especially on chromosomes 29–31, a relationship between the ploidy variation shown in Fig. 3 and the CNVs data shown in Fig. 4 is obvious, with no apparent repeats bias (Fig. 4A). Downing, evaluating seventeen L. donovani isolates with sensitive and resistant phenotypes to pentavalent antimonial (SSG), did not find significant association between the observed aneuploidy and SSG resistant phenotype20. However, Dumetz, evaluating the modulation of aneuploidy as a primary strategy to adapt to drug pressures in L. donovani, found a direct association between the chromosomes copy numbers, dosage and expression of specific genes66. Moreover, evidence of intrachromosomal amplification as mechanism to generate CNVs in New World Leishmania has also been reported en L. amazonensis67 and in the Viannia subgenus in L. guyanensis68 and in the L. braziliensis – L. peruviana complex69.
The highly conserved protein core (7157), common among L. mexicana, L. infantum, L. major, L. braziliensis and L. panamensis17, the few species-specific genes reported in studies of comparative genomics analysis17,19,70–72, and the results shown here, suggest that the virulence is probably influenced by differences in gene expression and dosage of this common conserved protein core in Leishmania spp. Different authors suggest that, in the absence of transcriptional control in Leishmania, it is possible that mechanisms such as amplification20,68,73–75, gene duplication76 or modulation of aneuploidy66 have evolved as mechanisms for altering mRNA levels, generating an extensive phenotypic heterogeneity that is subjected to selective forces such as drug resistance20,73,77,78, or as shown here, subjected to adaptation to animal models. In infections in hamster L. donovani is more disomic in parasites that infect the liver compared to the spleen, demonstrating that aneuploidy variation is also dependent of infected tissue32. Similarly, clones of L. donovani from Ethiopian patients with HIV co-infection isolated from the skin and spleen of the same patient showed different karyotypes64. Aneuploidy equally affects the gene dosage and the selection of beneficial alleles against different environmental changes.
Studies in L. donovani, found SNPs associated with drug resistance, with allelic frequencies that increased progressively with the concentration of Potassium Antimony Tartrate (PAT), evidencing selection of genotypes under pressure65. Assessing virulence in Viannia subgenus in an experimental setting is a difficult task that could only be done for a small number of strains. Analysis of protein evolution through the Ka/Ks statistic has been useful in other studies to assess positive selection in genes associated with virulence in Trypanosoma79, Plasmodium80, Fasciola81, Zika82, among others. The Ka/Ks analysis reveals that genes implicated in virulence such as ADP ribosylation factor, amastin like protein, aminopeptidase, ATG8, cysteine peptidases, elongation factors, folate/biopterin transporter, GP63, histones, HSPs, ppg3, among others, show allelic variability in L. panamensis even within the low number of sequenced samples (Supplementary Table S8). Thus, genomic adaptation strategies such as amplification of gene copy number or protein evolution through single nucleotide mutations can be a response to pressures in Leishmania spp. Further analyses with larger intraspecies genetic variability and other models could assess if the protein evolution is guided by positive selection to confer resistance to the host immune system. From a statistical perspective, these relatively orthologous sources of partial association to virulence could be interpreted as features and combined using one of the several well known machine learning techniques to try to build a model that allows to prioritize genes for further functional association studies (Supplementary Table S9). Following this approach, we predicted 230 genes as novel candidates for relation to virulence. However, some genes homologous to virulence factors (Supplementary Table S1) were included in the output dataset predicted by different machine learning approach (ribosomal proteins, amastin surface proteins, tubulins, elongation factors, folate/biopterin transporters, heat shock proteins, histones, kinesins, metallo-peptidases, paraflagellar rod protein, phosphoglycan beta 1,3 galactosyltransferases, proteophosphoglycans, surface antigen protein, tryparedoxin peroxidase, tuzin proteins, among others), showing the efficiency of the predictions. Of the 230 genes, 81 were annotated as hypothetical proteins. Being about 60% of the genes in Leishmania annotated as a hypothetical, the results shown here propose new targets to be prioritized for evaluation in the search for novel antiparasite therapies. Similarly, the present study shows how the machine learning strategies supported in biological traits generate data applicable to parasitological studies. Moreover, the annotation made to the new 81 hypothetical proteins possibly implicated in virulence demonstrates the need to review the annotation of the current Leishmania genomes.
Finally, this study highlights the importance of deep sequencing and genomic structural variation analyses in exploring the virulence of L. panamensis strains. Using a combination of studies in vivo, bioinformatics analyses and machine learning, we provide valuable insights indicating that the virulence of L. panamensis could be studied by the CNVs and SNPs. Different studies have shown that the subgenus Viannia species are similar in their genetic variability83 and genomes84. Thus, the findings shown here could also be applicable to others species of panamensis/guyanensis/shawi cluster. Studies evaluating the progressive genomic changes that mediate the adaptation of L. panamensis to BALB/c mice, that measure the CNVs and the allelic frequencies of SNPs during the course of the infection, and its comparison with previously reported in other species are necessary to contribute to the study of genomic instability as a possible mechanism that favours the adaptation to tissues and tropism, the susceptibility to drugs and virulence or degree of pathogenicity.
Our data provide the baseline to understanding the virulence of L. panamensis strains, and future studies will require a validation with a higher number of strains and functional genomic approaches are expected to complement the results shown here, for a better understanding of the mechanisms that control infections with different virulence.
Methods
Animals, parasites and DNA preparation
Female, 6–10 weeks old BALB/c mice (Charles River, USA) were maintained in a SPF animal facility at the Sede de investigación universitaria (SIU), Universidad de Antioquia. Ethical approval for all in vivo procedures was obtained by the Animal Ethics Committee of the Universidad de Antioquia, Colombia. All animals were handled in strict accordance with good animal practice as defined by the Colombian Code of practice for the care and use of animals for scientific purposes, established by Law 84 of 1989. Four L. panamensis strains, coded as UA140, UA946, UA1114 and UA1511, isolated at the “Programa de estudio y control de enfermedades tropicales, PECET (Universidad de Antioquia, Medellin, Colombia)” from patients suffering ACL, were used in this study. The UA140 strain is routinely kept by long term in vitro passages with occasional in vivo passages, and it is essentially avirulent in BALB/c, since no or very small self-limited lesions are typically observed in most of mice after infection. The UA946 strain has been adapted to grow in BALB/c mice for years and reproducibly induces large cutaneous ulcerative lesions with eventual necrosis and mutilation in infected mice. The UA1114 and UA1511 isolates have been kept in vitro cultures and used to infect BALB/c mice in several rounds of serial infections (3–5 in vivo passages). These two strains induce cutaneous lesions in a less reproducible manner in BALB/c mice, with variable amounts of mice exhibiting nodular or ulcerative lesions. The species identity of the four isolates was confirmed by mAb and isoenzymes. Promastigotes were grown at 26 °C in NNN or Schneider’s Drosophila medium (Sigma, USA) supplemented with 10% heat-inactivated FCS and 2% filtered sterile human urine. Five to six-day cultures (early stationary phase) were used for all purposes. The DNA of the four strains was extracted from 109 promastigotes harvested in early stat phase of growth, using the DNEasy DNA Purification Kit (Qiagen). DNA quantity and quality was assessed by Nanodrop.
Infections, follow up and parasite burden determination
5–7 animals per group were infected into the right ear (id) with 105 stationary promastigotes (in 20 uL sterile PBS). The measurement of the size of the lesion was made calculating the diameter of the lesion, provided an accurate and representative measurement of growth of the lesions during the course of the infection85–87. Lesion measurements were performed weekly, by registering the two crossed diameters of lesions, and calculating the lesion area (in mm2) with the formula . As a complementary clinical follow up, a severity “Score” (scaling from 0 to 4) was also implemented based on the appearance of the lesion, as follows: Score 0: no apparent lesion. Score 1: ear with small nodular lesion with no clear ulceration. Score 2: large nodular lesion or small nodular lesion that begins to ulcerate. Score 3: Frank ulcer. Score 4: large ulcers with necrotic areas and/or mutilation. For parasitic loads determination, mice were sacrificed at the 8th week post-infection and the infected ears removed to be utilized to quantify the amount of viable parasites by using a limiting dilution assay88. Clinical (size of the lesion and Score) and parasitological (number of viable parasites per ear) parameters were recorded in individual mice, and the mean, median and +/− SD were calculated and compared among groups. Promastigote cultures were also prepared from the ears of UA946-infected mice to be used for DNA preparation and genome sequencing by 454 technology. In a second in vivo experiment, similar samples were obtained from UA946-, UA140-, UA1114- and UA1511-infected ears for genome sequencing using Illumina technology.
Genome sequencing and assembly of the high virulent UA946 L. panamensis strain
Genome sequencing of the UA946 strain for de-novo assembly was performed on a combination of two different protocols for whole genome sequencing (WGS): paired end 454 GS FLX titanium Shotgun (8 kbp insert size, mean read length 450 bp), and paired end Illumina HiSeq (350 bp insert size, 100 bp read length), SRA accession SRP154327, BioProject: PRJNA481617. The quality of the reads was assessed by FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and using PRINSEQ program89, 6% of the reads having an average Phred quality score lower than 30 were removed. De novo assembly was carried out with the program NEWBLER v2.990. Sequencing data generated for UA946 strain genome assembly represented an expected 22-fold median coverage assuming a genome size of 31 Mbp. De-novo assembly produced a high quality genome with: 90 scaffolds, 31.2 Mb of total length and N50 of 620 kb (average length 345 kb). The maximum length of a scaffold was 1,534,379 bp. We also performed Illumina whole genome resequencing for UA946 (paired-end read 2 × 101 bp). 91.62% of these reads were aligned to the 454 assembly, and these were used to iteratively correct errors in the consensus sequence by iteratively mapping reads to the sequences using iCORN91 and close gaps using IMAGE92 through 24 iterations with different k-mers. ICORN corrected 1847 base errors and indels in 12 iterations and from the 1,130 gaps (1,298,188 bp) initially observed in the 454 assembly, the IMAGE closed 806 gaps (150,552 bp) and obtaining a final genome size of 31,390,823 bp (Supplementary File 1. Table 2).
Sixty-six of the 90 assembled scaffolds (31′312,814 bp) were oriented and assigned to the 35 chromosomes using MUMmer v3.2393 and ABACAS94 programs and confirmed by 25 PCR reactions of adjacent scaffolds. Additionally, through BLASTn itself, ACT95 and orientation of paired end reads, the orientation of contigs within the scaffolds was verified. Per-base quality of the assembly was evaluated using the software bowtie2 v2.2.396, and QualStats and CoverageStats commands of NGSEP v3.1.097. Even at 3′ end of reads, the percentage of different bases with respect to the genome assembly does not exceed 1% and 2% in 454 and Illumina reads, respectively (Supplementary File 1, Fig. 5A,B). Alignments of 454 reads achieved a 21-fold median coverage and Illumina reads achieved a 112-fold median coverage (Supplementary File 1. Fig. 5C,D). Analysis of paired-end reads that were not properly aligned in pair, and PCR assays to identify potential misassembles revealed only three cases that were manually corrected. A complete summary of the genomic characteristics of the strain UA946 of L. panamensis and its comparison with other species of Leishmania spp., is shown in Supplementary File 1. Table 3. The other three strains with reduced virulence were sequenced by Illumina protocols for HiSeq 2 × 101 bp paired-end reads with 350 bp insert length achieving a raw average read depth per sample larger than 85x.
Identification of repetitive regions for read alignment in the UA946 assembly
We performed two separate analyses to identify regions that can be considered as repetitive for short read alignment purposes: a self homology search with BLAST for repetitive regions of at least 500 bp and the clustering algorithm implemented in NGSEP based on multiple alignments of short Illumina reads97. Predictions in the first dataset were largely (97.4%) contained in the second dataset. While the first method reported 839 Kbp of repetitive sequence (2.7% of the total genome size), the second method reported 1.16 Mbp of repetitive sequence (3.7% of the total). Low percentages (<8%) of repetitive content were observed across the 35 mapped chromosomes and a high percentage (almost 50%) of repetitive content was only observed in the 78 Kbp of sequence not assigned to a chromosome (Supplementary File 1. Fig. 6).
Genome gene annotation of a high virulence L. panamensis strain
The annotated L. braziliensis (M2904)70 and L. panamensis (PSC-1)17 reference genomes were transferred to the assembled virulent L. panamensis UA946 draft genome based on sequence conservation and synteny with RATT98. Gene structure and functional annotation were manually inspected and edited using the Artemis program99. To improve the L. panamensis genome, new gene models were identified by using a combination of CodonUsage, Codon Adaptation Index (CAI)18, BLASTx and transcriptome mapping of promastigotes/amastigotes reads (454 shotgun protocol) on Open Reading Frames (ORFs) exceeding 200 bp in length. Several programs were used for functional annotation of the new gene models. We used the InterPro scan application of Blast2GO25 to search protein domains. TargetP 1.1100, SecretomeP 2.0101, SignalP 4.1102, TMHMM 2.0103, were used to evaluate the cellular localization of the proteins inferred from the annotation process and if these proteins are secreted or are transmembrane.
Whole genome comparison between the genomes of UA946, PSC-1 and M2904
Whole genome comparisons between the assemblies of the L. panamensis strains UA946, PSC-1 and between UA946 and the assembly of the L. braziliensis strain M2904 were performed using the package Mummer97. To validate the structural events, in particular the insertions within UA946, predicted by the whole genome alignments described above, we aligned to the UA946 assembly publicly available Illumina reads of M2904 and PSC-1, as well as the Illumina reads of UA946 sequenced for this study. Then, the average read depth within the predicted insertions with the average read depth across the genome was compared. Real DNA insertions in one strain relative to another can either be caused by new DNA segments only present in one assembly or by relocations of mobile or repetitive elements. Only the first case should be supported by a significant reduction in read depth for an alignment of reads taken from the strains not having the new DNA segment. In contrast, insertions of mobile repetitive elements should not produce a reduction of read depth within the region. In both cases, read pairs aligning at a distance significantly larger than the library average insert length should flank real insertions. Supplementary File 1. Fig. 7 shows an example of a 1 Kbp insertion within UA946 for which zero coverage and a large number of read pairs with abnormally large predicted insert length are observed flanking the region with the predicted insertion.
Read alignment and variants identification
Reference guided analysis of the Illumina data for each of the six samples included in this study was performed with the NGSEP pipeline v3.1.097.Reads for each sample were aligned to the UA946 assembly using bowtie2 v2.2.396,with default parameters for paired-end reads except for the maximum number of alignments to keep for each read (-k parameter), which was set to 3. Alignments were sorted by reference coordinates using picard (https://broadinstitute.github.io/picard/). Plots of differences against the reference genome per read position and read depth distribution were obtained running the commands QualStats and CoverageStats of NGSEP. The FindVariants command of NGSEP was then executed for each sample with the recommended parameters for Illumina WGS data: 1) Minimum genotype quality 40; 2) Minimum value allowed for a base quality score 30; and 3) Maximum number of 2 alignments allowed to start at the same reference site. Merging of variants from the 6 samples were performed following the recommendations available in the NGSEP documentation, including the command MergeVariants to obtain the set of variants across the samples, the command FindVariants to genotype the variants obtained in the previous step on each sample and the command MergeVCF to assemble the final dataset of variants genotyped in the six samples in variant call format (VCF). The command FindVariants also produce for each sample the calls of Copy Number Variation (CNVs) described in the results section. Merging of these calls into a consolidated set of 3,978 regions affected by CNVs was performed using the same heuristic clustering and genotyping procedure described for characterization of CNVs in common bean104.
The NGSEP commands Annotate, FilterVCF, and VCFDistanceMatrixCalculator were used respectively to perform functional annotation of variants, filtering and construction of distance matrices. The un-rooted dendrogram shown in Fig. 5 was built using the neighbour joining algorithm from a distance matrix calculated from a filtered VCF file having SNPs within L. panamensis in non-repetitive regions of the genome, and genotyped in the 5 L. panamensis strains. These filters were enforced using the following options of the FilterVCF command of NGSEP: “-saf” to provide the ids of the L. panamensis strains, “-fi” to filter invariant sites within L. panamensis, “-minI” to keep variants genotyped in the 5 strains, and “-frs” to remove repetitive regions. The Annotate and VCFDistanceMatrixCalculatorCommands were executed with default parameters.
Counts of synonymous and non-synonymous sites per gene and Ka/Ks values were estimated directly from the VCF file and the reference genome following an approximate approach implemented in a custom script. In brief, for each position of each codon, point mutations are simulated and counted as synonymous if the corresponding aminoacid does not change, or non-synonymous if the corresponding aminoacid changes. All changes were considered equally probable, so this does not account for transition/transversion ratio or for codon usage.
Machine learning model to predict Virulence genes
Based on literature review, we selected a set of 94 genes, which were validated as known virulence genes or implicated in amastigote stage (Supplementary Table S1). The other genes were distributed in 50 random groups of 160 genes per group. Then, we built a total of 450 different binary classification models to infer genes related to virulence using the software tool Weka105. Each model is built using one of the 50 random groups as a negative dataset for virulence and running one of the following machine learning approaches implemented in Weka: IBk, a J48 tree, K-Star, default naive Bayes (NBDef), naive Bayes with a kernel density estimator for numerical values (NBKernel), Random Forest (RF), the default support vector machine (SVMDef), a support vector machine with conjugate gradient descent (SVMConjugate) and the ZeroR method which is recommended as a lower bound for the other methods. Selected features for each gene include the number of non-synonymous substitutions and the estimated Ka/Ks between L. braziliensis and L. panamensis and the Ka/Ks ratio estimated from synonymous and non-synonymous substitutions, the number of non-synonymous polymorphisms within L. panamensis, the Ka/Ks ratio estimated from synonymous and non-synonymous polymorphisms, the raw prediction of copy number for UA946, UA1114, UA140 and UA1511 and also the number of heterozygous SNPs within the same four strains (12 features in total). Cross validation was performed in each model to assess its the predictive accuracy. Both SVM methods yielded identical results and were the most conservative across models with low false positive rate (below 0.05) but also low true positive rate (0.14 on average). The naive Bayesian approaches showed a larger but still small false positive rate (below 0.05) compared to the SVM, increasing the true positive rate over 0.25. The random forest approach increased the true positive rate to 0.42 but also increased the false positive rate to 0.16. Because the other methods do not seem to provide further improvement over the random forest, we decided to perform prediction of virulence considering only the models built using the SVM, the two Naive Bayesian approaches and the random forest (200 models). The complete set of genes obtained after excluding the 94 genes related to virulence was provided to Weka for discovery of new genes related to virulence using each model. A gene was predicted as related to virulence if at least 101 of the 200 models classify the gene as related to virulence. This corresponds to an ensemble model in which the decision on classification is taken by majority vote. The number of models that classify each gene as related to virulence is reported in Supplementary Table S9.
Electronic supplementary material
Acknowledgements
This work was supported by Colciencias grant 11551929249; Universidad de Antioquia UdeA, and by PhD Studentship to DU by Colciencias.
Author Contributions
O.T.C. conceived the study; O.T.C., J.R.R.P. and J.C.D. conceived and planned the experiments; D.U., J.D., H.I., J.A. and J.G., conducted all the genomics and bioinformatics analyses; J.R.R.P. conceived, designed and analysed the experiments in BALB/c mice; N.M. and J.V. performed the experiments in mice; J.D. and J.G., conducted the machine learning experiments; D.U., J.D., H.I., J.G., J.C.D, J.R.R.P. and O.T.C., discussed the results and contributed to the final manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-35778-6.
References
- 1.Reithinger R, et al. Cutaneous leishmaniasis. Lancet Infect Dis. 2007;7:581–596. doi: 10.1016/S1473-3099(07)70209-8. [DOI] [PubMed] [Google Scholar]
- 2.World Health, O. Control of the leishmaniases. World Heal. Organ Tech Rep Ser xii–xiii, 1–186, back cover (2010). [PubMed]
- 3.Pace D. Leishmaniasis. J Infect. 2014;69(Suppl 1):S10–8. doi: 10.1016/j.jinf.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 4.Zhang WW, Peacock CS, Matlashewski G. A genomic-based approach combining in vivo selection in mice to identify a novel virulence gene in Leishmania. PLoS Negl Trop Dis. 2008;2:e248. doi: 10.1371/journal.pntd.0000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang X, Crippen TL, Coates CJ, Wood TK, Tomberlin JK. Effect of quorum sensing by Staphylococcus epidermidis on the attraction response of female adult yellow fever mosquitoes, Aedes aegypti aegypti (linnaeus) (diptera: Culicidae), to a blood-feeding source. PLoS One. 2015;10:1–15. doi: 10.1371/journal.pone.0143950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raymond F, et al. Genome sequencing of the lizard parasite Leishmania tarentolae reveals loss of genes associated to the intracellular stage of human pathogenic species. Nucleic Acids Res. 2012;40:1131–1147. doi: 10.1093/nar/gkr834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gannavaram S, et al. Whole genome sequencing of live attenuated Leishmania donovani parasites reveals novel biomarkers of attenuation and enables product characterization. Sci Rep. 2017;7:4718. doi: 10.1038/s41598-017-05088-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang WW, et al. Genetic analysis of Leishmania donovani tropism using a naturally attenuated cutaneous strain. PLoS Pathog. 2014;10:e1004244. doi: 10.1371/journal.ppat.1004244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goodhead, I. et al. Whole-genome sequencing of Trypanosoma brucei reveals introgression between subspecies that is associated with virulence. MBio4 (2013). [DOI] [PMC free article] [PubMed]
- 10.Handler MZ, Patel PA, Kapila R, Al-Qubati Y, Schwartz RA. Cutaneous and mucocutaneous leishmaniasis: Clinical perspectives. J Am Acad Dermatol. 2015;73:897–908. doi: 10.1016/j.jaad.2014.08.051. [DOI] [PubMed] [Google Scholar]
- 11.Martinez JE, Travi BL, Valencia AZ, Saravia NG. Metastatic capability of Leishmania (Viannia) panamensis and Leishmania (Viannia) guyanensis in golden hamsters. J Parasitol. 1991;77:762–768. doi: 10.2307/3282713. [DOI] [PubMed] [Google Scholar]
- 12.Convit J, et al. The clinical and immunological spectrum of American cutaneous leishmaniasis. Trans R Soc Trop Med Hyg. 1993;87:444–448. doi: 10.1016/0035-9203(93)90030-T. [DOI] [PubMed] [Google Scholar]
- 13.Grimaldi G, Tesh RB. Leishmaniases of the New World: current concepts and implications for future research. Clin Microbiol Rev. 1993;6:230–250. doi: 10.1128/CMR.6.3.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bañuls AL, Hide M, Prugnolle F. Leishmania and the leishmaniases: a parasite genetic update and advances in taxonomy, epidemiology and pathogenicity in humans. Adv Parasitol. 2007;64:1–109. doi: 10.1016/S0065-308X(06)64001-3. [DOI] [PubMed] [Google Scholar]
- 15.Cunha J, et al. Characterization of the biology and infectivity of Leishmania infantum viscerotropic and dermotropic strains isolated from HIV+ and HIV− patients in the murine model of visceral leishmaniasis. Parasit Vectors. 2013;6:122. doi: 10.1186/1756-3305-6-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Saporito L, Giammanco GM, De Grazia S, Colomba C. Visceral leishmaniasis: host-parasite interactions and clinical presentation in the immunocompetent and in the immunocompromised host. Int J Infect Dis. 2013;17:e572–6. doi: 10.1016/j.ijid.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 17.Llanes A, Restrepo CM, Del Vecchio G, Anguizola FJ, Lleonart R. The genome of Leishmania panamensis: insights into genomics of the L. (Viannia) subgenus. Sci Rep. 2015;5:8550. doi: 10.1038/srep08550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharp PM, Li WH. The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rogers MB, et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011;21:2129–2142. doi: 10.1101/gr.122945.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Downing T, et al. Whole genome sequencing of multiple Leishmania donovani clinical isolates provides insights into population structure and mechanisms of drug resistance. Genome Res. 2011;21:2143–2156. doi: 10.1101/gr.123430.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sampaio MC, et al. A 245 kb mini-chromosome impacts on Leishmania braziliensis infection and survival. Biochem Biophys Res Commun. 2009;382:74–78. doi: 10.1016/j.bbrc.2009.02.128. [DOI] [PubMed] [Google Scholar]
- 22.Kimura M. Preponderance of synonymous changes as evidence for the neutral theory of molecular evolution. Nature. 1977;267:275–276. doi: 10.1038/267275a0. [DOI] [PubMed] [Google Scholar]
- 23.Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conesa A, et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 26.Loeuillet C, Banuls AL, Hide M. Study of Leishmania pathogenesis in mice: experimental considerations. Parasit Vectors. 2016;9:144. doi: 10.1186/s13071-016-1413-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Handman E, Elso C, Foote S. Genes and susceptibility to leishmaniasis. Adv Parasitol. 2005;59:1–75. doi: 10.1016/S0065-308X(05)59001-8. [DOI] [PubMed] [Google Scholar]
- 28.Kurey I, et al. Distinct genetic control of parasite elimination, dissemination, and disease after Leishmania major infection. Immunogenetics. 2009;61:619–633. doi: 10.1007/s00251-009-0392-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bretscher PA, Wei G, Menon JN, Bielefeldt-Ohmann H. Establishment of stable, cell-mediated immunity that makes ‘susceptible’ mice resistant to Leishmania major. Science (80-.). 1992;257:539–542. doi: 10.1126/science.1636090. [DOI] [PubMed] [Google Scholar]
- 30.Baldwin TM, Elso C, Curtis J, Buckingham L, Handman E. The site of Leishmania major infection determines disease severity and immune responses. Infect Immun. 2003;71:6830–6834. doi: 10.1128/IAI.71.12.6830-6834.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sterkers Y, Lachaud L, Crobu L, Bastien P, Pagès M. FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in Leishmania major. Cell Microbiol. 2011;13:274–283. doi: 10.1111/j.1462-5822.2010.01534.x. [DOI] [PubMed] [Google Scholar]
- 32.Prieto Barja P, et al. Haplotype selection as an adaptive mechanism in the protozoan pathogen Leishmania donovani. Nat Ecol Evol. 2017;1:1961–1969. doi: 10.1038/s41559-017-0361-x. [DOI] [PubMed] [Google Scholar]
- 33.Asadpour A, Riazi-Rad F, Khaze V, Ajdary S, Alimohammadian MH. Distinct strains of Leishmania major induce different cytokine mRNA expression in draining lymph node of BALB/c mice. Parasite Immunol. 2013;35:42–50. doi: 10.1111/pim.12018. [DOI] [PubMed] [Google Scholar]
- 34.Alimohammadian MH, Darabi H, Ajdary S, Khaze V, Torkabadi E. Genotypically distinct strains of Leishmania major display diverse clinical and immunological patterns in BALB/c mice. Infect Genet Evol. 2010;10:969–975. doi: 10.1016/j.meegid.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 35.Kebaier C, Louzir H, Chenik M, Ben Salah A, Dellagi K. Heterogeneity of wild Leishmania major isolates in experimental murine pathogenicity and specific immune response. Infect Immun. 2001;69:4906–4915. doi: 10.1128/IAI.69.8.4906-4915.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brittingham A, et al. Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J Immunol. 1995;155:3102–3111. [PubMed] [Google Scholar]
- 37.Thiakaki M, Kolli B, Chang KP, Soteriadou K. Down-regulation of gp63 level in Leishmania amazonensis promastigotes reduces their infectivity in BALB/c mice. Microbes Infect. 2006;8:1455–1463. doi: 10.1016/j.micinf.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 38.Joshi PB, Kelly BL, Kamhawi S, Sacks DL, McMaster WR. Targeted gene deletion in Leishmania major identifies leishmanolysin (GP63) as a virulence factor. Mol Biochem Parasitol. 2002;120:33–40. doi: 10.1016/S0166-6851(01)00432-7. [DOI] [PubMed] [Google Scholar]
- 39.Seay MB, Heard PL, Chaudhuri G. Surface Zn-proteinase as a molecule for defense of Leishmania mexicana amazonensis promastigotes against cytolysis inside macrophage phagolysosomes. Infect Immun. 1996;64:5129–5137. doi: 10.1128/iai.64.12.5129-5137.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen DQ, et al. Episomal expression of specific sense and antisense mRNAs in Leishmania amazonensis: modulation of gp63 level in promastigotes and their infection of macrophages in vitro. Infect Immun. 2000;68:80–86. doi: 10.1128/IAI.68.1.80-86.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sacks DL, et al. The role of phosphoglycans in Leishmania-sand fly interactions. Proc Natl Acad Sci USA. 2000;97:406–411. doi: 10.1073/pnas.97.1.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Descoteaux A, Luo Y, Turco SJ, Beverley SM. A specialized pathway affecting virulence glycoconjugates of Leishmania. Science (80-.). 1995;269:1869–1872. doi: 10.1126/science.7569927. [DOI] [PubMed] [Google Scholar]
- 43.Dobson DE, et al. Functional identification of galactosyltransferases (SCGs) required for species-specific modifications of the lipophosphoglycan adhesin controlling Leishmania major-sand fly interactions. J Biol Chem. 2003;278:15523–15531. doi: 10.1074/jbc.M301568200. [DOI] [PubMed] [Google Scholar]
- 44.Dobson DE, et al. Leishmania major survival in selective Phlebotomus papatasi sand fly vector requires a specific SCG-encoded lipophosphoglycan galactosylation pattern. PLoS Pathog. 2010;6:e1001185. doi: 10.1371/journal.ppat.1001185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Butcher BA, et al. Deficiency inbeta1,3-galactosyltransferase of a Leishmania major lipophosphoglycan mutant adversely influences the Leishmania-sand fly interaction. J Biol Chem. 1996;271:20573–20579. doi: 10.1074/jbc.271.34.20573. [DOI] [PubMed] [Google Scholar]
- 46.Remaley AT, et al. Leishmania donovani: surface membrane acid phosphatase blocks neutrophil oxidative metabolite production. Exp Parasitol. 1985;60:331–341. doi: 10.1016/0014-4894(85)90039-6. [DOI] [PubMed] [Google Scholar]
- 47.Katakura K, Kobayashi A. Acid phosphatase activity of virulent and avirulent clones of Leishmania donovani promastigotes. Infect Immun. 1988;56:2856–2860. doi: 10.1128/iai.56.11.2856-2860.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandes AC, Soares DC, Saraiva EM, Meyer-Fernandes JR, Souto-Padron T. Different secreted phosphatase activities in Leishmania amazonensis. FEMS Microbiol Lett. 2013;340:117–128. doi: 10.1111/1574-6968.12080. [DOI] [PubMed] [Google Scholar]
- 49.Cruz MC, et al. Trypanosoma cruzi: role of delta-amastin on extracellular amastigote cell invasion and differentiation. PLoS One. 2012;7:e51804. doi: 10.1371/journal.pone.0051804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cunningham ML, Titus RG, Turco SJ, Beverley SM. Regulation of differentiation to the infective stage of the protozoan parasite Leishmania major by tetrahydrobiopterin. Science (80-.). 2001;292:285–287. doi: 10.1126/science.1057740. [DOI] [PubMed] [Google Scholar]
- 51.Nare B, Hardy LW, Beverley SM. The roles of pteridine reductase 1 and dihydrofolate reductase-thymidylate synthase in pteridine metabolism in the protozoan parasite Leishmania major. J Biol Chem. 1997;272:13883–13891. doi: 10.1074/jbc.272.21.13883. [DOI] [PubMed] [Google Scholar]
- 52.Bello AR, Nare B, Freedman D, Hardy L, Beverley SM. PTR1: a reductase mediating salvage of oxidized pteridines and methotrexate resistance in the protozoan parasite Leishmania major. Proc Natl Acad Sci USA. 1994;91:11442–11446. doi: 10.1073/pnas.91.24.11442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ellenberger TE, Beverley SM. Biochemistry and regulation of folate and methotrexate transport in Leishmania major. J Biol Chem. 1987;262:10053–10058. [PubMed] [Google Scholar]
- 54.Besteiro S, Williams RA, Coombs GH, Mottram JC. Protein turnover and differentiation in Leishmania. Int J Parasitol. 2007;37:1063–1075. doi: 10.1016/j.ijpara.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brennand A, et al. Autophagy in parasitic protists: unique features and drug targets. Mol Biochem Parasitol. 2011;177:83–99. doi: 10.1016/j.molbiopara.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 56.Xie Z, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19:3290–3298. doi: 10.1091/mbc.e07-12-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 58.Kochl R, Hu XW, Chan EY, Tooze SA. Microtubules facilitate autophagosome formation and fusion of autophagosomes with endosomes. Traffic. 2006;7:129–145. doi: 10.1111/j.1600-0854.2005.00368.x. [DOI] [PubMed] [Google Scholar]
- 59.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6:463–477. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 60.Williams RA, Tetley L, Mottram JC, Coombs GH. Cysteine peptidases CPA and CPB are vital for autophagy and differentiation in Leishmania mexicana. Mol Microbiol. 2006;61:655–674. doi: 10.1111/j.1365-2958.2006.05274.x. [DOI] [PubMed] [Google Scholar]
- 61.Alexandratos A, et al. The loss of virulence of histone H1 overexpressing Leishmania donovani parasites is directly associated with a reduction of HSP83 rate of translation. Mol Microbiol. 2013;88:1015–1031. doi: 10.1111/mmi.12240. [DOI] [PubMed] [Google Scholar]
- 62.Papageorgiou FT, Soteriadou KP. Expression of a novel Leishmania gene encoding a histone H1-like protein in Leishmania major modulates parasite infectivity in vitro. Infect Immun. 2002;70:6976–6986. doi: 10.1128/IAI.70.12.6976-6986.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Smirlis D, et al. Leishmania histone H1 overexpression delays parasite cell-cycle progression, parasite differentiation and reduces Leishmania infectivity in vivo. Mol Microbiol. 2006;60:1457–1473. doi: 10.1111/j.1365-2958.2006.05205.x. [DOI] [PubMed] [Google Scholar]
- 64.Zackay A, et al. Genome wide comparison of Ethiopian Leishmania donovani strains reveals differences potentially related to parasite survival. PLoS Genet. 2018;14:e1007133. doi: 10.1371/journal.pgen.1007133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dumetz, F. et al. Molecular Preadaptation to Antimony Resistance in Leishmania donovani on the Indian Subcontinent. mSphere3 (2018). [DOI] [PMC free article] [PubMed]
- 66.Dumetz, F. et al. Modulation of Aneuploidy in Leishmania donovani during Adaptation to Different In Vitro and In Vivo Environments and Its Impact on Gene Expression. MBio8 (2017). [DOI] [PMC free article] [PubMed]
- 67.Do Monte-Neto RL, et al. Gene expression profiling and molecular characterization of antimony resistance in Leishmania amazonensis. PLoS Negl Trop Dis. 2011;5:e1167. doi: 10.1371/journal.pntd.0001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Monte-Neto R, et al. Intrachromosomal amplification, locus deletion and point mutation in the aquaglyceroporin AQP1 gene in antimony resistant Leishmania (Viannia) guyanensis. PLoS Negl Trop Dis. 2015;9:e0003476. doi: 10.1371/journal.pntd.0003476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Valdivia HO, et al. Comparative genomic analysis of Leishmania (Viannia) peruviana and Leishmania (Viannia) braziliensis. BMC Genomics. 2015;16:715. doi: 10.1186/s12864-015-1928-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Peacock CS, et al. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat Genet. 2007;39:839–847. doi: 10.1038/ng2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Real F, et al. The genome sequence of Leishmania (Leishmania) amazonensis: functional annotation and extended analysis of gene models. DNA Res. 2013;20:567–581. doi: 10.1093/dnares/dst031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tschoeke DA, et al. The Comparative Genomics and Phylogenomics of Leishmania amazonensisParasite. Evol. Bioinform. Online. 2014;10:131–53. doi: 10.4137/EBO.S13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ubeda JM, et al. Genome-wide stochastic adaptive DNA amplification at direct and inverted DNA repeats in the parasite Leishmania. PLoS Biol. 2014;12:e1001868. doi: 10.1371/journal.pbio.1001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leprohon P, et al. Gene expression modulation is associated with gene amplification, supernumerary chromosomes and chromosome loss in antimony-resistant Leishmania infantum. Nucleic Acids Res. 2009;37:1387–1399. doi: 10.1093/nar/gkn1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Victoir K, Dujardin JC. How to succeed in parasitic life without sex? Asking Leishmania. Trends Parasitol. 2002;18:81–85. doi: 10.1016/S1471-4922(01)02199-7. [DOI] [PubMed] [Google Scholar]
- 76.Mukherjee A, Langston LD, Ouellette M. Intrachromosomal tandem duplication and repeat expansion during attempts to inactivate the subtelomeric essential gene GSH1 in Leishmania. Nucleic Acids Res. 2011;39:7499–7511. doi: 10.1093/nar/gkr494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Laffitte MN, Leprohon P, Papadopoulou B, Ouellette M. Plasticity of the Leishmania genome leading to gene copy number variations and drug resistance. F1000Res. 2016;5:2350. doi: 10.12688/f1000research.9218.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mannaert A, Downing T, Imamura H, Dujardin JC. Adaptive mechanisms in pathogens: universal aneuploidy in Leishmania. Trends Parasitol. 2012;28:370–376. doi: 10.1016/j.pt.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 79.Llewellyn MS, et al. Deep sequencing of the Trypanosoma cruzi GP63 surface proteases reveals diversity and diversifying selection among chronic and congenital Chagas disease patients. PLoS Negl. Trop. Dis. 2015;9:e0003458. doi: 10.1371/journal.pntd.0003458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Otto TD, et al. Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts. Nat Commun. 2014;5:4754. doi: 10.1038/ncomms5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hacariz O, Akgun M, Kavak P, Yuksel B, Sagiroglu MS. Comparative transcriptome profiling approach to glean virulence and immunomodulation-related genes of Fasciola hepatica. BMC Genomics. 2015;16:366. doi: 10.1186/s12864-015-1539-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhu Z, et al. Comparative genomic analysis of pre-epidemic and epidemic Zika virus strains for virological factors potentially associated with the rapidly expanding epidemic. Emerg Microbes Infect. 2016;5:e22. doi: 10.1038/emi.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Boite MC, Mauricio IL, Miles MA, Cupolillo E. New insights on taxonomy, phylogeny and population genetics of Leishmania (Viannia) parasites based on multilocus sequence analysis. PLoS Negl Trop Dis. 2012;6:e1888. doi: 10.1371/journal.pntd.0001888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Coughlan S, et al. Leishmania naiffi and Leishmania guyanensis reference genomes highlight genome structure and gene evolution in the Viannia subgenus. R Soc Open Sci. 2018;5:172212. doi: 10.1098/rsos.172212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mitchell GF. Murine cutaneous leishmaniasis: resistance in reconstituted nude mice and several F1 hybrids infected with Leishmania tropica major. J Immunogenet. 1983;10:395–412. doi: 10.1111/j.1744-313X.1983.tb00351.x. [DOI] [PubMed] [Google Scholar]
- 86.Belkaid Y, et al. Development of a natural model of cutaneous leishmaniasis: powerful effects of vector saliva and saliva preexposure on the long-term outcome of Leishmania major infection in the mouse ear dermis. J Exp Med. 1998;188:1941–1953. doi: 10.1084/jem.188.10.1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Belkaid Y, et al. A natural model of Leishmania major infection reveals a prolonged “silent” phase of parasite amplification in the skin before the onset of lesion formation and immunity. J Immunol. 2000;165:969–977. doi: 10.4049/jimmunol.165.2.969. [DOI] [PubMed] [Google Scholar]
- 88.Lima HC, Bleyenberg JA, Titus RG. A simple method for quantifying Leishmania in tissues of infected animals. Parasitol Today. 1997;13:80–82. doi: 10.1016/S0169-4758(96)40010-2. [DOI] [PubMed] [Google Scholar]
- 89.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Otto TD, Sanders M, Berriman M, Newbold C. Iterative Correction of Reference Nucleotides (iCORN) using second generation sequencing technology. Bioinformatics. 2010;26:1704–1707. doi: 10.1093/bioinformatics/btq269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tsai IJ, Otto TD, Berriman M. Improving draft assemblies by iterative mapping and assembly of short reads to eliminate gaps. Genome Biol. 2010;11:R41. doi: 10.1186/gb-2010-11-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30:2478–2483. doi: 10.1093/nar/30.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics. 2009;25:1968–1969. doi: 10.1093/bioinformatics/btp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carver TJ, et al. ACT: the Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 96.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Duitama J, et al. An integrated framework for discovery and genotyping of genomic variants from high-throughput sequencing experiments. Nucleic Acids Res. 2014;42:e44. doi: 10.1093/nar/gkt1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Otto TD, Dillon GP, Degrave WS, Berriman M. RATT: Rapid Annotation Transfer Tool. Nucleic Acids Res. 2011;39:e57. doi: 10.1093/nar/gkq1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Carver T, et al. Artemis and ACT: viewing, annotating and comparing sequences stored in a relational database. Bioinformatics. 2008;24:2672–2676. doi: 10.1093/bioinformatics/btn529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 101.Bendtsen JD, Jensen LJ, Blom N, Von Heijne G, Brunak S. Feature-based prediction of non-classical and leaderless protein secretion. Protein Eng Des Sel. 2004;17:349–356. doi: 10.1093/protein/gzh037. [DOI] [PubMed] [Google Scholar]
- 102.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 103.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 104.Lobaton Juan David, Miller Tamara, Gil Juanita, Ariza Daniel, de la Hoz Juan Fernando, Soler Alvaro, Beebe Steve, Duitama Jorge, Gepts Paul, Raatz Bodo. Resequencing of Common Bean Identifies Regions of Inter–Gene Pool Introgression and Provides Comprehensive Resources for Molecular Breeding. The Plant Genome. 2018;11(2):0. doi: 10.3835/plantgenome2017.08.0068. [DOI] [PubMed] [Google Scholar]
- 105.Frank E, Hall M, Trigg L, Holmes G, Witten IH. Data mining in bioinformatics using Weka. Bioinformatics. 2004;20:2479–2481. doi: 10.1093/bioinformatics/bth261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.