

Abstract
Background and Purpose
Diethyldithiocarbamate (DDC) is a major metabolite of disulfiram that is a potential drug for alcoholism treatment. In the present study, we attempted to explore the possible effect of DDC on non‐alcoholic fatty liver disease (NAFLD) and related fibrosis in vivo.
Experimental Approach
C57BL/6 mice and Sprague Dawley (SD) rats received a methionine/choline‐deficient (MCD) diet to establish the model of NAFLD with or without DDC treatment. The livers and serum were assessed for histological changes and parameters related to lipid metabolism, liver injury, inflammation and fibrosis. Apoptosis and macrophage related markers were assessed by immunohistochemistry (IHC).
Key Results
DDC significantly reduced hepatic steatosis in rats with NAFLD, induced by the MCD diet. DDC reduced the oxidative stress and endoplasmic reticulum stress‐related parameters in mice with non‐alcoholic steatohepatitis, induced by the MCD diet. IHC for Bax and cleaved caspase‐3 showed that DDC inhibited the apoptosis of hepatocytes in the liver. DDC significantly reduced ballooning and Mallory−Denk bodies (MDB) in hepatocytes, accompanied by suppression of serum alanine aminotransferase, aspartate aminotransferase and MDB formation‐related genes. DDC significantly alleviated hepatic inflammation, accompanied by suppression of inflammation‐related genes. DDC suppressed the infiltration of macrophages, particularly inducible NOS‐positive pro‐inflammatory macrophages. In addition, DDC significantly alleviated liver fibrosis. Microarray analyses showed that DDC strongly affected lipid metabolism and oxidative stress‐related processes and pathways.
Conclusion and Implications
DDC improves hepatic steatosis, ballooning, inflammation and fibrosis in rodent models of NAFLD through modulating lipid metabolism and oxidative stress.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ATF4
activating transcription factor 4
- Bax
Bcl2‐associated X protein
- CYP2E1
cytochrome P450 2E1
- CHO
cholesterol
- CHOP
C/EBP homologous protein
- DDC
diethyldithiocarbamate
- DSF
disulfiram
- ER
endoplasmic reticulum
- Fas
fatty acid synthase
- iNOS
inducible NOS
- MCD
methionine choline deficient
- MCS
methionine and choline sufficient
- MDB
Mallory–Denk body
- NAFLD
non‐alcoholic fatty liver disease
- NASH
non‐alcoholic steatohepatitis
- Scd1
stearoyl‐CoA desaturase
- α‐sma
α‐smooth muscle actin
Introduction
Non‐alcoholic fatty liver disease (NAFLD) is characterized by excessive hepatic fat accumulation and defined by the presence of steatosis in >5% of hepatocytes. NAFLD includes two pathological conditions with different prognoses: non‐alcoholic fatty liver (NAFL) and non‐alcoholic steatohepatitis (NASH); the latter causes an elevated risk for cirrhosis and hepatocellular carcinoma (European Association for the Study of the Liver (EASL), et al. 2016). The prevalence of NAFLD is increasing annually worldwide and has become a major cause of end‐stage liver diseases and liver transplantation in western countries (Mittal et al., 2016; Vernon et al., 2011; Younossi et al., 2018). It is also increasingly recognized as an important aetiology of chronic liver diseases in China (Fan and Farrell, 2009).
Lifestyle modification consisting of diet, exercise, and weight loss has been advocated to treat patients with NAFLD (EASL, et al. 2016). However, no data are currently available on their long‐term effects on the natural history of NAFLD. Drug therapy should be indicated for progressive NASH but also for early‐stage NASH with increased risk of the development of fibrosis (Adams et al., 2005) or active NASH with high necroinflammatory activity (Sanyal et al., 2015). Up to now, no drugs have been approved for NASH by the regulatory agencies. Therefore, it is of immediate importance to identify effective therapeutic agents for NAFLD.
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7168 (DSF) is a drug that has been used for over six decades as a treatment for alcohol dependence (Diehl et al., 2010), with well‐established pharmacokinetics, safety and tolerance at the US Food and Drug Administration‐recommended dosage (Cvek, 2012). In the body, DSF is metabolized to dithiocarb [diethyldithiocarbamate (DDTC or DDC)], and sodium DDC was used to treat high‐risk breast cancer in a clinical trial (Dufour et al., 1993). A recent nationwide epidemiological study revealed that patients who continuously used DSF have a lower risk of death from cancer compared to those who stopped using the drug at their diagnosis (Skrott et al., 2017).
DDC or DSF is widely used for inhibiting cytochrome P450 2E1 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1330) in human studies because of its known selectivity and relative non‐toxicity (Pratt‐Hyatt et al., 2010). It has been reported that low doses of DDC protect against liver injury induced by many hepatotoxic agents via its inhibition of drug‐metabolizing enzymes and antioxidant effects in rats (Stott et al., 1997). Additionally, DDC is a well‐known NF‐κB inhibitor. DDC interferes with the NF‐κB pathway by inhibiting the nuclear translocation of NF‐κB (Matsuno et al., 2012). We previously demonstrated that DDC up‐regulates http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1628 in hepatic stellate cells via the http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514 and http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=285 signalling pathways in vitro (Liu et al., 2013; Liu et al., 2016). However, the possible effect of DDC on NAFLD and related fibrosis in vivo is still unknown.
In this study, the rats used were fed a methionine choline‐deficient (MCD) diet that induced NAFL with severe steatosis. The mice used were also fed a MCD diet, which induced NASH with severe hepatocyte ballooning, inflammation and fibrosis formation in these animals. We then investigated the effect of DDC on steatosis, hepatocyte ballooning, inflammation and fibrosis respectively.
Methods
Experimental animals
Animal experiments were approved by the Laboratory Animal Centre, affiliated with Beijing Friendship Hospital [SYXK (Jing) 2012‐0023], and were performed in accordance with institutional and national legal guidelines for animal protection. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010).
Eight‐week‐old male C57BL/6 mice and 6‐week‐old male Sprague Dawley (SD) rats were obtained from Beijing HFK Bio‐Technology, Beijing, China. All animals were housed in a 12 h light–dark cycle in a temperature‐controlled room and had free access to food and water. All animals were randomized into different groups. The investigator was not blind to the experimental groups.
NASH mouse model
The MCD diet is one of the best described dietary models for NAFL and NASH (Van Herck et al., 2017). C57BL/6 mice fed a MCD diet showed severe hepatic necroinflammation with a lesser degree of steatosis (Kirsch et al., 2003). Thus, in this study, we used mice fed the MCD diet to investigate the effect of DDC on hepatocyte ballooning, inflammation and fibrosis. The MCD and the methionine and choline‐sufficient (MCS; normal control) diets were purchased from MP Biomedicals (Solon, OH, USA). Eight‐week‐old male C57BL/6 mice were randomly assigned to three independent groups (group size: n = 8): MCS group, MCD group and MCD + DDC group. The MCS group were kept on MCS control diet for 8 weeks. The MCD group were kept on the MCD diet and treated with vehicle (1 × PBS) via daily gavage for 8 weeks. MCD + DDC group were kept on the MCD diet and treated with 450 mg·kg−1 DDC (D3506, CAS number: 20624‐25‐3, Sigma‐Aldrich, St. Louis, MO, USA) via daily gavage for 8 weeks by a technician blinded to the study design. At the end of the experiment, the mice were killed, and liver tissues and serum were obtained for analysis.
NAFL rat model
The liver injury and model of NASH induced by the MCD diet depends on the rodent species (Kirsch et al., 2003). Rats develop extensive steatosis when fed a MCD diet, while inflammation and necrosis are minor features, and fibrosis is absent. Therefore, in this study, we used rats fed a MCD diet to investigate the effect of DDC on steatosis. Six‐week‐old male SD rats were randomly assigned to three independent groups (group size: n = 10): MCS group, MCD group and MCD + DDC group. The MCS group were kept on the MCS control diet for 6 weeks. The MCD group were kept on the MCD diet and treated with vehicle (1 × PBS) via daily gavage for 6 weeks. The MCD + DDC group were kept on the MCD diet and treated with 312 mg·kg−1 DDC via daily gavage for 6 weeks by a technician blinded to the study design. At the end of the experiment, the rats were killed, and liver tissues and serum were obtained for analysis.
Serum analyses
Serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), total cholesterol (CHO), HDL and LDL were determined by the Central Laboratory of Deyi Diagnostics (Beijing, China) according to standardized and regularly validated criteria.
Histochemistry and immunohistochemical analyses
Formalin‐fixed liver samples were deparaffinized and stained with haematoxylin and eosin for determination of the NAFLD activity score including steatosis, ballooning and inflammation (Kleiner et al., 2005). Collagen was stained using Sirius Red, and hepatic lipid was stained with Oil red O. Sirius red‐stained sections were observed under both visible light and polarized light. Immunohistochemistry (IHC) was performed on formalin‐fixed sections. After rehydration of deparaffinized sections, antigens were retrieved at 95°C using citrate buffer (pH 6.0) for 30 min, followed by incubation overnight at 4°C using the following primary antibodies: mouse monoclonal antibody KP1against CD68 (Abcam Cat# ab955, RRID:AB_307338) at 1:500 dilution, rabbit polyclonal antibody against http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1250 (iNOS) (Abcam Cat# ab15323, RRID:AB_301857) at 1:100 dilution, rabbit polyclonal antibody against CD206 (Abcam Cat# ab64693, RRID:AB_1523910) at 1:1000 dilution and rabbit polyclonal antibody ACTA2 against α‐smooth muscle actin (α‐sma, Abcam Cat# ab5694, RRID:AB_2223021) at 1:500 dilution. Rabbit polyclonal antibody against BCl2‐associated X protein (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=910) (Cat# GB11007) at 1:500 dilution and rabbit polyclonal antibody against cleaved http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1619 (Cat# GB11009) at 1:500 dilution were purchased from Servicebio (Beijing, China). The primary antibodies were detected with corresponding HRP‐labelled secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA). The experiments were performed by a technician blinded to the experimental groups. Tissues were visualized in a Nikon Eclipse Ni‐U microscope (Nikon Corporation, Tokyo, Japan) with the appropriate filters. Representative images were taken with a Nikon DS‐Ri2 microscope camera (Nikon). Quantitative image analysis of staining was performed using Nikon NIS‐Elements Software.
Real‐time RT‐PCR
The total RNA was isolated from liver tissues using TRIzol® reagent (Invitrogen, Grand Island, NY, USA) according to the manufacturer's instructions. The total RNA yields were quantified, and the equivalent amounts of total RNA (2 μg) were reverse‐transcribed into single‐stranded cDNA. Equal amounts of cDNA were subjected to PCR in the presence of SYBR green dye using the ABI Power SYBR Green PCR Master Mix kit on an ABI Prism 7500 Sequence Detector (Applied Biosystems, Foster City, CA, USA). PCR without a template was used as the negative control; β‐actin mRNA was used as an internal control. The primers were shown in Supporting Information Tables S1 and S2. PCR was performed with 45 cycles of 15 s at 95°C and 60 s at 60°C after a 2 min initial denaturation at 95°C. Each sample was normalized according to the difference in the critical thresholds (CT) between the target gene and β‐actin and relative to the control. The amount of the target was calculated as 2−ΔΔCT. All experiments were performed independently three times, and the averages were used for the comparisons.
Western blot analysis
Samples of liver tissue (100 mg) were immediately solubilized in lysis buffer at 4°C for 30 min. Following microcentrifugation at 14 000× g for 5 min, the supernatants were transferred into a new tube, and the sample protein concentrations were determined using the Pierce Protein assay kit (Pierce, Rockford, IL, USA). The protein mixtures were loaded into each well and separated on 12% SDS‐PAGE. Following a 2 h run, the proteins were transferred onto nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ, USA). The membranes were blocked and subsequently incubated with rabbit polyclonal antibody against CYP2E1 (Abcam Cat# ab28146, RRID:AB_2089985) at 1:2000 dilution and mouse monoclonal antibody against β‐actin (Sigma‐Aldrich Cat# A5441, RRID:AB_476744) at 1:5000 dilution at 4°C overnight. After extensive washing, the membranes were incubated with the secondary antibody for 60 min followed by extensive washes. Specific antibody–antigen complexes were detected with ECL Western blot detection kits (Pierce, Rockford, IL, USA). All experiments were performed independently three times, and the averages were used for the comparisons. Protein expression was quantified via densitometric analyses of the immunoblots using the Quantity One software.
Double immunofluorescence
For immunofluorescence, formalin‐fixed liver samples were deparaffinized and rehydrated. Liver sections were immersed in EDTA antigen retrieval buffer (pH 8.0) and maintained at a sub‐boiling temperature for 15 min. After being washed three times with PBS (pH 7.4), the sections were blocked using 3% BSA for 30 min. The sections were incubated overnight at 4°C with rabbit monoclonal antibody EP1628Y against keratin 8 (K8) (Abcam Cat# ab53280, RRID:AB_869901) at 1:200 dilution and mouse monoclonal antibody P4D1 against ubiquitin (Santa Cruz Biotechnology Cat# sc‐8017, RRID:AB_628423) at 1:200 dilution. After being washed three times with PBS, the primary antibodies were detected with corresponding Alexa Fluor‐conjugated anti‐IgG, and the nuclei were counterstained with DAPI. The experiments were performed by a technician blinded to the experimental groups.
Microarray analyses of mouse liver samples
Total RNA was isolated from individual liver samples (n = 2 liver samples for MCS group, n = 3 liver samples for MCD group and n = 3 liver samples for MCD + DDC group) using TRIzol® reagent and were reverse‐transcribed into cDNA. cDNA gene expression analysis was performed on a mouse Oligo Microarray (Mouse OneArray Plus, Phalanx Biotech Group, Hsinchu, Taiwan) by BGI‐Beijing according to the manufacturer's instructions. The experiments were performed by a technician blinded to the experimental groups. The data were processed and analysed using Agilent 0.1 XDR software (Agilent Technologies, Santa Clara, CA, USA). Differentially expressed genes were mapped into biological processes or pathways using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes.
Statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018). All statistical analyses were performed using SPSS software, version 16.0. Data are expressed as mean ± SEM. One‐way ANOVA with post hoc Dunnett's multiple comparisons test was used for multiple groups. Post hoc tests were run only if F achieved P < 0.05, and there was no significant variance inhomogeneity. P < 0.05 was regarded as statistically significant.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Results
DDC attenuates hepatic steatosis in NAFL rats
We first examined the impact of DDC on MCD diet‐induced NAFL in rats. Compared to rats on MCS diet, rats on MCD diet for 6 weeks showed a pronounced hepatic steatosis. Treatment with DDC significantly attenuated MCD‐induced hepatic steatosis (Figure 1A, B). As shown in Figure 1C, MCD diet suppressed serum levels of CHO and HDL in rats, while DDC treatment could rescue the decrease of CHO and HDL. We then examined whether DDC regulated genes involved in fatty acid synthesis in rats. Similar to previous studies (Rinella et al., 2008), MCD diet significantly down‐regulated the expression of genes involved in fatty acid synthesis such as fatty acid synthase (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2608) and stearoyl‐CoA desaturase (Scd1). DDC treatment significantly up‐regulated the expression of Fas (Figure 1D).
Figure 1.
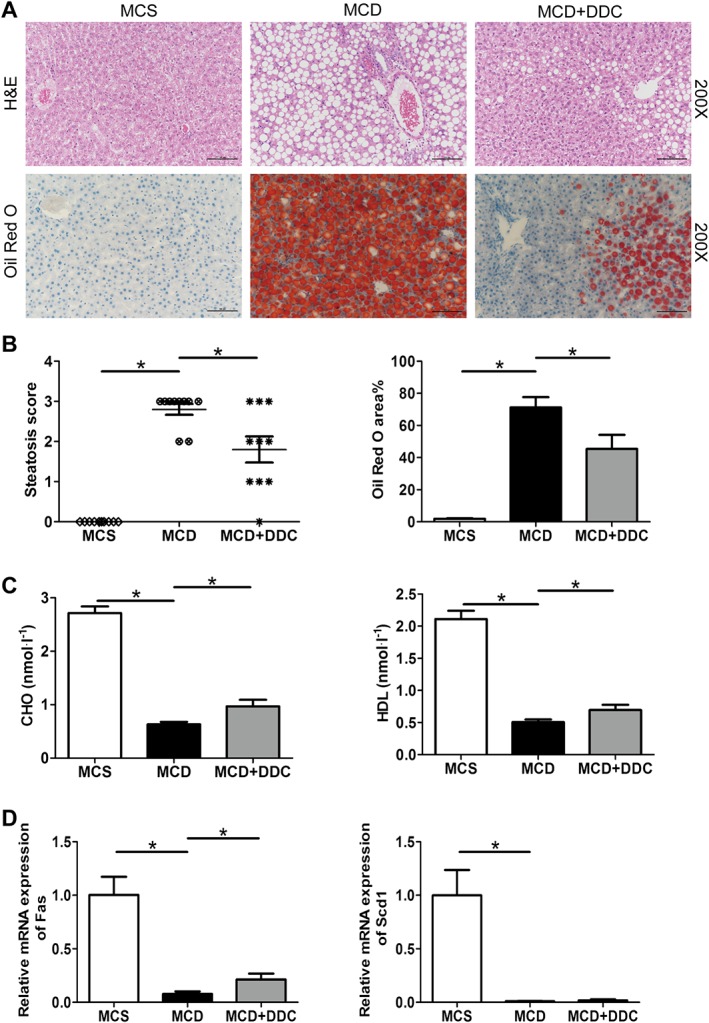
Treatment with DDC improves hepatic steatosis in rats fed a MCD diet for 6 weeks. Male SD rats were fed either MCD or MCS diets for 6 weeks. The MCD group were treated either with vehicle (1 × PBS) or 312 mg·kg−1 DDC via daily gavage for all 6 weeks. (A) H&E and Oil red O staining of representative liver sections (original magnification ×200) shows the distribution of lipid. (B) Scores of liver steatosis and the area of the Oil red O in liver sections were measured. (C) Serum CHO (mmol.L‐1) and HDL (mmol.L‐1) levels in rats. (D) The transcript levels of genes related to fatty acid synthesis (Fas and Scd1). Data are expressed as mean ± SEM; n = 10 rats per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
DDC modulates parameters related to lipid metabolism in NASH mice
Although DDC‐treated mice did not show significant histological changes of hepatic steatosis, parameters related to fatty acid metabolism in serum and the expression of fatty acid synthesis‐related genes in the liver changed significantly. As shown in Supporting Information Figure S1A–C, MCD diet suppressed serum levels of CHO, HDL and LDL in mice, while DDC treatment rescued the decrease of these parameters. MCD diet significantly down‐regulated the expression of genes involved in fatty acid synthesis including Fas, sterol regulatory element‐binding protein‐1 (Srebp‐1) and Scd1. Compared with MCD diet group, DDC treatment significantly up‐regulated the expression of Fas, while DDC group did not show significant changes in the expression of Srebp‐1 and Scd1 (Supporting Information Figure S1D–F).
DDC reduces oxidative stress, endoplasmic reticulum (ER) stress and apoptosis in the liver of NASH mice
We next investigated the effect of DDC on MCD diet‐induced oxidative stress and ER stress in the liver. As shown in Figure 2A, MCD diet induced the up‐regulation of CYP2E1 in mice liver, and DDC treatment significantly inhibited the increase of CYP2E1. As shown in Figure 2B–C, MCD diet induced the expression of ER stress‐related gene, C/EBP homologous protein (CHOP) and activating transcription factor 4 (ATF4), while DDC treatment significantly blocked the increase in CHOP and ATF4 in mice. These data indicate that DDC treatment could reduce the oxidative stress and ER stress induced by the MCD diet.
Figure 2.
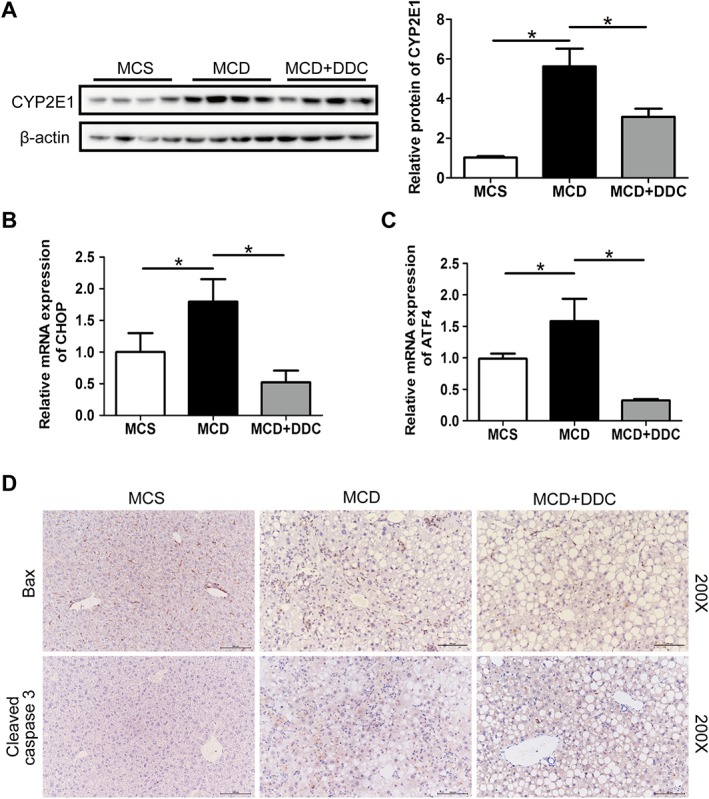
Treatment with DDC decreases oxidative stress, ER stress and apoptosis in the livers of mice fed a MCD diet for 8 weeks. Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. The MCD group were treated either with vehicle (1 × PBS) or 450 mg·kg−1 DDC via daily gavage for all 8 weeks. (A) Immunoblot analysis of CYP2E1 in liver lysates of mice. (B) The transcript levels of CHOP in the liver of mice. (C) The transcript levels of ATF4 in the liver of mice. Data are expressed as mean ± SEM; n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05. (D) Immunohistochemical staining for Bax and cleaved caspase‐3 in liver sections (original magnification 200×).
As shown in Figure 2D, the MCD diet induced the expression of pro‐apoptotic protein Bax and the activation of its downstream target caspase‐3 in the liver of mice. DDC treatment prevented the activation of Bax, which in turn resulted in decreased activation of caspase‐3. These findings indicate that DDC inhibited the apoptosis induced by MCD diet.
DDC attenuates MCD diet‐induced hepatic ballooning and Mallory–Denk body (MDB) formation in NASH mice
We then examined the impact of DDC on hepatic injury, including hepatic ballooning and MDB formation in mice liver. Compared to mice on MCS diet, mice on MCD diet for 8 weeks showed a severe hepatic ballooning. As shown in Figure 3A, there were many ballooned hepatocytes, which lost the cell shape and contained delicate strands of residual cytoplasmic material in MCD diet group. Treatment with DDC significantly attenuated MCD‐induced hepatic ballooning, and the score of ballooning significantly decreased (Supporting Information Figure S2A). The hepatocytes showed macrovesicular steatosis, where a single large fat droplet completely displaces the normal cytoplasm in mice treated with DDC (Figure 3A). As shown in Supporting Information Figure S2B–C, compared to mice on MCS diet, mice on MCD diet for 8 weeks showed high levels of serum ALT and AST, while DDC treatment suppressed the increase of ALT and AST. These data indicated that DDC ameliorated liver injury induced by MCD diet.
Figure 3.
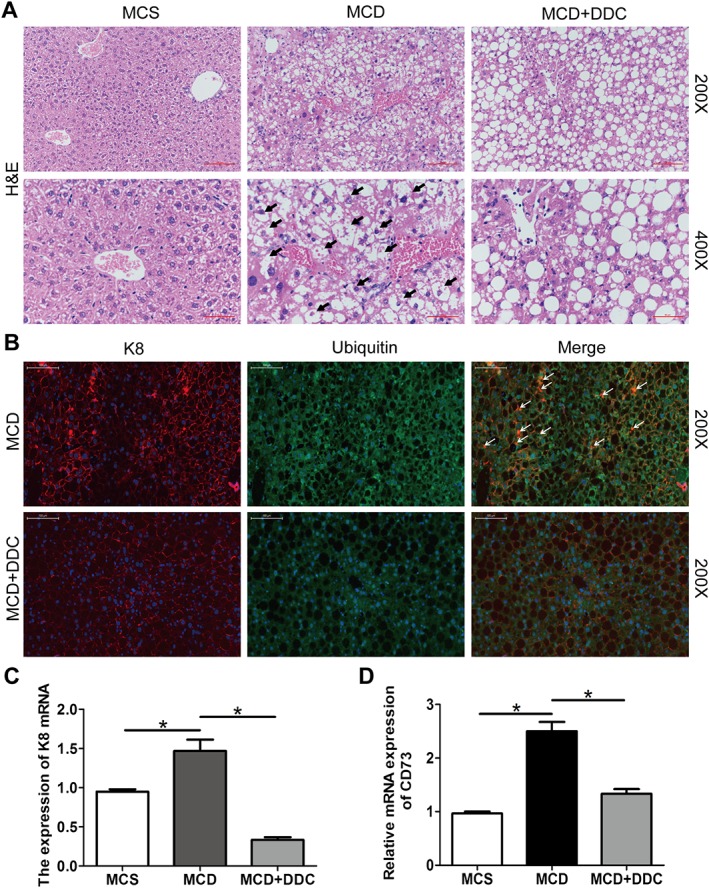
Treatment with DDC decreases ballooning and MDB in hepatocytes in mice fed a MCD diet for 8 weeks. Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. The MCD group were treated either with vehicle (1 × PBS) or 450 mg·kg−1 DDC via daily gavage for all 8 weeks. (A) H&E staining of representative liver sections (original magnification 200× and 400×) show the ballooning in hepatocytes (arrows), which lost their shape and contained delicate strands of residual cytoplasmic material in the MCD group. (B) Assessment of MDB formation using immunofluorescence staining for K8 and ubiquitin in livers. Livers were double‐labelled with antibodies to K8 (red) and ubiquitin (green). MDBs are seen as yellow clumps due to colocalization of keratins and ubiquitin (arrows) (original magnification 200×). (C) The transcript levels of K8 in the liver of mice. (D) The transcript levels of CD73 in the liver of mice. Data are expressed as mean ± SEM; n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
MDB formation in hepatocytes has been assessed by immunofluorescence staining for K8 and ubiquitin. As shown in Figure 3B, there was a significant increase in MDB numbers in MCD diet mice, and DDC treatment significantly reduced the number of MDBs. There was also an increase in K8 mRNA levels in MCD diet mice, and DDC treatment blocked the increase (Figure 3C). In addition, compared to mice on MCS diet, mice on MCD diet for 8 weeks induced the expression of CD73, which contributes to experimental MDB induction and is highly regulated in MDB associated liver injury in mice. DDC treatment inhibited the expression of CD73 (Figure 3D).
DDC suppresses hepatic inflammation and macrophages infiltration in NASH mice
Compared to mice on MCS diet, mice on MCD diet for 8 weeks showed a marked inflammatory cell infiltration both in the lobular and portal area of the liver. Treatment with DDC significantly attenuated MCD‐induced hepatic inflammation, and the score of inflammation significantly decreased (Figure 4A–B). Furthermore, the inflammation‐related genes (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5074, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=59 and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=62) increased in the MCD mice and were all normalized by DDC treatment (Figure 4C–E). These results suggested that DDC treatment results in a beneficial effect on hepatic inflammation.
Figure 4.
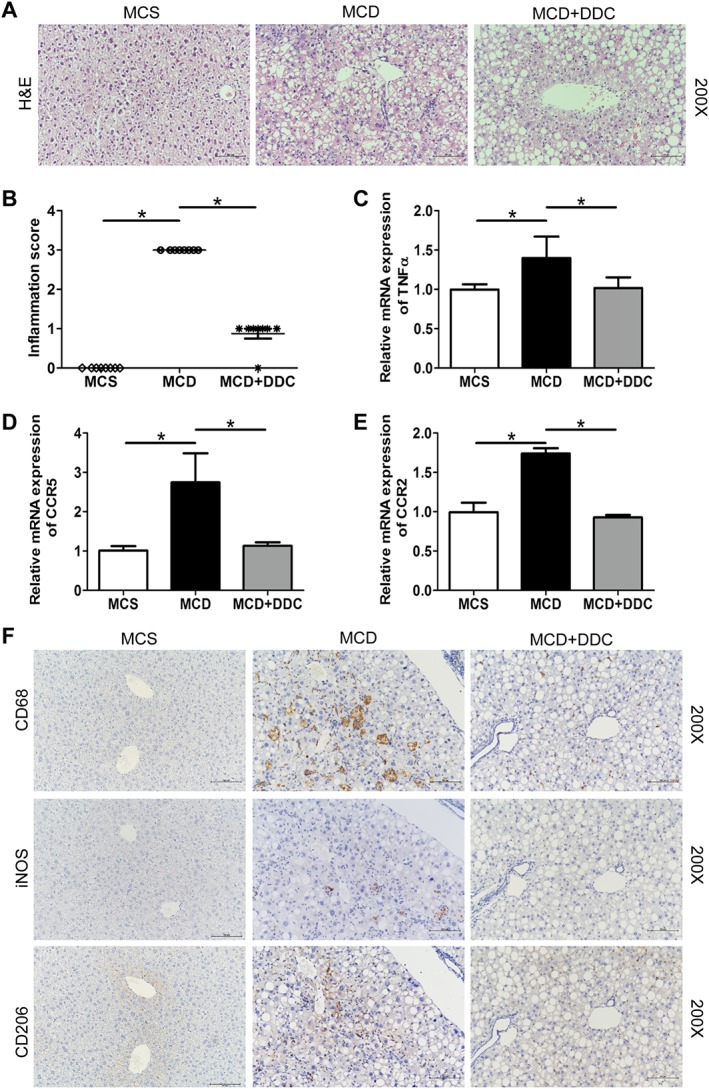
Treatment with DDC suppresses hepatic inflammation and macrophage infiltration in the livers of mice fed a MCD diet for 8 weeks. Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. The MCD group were treated either with vehicle (1 × PBS) or 450 mg·kg−1 DDC via daily gavage for all 8 weeks. (A) H&E staining of representative liver sections (original magnification 200×) showed hepatic inflammation. (B) Scores of hepatic inflammation in liver sections were measured. (C) The transcript levels of TNF‐α in the liver of mice. (D) The transcript levels of CCR5 in the liver of mice. (E) The transcript levels of CCR2 in the liver of mice. Data are expressed as mean ± SEM; n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05. (F) Immunohistochemical staining for CD68, CD206 and iNOS of continuous liver sections (original magnification 200×).
IHC analysis showed that the numbers of hepatic CD68+ (markers for general macrophages) cells, CD206+ (markers for M2 macrophages) cells and iNOS+ (markers for M1 macrophages) cells significantly increased in the liver of mice on MCD diet, and DDC inhibited these increases (Figure 4F). Dual immunofluorescence analysis showed that both CD68+iNOS+ M1 macrophages and CD68+CD206+ M2 macrophages significantly increased in the liver of mice on MCD diet, and DDC significantly attenuated MCD‐induced increase of M1 and M2 macrophages (Supporting Information Figure S3A–D). In addition, the suppression of CD68+iNOS+ M1 macrophages was more pronounced than CD68+CD206+ M2 macrophages in the liver after DDC treatment. In line with the data of IHC and immunofluorescence, the expression of CD206 and iNOS increased in MCD mice, and iNOS was suppressed by DDC treatment (Supporting Information Figure S3E–F). Taken together, DDC treatment mitigated the infiltration of macrophages and other myeloid cells and then inhibited the hepatic inflammation.
DDC suppresses MCD diet‐induced liver fibrosis in NASH mice
Since DDC treatment significantly attenuated MCD diet‐induced hepatic injury and inflammation, we next evaluated the effect of DDC on the development of hepatic fibrosis as a consequence of chronic liver injury. The changes of liver fibrosis were assessed by quantitative morphometric analyses for Sirius Red staining and α‐sma immunostaining of liver sections. Mice on MCD diet developed hepatic fibrosis with collagen deposition around the centrilobular vein with some central–central and central–portal bridges in MCD diet‐fed mice, and DDC treatment significantly attenuated MCD diet‐induced collagen deposition (Figure 5A–B). Equally, morphometry for α‐sma protein demonstrated a reduction of activated hepatic stellate cells (HSC) and myofibroblasts in MCD mice with DDC treatment (Figure 5C–D). These results uniformly showed that DDC significantly attenuated MCD diet‐induced hepatic fibrosis.
Figure 5.
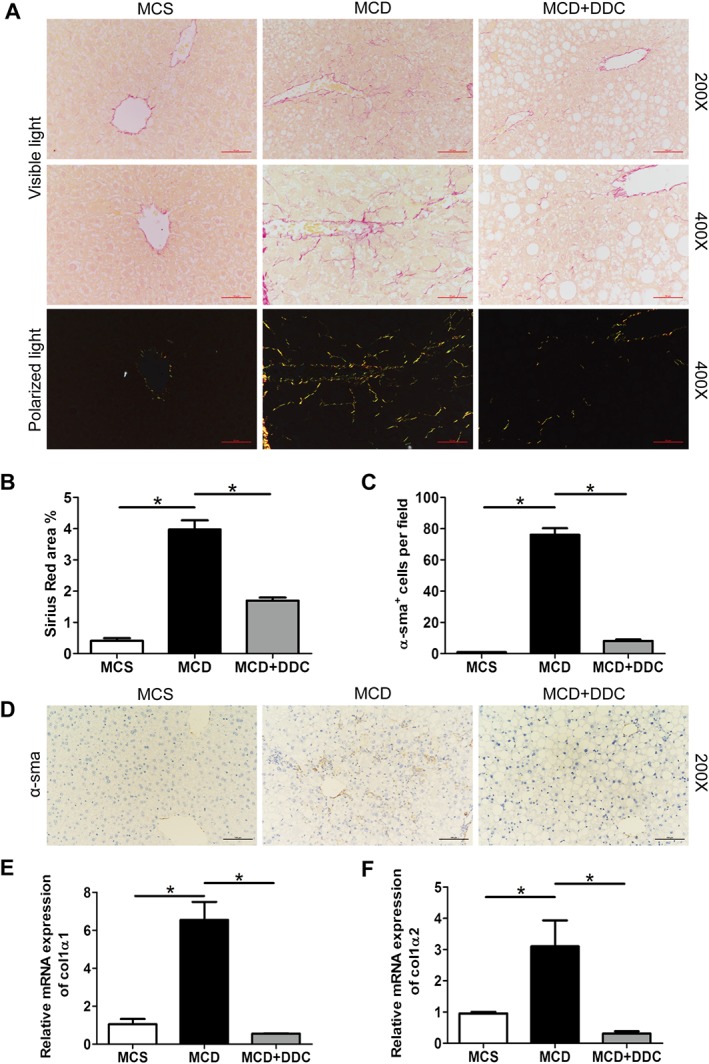
Administration of DDC attenuates hepatic fibrosis in the livers of mice fed a MCD diet for 8 weeks. Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. The MCD group were treated either with vehicle (1 × PBS) or 450 mg·kg−1 DDC via daily gavage for all 8 weeks. (A) Sirius Red staining of liver sections was observed under visible light and polarized light (original magnification 200× and 400×). Collagen type I presented a yellow, orange or red colour, while collagen type III appeared green under polarized light. (B) The area of Sirius Red in liver sections was measured. (C) The number of α‐sma‐positive cells was counted in 10 randomly selected microscopic fields (400×). (D) Immunohistochemical staining for α‐sma of liver sections (original magnification 200×). (E) The transcript levels of collagen 1α1 in the liver of mice. (F) The transcript levels of collagen 1α2 in the liver of mice. Data are expressed as mean ± SEM; n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
In agreement with the histological findings, the elevated expression of collagen 1α1 and collagen 1α2 in MCD‐fed mice was ameliorated by treatment with DDC (Figure 5E–F). Furthermore, as shown in Supporting Information Figure S4, mRNA expression of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5060 increased in MCD diet‐fed mice and was significantly suppressed by treatment with DDC.
DDC modulates lipid metabolism and oxidation–reduction process in NASH mice
To reveal the possible mechanisms of the protective effect of DDC on NAFLD, mice liver tissues from different groups were harvested, and the gene expression profile was analysed by microarray analysis (GSE113843). The top 296 differentially expressed genes were found to be highly associated with the protective effect of DDC on NAFLD, and the heat maps were shown in Figure 6A. Function enrichment analysis based on biological process identified the top 10 enriched GO terms (MCD + DDC group vs. MCD group) that were strongly affected by DDC treatment (Figure 6B). Among these biological processes, oxidation–reduction process, lipid metabolic process and fatty acid metabolic process were strongly affected by DDC treatment. The representative differentially expressed genes in oxidation–reduction and lipid metabolic processes were shown in Figure 7A–B. Pathway enrichment analysis identified the top 10 enriched pathways (MCD + DDC group vs. MCD group) that were strongly affected by DDC treatment (Figure 6C). Among these pathways, metabolic pathways, oxidative phosphorylation, NAFLD and PPAR signalling pathway were strongly affected by DDC treatment. The differentially expressed genes in NAFLD and PPAR signalling pathway were shown in Figure 7C–D. These data indicated that DDC might modulate MCD diet‐induced disorder of lipid metabolism and oxidative stress, which could lead to hepatic injury, inflammation and subsequent fibrosis (Figure 8).
Figure 6.
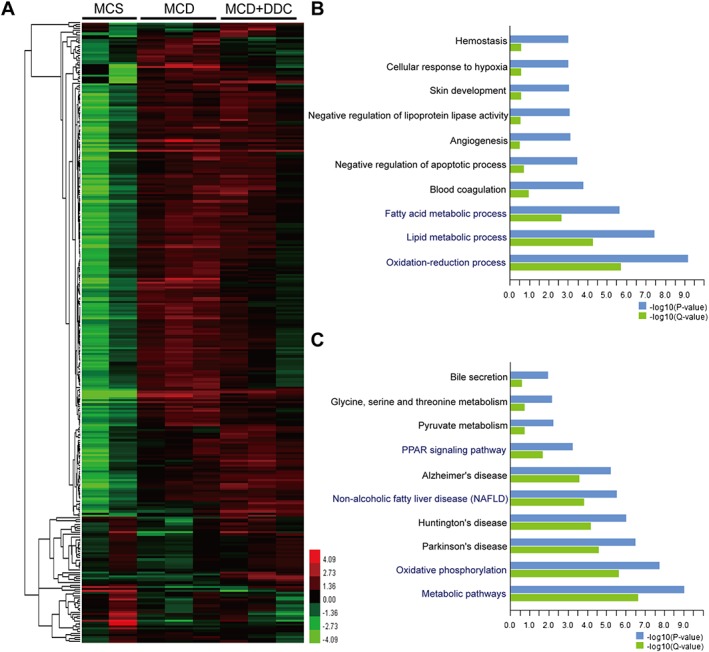
Microarray analyses of liver tissues in mice. MCD mice were treated either with vehicle (1 × PBS) or 450 mg·kg−1 DDC via daily gavage for all 8 weeks; liver tissues were harvested for microarray analysis. (A) Heat map of gene expression in liver tissues from different groups. (B) The top 10 enriched GO biological process terms affected by DDC treatment are plotted on the y‐axis versus a measure of significance (negative logarithm of the P‐value or Q‐value) on the x‐axis (MCD + DDC group vs. MCD group). (C) The top 10 enriched pathway terms affected by DDC treatment are plotted on the y‐axis versus a measure of significance on the x‐axis (MCD + DDC group vs. MCD group). The Q‐value is calculated by Benjamini.
Figure 7.
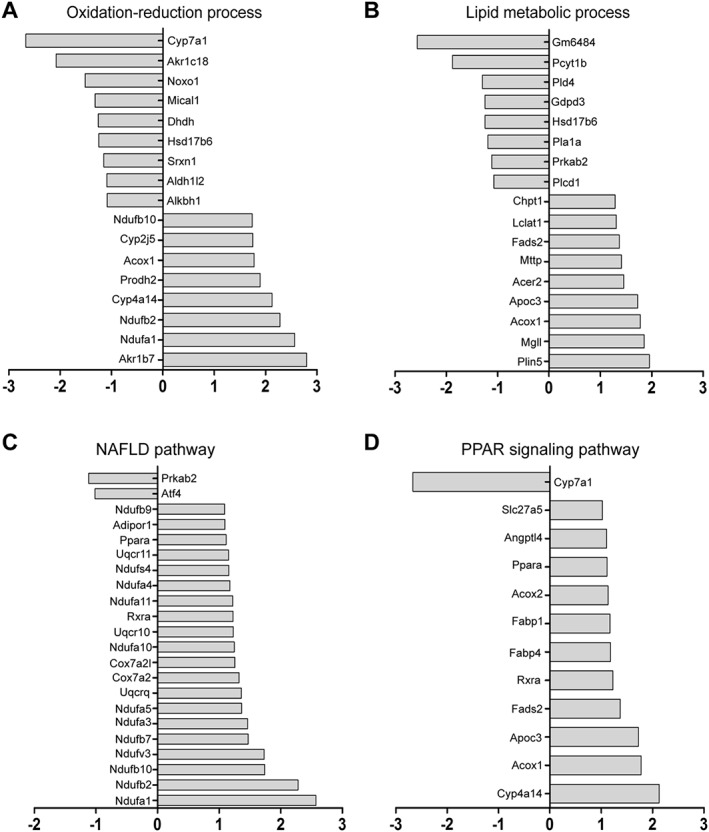
Expression of representative transcripts in mice liver in response to DDC treatment. MCD mice were treated either with vehicle (1 × PBS) or 450 mg·kg−1 DDC via daily gavage for all 8 weeks; liver tissues were harvested for microarray analysis. (A) Changes in the levels of representative oxidation–reduction process‐associated transcripts (MCD + DDC group vs. MCD group). (B) Changes in the levels of representative lipid metabolic process‐associated transcripts (MCD + DDC group vs. MCD group). (C) Changes in the levels of NAFLD pathway‐associated transcripts (MCD + DDC group vs. MCD group). (D) Changes in the levels of the PPAR signalling pathway‐associated transcripts (MCD + DDC group vs. MCD group).
Figure 8.
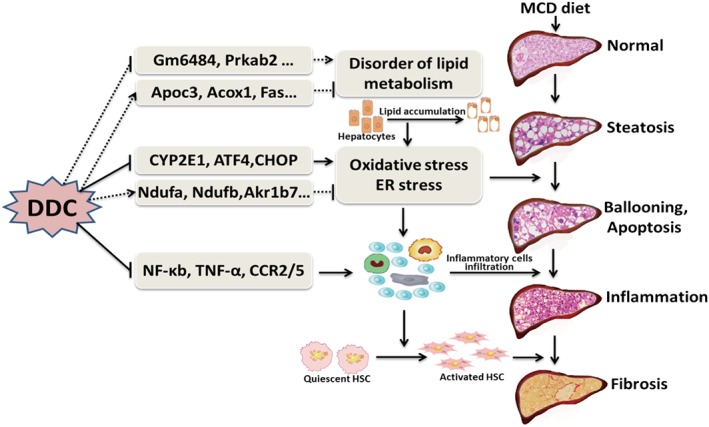
Possible mechanisms of DDC‐mediated amelioration of MCD diet‐induced NAFLD.
Discussions
As an old alcohol‐abuse drug, DSF and its active metabolite DDC have also been identified as a tumour suppressor (Cvek, 2012; Dufour et al., 1993; Skrott et al., 2017). Meanwhile, DDC is a NF‐κB inhibitor and has been widely used for inhibition of CYP2E1 (Pratt‐Hyatt et al., 2010; Matsuno et al., 2012). However, whether DDC has a role in NAFLD and related fibrosis has not been explored. In the present study, our data showed that DDC treatment significantly suppressed MCD diet‐induced steatosis in rats and oxidative stress, ER stress, hepatocyte ballooning, hepatic inflammation and subsequent liver fibrosis in mice.
NAFLD can be categorized histologically into NAFL and NASH. NAFL is defined as the presence of ≥5% hepatic steatosis without evidence of hepatocellular injury in the form of hepatocyte ballooning. NASH is defined as the presence of ≥5% hepatic steatosis and inflammation with hepatocyte injury (e.g. ballooning), with or without fibrosis (Chalasani et al., 2018). MCD diet is one of the best described dietary models for NAFL and NASH, and species of rodent influence liver injury in MCD nutritional model of NASH (Kirsch et al., 2003). Rats developed extensive steatosis when fed with MCD diet, while inflammation and necrosis were minor features, and fibrosis was absent. In contrast, C57BL/6 mice‐fed MCD diet showed severe hepatic necroinflammation and perisinusoidal fibrosis with a lesser degree of steatosis (Kirsch et al., 2003). In this study, we used rats fed with MCD diet to establish NAFL animal model and mice fed with MCD diet to establish NASH animal model.
In this study, we first investigated the possible effect of DDC on MCD diet‐induced hepatic steatosis in rats. Our data showed that treatment with DDC significantly attenuated MCD diet‐induced hepatic steatosis in rats. In agreement with previous studies (Rinella et al., 2008), MCD diet‐induced hypocholesterolaemia and decrease of HDL, while DDC treatment rescued hypocholesterolaemia and the decrease of HDL. In addition, DDC treatment could also correct the abnormal expression of fatty acid synthesis‐related gene Fas in MCD diet‐induced NAFL rats. These data indicated that the regulation of fatty acid synthesis by DDC might contribute to the suppression of MCD diet‐induced hepatic steatosis.
We next investigated the possible effect of DDC on MCD diet‐induced hepatic steatosis in mice. DDC treatment rescued hypocholesterolaemia and the abnormal of HDL and Fas in NASH mice. However, we did not observe significant changes of steatosis in histology, since C57BL/6 mice‐fed MCD diet for 8 weeks only showed severe hepatic necroinflammation and perisinusoidal fibrosis rather than steatosis in histology. Schwartz et al. (2013) have investigated the effect of DSF (Synonym: tetraethylthiuram disulfide) on NASH mice. In their model, significant steatosis and inflammation but no fibrosis were observed in mice liver, and they have not observed significant changes of hepatic steatosis in DSF‐treated mice. In their study, 20‐week‐old mice were used to establish NASH model induced by MCD diet for 6 weeks. The histology difference between this study and our study might be due to the age of mice and how long the mice were treated with MCD diet.
Excess lipid accumulation in the liver will result in oxidative stress and ER stress, which play important roles in the development and progression of NAFLD (Machado and Diehl, 2016). CYP2E1 is the major microsomal sources of oxidative stress in NAFLD (Robertson et al., 2001). MCD diet‐induced steatohepatitis is associated with up‐regulated hepatocyte microsomal CYP2E1 activity and increased hepatic lipid peroxidation (Weltman et al., 1996). Here, we showed that treatment with DDC significantly suppressed the increase of CYP2E1 in NASH mice. The unfolded protein response induced by hepatic lipid accumulation aggravates ER stress (Malhi and Kaufman, 2011), and persistent ER stress leads to hepatocyte injury, apoptosis and fibrosis (Gentile et al., 2011). Our data showed that MCD diet induced the expression of ER stress‐related gene, ATF4 and CHOP in mice liver, while DDC treatment blocked the increase. These data indicated that DDC treatment attenuated the oxidative stress and ER stress induced by MCD diet. Prolonged cellular stresses can cause serious damage, leading to apoptosis (Ashraf and Sheikh, 2015 ). Our data showed that MCD diet induced the expression of pro‐apoptotic protein Bax and the activation of its downstream target caspase‐3 in mice liver. DDC treatment inhibited the activation of Bax and caspase‐3.
Hepatocyte injury is also a defining lesion for NASH. Histologically, hepatocyte injury is indicated by hepatocyte ballooning that is characterized by an enlarged hyperchromatic nucleus and a foamy, pale cytoplasm (Brunt, 2011; Yeh and Brunt, 2014). In this study, treatment with DDC attenuated MCD induced hepatic ballooning. The formation of MDBs is one of the other histological features of hepatocyte injury in NASH (Zatloukal et al., 2007). MCD diet induced a significant increase in MDB numbers, while DDC treatment significantly reduced the number of MDBs. The abnormal expression of K8 is associated with the formation of MDB (Singla et al., 2012). We found that MCD diet induced an increase in K8 mRNA, and DDC treatment blocked the increase. It has been demonstrated that CD73 contributes to experimental MDB induction and is highly regulated in MDB‐associated liver injury in mice (Snider et al., 2013). In this study, mice on MCD diet for 8 weeks induced the expression of CD73, while DDC treatment inhibited the expression of CD73.
Hepatocyte injury will in turn trigger inflammatory and fibrogenic responses (Machado and Diehl, 2016). Histologically, inflammation in NASH is manifested by infiltration of inflammatory cells including macrophages, monocytes, neutrophils, and lymphocytes (Harmon et al., 2011). Pro‐inflammatory cytokines such as TNF‐α are secreted by injured hepatocytes and lead to increased macrophage infiltration (Harmon et al., 2011). In agreement with Schwartz's study (Schwartz et al., 2013), mice on a MCD diet showed a marked inflammatory cell infiltration in the liver, while DDC significantly attenuated hepatic inflammation. In addition, the inflammation‐related genes (TNF‐α, CCR2 and CCR5) increased in the MCD mice and were all normalized by DDC treatment. In Schwartz's study (Schwartz et al., 2013), DSF also reduced the expression of TNF‐α induced by the MCD diet. Imbalanced M1/M2 macrophages have emerged as a central mechanism underlying steatohepatitis. In NASH, macrophages are activated by various stimulants and shift to the pro‐inflammatory M1 phenotype (Smith, 2013). Our data showed that both CD68+iNOS+ M1 macrophages and CD68+CD206+ M2 macrophages significantly increased in the liver of mice on MCD diet. DDC significantly attenuated MCD‐induced infiltration of M1 and M2 macrophages. Furthermore, the suppression of M1 macrophages was more pronounced than M2 macrophages in the liver after DDC treatment. Taken together, data presented here showed that DDC treatment mitigated MCD diet‐induced hepatocyte injury and induced secretion of pro‐inflammatory cytokines and subsequent infiltration of inflammatory cells.
The mechanisms involved in the progression of steatosis to NASH are complex and not completely understood (Than and Newsome, 2015). The current view on the mechanisms underlying the development of NASH favours a model, in which steatosis and then steatohepatitis, induced by free fatty acid overload, insulin, oxidative stress, ER stress, hepatocyte lipoapoptosis, immune infiltration with activation of inflammatory cells and subsequent activation of HSC and myofibroblasts (Machado and Diehl, 2016). In this study, our data showed that DDC treatment attenuated hepatic steatosis, cell stress, hepatocyte ballooning and inflammation. In addition, DDC treatment significantly suppressed the expression of fibrogenesis‐related genes, the activation of HSC and myofibroblasts and subsequent collagen deposition in the liver of MCD‐induced NASH mice. Microarray analyses showed that DDC strongly affected oxidative stress and lipid metabolism‐related biological processes and pathways, including oxidation–reduction process, lipid metabolic process, fatty acid metabolic process, metabolic pathways, oxidative phosphorylation, NAFLD and PPAR signalling pathway. These data suggested that the modulation of lipid metabolism and oxidative stress by DDC might be involved in the prevention of MCD diet‐induced NAFLD.
However, the present study still has some limitations. Firstly, a MCD diet‐induced steatohepatitis model does not develop the typical metabolic abnormalities observed in human NASH, such as obesity, peripheral insulin resistance and hyperglycaemia. Further studies based on an animal model more closely resembling human NASH may be helpful to evaluate the applicability of DDC. Secondly, DDC was administered to the rats for all 6 weeks and the mice for all 8 weeks (early treatment group), and a later treatment schedule should be tried in the future. Thirdly, ATF4 and K8 mRNA levels showed a reduction in the MCD + DDC group compared to the MCS group. Whether this reduction is a side effect of the doses selected needs to be investigated in the future study. In addition, the detailed mechanism of DDC modulation on NAFLD and related fibrosis need to be elucidated in a future study.
In conclusion, the results presented here demonstrate that DDC attenuates MCD diet‐induced steatosis in NAFL rats and hepatocyte ballooning, inflammation and fibrosis in NASH mice. Treatment with DDC may have therapeutic potential for NAFLD and related fibrosis.
Author contributions
T.L., H.Y. and J.J. designed the study. T.L., P.W., M.C., X.Z., D.Z., H.X. and L.L. performed the experiments. T.L. and H.Y. analysed the data. T.L. wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstrac acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Treatment with DDC improves lipid metabolism related parameters in mice fed MCD diet for 8 weeks. Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. MCD group were treated either with vehicle (1×PBS) or 450 mg/kg DDC via daily gavage for all 8 weeks. (A) Serum CHO (mmol/L) levels of mice. (B) Serum HDL (mmol/L) levels of mice. (C) Serum LDL (mmol/L) levels of mice. (D) The transcript levels of Fas in the liver of mice. (E) The transcript levels of Srebp‐1 in the liver of mice. (F) The transcript levels of Scd1 in the liver of mice. Data are expressed as mean ± SEM. n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
Figure S2 Treatment with DDC suppresses hepatocyte ballooning and liver injury in the livers of mice fed MCD diet for 8 weeks.Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. MCD group were treated either with vehicle (1×PBS) or 450 mg/kg DDC via daily gavage for all 8 weeks. (A) Scores of hepatocyte ballooning in liver sections were measured. Data are expressed as mean ± SEM. n = 8 mice per group. *P < 0.05. (B) Serum ALT (U/mL) levels of mice. (C) Serum AST (U/mL) levels of mice. Data are expressed as mean ± SEM. n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
Figure S3 Treatment with DDC suppresses the number of M1 and M2 macrophages and the expression of related genesin mice liver.Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. MCD group were treated either with vehicle (1×PBS) or 450 mg/kg DDC via daily gavage for all 8 weeks. (A) Identification of CD68+iNOS+M1 macrophage subset using immunofluorescence staining. Liver sections were double‐labeled with antibodies to CD68 (green) and iNOS(red) (original magnification ×400). (B) Identification of CD68+CD206+M2 macrophage subset using immunofluorescence staining. Liver sections were double‐labeled with antibodies to CD68 (green) and CD206(red) (original magnification ×400). (C) The number of CD68+iNOS+cells was counted in ten randomly selected microscopic fields (×400). (D) The number of CD68+CD206+cells was counted in ten randomly selected microscopic fields (×400). (E) The transcript levels of iNOS in the liver of mice. (F) The transcript levels of CD206 in the liver of mice. Data are expressed as mean ± SEM. n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
Figure S4 Treatment with DDC suppresses the expression of TGF‐β in livers of mice fed MCD diet for 8 weeks. Male C57BL/6 mice were fed either MCD or MCSdiets for 8 weeks. MCD group were treated either with vehicle (1×PBS) or 450 mg/kg DDC via daily gavage for all 8 weeks. The transcript levels of TGF‐β in the liver of mice. Data are expressed as mean ± SEM. n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
Table S1 Primers for RT‐PCR detection (mouse).
Table S2 Primers for RT‐PCR detection (rat).
Acknowledgements
This study was supported by the National Natural Science Foundation of China (grant numbers 81770596, 81641022 and 81100287), the Wang Baoen Liver Fibrosis Foundation (grant numbers CFHPC20161019 and CFHPC20131032) and the Beijing Health System Talents Plan (grant number 2013‐3‐062).
Liu, T. , Wang, P. , Cong, M. , Zhao, X. , Zhang, D. , Xu, H. , Liu, L. , Jia, J. , and You, H. (2018) Diethyldithiocarbamate, an anti‐abuse drug, alleviates steatohepatitis and fibrosis in rodents through modulating lipid metabolism and oxidative stress. British Journal of Pharmacology, 175: 4480–4495. 10.1111/bph.14503.
References
- Adams LA, Sanderson S, Lindor KD, Angulo P (2005). The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol 42: 132–138. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The concise guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The concise guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide To PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashraf NU, Sheikh TA (2015). Endoplasmic reticulum stress and oxidative stress in the pathogenesis of non‐alcoholic fatty liver disease. Free Radic Res 49: 1405–1418. [DOI] [PubMed] [Google Scholar]
- Brunt EM (2011). Non‐alcoholic fatty liver disease: what's new under the microscope? Gut 60: 1152–1158. [DOI] [PubMed] [Google Scholar]
- Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M et al (2018). The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67: 328–357. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvek B (2012). Nonprofit drugs as the salvation of the world's healthcare systems: the case of Antabuse (disulfiram). Drug Discov Today 17: 409–412. [DOI] [PubMed] [Google Scholar]
- Diehl A, Ulmer L, Mutschler J, Herre H, Krumm B, Croissant B et al (2010). Why is disulfiram superior to acamprosate in the routine clinical setting? A retrospective long‐term study in 353 alcohol‐dependent patients. Alcohol Alcohol 45: 271–277. [DOI] [PubMed] [Google Scholar]
- Dufour P, Lang JM, Giron C, Duclos B, Haehnel P, Jaeck D et al (1993). Sodium dithiocarb as adjuvant immunotherapy for high risk breast cancer: a randomized study. Biotherapy 6: 9–12. [DOI] [PubMed] [Google Scholar]
- European Association for the Study of the Liver (EASL) , European Association for the Study of Diabetes (EASD) , European Association for the Study of Obesity (EASO) (2016). EASL‐EASD‐EASO Clinical Practice Guidelines for the management of non‐alcoholic fatty liver disease. J Hepatol 64: 1388–1402. [DOI] [PubMed] [Google Scholar]
- Fan JG, Farrell GC (2009). Epidemiology of non‐alcoholic fatty liver disease in China. J Hepatol 50: 204–210. [DOI] [PubMed] [Google Scholar]
- Gentile CL, Frye M, Pagliassotti MJ (2011). Endoplasmic reticulum stress and the unfolded protein response in nonalcoholic fatty liver disease. Antioxid Redox Signal 15: 505–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon RC, Tiniakos DG, Argo CK (2011). Inflammation in nonalcoholic steatohepatitis. Expert Rev Gastroenterol Hepatol 5: 189–200. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch R, Clarkson V, Shephard EG, Marais DA, Jaffer MA, Woodburne VE et al (2003). Rodent nutritional model of non‐alcoholic steatohepatitis: species, strain and sex difference studies. J Gastroenterol Hepatol 18: 1272–1282. [DOI] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW et al (2005). Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41: 1313–1321. [DOI] [PubMed] [Google Scholar]
- Liu T, Wang P, Cong M, Xu Y, Jia J, You H (2013). The CYP2E1 inhibitor DDC up‐regulates MMP‐1 expression in hepatic stellate cells via an ERK1/2‐ and Akt‐dependent mechanism. Biosci Rep 33: e00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Wang P, Cong M, Zhang D, Liu L, Li H et al (2016). Matrix metalloproteinase‐1 induction by diethyldithiocarbamate is regulated via Akt and ERK/miR222/ETS‐1 pathways in hepatic stellate cells. Biosci Rep 36: e00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado MV, Diehl AM (2016). Pathogenesis of nonalcoholic steatohepatitis. Gastroenterology 150: 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi H, Kaufman RJ (2011). Endoplasmic reticulum stress in liver disease. J Hepatol 54: 795–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuno T, Kariya R, Yano S, Morino‐Koga S, Taura M, Suico MA et al (2012). Diethyldithiocarbamate induces apoptosis in HHV‐8‐infected primary effusion lymphoma cells via inhibition of the NF‐κB pathway. Int J Oncol 40: 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal S, El‐Serag HB, Sada YH, Kanwal F, Duan Z, Temple S et al (2016). Hepatocellular carcinoma in the absence of cirrhosis in United States veterans is associated with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 14: 124–131.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt‐Hyatt M, Lin HL, Hollenberg PF (2010). Mechanism‐based inactivation of human CYP2E1 by diethyldithocarbamate. Drug Metab Dispos 38: 2286–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM (2008). Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline‐deficient diet. J Lipid Res 49: 1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Leclercq I, Farrell GC (2001). Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P‐450 enzymes and oxidative stress. Am J Physiol Gastrointest Liver Physiol 281: G1135–G1139. [DOI] [PubMed] [Google Scholar]
- Sanyal AJ, Friedman SL, McCullough AJ, Dimick‐Santos L, American Association for the Study of Liver Diseases , United States Food and Drug Administration (2015). Challenges and opportunities in drug and biomarker development for nonalcoholic steatohepatitis: findings and recommendations from an American Association for the Study of Liver Diseases‐U.S. Food and Drug Administration Joint Workshop. Hepatology 61: 1392–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz JJ, Emerson L, Hillas E, Phan A, Thiesset H, Firpo M et al (2013). Amelioration of hepatic inflammation in a mouse model of NASH using a dithiocarbamate derivative. Hepatol Int 7: 600–609. [DOI] [PubMed] [Google Scholar]
- Singla A, Moons DS, Snider NT, Wagenmaker ER, Jayasundera VB, Omary MB (2012). Oxidative stress, Nrf2 and keratin up‐regulation associate with Mallory–Denk body formation in mouse erythropoietic protoporphyria. Hepatology 56: 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrott Z, Mistrik M, Andersen KK, Friis S, Majera D, Gursky J et al (2017). Alcohol‐abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 552: 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K (2013). Liver disease: Kupffer cells regulate the progression of ALD and NAFLD. Nat Rev Gastroenterol Hepatol 10: 503. [DOI] [PubMed] [Google Scholar]
- Snider NT, Griggs NW, Singla A, Moons DS, Weerasinghe SV, Lok AS et al (2013). CD73 (ecto‐5′‐nucleotidase) hepatocyte levels differ across mouse strains and contribute to Mallory–Denk body formation. Hepatology 58: 1790–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott I, Murthy A, Robinson A, Thomas NW, Fry JR (1997). Low‐dose diethyldithiocarbamate attenuates the hepatotoxicity of 1,3‐dichloro‐2‐propanol and selectively inhibits CYP2E1 activity in the rat. Hum Exp Toxicol 16: 262–266. [DOI] [PubMed] [Google Scholar]
- Than NN, Newsome PN (2015). A concise review of non‐alcoholic fatty liver disease. Atherosclerosis 239: 192–202. [DOI] [PubMed] [Google Scholar]
- Van Herck MA, Vonghia L, Francque SM (2017). Animal models of nonalcoholic fatty liver disease–A starter's guide. Nutrients 9: E1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon G, Baranova A, Younossi ZM (2011). Systematic review: the epidemiology and natural history of non‐alcoholic fatty liver disease and non‐alcoholic steatohepatitis in adults. Aliment Pharmacol Ther 34: 274–285. [DOI] [PubMed] [Google Scholar]
- Weltman MD, Farrell GC, Liddle C (1996). Increased hepatocyte CYP2E1 expression in a rat nutritional model of steatosis with inflammation. Gastroenterology 111: 1645–1653. [DOI] [PubMed] [Google Scholar]
- Yeh MM, Brunt EM (2014). Pathological features of fatty liver disease. Gastroenterology 147: 754–764. [DOI] [PubMed] [Google Scholar]
- Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M et al (2018). Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 15: 11–20. [DOI] [PubMed] [Google Scholar]
- Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM et al (2007). From Mallory to Mallory–Denk bodies: what, how and why? Exp Cell Res 313: 2033–2049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Treatment with DDC improves lipid metabolism related parameters in mice fed MCD diet for 8 weeks. Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. MCD group were treated either with vehicle (1×PBS) or 450 mg/kg DDC via daily gavage for all 8 weeks. (A) Serum CHO (mmol/L) levels of mice. (B) Serum HDL (mmol/L) levels of mice. (C) Serum LDL (mmol/L) levels of mice. (D) The transcript levels of Fas in the liver of mice. (E) The transcript levels of Srebp‐1 in the liver of mice. (F) The transcript levels of Scd1 in the liver of mice. Data are expressed as mean ± SEM. n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
Figure S2 Treatment with DDC suppresses hepatocyte ballooning and liver injury in the livers of mice fed MCD diet for 8 weeks.Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. MCD group were treated either with vehicle (1×PBS) or 450 mg/kg DDC via daily gavage for all 8 weeks. (A) Scores of hepatocyte ballooning in liver sections were measured. Data are expressed as mean ± SEM. n = 8 mice per group. *P < 0.05. (B) Serum ALT (U/mL) levels of mice. (C) Serum AST (U/mL) levels of mice. Data are expressed as mean ± SEM. n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
Figure S3 Treatment with DDC suppresses the number of M1 and M2 macrophages and the expression of related genesin mice liver.Male C57BL/6 mice were fed either MCD or MCS diets for 8 weeks. MCD group were treated either with vehicle (1×PBS) or 450 mg/kg DDC via daily gavage for all 8 weeks. (A) Identification of CD68+iNOS+M1 macrophage subset using immunofluorescence staining. Liver sections were double‐labeled with antibodies to CD68 (green) and iNOS(red) (original magnification ×400). (B) Identification of CD68+CD206+M2 macrophage subset using immunofluorescence staining. Liver sections were double‐labeled with antibodies to CD68 (green) and CD206(red) (original magnification ×400). (C) The number of CD68+iNOS+cells was counted in ten randomly selected microscopic fields (×400). (D) The number of CD68+CD206+cells was counted in ten randomly selected microscopic fields (×400). (E) The transcript levels of iNOS in the liver of mice. (F) The transcript levels of CD206 in the liver of mice. Data are expressed as mean ± SEM. n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
Figure S4 Treatment with DDC suppresses the expression of TGF‐β in livers of mice fed MCD diet for 8 weeks. Male C57BL/6 mice were fed either MCD or MCSdiets for 8 weeks. MCD group were treated either with vehicle (1×PBS) or 450 mg/kg DDC via daily gavage for all 8 weeks. The transcript levels of TGF‐β in the liver of mice. Data are expressed as mean ± SEM. n = 8 mice per group. One‐way ANOVA followed by Dunnett's post hoc test for multiple comparisons. *P < 0.05.
Table S1 Primers for RT‐PCR detection (mouse).
Table S2 Primers for RT‐PCR detection (rat).