

Abstract
Aim
A Phase 1 study was performed to evaluate safety, pharmacokinetics (PK) and pharmacodynamics (PD) of the selective histamine H4 receptor antagonist SENS‐111, an oral small molecule.
Methods
One hundred healthy subjects were randomized in a placebo‐controlled, double‐blind study evaluating single‐ascending doses (SAD; 100–500 mg) and multiple‐ascending doses (MAD; 50–150 mg day−1, 4 days; 200–250 mg day−1, 7 days). Effects of SENS‐111 on nystagmus and vertigo induced by modified caloric tests were measured in the MAD studies. Population PK and PK/PD models were developed using a nonlinear mixed‐effects approach.
Results
SENS‐111 was well tolerated with mild to moderate events. No sedation was reported. A maximal tolerated dose was not reached. Dose‐proportional increases in concentrations were seen up to 200 mg and more than dose‐proportional thereafter, with mean half‐life between 24 and 56 h. The caloric test induced mild but measurable vertigo and nystagmus with large intra/inter‐individual variation for all parameters. SENS‐111 did not significantly impact nystagmus but significantly improved latency of vertigo appearance/disappearance, duration and European Evaluation of Vertigo questionnaire parameters vs. baseline. A two‐compartment model with first‐order absorption, distribution and elimination best fit the data. PK/PD indirect modelling applied to vertigo duration and latency of appearance indicated maximum activity between 100 and 500 ng ml−1 plasma concentrations, corresponding to 100 and 200 mg day−1, which are appropriate for clinical efficacy evaluations in vestibular diseases.
Conclusions
SENS‐111 is a well‐tolerated first‐in‐class H4 receptor antagonist with acceptable PK for oral daily dosing. PK/PD modelling determined plasma concentrations and doses for efficacy studies in patients with vertigo symptoms.
Keywords: nystagmus, pharmacokinetics/pharmacodynamics, type‐4 histamine receptor, vertigo, vestibular disorders
What is Already Known about this Subject
Current histamine antagonists have limited success for treating vestibular dysfunction.
The type‐4 histamine (H4) receptor is expressed in vestibular primary neurons and selective H4 receptor antagonists inhibit vestibular neuron activity in vitro and ex vivo, and alleviate vertigo symptoms in rats with unilateral vestibular lesions. However, PK/PD evaluations are lacking.
Preliminary preclinical studies of a novel selective H4 receptor antagonist SENS‐111 suggest that PK/PD evaluations in the clinical context of acute unilateral vestibulopathy are merited.
What this Study Adds
SENS‐111 is a well‐tolerated first‐in‐class H4 receptor antagonist with acceptable pharmacokinetics for oral daily dosing.
Population PK and PK/PD models were developed for SENS‐111 integrating modified caloric tests to evaluate nystagmus and vertigo as the primary signs of vestibular dysfunction, and these PK/PD analyses established criteria for dose selection for future clinical development.
The results support further clinical evaluation of SENS‐111 in patients with vertigo symptoms.
Introduction
Histamine receptors are potent molecular targets which play an important role in modulating vestibular functions.Histamine activation can be blocked by specific receptor antagonists 1. http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=262 antagonists, such as meclizine 2 are frequently used to treat vestibular dysfunction, including notably acute unilateral vestibulopathy (AUV) or motion sickness; however, their use is limited by the resulting sedation which reduces or delays central compensation. Betahistine acts against both H1 receptors and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=264 with a yet‐to‐be deciphered mechanism of action, accelerating vestibular compensation 3. Recent attention has focused on the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=265, which is expressed in mammalian vestibular primary neurons 4, 5, 6. A pronounced inhibitory effect on vestibular neuron activity was seen in vitro and ex vivo in the presence of two selective H4 receptor antagonists. Furthermore, when administered in vivo to rats suffering from unilateral vestibular lesions, these antagonists were found to alleviate vertigo symptoms. These data support the targeting of H4 receptor signalling as a potential avenue for treating vestibular disorders.
SENS‐111 (Seliforant; 2‐isobutyl‐6‐(3 methylamino) azetidin‐1‐yl) pyrimidin‐4‐amine), a novel oral small molecule, is a selective H4 receptor antagonist. To date, it has been administered in over 170 healthy volunteers. Here we report a clinical Phase 1 study carried out to determine the safety, tolerability and pharmacokinetics (PK) profile of SENS‐111 administered as single and multiple ascending doses in healthy volunteers. As a non‐sedative H4 receptor antagonist, it is anticipated that SENS‐111 can play a clinical role in the relief of vertigo symptoms in vestibular diseases, including AUV. In order to confirm data observed in vitro and in vivo, we conducted a clinical study where the effects of SENS‐111 on nystagmus and vertigo induced via caloric irrigation were evaluated in healthy subjects. Population PK and pharmacokinetic/pharmacodynamic (PK/PD) models were developed, and a PK/PD analysis was conducted to develop criteria for dose selection and to optimize the later‐phase clinical development plan.
Methods
Study design
A randomized placebo‐controlled, double‐blind study was performed in healthy volunteers with three sequential sub‐studies, a single‐ascending‐dose (SAD) and two multiple‐ascending‐dose (MAD) studies. The study was conducted at Eurofins Optimed (Gieres, France), and was registered as EudraCT No. 2014‐004056‐65. It was approved by the local ethics committee and carried out in accordance with the Declaration of Helsinki, Good Clinical Practice and national regulations. All subjects signed written informed consent prior to any study procedures.
Initially, single ascending doses of SENS‐111 (100, 200, 300, 400, 500 mg) or placebo were administered sequentially to cohorts of eight subjects (six test, two placebo per dose level). Two sequential multiple‐ascending‐dose studies of SENS‐111 or placebo were then performed in cohorts of 12 subjects (nine test, three placebo per dose level). In the first of these, SENS‐111 (50, 100, 150 mg day−1) or placebo was administered for four consecutive days, and in the second SENS‐111 (200, 250 mg day−1) or placebo was administered for seven consecutive days. Subjects were followed‐up for 7–9 days after the last dose. SENS‐111 and matched placebo were taken orally once daily in the morning, under fasted conditions. Subjects received standardized meals, exercise was restricted and smoking was prohibited. Adverse events (AEs) and physical examination evaluations were recorded throughout the study. Vital signs and ECG assessments were performed at baseline, on the day(s) of dosing (predose and ~2.5–3 h later for all doses, and 12 h later for the ≥300 mg single doses), and at the end of study. Laboratory parameters were evaluated at baseline, then every 2 days up to Days 4, 6 and 10 for the three sequential studies respectively, and at the end of study.
The decision to increase the dose to the next dose level was made after the Data Safety Monitoring Board (DSMB; consisting of sponsor representatives, investigators, pharmacovigilance and ENT specialists) performed a blinded review of safety/tolerability and PK data of at least six out of eight subjects of a given dose level for up to at least 48 h post‐dose. A decision could be made to add more subjects at the same dose level or investigate intermediate dose levels. Given that toxicity studies identified potential neurological toxic effects at very high doses and concentrations, the DSMB considered it safer to limit doses to maximum doses of 500 mg single administration and 250 mg once daily for 7 days, to maintain a large safety margin.
The primary objective was to establish the safety and tolerance profile of SENS‐111 when administered as single or multiple ascending doses. Secondary objectives were to characterize the PK properties of SENS‐111 and its metabolites, and the relationship between SENS‐111 exposure and its effect on nystagmus and vertigo (in the MAD studies).
Dose rationale
Previous in‐vitro experiments showed that SENS‐111 antagonizes H4 receptor activity in the inner ear at concentrations corresponding to human plasma concentrations of 60–150 ng ml−1. In‐vivo experiments in a range of pharmacologic models (mice, rat and monkey asthma models, rat vestibular deficiency model) showed that SENS‐111 is active at doses corresponding to human plasma concentrations of 100–500 ng ml−1. H4 blockade was previously evaluated in a first‐in‐human single‐dose study in healthy volunteers (via ex‐vivo measurement of eosinophil shape change after histamine stimulation) and correlated with SENS‐111 plasma concentrations, with SENS‐111 doses from 2 mg to 100 mg. Maximum PD effects were seen at 60–100 mg with mean plasma C max of 70–120 ng ml−1 respectively. Given that SENS‐111 concentrations in the inner ear are one‐third of plasma concentration in animal models, plasma concentrations of 210–360 ng ml−1 were expected to give significant H4 blockade in the inner ear. While single doses of 100 mg were safe in the previous clinical study, in the current SAD study, the 100 mg dose was repeated as a safety precaution, with planned escalation up to 400 mg. The 500 mg dose was added subsequently based on the safe profile at lower doses. The study design allowed for the planned MAD doses (50–150 mg day−1 in the 4‐day study and 200 mg day−1 in the 7‐day study per DSMB) to be refined according to the results of the SAD study. In the 7‐day MAD study, if the average exposure in the 200 mg cohort was lower than that observed in the 400 mg SAD cohort, a cohort of 250 mg day−1 was planned. The maximal 7‐day MAD dose tested was fixed at half the maximal tolerated dose in the SAD study.
Subjects
For all three SAD and MAD studies, men aged 18–45 years, non‐smokers or smokers of ≤5 cigarettes/day, with a body mass index (BMI) of between 18.5 and 28.5 kg m−2, normal dietary habits, and without any clinically relevant abnormalities, and using adequate contraceptive measures were eligible. Women aged 45–65 years who were not of childbearing potential and who fulfilled these criteria were eligible for the last cohort of SAD and the final two cohorts of the 7‐day MAD study. For all subjects, the main exclusion criteria were a history of ear/head trauma, ophthalmic surgery, history of or current deafness, family history of deafness, vestibular neuritis, vestibular migraine, Menière's disease and/or benign paroxysmal positional vertigo, current tympanic membrane perforation, acute or chronic otitis, strabismus, frequent headaches and/or migraine, recurrent nausea and/or vomiting, or any current or past acute or chronic disease or disorder.
PK sampling and bioanalytical analyses
For the SAD sub‐study, blood samples were collected pre‐dose, then 15 and 30 min post‐dose, and 1, 1.5, 2, 4, 6, 8, 12, 24, 48 and 72 h post‐dose. For the 4‐day MAD sub‐study, blood samples were collected pre‐dose on Day 1, then 30 min, 1, 1.5, 2, 4, 6, 8 and 12 h post‐dose, pre‐dose on Days 2 and 3, pre‐dose on Day 4 and 30 min, 1, 1.5, 2, 4, 6, 8, 12 h post‐dose, then 24, 48 and 72 h post‐dose (Days 5, 6 and 7). For the 7‐day MAD sub‐study, blood samples were collected pre‐dose on Day 1, 30 min, 1, 1.5, 2, 4, 6, 8, and 12 h post‐dose, post‐dose on Days 2–6, pre‐dose on Day 7 and 30 min, 1, 1.5, 2, 4, 6, 8 and 12 post‐dose, then 24, 48, 72, and 192 h post‐dose (Days 8, 9, 10 and 15). Blood was collected in polypropylene tubes containing lithium heparin and kept on ice, centrifuged immediately and stored at −80°C. Plasma concentrations of SENS‐111 and its two metabolites (UR‐64167 and UR‐64250) were determined using a validated LC–MS/MS analytical method (detection limits: 1–500 ng ml−1, CV between and within runs: 3.7% and 7.11% respectively, mean % deviation from nominal concentration: 1.6%).
Non‐compartmental PK analysis
Estimated parameters included maximum observed plasma concentration (C max), time to C max (t max), terminal phase elimination half‐life (t 1/2), apparent oral clearance (CL/F), apparent volume of distribution (Vd/F), area under the plasma concentration–time curve from time 0 to time t (AUC0–t) and to infinity (AUCinf). All below level of quantification (BLQ) values in the absorption phase were substituted by zero, except for BLQ values between quantifiable concentrations, which were treated as missing, before calculating PK variables. Terminal BLQ values were ignored. The elimination rate constant (K el) was determined by linear regression of the logarithmic concentration in plasma with time over the terminal phase. The best fit for each PK profile was automatically selected by the software. t 1/2 was calculated as ln2/K el, C max and t max were the observed values. AUCt was the last quantifiable concentration in plasma by the linear trapezoidal rule and was extrapolated to infinity using the terminal‐phase K el. AUC0–24 was calculated using the linear trapezoidal rule from 0 up to 24 h. Accumulation ratios were calculated as AUC0–24, Day ‘x’/AUC0–24, Day 1, where ‘x’ is Day 4 or Day 7. Dose proportionality was assessed by linear regression of dose vs. AUCinf and dose vs. C max using the parameter values estimated in the non‐compartmental analysis.
Vestibular dysfunction assessment
A modified caloric test was performed to evaluate the effects of SENS‐111 on nystagmus and vertigo, signs of vestibular dysfunction. The right ear was irrigated with water (25°C, 250 ml min−1) until a nystagmus peak slow phase velocity (SPV) of at least 20° s−1 was reached during the culmination phase.
The caloric test was performed for the MAD sub‐study, three times at 3‐h intervals on Day −1 (baseline). It was performed only once daily 2–3 h post‐dose from Days 1 to 4, and at 2 h post‐dose on Day 7 for the 7‐day MAD study to record vertigo latency (appearance and disappearance) and duration.
Nystagmus was recorded by videonystagmography using a mono‐camera videonystagmograph. Culmination was considered the maximum SPV value. Online automatic detection of maximal SPV was deduced from the slow phase cumulative curve (desaccaded nystagmus) over 2 s, according to the formula SPV (t) = (Cum [+1 s] – Cum [−1 s])/2. Subjects were required to look directly ahead and wore an eye mask to avoid visual fixation and counted down during recording to standardize their level of vigilance. Recording lasted up to 5 min after irrigation and was stopped earlier if eye movement speed was 0 for 2 min.
Vertigo intensity was measured using a visual analogue scale (VAS; 0–10, 0 being none) within 5 min after the end of the caloric irrigation. Vestibular syndrome was evaluated in terms of illusion of movement, duration of the illusion, motion intolerance, neurovegetative signs and instability using the physician‐administered European Evaluation of Vertigo (EEV) questionnaire (0–4, 0 being none) 7, 5 min after the end of the caloric irrigation.
As there was a marked intraindividual variability (for example, SPV had an intraindividual variability at baseline ranging from 2% to 84% in healthy subjects), the mean value of the three assessments was used as the baseline value for all nystagmus and vertigo variables.
Population PK and PK/PD modelling
A nonlinear mixed‐effects approach was used to develop population PK and PK/PD models, to evaluate the relationship between exposure and PD markers (nystagmus and vertigo parameters) and provide a rationale for dose selection in late‐phase clinical development.
Model development strategy
Model development was based on two sequential steps: (1) development of a population PK model exploring alternative absorption and disposition modelling approaches to account for single and repeated dose administrations, and (2) exploration of alternative population PK/PD models for characterizing the exposure–response relationship between SENS‐111 and PD marker outcomes of SPV peak at culmination and nystagmus frequency, latency of nystagmus culmination, latency of onset of a nystagmus (defined by SPV >2° s−1), duration of nystagmus, latency of onset/offset and duration of vertigo sensation, vertigo intensity and EEV.
Population PK model building
Different models (including first and zero order processes) were evaluated to characterize SENS‐111 absorption, and the first‐order process was the best performing model. The inter‐individual variability (IIV) model (random effect) described the unexplained random variability in individual values of structural model parameters (fixed effect). It was assumed that the IIV of the PK parameters was log‐normally distributed (random effect).
A two‐compartment model was ultimately retained. It was characterized by a central compartment, and peripheral distribution compartments with first‐order absorption, distribution and elimination rate constants described the PK of SENS‐111. The model parameters were clearance (estimated assuming bioavailability of 1), central volume (estimated assuming bioavailability of 1), inter‐compartmental clearance, peripheral volume, absorption rate constant and absorption lag time.
The covariate analysis was conducted to evaluate the potential impact of dose and time on clearance (CL) and volume (V). The following models were evaluated:
where CL and V are for the dose of 50 mg, and at Day 1, DosCL and TimCL and DosV and TimV are parameters characterizing the change in CL and V as a function of the dose and time, respectively.
Population PK/PD model building
An indirect modelling approach was used to characterize the exposure–response relationship, assuming that a measured response (R) may be produced by indirect mechanisms; for example, factors controlling the input or production (k in) of the response variable may be either inhibited or stimulated, or determinants of loss (k out) of the response variable may be inhibited or stimulated.
The rate of change of the response (R) over time for subjects treated with placebo was described by:
where k in represents the zero‐order constant for production of the response and k out defines the first‐order rate constant for loss of the response.
As the system is assumed to be stationary, the response variable (R) begins at a predetermined baseline value (Bas), changes with time and returns to Bas. Thus:
In the presence of SENS‐111 the PD response was described by:
where F1 and F2 are the drug‐related model components affecting the k in and k out values.
For latency of vertigo appearance, SENS‐111 was assumed to stimulate k in (positive response) and k out (negative response) at different levels of exposure according to the following functions:
where Cp represents the model‐predicted time‐varying plasma concentration of SENS‐111.
For duration of vertigo and latency of vertigo disappearance, SENS‐111 was assumed to inhibit k in and k out at different levels of exposure according to the following functions:
The indirect‐response model parameters describing the placebo and SENS‐111 responses were simultaneously estimated by jointly fitting the placebo and the drug‐related PD measurements for each PD measurements considered.
Modelling options
The relationship between a PK parameter (P) and its variance can be expressed as:
where P j is the value of the PK parameter for the jth individual, P TV is the typical value of P for the population, and η p denotes the difference between P j and P TV, independently, which was identically distributed with a mean of zero and variance of ωp2.
The residual variability, which comprised, but is not limited to, IIV, experimental errors, process noise and/or model misspecifications, was modelled using a combined error structures as: y ij = y tij(1 + ε1ij) + ε2ij. y ij represents the jth observation in the ith individual, y tij represents the corresponding model prediction, and ε1ij (or ε2ij) is a normally distributed random error with a mean of zero and a variance of σ2.
Evaluation of population models
Model selection was based on physiological and pharmacological rationale and the principle of parsimony. Model discrimination was based on several criteria. For each analysis, the improvement in model goodness‐of‐fit and significance of additional parameters were assessed with: (i) the likelihood ratio test (LRT). Significant improvement in model fit was assessed by the change in the minimum objective function values (OFV). Minimization of the test and the reference models OFV (i.e., ∆OFV = [OFVtest – OFVreference]) is proportional to a change in −2 log likelihood and is chi‐squared (χ2) distributed. The LRT was used to evaluate the statistical significance of nested models; and (ii) the Akaike Information Criterion (AIC).
Model performance
Model performance/validation and stability were assessed using goodness‐of‐fit plots, and visual predictive checks (VPC). Goodness‐of‐fit plots were generated including observed data vs. individual and model‐predicted concentrations, absolute‐weighed residuals vs. individual predictions, and conditional‐weighted residuals vs. time. Adequacy of the final model was evaluated with VPC, including the effects of statistically significant covariates.
Two hundred replicates of the original dataset were simulated based on the final model. The 5th, 50th and 95th percentiles of the observed data were computed using the simulated data together with their 95% confidence intervals. Statistics were calculated from simulated and observed data for comparison. Distributions of quantiles of simulated data were compared to observed data quantiles.
Simulation of effective dose
Simulations exploring three dose regimens for a relevant clinical response were conducted for 100 mg and 200 mg at time 0, 12, 24, 48, 72 and 96 h, and for 300 mg at time 0, then for 200 mg at 24, 48, 72 and 96 h. The population PK/PD model for vertigo duration and latency of vertigo appearance and disappearance was used to estimate response by simulating the treatment response using Monte Carlo simulation. Plots of the median SENS‐111 concentration with the prediction intervals and median change from baseline of the duration of vertigo and the latency of vertigo appearance with the prediction intervals were generated.
Statistical methods
Safety was analysed in all subjects who received at least one dose of SENS‐111. AEs were coded using MedDRA, v17. Changes of SPV peak at culmination, frequency and latency of nystagmus appearance, latency of nystagmus culmination and disappearance and duration from baseline to 2‐h post‐treatment for Days 1–4 (and Day 7 for the 7‐day MAD study) were analysed by analysis of covariance. Active doses were compared from the mixed linear model using a Tukey adjustment 8. A repeated‐measures mixed linear model was used with treatment/dose group, time and baseline value as fixed effects and subject as a random effect. If the residual distribution was not normal (Shapiro–Wilk test), log transformation was applied (before change). If a normal hypothesis was not demonstrated, rank data were retained. For vertigo, latency, duration, and intensity were analysed with the same caloric test. The EEV was analysed with a GENMOD generalized linear model (five items with five categorical modalities each), and the total EEV score was analysed by a repeated measures analysis of variance model. A significance threshold of P = 0.05 was used. Non‐compartmental PK analyses were performed with Kinetica (v4.3), PK/PD population analyses using R (v3.2.2), SAS (v9.3), NONMEM software (v7.3, ICON Development Solutions), R‐based package Xpose (v4.3) and Perl‐based software Perl‐speaks‐NONMEM (PsN) (v3.4.2) 9.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 10, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 11.
Results
Subject population
Overall, 100 subjects were randomized and received at least one dose of SENS‐111 (n = 75) or placebo (n = 25) and completed the study; 40 subjects (30 SENS‐111, 10 placebo) including 28 males were treated in the SAD study, 36 (n = 27 SENS‐111, n = 9 placebo) in the 4‐day MAD all of whom were male, and 24 (n = 18 SENS‐111, n = 6 placebo) in the 7‐day MAD including 12 males. Demographics were comparable between the three sub‐study populations; mean age was between 29 and 44 years (range 19–65), mean BMI was between 23.1 and 24.1 kg m−2 (range 18.9–29.0), and most subjects were Caucasian (81 subjects; 81%) or black (17 subjects; 17%).
Safety
Single and multiple doses of SENS‐111 up to 250 mg were well tolerated and a maximal tolerated dose was not reached. AEs were reported in nine subjects (23%, four of whom received placebo) in the SAD study, seven subjects (19%, two of whom received placebo) in the 4‐day MAD study, and three subjects (13%, one of whom received placebo) in the 7‐day MAD study (Table 1). All AEs were mild to moderate, and resolved before study end. Few AEs were considered related; in subjects receiving SENS‐111, single episodes of dyspepsia and dry mouth were considered possibly related. Among placebo‐treated subjects, myalgia and headache were reported as possibly related. Other events were considered as unlikely or not related. There was no indication of dose‐dependency for any AEs. Abnormal laboratory values and changes in vital signs and electrocardiogram parameters occurred sporadically, but none considered clinically relevant. Minor deviations in ECG and vital sign assessments were reported that did not affect the quality and validity of the data. There were no deaths, serious AEs or discontinuations due to AEs.
Table 1.
All adverse events in the SAD and MAD studies, by subject
| Single‐dose study | Multiple‐dose studies | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n = 10) | SEN‐111 | Placebo (n = 9) | SEN‐111 (4‐days) | Placebo (n = 6) | SEN‐111 (7‐days) | ||||||||
| 100 mg (n = 6) | 200 mg (n = 6) | 300 mg (n = 6) | 400 mg (n = 6) | 500 mg (n = 6) | 50 mg (n = 9) | 100 mg (n = 9) | 150 mg (n = 9) | 200 mg (n = 9) | 250 mg (n = 9) | ||||
| Palpitations | 1 | ||||||||||||
| Dyspepsia | 1 | ||||||||||||
| Hypoglycaemia | 1 | ||||||||||||
| Back pain | 2 | 1 | 1 | ||||||||||
| Myalgia | 1 | ||||||||||||
| Headache | 2 | 1 | |||||||||||
| Erythema | 1 | ||||||||||||
| Vascular pain | 1 | ||||||||||||
| Cerumen impaction | 1 | 1 | |||||||||||
| Blood CPK increased | 1 | ||||||||||||
| Pre‐syncope | 1 | ||||||||||||
| Sciatica | 1 | ||||||||||||
| Cough | 1 | ||||||||||||
| Allergic rhinitis | 1 | ||||||||||||
| Dry mouth | 1 | ||||||||||||
| Total (%) | 4 (40%) | 1 (17%) | – | – | 2(34%) | 2(34%) | 2 (22%) | – | 2 22%) | 3 (33%) | 1 (33%) | 1 (11%) | 1 (11%) |
Non‐compartmental PK
Estimated PK parameters of the SAD and MAD studies are summarized in Tables S1 and S2 respectively. Single doses of SENS‐111 were absorbed relatively rapidly with a median t max ranging from 0.5 to 1.8 h. Plasma concentrations increased dose‐proportionally up to 200 mg and more than dose‐proportionally at higher doses. Mean plasma concentration–time curves (Figure S1A) showed moderate IIV (%CV 23.8–59.8%). Mean AUCt ranged from 1935 to 15 361 ng ml−1 h−1 with moderate IIV (%CV 10.0–26.5%). Mean t 1/2 ranged from 22 to 30 h. Vd/F was relatively large, ranging from 1068 to 1922 l.
Results of the MAD studies were consistent with the SAD studies. C max were observed after 1–8 h. Plasma concentrations again increased dose‐proportionally up to 200 mg after 4‐ and 7‐day repeat doses (Figure S1B and C). IIV was moderate for both C max and AUC values (from approximately 20% to 35%). t 1/2 after 4 days was similar to that of single dosing (25–28 h) but increased after 7 days of dosing (48–56 h). Some accumulation was observed, with mean AUC0–24 accumulation ratios of 1.8–2.0 in the 4‐day study and 2.6–2.7 for the 7‐day study, after 4 and 7 days respectively.
For both single and repeat dosing, plasma concentrations of the SENS‐111 metabolites UR‐64167 and UR‐64250 were quantifiable at all doses (Tables S3 and S4). For all studies, UR‐67147 was present at up to 5% of the parent compound SENS‐111 with similar t max and t 1/2, and concentrations of UR‐64250 were up to 1%, with a longer t max and a shorter t 1/2 at 7 days. No associated dose‐dependency was apparent.
Functional impact of SENS‐111 on nystagmus and vertigo
In both MAD studies, the caloric test induced vertigo and nystagmus at baseline almost systematically, which was mild but measurable but with large inter‐ and intra‐individual variability (15–47% mean of three values per subject). A summary of baseline parameters is provided in Tables 2 and 3. Inter‐ and intra‐individual variability was high for all parameters; for nystagmus, PSV peak was 35% (mean 23.6° s−1, range 6.1–63° s−1) and 1.2–83.7% respectively, for latency of culmination 17% (mean 78.5 s, range 44–121 s) and 1.2–48.2% respectively, and for nystagmus duration 27% (mean 88 s, range 0–403 s) and 1.6–38.1% respectively. Likewise, in terms of vertigo, inter‐ and intra‐individual variability for latency of appearance were 39% (mean 33 s, range 0–120 s) and 0–52.9% respectively, for duration they were 32% (mean 110 s, range 0–154 s) and 1.1–42.1% respectively, and for latency of disappearance they were 23% (mean 145 s, range 0–350 s) and 0.7–27.7% respectively.
Table 2.
Baseline mean ± SD for nystagmus parameters for the overall populations in the MAD studies
| Day 4 (n = 36) | Day 7 (n = 24) | |
|---|---|---|
| SPV peak (° s −1 ) | 26.10 ± 8.06 | 26.49 ± 10.60 |
| Nystagmus frequency (Hz) | 2.71 ± 0.54 | 2.55 ± 0.38 |
| Latency of nystagmus appearance (s) | 23.1 ± 4.7 | 23.1 ± 6.1 |
| Latency of nystagmus culmination (s) | 77.3 ± 12.8 | 74.1 ± 12.7 |
| Latency of nystagmus disappearance (s) | 224.7 ± 39.3 | 205.8 ± 64.3 |
| Duration of nystagmus (s) | 201.6 ± 40.5 | 182.6 ± 64.8 |
SPV, slow phase velocity
Table 3.
Baseline mean ± SD for vertigo parameters for the overall populations in the MAD studies
| Day 4 (n = 36) | Day 7 (n = 24) | |
|---|---|---|
| Latency of vertigo appearance (s) | 32.8 ± 15.4 | 32.2 ± 8.5 |
| Latency of vertigo disappearance (s) | 150.3 ± 24.7 | 141.9 ± 46.4 |
| Duration of the vertigo (s) | 117.5 ± 29.2 | 109.6 ± 45.1 |
| Intensity sensation VAS (mm) | 57.8 ± 18.3 | 54.7 ± 24.0 |
| Illusion of movement, 2/3/4, n (%) a | 9 (25.0) /10 (27.8) /17 (47.2) | 7 (29.2) /7 (29.2) /10 (41.7) |
| Duration of illusion, <1 min/ ≥1 min −1 h −1 , n (%) | 20 (55.6) /16 (44.4) | 12(50.0) /12 (50.0) |
| Motion intolerance, 0/1/2/3 b | 18 (50.0) /11 (30.6) /6 (16.7) /1 (2.8) | 16 (66.7) /3 (12.5) /4 (16.7) /1 (4.2) |
| Neurovegetative signs, 0/1/2 c , n (%) | 30 (83.3) /4 (11.1) / 2(5.6) | 24 (100.0) /0/0 |
| Instability, 0/1/2/3 d , n (%) | 23(63.9) /12(33.3) /1(2.8) | 17(70.8) /6(25.0) /0/1(4.2) |
| Total EEV score (0 to 20) | 5.9 ± 2.0 | 5.6 ± 2.0 |
2 = Feeling of swaying to the right or left; 4 = Impression of spinning
0 = No motion intolerance; 1 = Rarely or few; 2 = Sometimes or moderate; 3 = Often or marked
0 = No signs; 1 = Nausea uncorrelated with attacks of vertigo; 2 = Nausea correlated with attacks of vertigo
0 = No instability; 1 = Instability but no falls and no interference with daily activity; 2 = Instability without falls, but interferences with daily activity; 3 = Instability with occasional falls
For nystagmus, no significant changes were seen over time in subjects treated with SENS‐111. For vertigo, significant improvements (P < 0.05) were seen with SENS‐111 for all time points grouped vs. baseline for several parameters; latency of vertigo appearance was longer in the 150 mg group, latency of vertigo disappearance was shorter at all dose levels from 50 to 200 mg, duration of vertigo was shorter at dose levels 50, 100, 150 and 250 mg, and vertigo intensity sensation was lower at 100, 150 and 200 mg. For the EEV questionnaire, significant improvements were seen vs. baseline for movement (100, 150 and 200 mg), motion intolerance (150 mg), instability sensation (150, 200 and 250 mg) and total EEV score (50, 100, 150 and 200 mg).
Population PK model
The potential impact of dose and time on clearance and volume was evaluated using a step‐wise approach: at each step, one parameter (clearance or volume) was assumed to be affected by one covariate (dose or time). If the LRT indicated a significant contribution of the selected covariate, another covariate was included in the model. The final results of the analysis indicated that both dose and time significantly affected SENS‐111 clearance and volume, with clearance and volume expected to decrease with dose increases and with time. The population PK parameter estimates are presented in Table 4.
Table 4.
Population PK parameter estimates
| Parameter | Estimate | SE | RSE | |
|---|---|---|---|---|
|
Fixed
effect |
KA (h−1) | 2.400 | 0.356 | 14.80% |
| CL (l h−1) | 52.400 | 2.420 | 4.60% | |
| VC (l) | 1150.000 | 70.200 | 6.10% | |
| Q (l h−1) | 24.900 | 3.130 | 12.60% | |
| V3(l) | 539.000 | 20.800 | 3.90% | |
| LAG (h) | 0.244 | 0.0016 | 0.70% | |
| DayV | 0.079 | 0.0129 | 16.40% | |
| DoseV | 0.002 | 0.0004 | 14.60% | |
| DayCL | 0.056 | 0.0065 | 11.60% | |
| DoseCL | 0.002 | 0.0002 | 14.90% | |
| Add error | 1.270 | 0.418 | 32.90% | |
| Prop error | 0.200 | 0.0084 | 4.20% | |
|
Random
effect a |
KA | 1.220 | 0.328 | 26.90% |
| CL | 0.077 | 0.013 | 16.60% | |
| VC | 0.093 | 0.017 | 18.00% | |
| V3 | 0.025 | 0.010 | 38.20% | |
| LAG | 0.0004 | 0.0002 | 63.50% |
SE, standard error; RSE, relative standard error
The random effect values represent the IIV variance
Overall, the goodness‐of‐fit plots (Figure S2) showed no apparent bias suggesting that this model was adequate for describing SENS‐111 PK, which was assessed with VPCs using dose‐normalized concentrations of SENS‐111 (Figure 1). In addition to the dose‐normalized VPCs, additional VPCs were generated (Figure S3) on the untransformed data to evaluate the adequacy of the model to estimate the time‐varying change in exposure (VPCs of 200 mg on Day 1 vs. Day 7) and to estimate the dose‐dependent exposure (VPCs of 100 mg and 500 mg on Day 1).
Figure 1.
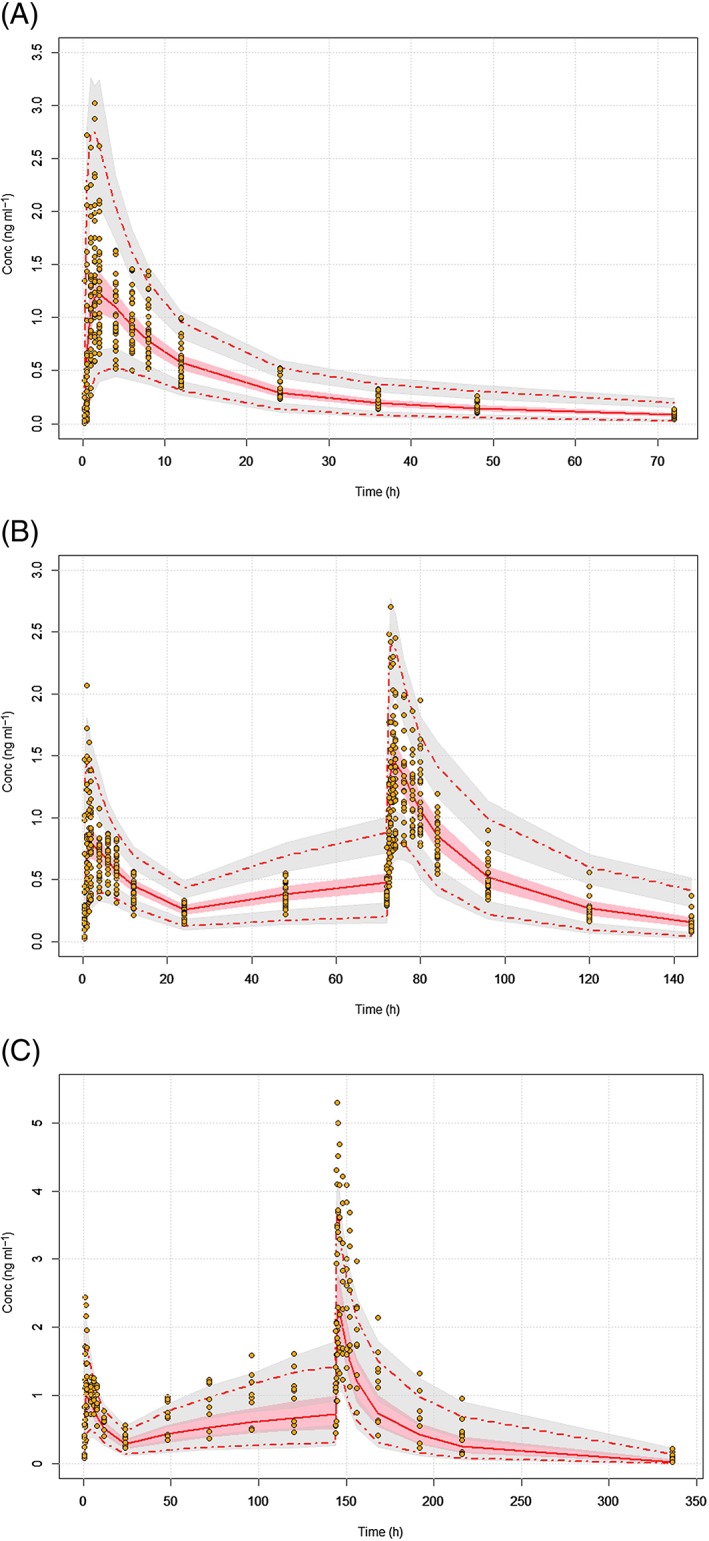
Visual predictive checks (VPCs) on dose‐normalized concentrations for (A) single ascending doses (B) and 4‐day and (C) 7‐day multiple ascending doses. The red lines represent the 5th, 50th, and 95th percentiles of the observed data (orange circles), the shaded grey areas are the 95% confidence intervals of the 5th and 95th percentiles of the simulated data, and the pink shaded areas are the 90% confidence intervals of the 50th percentiles of the simulated data
Population PK/PD model
A descriptive analysis of the relationship between PD measurements and individual level of SENS‐111 exposure was conducted prior to formal PK/PD modelling. Placebo data used to develop the PK/PD model are shown for latency of vertigo appearance and disappearance as well as duration of vertigo in Figure S4. A low and high level of SENS‐111 exposure were considered for duration of vertigo (low exposure from 50 to 550 ng ml−1 and high exposure >700 ng ml−1), latency of vertigo appearance (low exposure from 100 to 500 ng ml−1 and high exposure >600 ng ml−1), latency of vertigo disappearance (low exposure from 100 to 800 ng ml−1 and high exposure >800 ng ml−1). This showed a clear differential response level at low vs. high SENS‐111 concentrations (Figure S5), with the duration of vertigo reduced from 50 to 550 ng ml−1 and increased from 700 ng ml−1, the latency of vertigo appearance increased from 100 to 500 ng ml−1 with a reduction from 600 ng ml−1, and the latency of vertigo disappearance decreased in the range 100–800 ng ml−1. This behaviour, consistent with preclinical findings 12, indicated a bell‐shaped exposure–response relationship (Figure 2). The estimated population PK/PD parameters for the latency of vertigo appearance, duration of vertigo and latency of vertigo disappearance are presented in Table 5.
Figure 2.
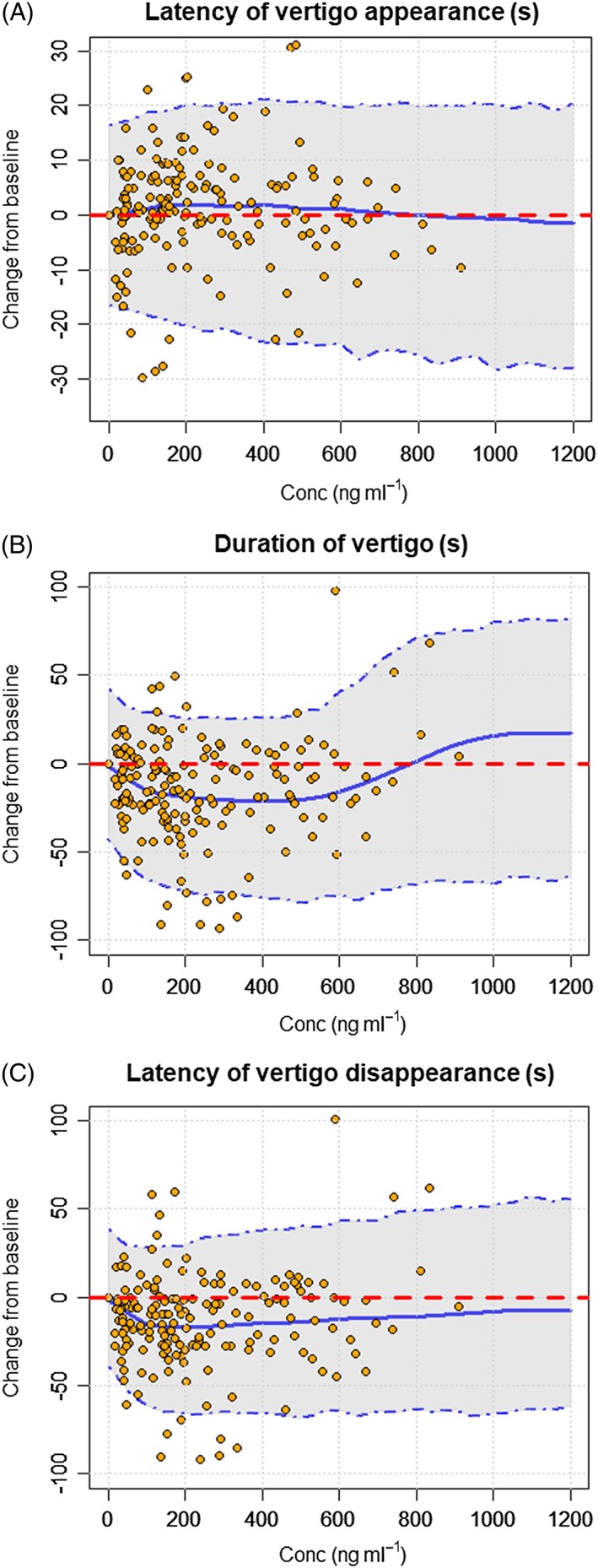
Visual predictive checks (VPCs) of the PK/PD model for latency of vertigo appearance (A), duration of vertigo (B) and latency of vertigo disappearance (C). The blue lines represent the 5th, 50th and 95th percentiles of the simulated data and the orange circles represent the observed data
Table 5.
Population PK/PD parameter estimates
| Parameter | Estimate | SE | RSE | |
|---|---|---|---|---|
| Latency of vertigo appearance | ||||
| Fixed effect | EMAX1 | 0.24 | 0.011 | 4.60% |
| EC501 | 138.00 | 10.900 | 7.90% | |
| EMAX2 | 2.61 | 0.085 | 3.30% | |
| EC502 | 15200.00 | 4270.000 | 28.10% | |
| K out | 0.36 | 0.067 | 18.60% | |
| Pop error | 0a | |||
| Add error | 10.2 | 0.997 | 9.80% | |
| Random effect b | EC501 | 0.133 | 0.006 | 4.30% |
| EC502 | 4.46 | 0.005 | 0.10% | |
| Duration of vertigo | ||||
| Fixed effect | EMAX1 | 0.22 | 0.012 | 5.60% |
| EC501 | 51.90 | 0.058 | 0.10% | |
| EMAX2 | 0.36 | 0.002 | 0.60% | |
| EC502 | 720.00 | 4.590 | 0.60% | |
| K out | 0.83 | 0.005 | 0.60% | |
| ga2 | 22.90 | 0.043 | 0.20% | |
| ga1 | 0.68 | 0.008 | 1.10% | |
| Pop error | 0.20 | 0.006 | 3.00% | |
| Add error | 16.70 | 0.206 | 1.20% | |
| Random effect b | EMAX1 | 0.62 | 0.481 | 78.10% |
| EC502 | 0.04 | 0.002 | 4.30% | |
| Latency of vertigo disappearance | ||||
| Fixed effect | EMAX1 | 0.26 | 0.009 | 3.40% |
| EC501 | 21.10 | 7.050 | 33.40% | |
| EMAX2 | 0.81 | 0.036 | 4.50% | |
| EC502 | 8960.00 | 2060.000 | 23.00% | |
| K out | 0.66 | 0.167 | 25.50% | |
| ga2 | 0.50 | 0.001 | 0.20% | |
| ga1 | 1a | |||
| Pop error | 0.20 | 0.002 | 0.80% | |
| Add error | 10.90 | 0.165 | 1.50% | |
| Random effect b | EMAX1 | 0.27 | 0.001 | 0.30% |
| EC502 | 1.78 | 0.001 | 0.00% | |
fixed; SE, standard error; RSE, relative standard error
The random effect values represent the IIV variance
The analysis of the EC501 and EC502 parameters provided some insight into the indirect response mechanism: the estimated values of these parameters indicated that for the latency of vertigo the most robust effect was on the simulation of k in that occurred at much lower exposure than the simulation of k out. For duration and latency of vertigo, the most robust effect was associated with the inhibition of k in that occurs at much lower exposure than the inhibition of k out for the two PD parameters.
The model showed significance (P < 0.05) for three vertigo parameters. Duration of vertigo was reduced by 27% to 30% (up to 27–28 s) from 50 to 550 ng ml−1 and increased from 700 ng ml−1. Latency of vertigo appearance increased by 10% to 13% (up to 3 to 4 s) from 100 to 500 ng ml−1 with a reduction from 600 ng ml−1. Latency of vertigo disappearance decreased by 15% to 20% (up to 20 to 25 s) from 100 to 800 ng ml−1 with no deleterious effects at high concentrations. Finally, simulations of the effective dose and dose regimen for clinical trial validation of the dose regimen are provided for three potential regimens in Figure 3, showing that twice daily (b.i.d.) administration of SENS‐111 on Day 1 achieved steady state rapidly.
Figure 3.
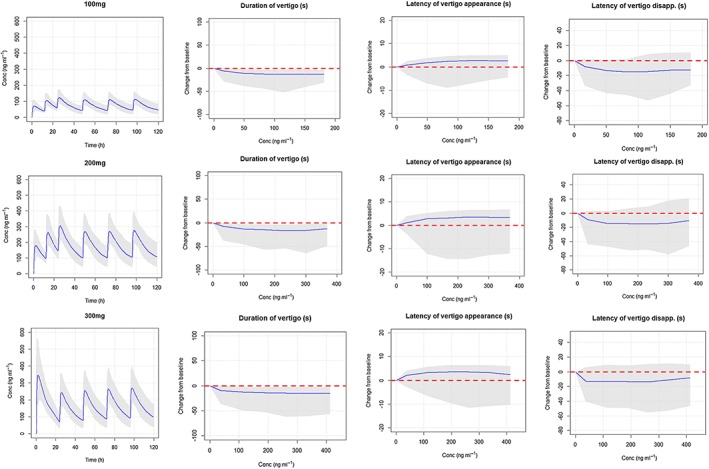
Simulated SENS‐111 concentrations and change from baseline of the duration of vertigo and the latency of vertigo appearance/disappearance, for three potential dose regimens. The solid blue lines represent the median predictions and the grey shaded areas the 5th and the 95th percentiles of the prediction interval
Discussion
Symptomatic treatment to mitigate vertigo, dizziness, nausea and vomiting is the cornerstone of management of acute vestibular disorders and notably of AUV, while physical therapy is used to improve central compensation 13. Anticholinergics, antihistamines and benzodiazepines are administered to counter vertigo and suppress vestibular responses, and antiemetics are used to combat nausea and vomiting 14. These agents act primarily through sedation or reduction of nausea and are associated with difficulties regulating central compensation. In this setting, the benefits of causal treatment with corticosteroids appear to be limited to accelerating patients' mid‐term recovery 15, without any proven long‐term benefit 16. In this Phase 1 clinical study in healthy men and women volunteers, oral SENS‐111 was generally well tolerated at single doses up to 500 mg and with multiple doses up to 250 mg daily for 7 consecutive days. No safety concerns were raised, with all AEs being mild to moderate and resolving before the end of the study, and no apparent dose dependency. Importantly, no sedation or somnolence was reported, even at the highest doses.
The PK profile of SENS‐111 and its two metabolites is linear after both single and repeated administrations from 50 to 200 mg but is non‐linear (increasing more than dose‐proportionally) for doses over 250 mg. Moderate IIV is apparent for C max, and AUC of SENS‐111 mostly between 20% and 35%, as well as for t max with highly divergent peaks ranging from 0.5 to 8 h across the three sub‐studies. Mean t 1/2 for SENS‐111 is relatively long, ranging from 25 to 56 h for multiple doses, giving accumulation ratios around 2.0 after 4‐day dosing and 2.7 after 7‐day dosing. Steady state is achieved after 7‐day dosing but is not apparent after 4 days. UR‐64167 and UR‐64250 exposure correspond to about 5% and 1% of SENS‐111 exposure respectively, did not increase or decrease with SENS‐111 dose or after repeated administrations, and are not anticipated to have a noticeable clinical impact. Preclinical studies have shown that SENS‐111 and its metabolites are excreted in urine and to a lesser extent in bile.
Caloric inductions with warm and cold water (44°C and 30°C) or air (50°C and 24°C) are often used in clinical practice by ENT doctors. This is a diagnostic tool whose primary goal is to assess whether the response to an identical cold or hot stimulus is symmetrical between ears and to evaluate the severity of vestibular dysfunction in the case of imbalance. The caloric test has rarely been used as a pharmacodynamic test either in healthy volunteers or in patients, and is thus not standardized. The effects of drugs used as standard of care in vertigo symptoms such as H1 histamine receptors antagonists (meclizine, dimenhydrinate) or agonists (betahistine), H3 receptor antagonists (betahistine) or anticholinergic agents (scopolamine) on parameters related to duration of induced vertigo symptoms (by modified calorics or more cumbersome tests such rotary chair, torsion or parallel swing) including nystagmus, are typically modest (less than 30% improvement) with large variability 17, 18, 19, 20, 21; specifically, effects on nystagmus PSV are either mild or absent. In our study, the caloric test was done in healthy volunteers with colder water than usual (25°C instead of 30°C) in only one ear and induced only mild but still measurable nystagmus and vertigo. The use of even colder water (20°C) and/or the repetition of three caloric tests at each time point may enhance vestibular imbalance and reduce the variability in otherwise healthy volunteers.
In our study, despite large inter‐ and intra‐individual variability for all nystagmus and vertigo parameters, as is commonly reported for healthy volunteers 22, 23, significant improvements were seen for vertigo appearance, sensation and disappearance with up to 7 days of repeated SENS‐111 administrations compared to baseline for most doses evaluated, although no significant changes were seen for nystagmus over time. The absence of an apparent impact for nystagmus may be accounted for by the very high variability and by the fact that this measurement typically only takes into account a few beats around PSV, whereas the other measures are more continuous.
The modest improvement (20–30%) in vertigo parameters obtained in healthy volunteers with our modified caloric test is consistent with that observed with standard of care treatments 17, 18, 19, 20, 21 and with non‐clinical data in rats with severe vestibular dysfunction induced by kainate or arsanilate 12. A moderate effect on acute vertigo symptoms, such as in AUV, is usually recommended so as to avoid jeopardizing central compensation which is a critical component of effective recovery after acute peripheral loss. In fact, a marked suppression of vertigo symptoms would prevent central compensation and could increase the risk of lasting dizziness and imbalance.
The analysis of the exposure–response relationship shows that the SENS‐111 activity is related to plasma concentrations; displaying a bell‐shaped curve with a maximum effect at exposures of 200–400 ng ml−1 for latency of vertigo appearance and disappearance and at exposure of 200–600 ng ml−1 for duration of vertigo, with no effect at higher exposure. The bell‐shaped response observed for the clinical endpoints was factored into the model, assuming a dual mechanism of action. The initial mechanism, associated with a positive response, was activated at low level of exposure (<600 ng ml−1), while the second mechanism accounted for the lack of response at higher exposure. It can be assumed that at high concentrations, the selectivity of the drug for the peripheral H4 receptor is reduced due to a central effect on vestibular nuclei neurons in the brainstem, which integrate and relay the signal from the peripheral vestibular system through the abducens and oculomotor nuclei to the eye muscles.
The most consistent PD effects are observed from 100 to 200 mg day−1, corresponding to a mean C max of ~100 ng ml−1 at Day 1 and 170 ng ml−1 at Day 4 with 100 mg, and 280 ng ml−1 at Day 1 and 590 ng ml−1 at Day 7 with 200 mg. PK simulations for potential clinical dose regimens show that steady state is more rapidly obtained when SENS‐111 is administered b.i.d. on the first day of treatment. Thus, a treatment regimen of 100 mg or 200 mg SENS‐111 b.i.d. on the first day of treatment, followed by once daily administration of the corresponding dose thereafter, should result in the targeted plasma concentrations.
The two main limitations of the study are the large inter‐ and intra‐individual variability and the small effect of the caloric test on nystagmus or vertigo parameters; vertigo induced by the caloric test only lasted 110 s and was rated as mild by subjects, which may weaken the translatability of these data to patients suffering from severe vertigo, although the magnitude and effective concentrations correspond to effects seen in a preclinical rat model of severe vertigo 12. As the use of placebo in this study was only designed to ensure that subjects and the investigator were blinded, the sample size of placebo‐treated subjects is too small and the IIV too large to adequately compare SENS‐111 vs. placebo. Despite these limitations, the PK/PD relationship identified offers some degree of confidence in terms of the robustness of the data, which were consistent with those obtained in animal models.
In conclusion, oral SENS‐111 is safely administered clinically as single and once daily repeated doses up to 250 mg day−1 for 7 days. The absence of sedation or somnolence combined with the PK/PD modelling and simulation identifying the 100 and 200 mg day−1 repeated doses as suitable dose regimens, support the evaluation of SENS‐111 as symptomatic treatment of vestibular disorders, notably in patients suffering from AUV, for whom no effective non‐sedative treatments are available.
Competing Interests
F.V., R.G., S.P. and S.S. received financial remuneration from Sensorion for their participation in the study. P.A. is a shareholder and employee of Sensorion. E.W. is a shareholder and former employee of Sensorion.
Funding for this study was provided by Sensorion. We thank Optimed/Eurofins who carried out the clinical phase studies and some of the statistical analyses, Atlanbio (J. Girault and N. Ripoche) who performed the PK sampling bioanalysis, Sarah MacKenzie, PhD for medical writing support (all funded by Sensorion), and Sensorion who sponsored the trial.
Contributors
The study was performed in a Phase 1 unit and the protocol and caloric test were designed by F.V. and S.S. who also supervised the analysis.
Supporting information
Table S1 Non‐compartmental PK parameters of oral SENS‐111 in the SAD study, arithmetic mean ± SD
Table S2 Non‐compartmental PK parameters of oral SENS‐111 in the MAD studies, arithmetic mean ± SD
Table S3 Non‐compartmental PK parameters of SENS‐111 metabolites in the SAD study, arithmetic mean ± SD
Table S4 Non‐compartmental PK parameters of SENS‐111 metabolites in the MAD study, arithmetic mean ± SD
Figure S1 Mean (SD) plasma concentration–time profiles for SENS‐111. (A) for the SAD study on Day 1; (B) for the 4‐day MAD study after 1 day (dotted lines) and 4 days (solid lines); and (C) for the 7‐day MAD study after 1 day (dotted lines) and 7 days (solid lines)
Figure S2 Goodness of fit plots for SENS‐111
Figure S3 Visual predictive checks (VPCs) using untransformed data to estimate the time‐varying change in SENS‐111 exposure (200 mg on Day 1 vs. Day 7) and dose‐dependent SENS‐111 exposure
Figure S4 Mean absolute and relative change from baseline over time with placebo treatment are shown for duration of vertigo, and latency of appearance and disappearance
Figure S5 Boxplots showing the relationship between vertigo duration, and latency of appearance and disappearance with level of SENS‐111 exposure
Venail, F. , Attali, P. , Wersinger, E. , Gomeni, R. , Poli, S. , and Schmerber, S. (2018) Safety, tolerability, pharmacokinetics and pharmacokinetic‐pharmacodynamic modelling of the novel H4 receptor inhibitor SENS‐111 using a modified caloric test in healthy subjects. Br J Clin Pharmacol, 84: 2836–2848. 10.1111/bcp.13744.
References
- 1. Housley GD, Norris CH, Guth PS. Histamine and related substances influence neurotransmission in the semicircular canal. Hear Res 1988; 35: 87–97. [DOI] [PubMed] [Google Scholar]
- 2. Cohen B, DeJong JM. Meclizine and placebo in treating vertigo of vestibular origin. Relative efficacy in a double‐blind study. Arch Neurol 1972; 27: 129–135. [DOI] [PubMed] [Google Scholar]
- 3. Redon C, Lopez C, Bernard‐Demanze L, Dumitrescu M, Magnan J, Lacour M, et al Betahistine treatment improves the recovery of static symptoms in patients with unilateral vestibular loss. J Clin Pharmacol 2011; 51: 538–548. [DOI] [PubMed] [Google Scholar]
- 4. Desmadryl G, Gaboyard‐Niay S, Brugeaud A, Travo C, Broussy A, Saleur A, et al Histamine H4 receptor antagonists as potent modulators of mammalian vestibular primary neuron excitability. Br J Pharmacol 2012; 167: 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Takumida M, Takumida H, Anniko M. Localization of histamine (H1, H2, H3 and H4) receptors in mouse inner ear. Acta Otolaryngol 2016; 136: 537–544. [DOI] [PubMed] [Google Scholar]
- 6. Wersinger E, Gaboyard‐Niay S, Travo C, Soto E, Baez A, Vega R, et al Symptomatic treatment of vestibular deficits: therapeutic potential of histamine H4 receptors. J Vestib Res 2013; 23: 153–159. [DOI] [PubMed] [Google Scholar]
- 7. Mègnigbêto CA, Sauvage JP, Launois R. The European Evaluation of Vertigo (EEV) scale: a clinical validation study. Rev Laryngol Otol Rhinol (Bord) 2001; 122: 95–102 [in French]. [PubMed] [Google Scholar]
- 8. Tukey JW. The problem of multiple comparisons. Princeton, NJ: Department of Statistics, Princeton University, 1953. [Google Scholar]
- 9. Lindbom L, Pihlgren P, Jonsson EN, Jonsson N. PsN‐Toolkit – a collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 2005; 79: 241–257. [DOI] [PubMed] [Google Scholar]
- 10. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dyhrfjeld‐Johnsen J, Wersinger E, Petremann M, Brieuc D, Challuau D, Gueguen C, et al Translational predictivity of preclinical model studies of the anti‐vertigo drug SENS‐111 for clinical PK/PD relationships. Abstract PD170. Association for Research in Otolaryngology (ARO), 40th MidWinter Meeting, February 11‐15, 2017, Baltimore MD.
- 13. Hillier S, McDonnell M. Is vestibular rehabilitation effective in improving dizziness and function after unilateral peripheral vestibular hypofunction? An abridged version of a Cochrane Review. Eur J Phys Rehabil Med 2016; 52: 541–556. [PubMed] [Google Scholar]
- 14. Hain T. Drug treatment of Vertigo. Available at https://www.dizziness-and-balance.com/treatment/drug/drugrx.html (last accessed September 2017).
- 15. Shupak A, Issa A, Golz A, Kaminer M, Braverman I. Prednisone treatment for vestibular neuritis. Otol Neurotol 2008; 29: 368–374. [DOI] [PubMed] [Google Scholar]
- 16. Goudakos JK, Markou KD, Psillas G, Vital V, Tsaligopoulos M. Corticosteroids and vestibular exercises in vestibular neuritis. Single‐blind randomized clinical trial. JAMA Otolaryngol Head Neck Surg 2014; 140: 434–440. [DOI] [PubMed] [Google Scholar]
- 17. Mangabeira‐Albernaz PL. Calcium antagonists as a peripherally acting labyrinthine suppressant in humans. Acta Otolaryngol Suppl 1988; 460: 99–103. [DOI] [PubMed] [Google Scholar]
- 18. Gutner LB, Gould WJ, Batterman RC. Action of dimenhydrinate (dramamine) and other drugs on vestibular function. AMA Arch Otolaryngol 1951; 53: 308–315. [DOI] [PubMed] [Google Scholar]
- 19. Philipszoon AJ. Influence of cinnarizine on the labyrinth and on vertigo. Clin Pharmacol Ther 1962; 3: 184–190. [DOI] [PubMed] [Google Scholar]
- 20. Martin N, Oosterveld WJ. The vestibular effects of meclizine hydrochloride‐niacin combination (Antivert). Acta Otolaryngol 1970; 70: 6–9. [DOI] [PubMed] [Google Scholar]
- 21. Kiroglu MM, Dagkiran M, Ozdemir S, Surmrlioglu O, Tarkan O. The effects of betahistine and dimenhydrinate on caloric test parameters; slow‐phase velocity of nystagmus. J Int Adv Otol 2014; 10: 68–71. [Google Scholar]
- 22. de Barros ACMP, Caovilla HH. From nystagmus to the air and water caloric tests. Braz J Otorhinolaryngol 2012; 78: 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strupp M, Magnusson M. Acute unilateral vestibulopathy. Neurol Clin 2015; 33: 669–685. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Non‐compartmental PK parameters of oral SENS‐111 in the SAD study, arithmetic mean ± SD
Table S2 Non‐compartmental PK parameters of oral SENS‐111 in the MAD studies, arithmetic mean ± SD
Table S3 Non‐compartmental PK parameters of SENS‐111 metabolites in the SAD study, arithmetic mean ± SD
Table S4 Non‐compartmental PK parameters of SENS‐111 metabolites in the MAD study, arithmetic mean ± SD
Figure S1 Mean (SD) plasma concentration–time profiles for SENS‐111. (A) for the SAD study on Day 1; (B) for the 4‐day MAD study after 1 day (dotted lines) and 4 days (solid lines); and (C) for the 7‐day MAD study after 1 day (dotted lines) and 7 days (solid lines)
Figure S2 Goodness of fit plots for SENS‐111
Figure S3 Visual predictive checks (VPCs) using untransformed data to estimate the time‐varying change in SENS‐111 exposure (200 mg on Day 1 vs. Day 7) and dose‐dependent SENS‐111 exposure
Figure S4 Mean absolute and relative change from baseline over time with placebo treatment are shown for duration of vertigo, and latency of appearance and disappearance
Figure S5 Boxplots showing the relationship between vertigo duration, and latency of appearance and disappearance with level of SENS‐111 exposure